CHEMICAL BIOLOGY
Proteins: Computational Analysis of Structure, Function, and Stability
M. Michael Gromiha, National Institute of Advanced Industrial Science and Technology, Tokyo, Japan
S. Selvaraj, Bharathidasan University, Tiruchirappalli, India
doi: 10.1002/9780470048672.wecb495
Proteins are biological macromolecules synthesized with genetically determined sequences of amino acids by the formation of peptide bonds through the removal of water molecules. The chemical nature of amino acids is important for the folding and stability of protein structures. In this article, we give an overview of the computational analysis of protein structure and function from the viewpoint of chemical biology. The biological insights obtained from the analysis of known protein structures and their applications to protein structure prediction and folding rates are outlined. The computational analysis needed for understanding the functions and stability of proteins and for predicting the functionally important residues is described. The methods developed for understanding the factors that influence the stability of proteins and predicting protein stability during mutation are explored. Furthermore, the current status of computational protein design and the approaches to understanding the recognition mechanism of protein complexes are discussed.
The formation of stable secondary structures and a unique tertiary structure of proteins are dictated by the interactions between constituent amino acid residues along the polypeptide chain and by their interactions with the surrounding medium. During the process of protein folding, the hydrophobic force drives the polypeptide chain to the folded state and overcomes the entropic factors while hydrogen bonds, ion pairs, disulfide bonds, and van der Waals interactions define the shape and keep it from falling apart. The structure of a protein mainly dictates its function, and the attainment of stable conformation is essential for proper function. Hence, many methods have been developed to determine the three-dimensional structures of proteins experimentally.
X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy are widely used to determine the structures of proteins and their complexes. These structures have been deposited in Protein Data Bank (PDB) and are available at http://www.rcsb.org/ (1). Currently, the PDB has nearly 50,000 structures, and the wealth of data provide valuable information on relating the structures of proteins with their functions, interactions, and evolution. For example, the structure of a protein explicitly reveals the presence of various interactions, cavities, clefts, active sites, binding regions, and so forth. Figure 1 shows the crystal structure of X-linked inhibitor of apoptosis (XIAP) binding with small-molecule inhibitor, which helps to restore apoptosis and offset the cancerous state. The availability of many protein structures has enabled researchers to perform various computational analysis from different perspectives such as structure, function, and stability.
The applications of the chemistry of amino acids to the biological problem, protein structure and function, and folding and stability are the main focus of this article. The article is divided into five main sections that include the biological insights on protein structures, chemical applications including protein functions, thermodynamics of proteins, protein interactions, and computational protein design.

Figure 1. Three-dimensional structure of XIAP protein binding with small-molecule inhibitor (PDB code: 1TFQ).
Biological Insights
The computational studies on protein structures have been carried out from different perspectives in biology. These perspectives include 1) comparison of protein structures, 2) structural analysis, and 3) protein structure prediction. Table 1 lists the online resources that are providing such services.
Table 1. Online resources for protein structure analysis and prediction
|
Protein Data Bank |
|
|
PDB |
http://www.rcsb.org/pdb/home/home.do |
|
Protein classification |
|
|
SCOP |
http://scop.mrc-lmb.cam.ac.uk/scop/ |
|
CATH |
http://cathwww.biochem.ucl.ac.uk/latest/index.html |
|
Protein structure comparison |
|
|
DALI |
http://ekhidna.biocenter.helsinki.fi/dali/start |
|
CE |
http://cl.sdsc.edu/ce.html |
|
PRIDE |
http://hydra.icgeb.trieste.it/pride/ |
|
MATRAS |
http://biunit.aist-nara.ac.jp/matras/ |
|
TOPS |
http://www.tops.leeds.ac.uk/ |
|
Protein three-dimensional structure prediction |
|
|
FRankenstein |
http://genesilico.pl/meta/ |
|
PROSPECTOR |
http://128.205.242.l/current_buffalo/skolnick/prospector.html |
|
MODELLER |
http://salilab.org/modeller/modeller.html |
|
GenTHREADER |
http://bioinf.cs.ucl.ac.uk/psipred/psiform.html |
|
ROBETTA |
http://robetta.bakerlab.org/ |
|
FORTE |
http://www.cbrc.jp/htbin/forte-cgi/forteJorm.pl |
|
I-TASSER |
http://zhang.bioinformatics.ku.edu/I-TASSER |
|
Computation of solvent accessibility |
|
|
ACCESS |
http://www.csb.yale.edu/ |
|
ASC |
http://mendel.imp.univie.ac.at/mendeljsp/studies/asc.jsp |
|
DSSP |
http://www.cmbi.kun.nl/gv/dssp/ |
|
|
ftp://ftp.cmbi.kun.nl/pub/molbio/data/dssp/ |
|
GETAREA |
http://www.scsb.utmb.edu/getarea/ |
|
NACCESS |
http://wolf.bms.umist.ac.uk/naccess/ |
|
POPS |
http://mathbio.nimr.mrc.ac.uk/~ffranca/POPS |
|
Amino acid index database |
|
|
AAindex |
http://www.genome.ad.jp/aaindex/ |
|
Computation of hydrogen bonds |
|
|
HBPLUS |
http://www.biochem.ucl.ac.uk/bsm/hbplus/home.html |
Comparison of protein structures
The function of a protein mainly depends on its structure, and proteins of similar structures perform similar functions. Structure comparison is the process of analyzing two or more structures to evaluate the extent of similarity in their three-dimensional structures. The comparison of protein structures is very important in the design of algorithms that predict protein function and is expected to play a major role in structural genomics projects. The comparison also enables one to classify the protein structures into different structural classes, folds, families, and superfamilies. Typically, the structures being compared are superposed, and the measure, root mean square deviation (RMSD), is widely used to characterize structural relatedness among the proteins being compared. It has been reported that the method PRIDE (2), based on the distribution of amino acid Ca atoms, is fast and can handle large amounts of data and vast structural databases. However, it has many false positives and negatives. Yet, the method combinatorial extension (3), based on aligned fragment pairs, is an accurate one, but it is slow for comparison. In addition, comparison of protein structures is helpful to get the biological information in distantly related protein structures to detect the similar and dissimilar regions between protein structures.
Analysis of protein structures
The computational analysis of unrelated protein structures (nonredundant set) enables one to identify the fundamental principles that govern their folding and stability. Different types of computational analyses such as solvent accessibility; surrounding hydrophobicity; flexibility; thermodynamic parameters; average medium- and long-range contacts; preference of amino acid residues in α-helical, β-strand, and coil regions; and so forth have been carried out. In these calculations, either the coordinates of α-carbon atoms or all atoms in protein structures have been used. Solvent accessibility is a measure to identify the location of amino acid residues (inside the protein core or on the surface) in protein structures (4). It is defined as the locus of the center of the solvent molecule (usually a sphere of water with a radius 1.4 A) as it rolls over the van der Waals surface of the protein (Fig. 2). The concept of solvent accessibility is used to compute the hydrophobic or solvation free energy. Surrounding hydrophobicity of a residue in protein structure is the sum of the experimental hydrophobic indices of the residues that are located within a distance of 8A (5). The short-, medium-, and long-range contacts are computed with the information about the contacting residues in space (less than 8A) and their respective locations with the intervals of 1-2, 3-4 and > 4 residues, respectively in the sequence (Fig. 3). The conformational parameters have been derived by the ratio between the frequency of occurrence of a specific amino acid residue (say Ala) in a particular conformation (say a-helix) and the occurrence of the same residue in the whole protein (6). These parameters derived from the computational analysis of protein structures have been used extensively to predict the secondary structures, solvent accessibility, and residue contacts from amino acid sequence.
Furthermore, the parameters derived from protein threedimensional structures characterize the topology of proteins, which play an important role in kinetics of protein folding. For example, contact order (7) and long-range order (8) are derived from the information about residue contacts in protein structures. Contact order reflects the relative importance of local and nonlocal contacts to the native structure of a protein, whereas long-range order accounts for the contacts between two residues that are close in space and far in the sequence in protein structure. These parameters have shown a strong correlation with protein folding rates, which is a measure of slow/fast folding of proteins from the unfolded state to their native three-dimensional structures. The amino acid properties and the topological parameters are successfully used to predict protein folding rates from amino acid sequence and structure, respectively.
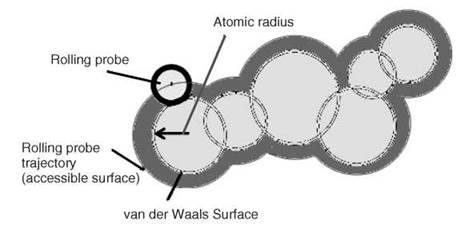
Figure 2. A cross section of a part of a macromolecule in space (atoms are shown as black circles); rolling probe, van der Waals, and accessible surface areas are indicated in the figure.

Figure 3. Representation of short-, medium-, and long-range contacts in protein structures. A typical example for the contacting residues of Thr152 in T4 lysozyme within 8A is shown. (s: short-range contacts; m: medium-range contacts; and l: long-range contacts.)
Prediction of protein structures
Predicting the three-dimensional structure of a protein from its amino acid sequence, known as the protein folding problem, is a challenging task. Considering the rapid growth of amino acid sequences against known structures, prediction of protein structures from sequences is necessary in structural and functional genomics. As an intermediate step, several methods have been proposed for predicting protein secondary structures and solvent accessibility. These methods are based on statistical analysis, knowledge-based methods, hidden Markov models, alignment profiles, and machine learning techniques. Recent studies showed the highest accuracy of 80% in predicting the secondary structures of proteins (9) and 85% in predicting two-state solvent accessibility (10). Three-dimensional structure prediction is more challenging, and the performance of different methods is tested with blind predictions of proteins, which are assessed by the forum, Critical Assessment of Techniques for Protein Structure Prediction (CASP) (11). The measures, GDT_TS, related to the difference in position of main chain C, atoms between a model and corresponding experimental structure and AL0 about the assignment of amino acid positions with experimental structures are used for evaluating the accuracy. Currently, the most promising approach is based on the combination of fold recognition using threading techniques and fragment assembly (12). Furthermore, predictions are made using different approaches such as comparative modeling to find the most probable structure for a sequence using its alignment with related structures, new folds, profiles, and ab initio structure prediction using an all-atom model.
Chemistry
Protein function
The functions of proteins are dictated with the importance of specific amino acid residues that are acting as catalytic site residues and binding site residues, which are capable of binding small molecules (ligands) and interacting with macromolecules. The chemistry of amino acids plays a major role in developing computational models for characterizing the functionally important residues in proteins. Thornton’s group (13) collected the catalytic site residues in protein structures and developed a database, Catalytic Site Atlas (CSA), which is available at http://www.ebi.ac.uk/thornton-srv/databases/CSA/. The catalytic site residues have been analyzed with the pattern of hydrogen bonding, location of amino acids in proteins, conservation of residues, ability to possess binding pockets, and so forth. Furthermore, the catalytic mechanisms of hydrolysis and transfer reactions have been compared with the aid of a database of enzyme catalytic mechanisms (http://mbs.cbrc.jp/EzCatDB/).
Functional residues identified by site-directed mutagenesis
The site-directed mutagenesis experiments provided a large amount of data about the functional importance of amino acid residues in a protein. Systematic studies on p53 tumor suppressor protein showed that the mutation of each residue leads to different kinds of tumors. Furthermore, several residues are identified as important for drug resistance, protein translocation, substrate binding, increased affinity and activity, and so forth. The information about human genes, genetic disorders, and the effects of mutation has been collected in the form of databases, Online Mendelian Inheritance in Man (OMIM) and Human Gene Mutation Database (HGMD). The combined information about the mutational effects of protein stability and diseases along with sequence and structural details is available at http://gibk26.bse.kyutech.ac.jp/jouhou/3dinsight/disease.html. These resources will be helpful in predicting the functionally important residues, which is eventually useful for protein engineering experiments.
Prediction of functionally important residues
The functionally important residues are dispersed in a protein, which makes it difficult to map them on structures. However, the comparison of protein structures provides useful information for predicting protein function. Valencia’s group (14) proposed a method based on structural templates in the form of multiple alignments with structures and local conservation of residues for predicting the functionally important residues (e.g., catalytic sites and residues that are involved in substrate or cofactor binding). Generally, a database will set up the functional residues, and their characteristic features will be analyzed based on available sequence and structure analysis tools. These parameters are used for predicting the functionally important residues. Fisher et al. (15) analyzed the conservation of functional residues and their neighboring residues, the distribution of amino acid residues, and the predicted secondary structure and solvent accessibility and developed a method for predicting functionally important residues.
Protein Thermodynamics
Protein stability is the free energy difference (AG) between the folded and unfolded states at physiological conditions, and it is in the range of 5-25 kcal/mol. Site-directed mutagenesis experiments provided a wealth of data for understanding the importance of chemical interactions for the stability of proteins during amino acid substitutions. Protein stability is experimentally measured with differential scanning calorimetry, circular dichroism, fluorescence spectroscopy, and so forth. The availability of such data in an electronically accessible database would be a valuable resource for the analysis and prediction of protein mutant stability.
Thermodynamic database for proteins and mutants
ProTherm (16) is a large collection of thermodynamic data on protein stability, which has information on 1) protein sequence and structure (2) mutation details (wild-type and mutant amino acid: hydrophobic to polar, charged to hydrophobic, aliphatic to aromatic, etc.), 3) thermodynamic data obtained from thermal and chemical denaturation experiments (free energy change, transition temperature, enthalpy change, heat capacity change, etc.), 4) experimental methods and conditions (pH, temperature, buffer and ions, measurement and method, etc.), 5) functionality (enzyme activity, binding constants, etc.), and 6) literature.
Amino acid properties and protein stability
Analysis of the relationship between amino acid properties and protein stability showed that hydrophobicity is the major factor for the stability of proteins during substitution of amino acids in the interior of the protein. The stability of protein mutants is attributed to the number of carbon atoms, which shows the direct relationship between hydrophobicity and stability.
For the mutations on the surface of the protein, the classifications based on the chemical nature of amino acids, such as hydrophobic amino acids (Ala, Cys, Phe, Gly, Ile, Leu, Met, Val, Trp, and Tyr), amino acid side chains that can form hydrogen bonds (Asp, Cys, Glu, His, Lys, Met, Asn, Gln, Arg, Ser, Thr, Trp, and Tyr), and so forth, improved the correlation between amino acid properties and protein mutant stability. Furthermore, the inclusion of neighboring and surrounding residues remarkably improved the correlation in all the subgroups of mutations. This result indicates that the information from nearby polar/charged amino acid residues and/or the aliphatic and aromatic residues that are close in space is important for the stability of exposed mutations.
Empirical potentials and protein stability
The information about the preference of amino acid residues for contact with each other in protein three-dimensional structures has been used to derive empirical potentials for understanding protein folding and stability. The most widely used approach is the computation of the frequencies of sequence and structure features and the conversion of the frequencies into free energies (17). The potentials based on torsion angles and contacting residues have been used for predicting protein mutant stability (18) Furthermore, free energy approach has also been used to estimate the stability change between wild-type and mutant proteins by Serrano’s group (19). In this method, all the free energy components that are important for stability have been systematically considered to compute the stability, especially, the van der Waals contributions of all atoms, solvation free energy between polar and nonpolar groups, formation of intra- and inter-molecular hydrogen bonds, water bridges, electrostatic interactions, and cost of entropy for fixing backbone and side chains in the folded state. In addition, machine learning techniques have been developed for predicting protein mutant stability. In Table 2, we list the online resources for predicting the stability of proteins during amino acid substitutions.
Table 2. Web servers for protein stability prediction
|
Thermodynamic database for proteins and mutants |
|
|
ProTherm |
http://gibk26.bse.kyutech.ac.jp/jouhou/protherm/protherm.html |
|
Stabilizing residues in protein structures |
|
|
SRide |
http://sride.enzim.hu |
|
Prediction of protein mutant stability |
|
|
FOLD-X |
http://fold-x.embl-heidelberg.de |
|
CUPSAT |
http://cupsat.tu-bs.de/ |
|
I-Mutant2.0 |
http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant2.0/I-Mutant2.0.cgi. |
|
MUpro |
http://www.igb.uci.edu/servers/servers.html |
|
iPTREE-STAB |
http://bioinformatics.myweb.hinet.net/iptree.htm |
Stabilizing residues in protein structures
The key residues that are important for the stability of protein structures have been delineated from a consensus approach (20). This method includes the concept of long-range interactions, hydrophobicity, and conservation of amino acid residues in homologous sequences. It shows good agreement with experimental thermodynamic data of globular proteins, and the consensus method could be used to identify potential candidates for protein engineering.
Protein Interactions
Protein-Protein Interactions
Protein interactions play a key role in many cellular processes. Despite the fact that protein interactions are remarkably diverse, all protein interfaces share certain common properties (21). The structural analysis of protein-protein complexes showed that a specific preference for amino acid residues is found at the interface, and most interfaces consists of completely buried cores surrounded by partially accessible rims. The binding sites consist of a few highly packed regions, which contribute significantly to the free energy of binding. In protein-protein complexes, contacts between hydrophobic residues were favorable, pairs of hydrophilic and polar residues were unfavorable, and the charged residues tended to pair subject to charge complementarily. The buried interface is mainly created by nonpolar (carbon-containing) groups followed by polar (N-, S-, and O-containing) groups. Furthermore, hydrogen bonds and salt bridges are found to be important for protein-protein interactions.
Protein-protein interactions represent a highly populated class of targets for drug discovery (22). Integrins are cell surface receptors and are the first example of drug-like protein-protein inhibitors. They bind various protein ligands, such as fibronectin and vitronectin, which possess an Arg-Gly-Asp motif. Furthermore, inhibitors of integirn receptors from various chemical classes, such as benzodiazepine, pyrrolo-benzodiazepine-dione, oxoisoquinoline, and so forth, have been reported. The binding of X-linked inhibitor of apoptosis with small-molecule inhibitor helps to restore apoptosis and offset the cancerous state (Fig. 4). The analysis of several protein-protein complexes showed that the structural characteristics of protein binding sites and the attributes of small-molecule ligands share a common mechanism in drug discovery.
However, the fast development of mass spectroscopy and other experimental techniques for studying protein interactions has enabled the construction and analysis of protein interaction networks. The interaction maps obtained for one species can be used to predict interaction networks in other species, which is useful for identifying the functions of unknown proteins. The protein interaction databases provide a wealth of information regarding the interacting pairs of protein-protein interactions.

Figure 4. XIAP-bound positions of a tetrapeptide fragment, Ala-Val-Pro-Ile (22). The surface of XIAP is shown in grey-white; the peptide is colored dark black; and the small-molecule inhibitor is colored with black and white, and depicted in stick format.
Protein-nucleic acid interactions
Protein-nucleic acid interactions play a key role in many vital processes, including regulation of gene expression, DNA replication and repair, and packaging. The remarkable specificity with which proteins recognize target DNA sequences is of considerable theoretical and practical importance, and its basis has been demonstrated through structural analysis of many protein-DNA complexes. The structural analysis of protein-DNA interactions showed that hydrogen bonds are the most frequent interactions followed by van der Waals, hydrophobic, and electrostatic interactions. The electronegative atoms in protein and DNA make many hydrogen bonds in the major groove of DNA, whereas hydrophobic interactions are dominant in the minor groove. The protein-DNA recognition is characterized by both nonspecific and specific interactions via the contacts between the atoms in amino acids and the phosphate/sugar chains of DNA and between the side chains of amino acids and the bases of DNA. In specific interactions, the positive-charged amino acids, Arg and Lys, are dominant in the binding sites of proteins. The main contributor from DNA is thymine with Lys and guanine with Arg for specific interactions; nonspecific contacts are mainly coordinated with phosphate groups of DNA. In addition, the weak interactions including CH...O contacts that involve the thymine methyl group and position C5 of cytosine are comparable to the number of protein-DNA hydrogen bonds. Furthermore, the chemical and physical properties, such as polarity, size, shape, and packing; conformational changes in DNA (bending and flexibility); and water molecules bridging amino acids and bases, are important for the specificity of protein-DNA complexes.
The free energy of binding is a measure for understanding the importance of specific residues/bases for binding, and the experimentally measured specificities of protein-nucleic acid interactions have been compiled in ProNIT database (http://gibk26.bse.kyutech.ac.jp/jouhou/pronit/pronit.html). The binding of protein with DNA is also characterized by the preference of secondary structures in proteins as well as specific motifs, such as helix-turn-helix, leucine zipper, zinc finger, helix-loop-helix, and so forth. Recently, for the first time, the thermodynamic characterization of the stability and specific DNA binding of a full-length gene product of the Myc/Mad/Max family, namely, Max protein isoform p21 (Max p21) with helix-loop-helix-leucine zipper motif, have been reported (23). They showed that the association is driven by a large exothermic effect, which is partly compensated for by entropic factors.
In protein-RNA interactions, the recognition frequently occurs in noncanonical and single-strand-like structures that allow interactions to occur from a much wider set of geometries and make use of unique base shapes and hydrogen-bonding ability. Although it forms a glove-like tight binding pocket around RNA bases, the size, shape, and nonpolar binding patterns differ between specific RNA bases. Adenine is distinguishable from guanine based on the size and shape of the binding pocket and steric exclusion of the guanine N2 exocyclic amino group. The unique shape and hydrogen-bonding pattern for each RNA base allow proteins to make specific interactions through a very small number of contacts. Furthermore, knowledge-based potential functions are shown to be useful for predicting the specificity and binding free energy of protein-RNA complexes.
Protein-ligand interactions
The biological functions of proteins depend on their direct physical interactions with other molecules. Specificity of such interactions is crucial so that each protein must interact only with theappropriate molecule and not with any others present in the cell. A molecule of any type that interacts with a protein is designated as a ligand. The affinity between a protein (P) and a ligand (L) is measured by the association constant (Ka), and for the binding reaction at equilibrium, Ka = [P.L]/[P][L]. The inverse of Ka is dissociation constant (Kd), and the free energy of binding, ∆Gbind = —RT lnKa = RT lnKd.
Protein-ligand interactions are important for finding new pharmaceuticals, industrial compounds, and functional molecules for food products. It has been shown that a ligand’s partial charges are critically important for estimating the electrostatic interactions in ligand-receptor interactions. Furthermore, the van der Waals energy between proteins and ligands and solvation energy based on the interactions of chemical groups with aqueous medium are used to identify the ligand binding sites.
Prediction of ligand binding sites is an essential part of the drug discovery process. Generally, the binding sites are predicted with bioinformatics approaches, whereas the binding free energy is calculated with several interacting terms such as electrostatics, van der Waals, hydrophobicity, and conformational entropy. Systematic energy calculations provide the knowledge about the binding affinity of protein-ligand complexes, which may lead to the identification of the proper target for drug discovery. However, ligand binding sites have been identified from protein-ligand complexes by setting a minimum distance between the protein atoms and ligand. Using proper training and test procedures, researchers can predict the binding sites using the information about binding site residues, neighboring residues, location based on secondary structure and solvent accessibility, and so forth.
Several databases have been developed to accumulate the protein-ligand complexes and their binding sites. They are derived mainly from the protein data bank. In addition, databases are also available for the binding free energy of different protein-ligand complexes.
Computational Protein Design
Computational protein design is a practical option for solving problems in protein engineering. Several advances have been made in this area that include the development of new potential functions, efficient ways of computing free energies, flexible treatments of solvent and solvent-mediated effects (pairwise approximation to continuum electrostatics implemented with the finite-difference Poisson-Boltzmann model), energy function approximations, ensemble-based approaches for inclusion of entropic effects, improvements to stochastic search techniques, and methods to design combinatorial libraries for screening and selection. These new approaches have several applications such as the successes in the design of specificity for protein folding, binding, and catalysis, in the redesign of proteins for enhanced binding affinity, and in the application of design technology to study and alter enzyme catalysis (24).
Computational protein design is also applied for redesigning a whole protein. Mayo’s group (25) redesigned a protein, Drosophila melanogaster engrailed homeodomain, which has a sequence of 51 amino acid residues. They used dead-end elimination theorem to overcome the complexity of calculations including the energy functions for simultaneously modeling the residues in solvent-exposed protein surface and in hydrophobic core. Two of the designed sequences were evaluated experimentally, and those two sequences differ by 11 mutations and share 22% and 24% sequence identity with the wild-type protein. Figure 5 shows the overlay of backbones of the designed protein and the wild-type protein. Interestingly, computationally designed proteins were considerably more stable than the naturally occurring protein.

Figure 5. The overlay of the backbones of designed protein (black) and wild-type (gray) engrailed homeodomain, pdb code 1enh (25).
Conclusions
The chemical nature of amino acids mainly dictates the contacts between them in protein structures in terms of hydrophobic, electrostatic, hydrogen bonding, disulfide bonds, and van der Waals interactions, which are key determinants for the structure, folding, stability, and functions of proteins. The computational analyses of protein structures, such as comparison, prediction, and development of structure-based parameters, can identify the structurally similar regions, functionally important residues, and modeling and folding rates of proteins. The functionally important residues have been identified with catalytic sites and substrate binding sites and are important to protein transport, signaling, channels, enhance activity, affinity, and so forth. These residues are predicted with structurally similar regions, conservation of residues, and physico-chemical properties.
The relationship between amino acid properties and protein stability revealed that the number of carbon atoms (methyl and methylene groups) that reflects the property hydrophobicity has a strong relationship with protein stability for the mutations in the interior of the protein. Yet, hydrophobic, hydrogen bond, electrostatic, and other polar interactions are important for the stability of mutation at the surface of the protein. The atom pair potentials set up on the basis of chemical nature and connectivity successfully could predict protein stability during amino acid substitution.
The structural analysis of protein complexes showed that the chemical behavior of the interacting partners is important for understanding the recognition mechanism. The binding of small molecules with proteins provides deep insights for drug discovery. In addition, computational approaches have been used effectively to design stable proteins and protein engineering.
References
1. Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007; 35:D301-D303.
2. Carugo O, Pongor S. Protein fold similarity estimated by a probabilistic approach based on C(alpha)-C(alpha) distance comparison. J. Mol. Biol.. 2002; 315:887-898.
3. Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998; 11:739-747.
4. Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971; 55:379-400.
5. Manavalan P, Ponnuswamy PK. Hydrophobic character of amino acid residues in globular proteins. Nature 1978; 275:673-674.
6. Chou PY, Fasman GD. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry 1974; 13:211-222.
7. Plaxco KW, Simons KT, Baker, D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998; 277,985-994.
8. Gromiha MM, Selvaraj S. Comparison between long-range interactions and contact order in determining the folding rates of two-state proteins: application of long-range order to folding rate prediction. J. Mol. Biol. 2001; 310:27-32.
9. Yao XQ, Zhu H, She ZS. A dynamic Bayesian network approach to protein secondary structure prediction. BMC Bioinformatics. 2008; 9:49.
10. Pollastri G, Martin AJ, Mooney C, Vullo A. Accurate prediction of protein secondary structure and solvent accessibility by consensus combiners of sequence and structure information. BMC Bioinformatics. 2007; 8:201.
11. Moult J, Fidelis K, Kryshtafovych A, Rost B, Hubbard T, Tramontano A. Critical assessment of methods of protein structure prediction-Round VII. Proteins. 2007; 69:3-9.
12. Zhang Y. Template-based modeling and free modeling by ITASSER in CASP7. Proteins. 2007; 69:108-117.
13. Porter CT, Bartlett GJ, Thornton JM. The catalytic site atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004; 32:D129-133.
14. Lopez G, Valencia A, Tress ML. Firestar-prediction of functionally important residues using structural templates and alignment reliability. Nucleic Acids Res. 2007; 35:W573-577.
15. Fischer JD, Mayer CE, Soding J. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics. 2008; 24:613-620.
16. Gromiha MM, An J, Kono H, Oobatake M, Uedaira H, Sarai A. ProTherm: Thermodynamic Database for Proteins and Mutants. Nucleic Acids Res. 1999; 27:286-288.
17. Sippl M.J. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 1990; 213,859-883.
18. Parthiban V, Gromiha MM, Hoppe C, Schomburg D. Structural analysis and prediction of protein mutant stability using distance and torsion potentials: role of secondary structure and solvent accessibility. Proteins. 2007; 66:41-52.
19. Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002; 320:369-387.
20. Gromiha MM, Pujadas G, Magyar C, Selvaraj S, Simon, I. Locating the stabilizing residues in (alpha/beta)8 barrel proteins based on hydrophobicity, long-range interactions, and sequence conservation. Proteins. 2004; 55:316-329.
21. Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part I. Experimental techniques and databases. PLoS Comput Biol. 2007; 3:e42.
22. Fry DC. Protein-protein interactions as targets for small molecule drug discovery. Biopolymers. 2006; 84:535-552.
23. Meier-Andrejszki L, Bjelic S, Naud JF, Lavigne P, Jelesarov I. Thermodynamics of b-HLH-LZ protein binding to DNA: the energetic importance of protein-DNA contacts in site-specific E-box recognition by the complete gene product of the Max p21 transcription factor. Biochemistry. 2007; 46:12427-12440.
24. Lippow SM, Tidor B. Progress in computational protein design. Curr. Opin. Biotechnol. 2007; 18:305-311.
25. Shah PS, Hom GK, Ross SA, Lassila JK, Crowhurst KA, Mayo SL. Full-sequence computational design and solution structure of a thermostable protein variant. J. Mol. Biol. 2007; 372:1-6.
Further Reading
Branden, C, Tooze, J. Introduction to Protein Structure. 1999. Garland, New York.
Carugo O. Recent progress in measuring structural similarity between proteins. Curr. Protein Pept. Sci. 2007; 8:219-241.
Chakrabarti P, Bhattacharyya R. Geometry of nonbonded interactions involving planar groups in proteins. Prog. Biophys. Mol. Biol. 2007; 95:83-137.
Creighton TE. Protein Folding. 1992. WH Freeman, New York.
Dill KA. Dominant forces in protein folding. Biochemistry. 1990; 29:7133-7155.
Gromiha MM, Selvaraj S. Inter-residue interactions in protein folding and stability. Prog. Biophys. Mol. Biol. 2004; 86:235-277.
Gromiha MM, Selvaraj S, eds. Recent Research Developments in Protein Folding, Stability and Design. 2002. Research Signpost, Trivandrum, India.
Kawashima S, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 2000; 28:374.
Matthews BW. Studies on protein stability with T4 lysozyme. Adv. Protein Chem. 1995; 46:249-278.
Melo F, Feytmans E. Novel knowledge-based mean force potential at atomic level. J. Mol. Biol. 1997; 67:207-222.
Pace CN. Conformational stability of globular proteins. Trends Biochem. Sci. 1990; 15:14-17.
Pain RH. Mechanisms of Protein Folding. 2000. Oxford University Press, New York.
Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006; 34:W239-242.
Pfeil, W. Protein Stability and Folding: A Collection of Thermodynamic Data. 1998. Springer, New York.
Sarai A, Kono H. Protein-DNA recognition patterns and predictions.
Annu. Rev. Biophys. Biomol. Struct. 2005; 34:379-398.
Sippl MJ. Knowledge-based potentials for proteins. Curr. Opin. Str. Biol. 1995; 5:229-235.
Sorai M, ed. Comprehensive Handbook of Calorimetry and Thermal Analysis. 2004. Wiley, Hoboken, NJ.
See Also
Chemistry of Amino Acids
Computational Chemistry in Biology
Energetics of Protein Folding
Protein Informatics
Physical Chemistry in Biology