CHEMICAL BIOLOGY
Synthetic Sphingolipids as Bioactive Molecules: Roles in Regulation of Cell Function
Robert Bittman, Queens College of The City University of New York, Flushing, New York
doi: 10.1002/9780470048672.wecb576
Sphingolipids are a highly diverse class of lipids, which encompasses thousands of different structural combinations of the polar head group, amino group or amide chain, and sphingoid long-chain base. Initially regarded as inert structural components of eukaryotic cell membranes and plasma lipoproteins, sphingolipids now are recognized as key participants in the life of virtually all cell types. They influence a myriad of biological processes, including cell growth, cell death, and susceptibility to inflammation and infections. The number of known sphingolipid-dependent cell signaling systems is increasing rapidly as research efforts in this field progress. Sphingolipids stabilize transient microdomains in membranes known as ''rafts.'' Signaling proteins and receptors partition into membrane rafts, and some toxins, pathogens, and viruses bind to sphingolipids that are concentrated in rafts and subsequently undergo endocytosis (see also Extracellular Lipid Signals and Lipid Rafts). Synthetic, non-natural analogs of sphingolipids hold promise as drug candidates for treatment of various disease states via their ability to perturb cell signaling, membrane trafficking, and binding of infectious agents. The repertoire of synthetic sphingolipids includes analogs that target enzymes in the sphingolipid biosynthetic pathway and those that act as antiproliferative agents, immunomodulators, and regulators of lipid raft formation (and resultant receptor function). This review is intended to bring attention to selected synthetic analogs of sphingosine, ceramide, and glycosphingolipids that may lead to therapeutic intervention in certain patho-physiologic conditions.
Sphingolipids mediate a broad range of intracellular actions, including cell proliferation, cell survival, thermotolerance, vascular maturation, angiogenesis, inflammation, and immunomodulatory activity. Many intracellular targets of sphingolipids now are well established, and others currently are being investigated. In addition, some sphingolipids are ligands for G-protein-coupled receptors on the cell surface.
Chemical Structures of Sphingoid Long-Chain Bases and Their Glycosylated and Phosphorylated Derivatives
Sphingoid long-chain bases
Sphingolipids contain a sphingoid base backbone, which often is referred to as the long-chain base. The long-chain base is an amino alcohol with a long aliphatic chain such as sphingosine, dihydrosphingosine, and phytosphingosine (compounds 1-3, Fig. 1). Compound 1 is (2S,3R)-2-amino-4E-octadecene-1,3-diol, commonly known by the names (2S,3R)-sphingosine, D-erythro-sphingosine, sphingenine, and d18:1∆4. In the latter nomenclature, “d” designates the presence of a dihydroxy-containing sphingoid base, 18 refers to the number of carbon atoms in the long-chain base, 1 refers to the number of double bonds, and ∆4 indicates the position of the double bond (as in the shorthand nomenclature used for fatty acids). This lipid with an 18-carbon aliphatic chain and a C-4,C-5-trans-double bond is the major constituent of the backbone of sphingolipids found in most mammalian cells. Many structural variations of compound 1 are found in the natural sphingolipids that are distributed widely in eukaryotic cell membranes. Modifications occur in the length of the aliphatic chain and in the degrees of unsaturation, methyl branching, and hydroxylation. D-erythro-Dihydrosphingosine (compound 2, also known as (2S,3R)-sphinganine and d18:0) and D-ribo-phytosphingosine (compound 3) are the two major sphingoid long-chain bases of fungi and plants. Phytosphingosine does not possess a 4,5-trans-double bond. The “t” indicates that it is a trihydroxy-containing sphingoid base; the additional hydroxy group is linked to C-4 of the aliphatic chain. Phytosphingosine forms the backbone of many glycosphingolipids (1, 2).
Sphingadienes are found in the long-chain base of some glucosylceramides of plants, fungi, and marine sources. An example is 4,8-octadecadiene-1,3-diol (also known as 4,8-sphingadiene or d18:2∆4∆8), which has a C-8,C-9-double bond in addition to the C-4,C-5-trans-double bond (compound 4, Fig. 1). In the predominant sphingoid base of wheat and soybean cerebrosides, the stereochemistry of the C-8,C-9 double bond of 4,8-sphingadiene is 65% trans and 35% cis, and the amide chain is predominantly a-hydroxypalmitic acid (3). The sphingosine and sphingadiene bases of the glycosphin- golipids of some organisms bear a methyl branch. For example, a sea anemone has a 4,8-sphingadiene backbone that contains 18 carbon atoms with a methyl branch at C-9 (4), and the nematode Caenorhabditis elegans has a sphingosine backbone that contains 16 carbon atoms with an iso branch, for example, a methyl group at C-15 (5). The sphingoid base backbone of sphingomyelin in the tobacco hornworm moth Manduca sexta is 4E,6E-sphingadiene (6). The glycosphingolipid of a marine annelid contains a 4E,8E-sphingadiene backbone with a phosphocholine esterified to the C-6 position of galactose (7). Glycosphingolipids isolated from a starfish contained a 4E,13Z-sphingadiene (8) and a 4E,15Z-sphingadiene backbone (9).
Sphingatrienes also occur naturally but in minor amounts. Sphingomyelin isolated from squid nerve and from starfish contains a branched, triunsaturated sphingoid base, 2-amino-9- methyl-4E,8E,10E-octadecatriene-1,3-diol (10), and glycosphingolipids from starfish contain the same long-chain base without the methyl branch (9).
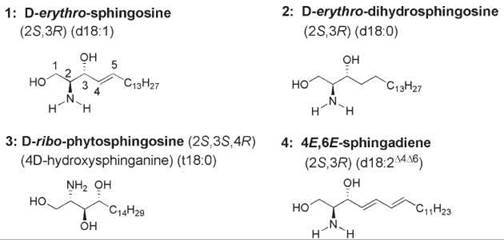
Figure 1. The structural feature common to all sphingolipids is a long-chain sphingoid base. Sphingoid bases are 2-amino-1,3-diols that bear an aliphatic chain. The chain length varies from about 14 to 24 carbon atoms, and the extent of unsaturation, hydroxylation, and methyl-branching in the chain also varies. 1, (2S,3R,4E)-sphingosine; 2, (2S,3R)-dihydrosphingosine; 3, (25,35,4R)-phytosphingosine; 4, 4E,6E-sphingadiene.
Sphingolipids with modifications at C-1 and C-2
Many examples of sphingolipids exist in which the C2-amino and/or terminal hydroxy group is modified. N,N-Dimethyl-D-erythro-sphingosine is a naturally occurring sphingolipid that, like D-erythro-sphingosine, inhibits the ubiquitous signal transducer protein kinase C (PKC) (11). At high concentrations (10 pM), N,N-dimethylsphingosine also inhibits sphingosine kinases, the enzyme that is responsible for the formation of sphingosine 1-phosphate (S1P), but at low concentrations (<1 μM) it activates sphingosine kinase in the cytosol via a PKCε-dependent mechanism (12).
The C-1 hydroxy group of sphingosine is replaced by a hydrogen in 1-deoxy-D-erythro-sphingosine [(2S,3R)-2-amino-3-octadecanol]. Because no functional group is present at the terminal carbon, chemical/biochemical modification at that site is precluded. This naturally occurring sphingosine analog, which is called spisulosine or ES-285, was isolated from the clam Mactromeris polynima and was found to induce an atypical form of apoptosis in murine leukemia cells (13). A cyclodextrin-based formulation of spisulosine is being used in Phase I clinical trials as an anticancer agent (14).
Sphingolipids with a sulfate group esterified to the C-1 position were isolated from marine invertebrates and shown to possess various bioactivities, such as the inhibition of neuraminidase and telomerase activity (15).
Ceramides are N-acyl-D-erythro-sphingosines (compound 1, Fig. 2). Thus, ceramide is a 2-amido-1,3-diol. The fatty amide group in ceramide is structurally heterogeneous, differing in the length and extent of saturation and hydroxylation of the aliphatic chain. In some species the fatty amide bears an α-hydroxy group. Typical fatty acids that are prevalent in the fatty amide linkage of natural ceramides are C16:0, C18:0, C20:0, C22:0, and C24:1. The structural features that comprise the ceramide molecule are depicted in Fig. 2. These features include the two stereocenters of the sphingosine backbone (at C-2 and C-3), the amide group, the aliphatic chain with the trans-double bond, and the two hydroxy groups. Many of these sites have been altered by chemical synthesis in efforts to establish structure-function relationships (16).
In human skin, ceramides are present in lamellar sheets in intercellular spaces of the stratum corneum (the outermost layer of skin). Together with cholesterol and saturated free fatty acids, ceramides maintain the water permeability barrier of skin and block transepidermal water loss (17, 18). The stratum corneum also contains free sphingosine, sphinganine, and 6-hydroxysphingosine, which may be formed by ceramidase action on ceramides (19). The three long-chain bases found in skin ceramides are sphingosine, 6R-hydroxy-D-erythro-sphingosine, and phytosphingosine. Ceramides in skin are classified according to whether their N-acyl chain bears an ω-hydroxy group, which allows covalent bonding to take place with proteins and fatty acids via an ester link. Compound 2 (Fig. 2) is known as “ceramide B,” which constitutes about 25% of the dry weight of the human stratum corneum tissue and is protein bound (20, 21). In D-erythro-30’-(linoleoyloxy)triactanoyl-4-sphingenine, a 30-carbon saturated fatty amide chain is esterifed to linoleic acid and forms a wax-like compound.
Other examples of sphingolipids with modifications at C-1 are glycosylated and the following phosphorylated derivatives.

Figure 2. The structures of ceramide: 1, a schematic structure of ceramide that illustrates the sites at which structural modifications may be introduced by chemical synthesis, and the structure of a representative ceramide found in human skin (2). Structural changes in the ceramide molecule have been introduced at many sites. Alterations include the configuration at C-2 and C-3; the lengths of the fatty amide chain and of the aliphatic chain attached to C-5; the positions of the unsaturation and the secondary hydroxy group; the replacement of the hydroxyl groups with other atoms (hydrogen or fluorine) or functionalities (methoxy, methylthio, and keto); the incorporation of aromatic, heteroaromatic, and other rings in place of the alkenyl side chain of the sphingoid base; and the replacement of the carbonyl group of the carboxamide group. In addition, the 2-amino-1,3-diol functionalities have been incorporated into cyclic structures.
Phosphorylated sphingolipids
Sphingomyelin (ceramide 1-phosphorylcholine, compound 1, Fig. 3) is a phospholipid in which the terminal hydroxy group of ceramide is esterified to phosphocholine. Sphingomyelin and the glycerophospholipid phosphatidylcholine have the same polar head group; together, the two choline-containing phospholipids account for >50% of the total phospholipids content of many mammalian membranes. Sphingomyelin is one of the most abundant phospholipids in the plasma membranes of mammalian cells and plasma lipoproteins, and its metabolism provides a source of sphingolipid second messengers. Sphingomyelinase C catalyzed hydrolysis of the phosphodiester bond yields ceramide and phosphocholine (Fig. 3). Hydrolysis catalyzed by sphingomyelinase D cleaves the phosphodiester bond on the choline side of the molecule and forms ceramide 1-phosphate and choline. Autotaxin (ATX), an enzyme that promotes cancer cell invasion, cell migration, metathesis, and angiogenesis, catalyzes the hydrolysis of lysosphingomyelin to sphingosine 1-phosphate in addition to hydrolyzing lysophosphatidylcholine to the lipid mediator lysophosphatidic acid. ATX is subject to product inhibition by two important lysophospholipid signaling molecules, lysophosphatidic acid and sphingosine 1-phosphate (22). Therefore, this enzyme is an attractive drug target that may be inhibited by synthetic lyso sphingo- and/or glycerophospholipids.

Figure 3. The structures of sphingomyelin (1), ceramide aminoethylphosphonate (2), and inositol phosphorylceramide (InsPCer) (3).
In mammalian cells, the predominant long-chain base in sphingomyelin is D-erythro-C18-sphingosine. Dihydrosphingosine is a minor long-chain base of sphingomyelin of most mammalian membranes; for example, the dihydrosphingosine content of egg (chicken) and milk (bovine) sphingomyelin is —1% and 15-20%, respectively (23). An exception, however, is the adult human eye lens membrane, in which dihydrosphingosine comprises about 50% of the total phospholipid and about 80% of the total sphingomyelin content (24). The absence of unsaturation in the long-chain base results in modified physical properties. The double bond of sphingomyelin is essential in maintaining the transbilayer asymmetry of a glycosphingolipid in phospholipid vesicles (25) and in inhibiting protein-mediated glycosphingolipid transfer between membranes (26), as demonstrated by replacing sphingomyelin with dihydrosphingomyelin in bilayer membranes.
The C-3-hydroxy group and the C-4,C-5-trans-double bond of sphingomyelin and other sphingolipids are located in the lipid-water interfacial region. These groups play a role in organizing interfacial water via hydrogen bonding with the surrounding water molecules and neighboring lipids. Intramolecular hydrogen bonding (with the participation of strongly bound water molecules) between the C-3-hydroxy group of the long-chain base and the bridging oxygen of the phosphate results in restricted polar head group mobility (27). The trans -double bond of the sphingoid base plays a role in the extent of hydration of the interfacial region of sphingomyelin-containing bilayers. Dihydrosphingomyelin has been proposed to form stronger intermolecular hydrogen bonds with neighboring dihydrosphingomyelin, cholesterol, and other lipid molecules than does sphingomyelin (28), whereas sphingomyelin may form more stable intramolecular hydrogen bonds (27). Dihydrosphingomyelin has a higher affinity for cholesterol than sphingomyelin and protects cholesterol and unsaturated phosphatidylcholines from oxidation better than sphingomyelin (29).
A heterogeneous mixture of fatty amide chains often is found in natural sphingomyelins and other sphingolipids, varying with the source and age (30). The fatty acyl chain content of milk sphingomyelin, for example, is ~33% C23:0, and ~20% each of C16:0, C22:0, and C24:0. About 50% of the N-acyl chain composition of bovine brain sphingomyelin is C18:0, with about 20% C24:1 and 8% C22:0. Egg sphingomyelin is an exception, being relatively homogeneous with respect to N-acyl content—largely C16:0 (~80-85%) and C18:0 (~12%). An abundance of long, saturated amide chains exists in sphingomyelin compared with the fatty acyl chains of glycerophospholipids, which provides a marked disparity in the intramolecular chain lengths of the sphingoid chain and the N-acyl chain. A long, saturated N-acyl chain in sphingomyelin is conducive to tight packing between sphingomyelin and cholesterol, which may result in the formation of ordered microdomains in bilayer membranes. Synthetic sphingomyelin bearing a N-oleoyl chain is not incorporated into cholesterol-rich domains (31), probably because of its high miscibility with phosphatidylcholine (32).
The lengths of both the long-chain base and fatty amide chain tend to be shorter in the sphingolipids isolated from invertebrate sources than from mammalian sources. For example, the major long-chain base of the sphingolipids of Drosophila (33), the moth Manduca sexta (6), and honey bees (34) is C14-D-erythro-sphingosine (the 14-carbon homolog of the long-chain base, with smaller amounts of the C15-C17 sphingoid bases), and the N-acyl chains of their sphingolipids are also shorter (35). Incidentally, the fatty acyl chains of their glycerophospholipids are also shorter than those of glycerophospholipids in mammalian cell membranes; thus the temperature of the main gel to liquid-crystalline phase transition is lower in invertebrate membranes than in mammalian membranes.
Invertebrates also contain ceramide phosphoethanolamine, an analog of sphingomyelin in which the choline is replaced by ethanolamine, as the major sphingophospholipid constituent of their membranes. The same enzyme that catalyzes the transfer of phosphocholine from phosphatidylcholine to ceramide (SM synthase) also transfers phosphoethanolamine from phos- phatidylethanolamine. Ceramide phosphoethanolamine seems to have an antioxidant function in Drosophila (33) similar to sphingomyelin in mammalian cells (36, 37). Little information is available regarding the other functions of ceramide phoshoethanolamine because most physical studies of sphingophospholipids have been focused on sphingomyelin. Sphingomyelin isolated from natural sources partitions efficiently into highly ordered lateral domains of lipid bilayers enriched in cholesterol and interacts preferentially with cholesterol (38), but ceramide phosphoethanolamine seems to form less tightly packed bilayers with cholesterol (39).
Sphingophosphonolipids are another type of sphingophospholipid in which the polar head group contains a carbon-phosphorus bond. Ceramide 2-aminoethylphosphonate is a typical example (compound 2, Fig. 3). In some species, the head group also bears a 1-hydroxy group or an N-methyl group. Sphingophosphonolipids are minor constituents of mammalian cells but are abundant in the bacterium Bacteriovorax stolpii, in which the long-chain bases are mostly C17 isobranched phytoceramide and dihydroceramide (40, 41).
The principal sphingosylphosphatide component of yeast, fungi, and slime molds is inositol phosphorylceramide (InsPCer, compound 3, Fig. 3), which has a phytoceramide or dihydrosphingosine backbone and an inositol phosphate head group. InsPCer is formed in a reaction catalyzed by InsPCer synthase, which transfers inositol phosphate from phosphatidylinositol to the C-1 hydroxy group of dihydroceramide or phytoceramide and therefore liberates diacylglycerol (42). Glycosylated derivatives of InsPCer constitute the lipid moiety of most glycosylphosphatidylinositol-(GPI)-anchored membrane proteins of eukaryotic cell membranes (43, 44). As no mammalian analog of InsPCer synthase exists, this enzyme is a potential therapeutic target for the development of selective antifungal and antiprotozoal agents that interfere with sphingolipid biosynthesis in fungal and protozoan cells but not in mammalian cells (45, 46). Microbial natural products that inhibit InsPCer synthase include the cyclic peptide aureobasidin A, an amphipathic lipid known as khafrefungin, and a macrolide called galbonolide A or rustmicin (46, 47). The structures of the latter two compounds are shown in Fig. 4.
The phosphorylated metabolites of sphingomyelin are sphingosylphosphocholine (lyso-sphingomyelin, the N-deacylated derivative of sphingomyelin), sphingosine 1-phosphate, and ceramide 1-phosphate. These sphingolipid metabolites are signaling molecules that regulate diverse cellular functions (48-53).
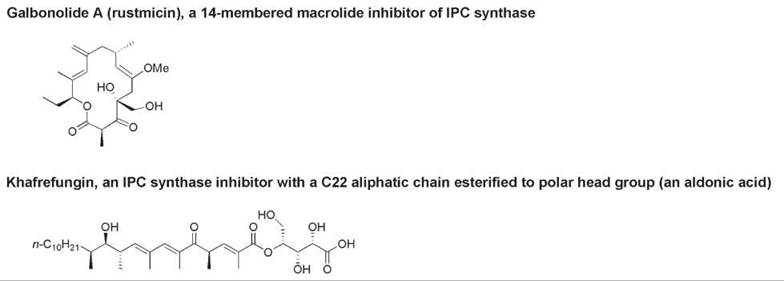
Figure 4. The structures of natural inhibitors of InsPCer synthase: galbonolide A and khafrefungin.
Glycosylated sphingolipids
The terminal hydroxy group of sphingosine is linked to at least one carbohydrate moiety in glycosphingolipids. In mammalian cells, the sugar moiety is in a β-glycosidic link to the sphingoid base, as in β-glucosylceramide (Fig. 5). Cerebrosides are one subgroup of glycosphingolipids in which a galactose or glucose is linked to ceramide (compound 1, Fig. 5). Sulfatides are a subgroup of galactocerebrosides in which a sulfate group is esterified to the C-3 position of the sugar; the sulfate group of 3'-phosphoadenosine 5'-phosphosulfate is transferred to the galactocerebroside in a reaction catalyzed by a sulfotransferase. β-Galactosylceramide and its 3-sulfate ester bind to the gp-120 envelope protein of HIV-1; this interaction allows HIV-1 to infect CD4-negative cells (54). Globosides have at least two saccharide units linked by the anomeric hydroxy group of one sugar to the terminal hydroxy group of the sphingoid base. Plants and fungi contain inositol-linked phosphophytoceramides (InsPCer) such as compound 3 (Fig. 3), a major glycophosphosphingolipid of yeast, and mannosylated and dimannosylated derivatives.
Gangliosides (compound 2, Fig. 5) are ubiquitous components of vertebrate cells and are abundant in nervous tissues. They are localized in the outer leaflet of plasma membranes and form clusters in lipid microdomains, where they participate in various recognition and cell signaling processes (55). The oligosaccharide head group of gangliosides contains at least one N-acetylneuraminic acid residue. This monosaccharide, which is a sialic acid and is abbreviated as NANA, NeuAc, or Neu5Ac, is recognized (together with uncharged saccharide units) by many carbohydrate-related proteins (56). Thus gangliosides are sialylated glycosphingolipids. The predominant N-acyl chain of the ceramide backbone in gangliosides is stearoyl (C18:0). Figure 5 shows the structures of the gangliosides GM1 and GM3. In ganglioside nomenclature, the subscript letter “M” refers to “mono,” which is the number of NANA residues in GM1. The difference between 5 and the number after the “M” indicates the number of uncharged sugar residues present in the ganglioside: four in GM1 and two in GM3. GM3 is the principal ganglioside found in human T lymphocytes. GM1 is a principal ganglioside in the brain but is also present in intestinal epithelial cell membranes where its physiologic function is not known. Gangliosides affect a multitude of cellular processes, including binding specifically to viruses and many bacterial toxins. A well-known example is the binding of cholera toxin, the enterotoxin produced by Vibrio cholerae, to GM1 on the surface of human intestinal epithelial cells (57). The development of artificial receptors for the toxin may form the basis for the design of a potential anticholera drug (58). Another example of a specific interaction between a protein and a ganglioside is the binding of GM1 to α-synuclein, a presynaptic protein with a propensity to form fibrils and aggregates that have been implicated in Parkinson’s disease and other neurodegenerative diseases (59). Ganglioside GM3 binds to insulin receptors; in fact, the accumulation of GM3 in membrane microdomains (rafts) of adipocytes results in the development of insulin resistance, apparently because of the dissociation of the insulin receptor from its complex with caveolin-1 in rafts (60). Because GM3 is a negative regulator of insulin signaling, the inhibition of GM3 biosynthesis may offer a novel therapeutic approach for insulin-resistant type 2 diabetes.

Figure 5. The structures of (1) glycosphingolipids and (2) the gangliosides GM3 and GM1. GM3 contains ceramide, one glucose residue, one galactose residue, and one NANA residue. GM1 is a more complex ganglioside that contains an N-acetylgalactosamine residue and a galactose residue in addition to the components of GM3.
Role of Chemical Structure in the Biological Activity or Biophysical Behavior of Ceramide
The presence of two chiral carbon atoms in sphingosine results in the possible formation of four stereoisomers (Fig. 6). Only the D-erythro stereoisomer occurs naturally, and this stereoisomer is the only form that is used efficiently as the substrate for sphingolipid-using enzymes such as ceramidase (61, 62) and protein phosphatases (63). The “D” nomenclature is based on the Fischer projection and its relationship to the Fischer projection of D-glyceraldehyde (Fig. 6). Many studies have been carried out to assess how the various bioactivities of the unnatural stereoisomers of ceramides differ from that of D-erythro-ceramide. Short-chain ceramides (having a C2 to C8 N-acyl chain) were used in many of these studies because they are water soluble and cell permeable. In general, the D-erythro form was found to be the most effective with regard to bioactivity. For example, only the D-erythro isomer of C8-ceramide was capable of promoting the fusion of an alpha virus with target liposomes; the other three isomers were inactive (64). However, in some studies the D-threo isomer had equal or higher activity than the D-erythro isomer (65), and in still other studies both the D-threo and L-threo isomers had higher activity than did the D-erythro isomer (66). In other studies, a lack of stereospecificity was found, as all four stereoisomers exhibited approximately equal activity (67-69).
Short-chain analogs of D-erythro-ceramide trigger apoptosis, thereby mimicking the activity of endogenous ceramide. However, biophysical studies indicate that short-chain ceramides perturb the physical properties of model membrane bilayers differently than their long-chain counterparts. For example, they did not form the ceramide-rich domains that are observed characteristically with C16:0- and C18:0-ceramide (70). A study of the ability of ceramide analogs to displace sterols from rafts indicated that C12:0- and C16:0-ceramides are faithful mimics of natural ceramides in model bilayers (71).
The presence of the trans-double bond between C-4 and C-5 contributes to the close packing of D-erythro-ceramide molecules at the lipid-water interface (72). The unsaturation is required for most bioactivities of ceramide that have been studied, as dihydroceramide generally exhibits much lower bioactivity. Studies with the cis and triple bond derivatives of ceramide have provided different results; one study found a high activity of these unsaturated ceramide analogs (73), but other studies found markedly reduced bioactivities (69, 74). A synthetic analog of ceramide in which the unsaturation remains at C-4 but is in an exo-methylene group was also active in inducing apoptosis in mouse embryonic fibroblasts (75). The incorporation of the trans-double bond into an aromatic or heteroatomic ring also provided bioactive ceramide analogs (76, 77).
Dihydroceramide exhibits different physical and biochemical effects compared with the corresponding ceramide bearing the same N-acyl chain. For example, channel formation in mitochondrial membranes (78) and protein phosphatase activity in pancreatic beta cells are inhibited by dihydroceramide but are activated by ceramide (63, 79). Dihydroceramide is less effective than ceramide in inducing apoptosis (68).
The terminal hydroxy group of ceramide has also been modified. 1-O-Methoxyceramide did not inhibit mitochondrial ceramidase (69), and 1-fluoroceramide was a weak inhibitor of glucosylceramide formation in cultured murine neuronal cells (80). The 1-methylthio analog of dihydroceramide activated sphinganine kinase and disrupted neuron axonal growth in cell cultures (81).
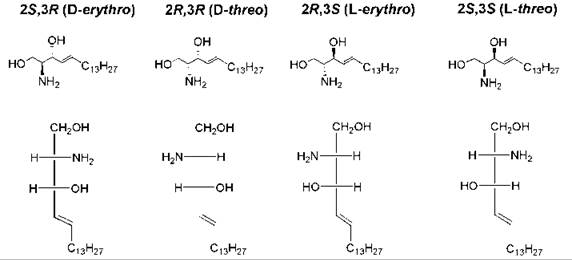
Figure 6. Structures of the four stereoisomers of sphingosine. Sphingosine has two chiral carbon atoms (C-2 and C-3). The Fischer projection formula of each structure is also shown, with C-1 at the top, to illustrate the D/L and erythro/threo stereochemical nomenclature. C-3 has an erythro or threo configuration as it relates to C-2, depending on whether the similar groups (amino and hydroxy) are on the same or opposite side of the Fischer projection. D versus L refers to the configuration at C-2 relative to the configuration of D-glyceraldehyde versus L-glyceraldehyde.
Biological Background
In the anabolic pathway of ceramide biosynthesis, N-acylation of a sphingoid long-chain base takes place in the endoplasmic reticulum and Golgi apparatus. In the catabolic pathway of ceramide biosynthesis, degradation of sphingomyelin and glycosphingolids takes place at the plasma membrane and lysosomes, respectively. The enzymes in both the anabolic and catabolic pathways of sphingolipid metabolism are important determinants of cell survival, growth, and differentiation. Because sphingolipids have been implicated in a wide range of human diseases and neurological syndromes, sphingolipid-using enzymes represent potential targets for therapy of a variety of diseases characterized by an accumulation of sphingolipids or an improper balance among growth-supporting (such as sphingosine 1-phosphate, ceramide 1-phosphate, and glycosylceramides) and growth-inhibiting sphingolipids (such as ceramide and sphingosine).
A defect in a single lysosomal enzyme or cofactor results in substrate accumulation in one or more organs. This imbalance between the formation and breakdown of glycosphingolipids is associated with neurodegeneration and high mortality (see also Lipid Homeostasis, Chemistry of). Partial reduction in the rate of biosynthesis of the glycolipid that accumulates in a lipid storage disease is known as substrate reduction therapy, which is an alternative therapeutic approach to enzyme replacement. An inhibitor of an enzyme in an anabolic glycosphingolipid pathway is used to reduce a key glycosphingolipid partially to a level at which the residual catabolic activity is capable of preventing excessive substrate accumulation. The imino sugar N-butyldeoxynojirimycin (NB-DNJ), which is marketed as miglusatat or Zavesca, is a synthetic sphingolipid derivative that selectively modulates cellular glycosphingolipid levels. It inhibits the ceramide-specific glycosyltransferase that catalyzes the first committed step in glycosphingolipid synthesis and shows promising results in the management of some animal models of glycosphingolipid storage diseases because it offsets the accumulation of glucosylceramide and more complex glycosphingolipids (82).
The sphingolipid pathway has many sites at which inhibitors may be directed to bring about the pharmacologic manipulation of sphingolipid metabolism. As ceramide has a central role in a myriad of cell signaling events, a great deal of attention has been devoted to the study of ceramide metabolism and function in the past two decades. The direct targets of ceramides that have been identified are the protease cathepsin D, ceramide-activated protein phosphatases, ceramide-activated protein kinases, the kinase suppressor of Ras (KSR), isoforms of protein kinase C (PKC), c-Jun N-terminal kinase (JNK), Akt, and PTEN (83, 84). The phosphorylation state of proteins with pro- and antiapoptotic activities is modulated by intracellular ceramide levels.
Regulation of the endogenous levels of ceramide with naturally occurring inhibitors of sphingolipid-using enzymes
The structures of some naturally occurring compounds that act on sphingolipid-metabolizing enzymes are shown in Fig. 7. These compounds include 1) sphingols, which inhibit serine palmitoyl-CoA transferase, 2) fumonisins and australifungin, which inhibit (dihydro)ceramide synthase, 3) scyphostatin, which inhibits the neutral-pH optimum isozyme of sphingomyelinase, 4) 2-acetyl-4-tetrahydroxybutylimidazole (THI), which inhibits S1P lyase, and 5) aureobasidin A, khafrefungin, and galbonolide A, which inhibit InsPCer synthase (Fig. 4).

Figure 7. The structures of various naturally occurring inhibitors of enzymes in the sphingolipid pathway.
Overview of sphingolipid biosynthesis
The de novo synthesis of ceramide takes place in the endoplasmic reticulum and begins with the decarboxylative condensation of L-serine with palmitoyl-CoA, catalyzed by serine palmitoyl-CoA transferase (SPTase) (Fig. 8). As the name of the enzyme implies, palmitoyl-coenzyme A is recognized preferentially by mammalian SPTase, and the palmitoyl chain is transferred to the amino group of L-serine with the formation of 3-ketosphinganine. In this reaction, carbon dioxide is released from serine, with the other two carbon atoms of serine providing the source of C-1 and C-2 of the long-chain base. Therefore, the 18-carbon long-chain base predominates in the sphingolipids of mammalian cells. Although most prokaryotic cells do not contain sphingolipids, some bacteria such as the Sphingomonas species synthesize glycosphingolipids. The SPTase of these bacteria is a cytoplasmic homodimer, in contrast to the membrane-bond heterodimeric form found in eukaryotes (85); as mentioned above their sphingoid long-chain bases tend to have fewer than 18 carbons, which implies that their SPTase prefers an acyl-CoA with fewer than 16 carbons in the acyl chain.
Inhibition of the first step in sphingolipid biosynthesis with sphingols, L-cycloserine, or β-chloro-L-alanine results in the depletion of all sphingolipids. Sphingols such as myriocin (also known as ISP-1, Fig. 9) and sphingofungins (Fig. 9) have been isolated from fungal cultures and often are used as biochemical tools to examine the roles of sphingolipids in vitro. Growth inhibition of cultured cells by myriocin and sphingofungin B was rescued by exogenous sphingolipids (86). Myriocin treatment of apoE1 knockout mice decreased the sphingomyelin content of lipoproteins and lowered the levels of plasma cholesterol and triacylglycerols, which indicates that sphingolipid biosynthesis may be a therapeutic target for treating dyslipidemia and atherosclerosis (87).
Myriocin is a competitive inhibitor of SPTase, which forms an aldimine adduct with the active site of the enzyme. However, cycloserine and β-chloro-L-alanine inhibit SPTase by a different mechanism; they react with the coenzyme of SPTase, pyridoxal 5'-phosphate, inhibiting the enzyme irreversibly and thereby downregulating the biosynthesis of sphinganine (88). As shown in Fig. 9, myriocin and sphingofungin B (both of which inhibit SPTase in the nanomolar range) bear a structural resemblance to sphingosine and its biosynthetic intermediates and may also mimic the transition state of the SPTase-catalyzed reaction. The configurations at C-2, C-3, C-4, and C-5 of sphingofungin B are important for its inhibitory potency on SPTase; however, the 14-hydroxy epimer of sphingofungin B is as potent as the natural 14 S stereoisomer, which indicates that the configuration at C-14 is not critical (89).

Figure 8. Outline of the sphingolipid biosynthetic pathway. Natural and synthetic inhibitors of sphingolipid-metabolizing enzymes are shown adjacent to the inverted T bars.

Figure 9. Structures of naturally occurring inhibitors of SPTase.
Inhibition of other enzymes in the sphingolipid metabolic pathway by natural products
In the second step of the sphingolipid biosynthetic pathway, the carbonyl group of 3-ketosphinganine is reduced with NADPH to form D-erythro-sphinganine. No naturally occurring inhibitors of 3-ketosphinganine reductase have been reported yet.
The N-acylation of sphinganine with a fatty acyl-CoA is catalyzed by acyl-CoA-dependent ceramide synthases (also called dihydroceramide synthases; at least six genes for this enzyme exist in mammalian cells), which produces dihydroceramide. The fungal natural products fumonisin and australifungin (Fig. 7) are specific inhibitors of N-acylsphinganine transferase activity in mammalian and fungal cells, respectively. The structure of fumonisin resembles the structures of both the sphingosine base and fatty acyl-CoA cosubstrates of dihydroceramide synthase. Therefore, the aminopentol portion of fumonisin may compete with the long-chain base for binding to the enzyme, and the polar region that contains the two TCA substituents may block binding of the fatty acyl-CoA. Fumonisin-induced disruption of sphingolipid metabolism resulted in neurotoxicity, birth defects, cancer, and renal and liver failure (90, 91). γ-Tocopherol, the predominant form of dietary vitamin E, also raises the levels of dihydroceramide and dihydrosphingosine in human prostate cancer cell lines (92). Although the mechanism by which these sphingolipids are elevated has not been established, this result is another example of how an interruption in the de novo sphingolipid pathway can culminate in cell death.
Dihydroceramide desaturase catalyzes the last enzyme in the de novo biosynthetic pathway, which is the introduction of the C-4,C-5-trans-double bond into the long-chain base. No naturally occurring inhibitors of this enzyme have been reported yet. In plants and fungi, dihydroceramide is hydroxylated at C-4 instead of undergoing dehydrogenation. The hydroxylated product, phytoceramide, is the precursor of complex sphingolipids such as InsPCer and various glycosyl-InsPCer derivatives such as mannose-IPC, mannose diinositolphosphorylceramide, dimannose inositolphosphorylceramide, and galactose-dimannose inositolphosphorylceramide.
Metabolism of ceramide
Ceramide formed in mammalian systems is metabolized to sphingomyelin, glycosphingolipids, and ceramide 1-phosphate by the actions of sphingomyelin synthase, glucosylceramide synthase, and ceramide kinase on ceramide, respectively. Ceramide synthesized in the endoplasmic reticulum is transported to the Golgi apparatus for synthesis of complex sphingolipids (93). Sphingomyelin is synthesized from ceramide by transfer of the phosphocholine head group of phosphatidylcholine. As mentioned above, sphingomyelin synthase catalyzes this reaction, and diacylglycerol (an activator of PKC) is released. InsPCer synthase carries out the analogous process in yeast and plants, transferring inositol phosphate to phytoceramide or dihydroceramide. Cerebrosides and gangliosides are synthesized from ceramide and nucleotide sugars in the presence of glycosyl transferases.
Hydrolysis of the amide bond of ceramide by ceramidases generates sphingosine, which has broad antibacterial and antifungal activity in addition to its PKC inhibitory activity (94). Phosphorylation of sphingosine at the terminal hydroxy group by sphingosine kinases affords S1P. S1P acts both as an intracellular second messenger by activating internal cell targets and extracellularly as an agonist of G-protein coupled receptors by mediating a diverse range of cellular effects (51, 52, 95). Sphingosine kinases are activated by several growth- and survival-promoting agents.
S1P lyase, a pyridoxal 5'-phosphate dependent enzyme that has a potential role in controlling cell fate and stress responses (96, 97), cleaves S1P between C-2 and C-3 to produce an α,β-unsaturated fatty aldehyde and phosphoethanolamine. Pyridoxal 5'-phosphate forms a Schiff base with the amino group of S1P. S1P lyase plays a role in determining the balance between the intracellular levels of S1P and ceramide.
S1P is dephosphorylated by S1P phosphatases. The S1P phosphatase-1 isozyme decreases the transport of ceramide from the endoplasmic reticulum to the Golgi, as determined by the visualization of a fluorescent analog of ceramide that accumulates in the ER (98). S1P phosphatase type-2 is upregulated by inflammatory stimuli (99).
Natural products that interfere with ceramide metabolism
Ceramide also is formed by sphingomyelinase (SMase)-catalyzed hydrolysis of sphingomyelin. Acidic and neutral SMases are activated by stress stimuli and external agents such as the Fas ligand (FasL), tumor necrosis factor-α (TNF-α), growth factors, and chemotherapeutic agents, which produces a transient rise in intracellular ceramide levels by hydrolysis of the phosphocholine moiety of sphingomyelin. SMases play important roles in cell signaling pathways that regulate cell growth, differentiation, cell cycle arrest, and apoptosis. A secretory form of acidic SMase is important in sphingomyelin catabolism, generating ceramide from sphingomyelin in plasma membrane domains. The resulting coalescence of ceramide-rich domains leads to the initiation of apoptosis, internalization of pathogens, and secretion of cytokines (100, 101).
The fungal metabolite scyphostatin (Fig. 7) is a potent, reversible inhibitor of membrane-bound Mg2+-dependent neutral sphingomyelinase (nSMase), thereby interfering with the generation of ceramide (102, 103). The activity of nSMases is sensitive to the cellular redox state, for example, the ratio of glutathione in the reduced versus the oxidized form (104, 105). The farnesyltransferase inhibitor manumycin A, a polyenamide produced by the Streptomyces species, also inhibits nSMase irreversibly (106).
The lysosomal acid sphingomyelinase (aSMase) isoform is inhibited by L-carnitine, a cofactor of acyl-coenzyme A transport in mitochondria (107, 108), and by phosphatidylinositol polyphosphates, which occur in plant, yeast, and mammalian cells (109).
A caramel food colorant and component of coffee, 2-acetyl-4-tetrahydroxybutylimidazole (THI, Fig. 7), inhibits S1P lyase, which causes S1P to accumulate in the lymph nodes (110).
Interrelationships Among Sphingolipid Metabolites
Figure 10 shows interrelationships among the sphingolipid metabolites. Ceramide is converted to glycosylceramides and more complex glycosphingolipids and to 1-O-acylceramide by glycosylceramide synthases; it is converted to ceramide 1-phosphate by ceramide kinase and to sphingosine by ceramidases (Fig. 10a). The interplay between the kinases and phosphatases may control the concentrations of these sphingolipid mediators. Figure 10b shows the interconversions among the sphingolipid metabolites, together with the natural and synthetic inhibitors of enzymes in the sphingolipid pathway.

Figure 10. (a) Outline of the sphingolipid metabolic pathway with an emphasis on the steps involved in the conversion of sphingomyelin to ceramide, ceramide to sphingosine, and sphingosine to sphingosine 1-phosphate. The inverted T bars indicate the natural and synthetic inhibitors of the enzymes in the pathway. (b) Interconversions among sphingolipid metabolites with opposing activities affect cell homeostasis. The balance between sphingoid bases plays an important role in the control of cell fate. For example, although ceramide plays a key role in the cellular stress response and the induction of apoptosis, it may be metabolized to S1P, which inhibits apoptosis and promotes DNA synthesis, angiogenesis, and cell migration. Tipping the balance between the accumulation of a pro-apoptotic and an anti-apoptotic sphingolipid may result in metabolic dysfunctions.
Elevating the ceramide content of cells
Stimulating ceramide biosynthesis and inhibiting the conversion of endogenous ceramide to other sphingolipid metabolites is a strategy that may be applied to block key steps in cancer progression such as cell growth and cell survival. The intracellular level of ceramide can be elevated by 1) activating neutral and acid SMases, 2) stimulating the de novo synthesis of ceramide, and 3) blocking the metabolic conversion of ceramide to other sphingolipids (for example, by inhibiting enzymes that use ceramide as a substrate, such as sphingomyelin synthase, glycosylceramide synthases, ceramidases, ceramide kinase, and O-acyl transferases).
Lowering the ceramide content of cells
Inhibitors of the enzymes that participate in the de novo biosynthesis of ceramide may be used to treat pathologies associated with elevated intracellular ceramide levels, such as chemotherapy-induced cell death and insulin resistance in muscle.
Designing Sphingolipid Analogs to Modulate Sphingolipid Metabolism
Synthetic analogs of sphingolipids offer advantages over natural sphingolipids
1. The extensive hydrophobicity of some natural compounds can be reduced by synthesizing compounds with shorter hydrocarbon chains or with more polar groups, thereby enhancing the aqueous solubility and cellular uptake (which may be directed to different intracellular compartments). Many cell- permeable analogs of ceramide mimic the effects of TNF-α, chemotherapeutic agents, oxidants, and ionizing radiation, which activate SMases to produce ceramide. Moreover, the subcellular localization of the analog may be dependent on the lipophilicity and/or net charge of the analog. N-Hexanoyl- and N-octanoyl-D-erythro-ceramide are examples of cell-permeable analogs of natural ceramide that induce cell cycle arrest or apoptosis in A549 cells (111, 112).
2. As discussed above, altering the stereochemistry of the sphingosine backbone has produced, in many instances, stereoisomers with altered bioactivity compared with the natural stereoisomer, for example, in activating the catalytic subunit of serine-threonine protein phosphatase 2A (63) or in inhibiting mitochondrial ceramidase (69).
3. The chemical and metabolic stability of labile natural compounds can be enhanced by incorporating stabilizing groups such as a phosphonate in place of a phosphodiester bond or a C -glycoside in place of an O-glycosidic bond, thereby creating new agents that resist phosphatase (113-115) and glycosidase action in cells (116).
4. The toxicity and bioavailability can be varied by conjugation to form new derivatives such as glucuronides that are metabolized differently than the parent compound. An example is a glucuronide derivative of the aminophenol amide of all-trans retinoic acid, fenretinamide (N-(4-hydroxyphenyl)-retinamide, 4-HPR) (117).
5. Structure-activity relationships may be established if several analogs are available. This step is a necessary step in drug design that leads to optimizing the properties of the therapeutic agent and developing a lead compound.
Many non-natural sphingolipid analogs have been synthesized with the aim of achieving a potential therapeutic advantage with respect to one of the following bioactivities: 1) to manipulate the activity of enzymes in the sphingolipid biosynthetic pathway and thus alter the balance between the pro-survival and antiproliferative sphingolipid metabolites, 2) to alter the stability of lipid rafts and cause aberrant localization of signaling molecules, 3) to produce immunosuppression via alteration of the number of circulating lymphocytes, or 4) to activate natural killer T cells to produce a desired array of cytokines.
Elevating Intracellular Ceramide Levels with Unnatural Sphingolipid Analogs
Irradiation (UV and y rays), chemotherapy, pathogenic infections, and many other external stress stimuli activate SMases and elevate the endogenous levels of ceramide in tumor cells, which promotes apoptosis. However, tumor cells may counteract these treatments by activation of ATP-dependent efflux proteins and by converting endogenous ceramide to other sphingolipid metabolites, which thereby evades apoptosis.
Use of unnatural sphingolipid analogs to elevate endogenous ceramide levels in tumor cells by interfering with ceramide trafficking
After the de novo synthesis of ceramide in the endoplasmic reticulum, additional biosynthesis of sphingolipids continues in the Golgi apparatus and plasma membrane. Sphingomyelin synthase uses ceramide as a substrate for sphingomyelin production in the lumen of the Golgi. A cytosolic ceramide transfer protein, CERT, transports ceramide in a nonvesicular manner from the endoplasmic reticulum to the Golgi complex (118). Ceramide transport is required for the synthesis of sphingomyelin and presumably other sphingolipids. A Drosophila mutant that lacked the functional CERT gene had a depressed content of ceramide phosphoethanolamine (the sphingomyelin analog in Drosophila) and ceramide, which resulted in altered membrane permeability behavior and an enhanced susceptibility to oxidation of cellular components (33). A synthetic analog that resembles ceramide with respect to structure and stereochemistry, (1R,3R)-A-(3-hydroxy-1-hydroxymethyl-3-phenylpropyl)dodecamide (1R,3R)-HPA-12 (Fig. 11), inhibited sphingomyelin biosynthesis by blocking ceramide trafficking (119).

Figure 11. Structures of synthetic inhibitors of ceramide biosynthesis.
Use of synthetic sphingolipid analogs to modulate endogenous ceramide levels
1. Inhibition of neutral SMases. Mg2+-dependent nSMases in plasma membranes hydrolyze sphingomyelin to yield ceramide and phosphocholine. As ceramide is a key signaling molecule in the apoptotic and inflammation response to stress signals, inhibitors of SMase isoforms may provide lead compounds for the treatment of inflammation, ischemia, neurodegenerative diseases, and infarction. The following examples are representative of efforts to develop synthetic sphingophospholipid analogs with antiapoptotic activity. 3-O-Methyl- and 3-O-ethyl-sphingomyelin (Fig. ll) inhibited nSMase without markedly affecting the activity of the acid isoform (120). Inhibition of nSMase also was observed with hydrolytically stabilized tertbutylcarbamate and urea derivatives of sphingomyelin, which also prevented apoptotic neuronal cell death in an ischemic model (121, 122). S1P analogs with a difluoromethylenephosphonate link instead of the phosphate group (SMA-3 and SMA-7, Fig. 11) inhibited nSMase in pheochromocytoma PC-12 cells and inhibited cerebral infarct in mice (123). Phosphonocholine analogs of sphingomyelin, in which an oxygen atom in the phosphate ester head group was replaced by a carbon, nitrogen, or sulfur atom, were also inhibitors of nSMase (124) as was a lactone derivative of ceramide (125) (Fig. 11). A stable desepoxy analog of scyphostatin inhibited nSMase in monocytes, macrophages, and hepatocytes, and inhibited apoptosis (126) (Fig. 11).
2. Inhibition of SM synthase. Tricyclodecan-9-yl xanthogenate (D609, Fig. 12) is a known inhibitor of phospholipase C (which hydrolyzes phosphatidylcholine) (127). It inhibited SM synthase and induced apoptosis in U937 human monocytic leukemia cells (128) and rat PC12 cells (129).
3. Inhibition of glucosylceramide (GlcCer) synthase. This enzyme incorporates a glucosyl residue from UDP-glucose into ceramide to form β-glucosylceramide, which is a precursor of complex glycosphingolipids that participate in many physiologic and pathophysiologic processes. Therefore, this enzyme is a potential drug target. NB-DNJ (Fig. 12) is a competitive inhibitor of GlcCer synthase with respect to ceramide and is a noncompetitive inhibitor with respect to UDP-glucose; molecular modeling studies indicated that NB-DNJ is structurally similar to ceramide but not to glucose (130). NB-DNJ reduced the level of GlcCer that accumulates in Gaucher disease and also acted as a chemical chaperone for the acid β-glucosidase that is defective in this disease.
Other competitive inhibitors of GlcCer synthase are ceramide analogs with a cyclic amino head group such as morpholine or pyrrolidine instead of the primary hydroxy group at C-1. They also have a phenyl group in place of the aliphatic chain of the sphingosine backbone. A lead compound that was developed to inhibit GlcCer synthase is (1R,2R)-1-phenyl-2-aminodecanoyl-3-morpholino-1-propanol (D-threo-PDMP, Fig. 12), which has an N-decanoyl amide chain (131). This ceramide analog leads to the accumulation of ceramide in neuroblastoma and other cell types and also possesses antitumor activity (132). The inhibitory potency on GlcCer synthase was enhanced by the elongation of the N-acyl group from C10 to C16, by the introduction of electron-rich aromatic substituents, by the replacement of the morpholino with a pyrrolidino head group, and by the addition of water-soluble links. In addition to inhibiting the activity of GlcCer synthase, D-threo-PDMP exerts several other effects, including the alteration of the structure and function of membrane domains in late endosomes, the inhibition of LDL degradation, and the enhanced uptake of paclitaxel (133). Administration of high doses of L-PDMP increases GM1 synthesis in the brain (134).
4. Inhibition of ceramidases. Ceramidases specifically hydrolyze the amide bond of ceramide to form sphingosine and a fatty acid without acting on the amide bond of other sphingolipids. Enzymatic forms of ceramidase with acidic, neutral, and alkaline activities have been studied. The enzyme with an acidic pH optimum is localized primarily in lysosomes and thus is important in catabolism. It is activated by the glycoprotein sphingolipid activator protein D (SAP-D) (135, 136). N-Oleoylethanolamine (Fig. 12), the first known inhibitor of this enzyme, has a low potency (IC50 > 0.5 mM) and a low specificity (137, 138). Point mutations in the gene encoding acid ceramidase lead to the lysosomal storage disease known as Farber disease. As acid ceramidase is overexpressed in several human cancers, currently a great deal of interest exists in developing new inhibitors of this enzyme to induce an accumulation of endogenous ceramide and a suppression of cell growth (139). Ceramidase also catalyzes the reverse of the hydrolysis reaction, for example, the condensation of sphingosine with a fatty acid without the participation of cofactors (140). N-(2-Oxo)-acylsphingosines inhibited acidic ceramidase with a low selectivity and low potency (138). D-erythro-2-(N-Myristoylamino)-1-phenyl-1-propanol (D-e-MAPP, Fig. 12) inhibited alkaline ceramidase, which raises the intracellular levels of ceramide and arrests cell growth (141). A ceramide analog in which a 4-nitrophenyl group replaces the aliphatic chain (denoted as B13, Fig. 12) is a potent inhibitor of acid ceramidase; it induced apoptosis in prostate and colon cancer cell lines (142, 143). A urea derivative of ceramide also inhibited mitochondrial ceramidase (69). Analogs of D-e-MAPP and B13 affect the levels of ceramide, sphingosine, and S1P in MCF7 cells (144). Examples of other potent and specific inhibitors of acid ceramidase are LCL-204 (Fig. 12) and LCL-102, which are lysosomotropic analogs of B13.
5. Inhibition of ceramide synthase. A short-chain cyclopropenyl-containing ceramide analog called GT-11 (145) (also called C8-cyclopropenylceramide) (146) (Fig. 12) inhibits ceramide synthase.
6. Inhibition of dihydroceramide desaturase. The last enzyme in the de novo biosynthesis of ceramide is inhibited by GT-11, but at high concentrations this compound also inhibited S1P lyase and SPTase (147, 148). Fenretinide (4-HPR, Fig. 12) also inhibited dihydroceramide desaturase (147, 148). This synthetic retinoid and its analogs have apoptogenic activity, which elevates intracellular ceramide levels and induces cell death in a variety of cell types in vitro and in vivo by multiple mechanisms (117, 149, 150).
7. Inhibition of S1P lyase. S1P lyase is inhibited by sulfhydryl reagents, FTY720 (151), and by racemic 2-vinyldihydrosphingosine 1-phosphate (152) (Fig. 12). When S1P lyase is overexpressed, ceramide levels are increased and apoptosis is induced (153).

Figure 12. Structures of synthetic inhibitors of sphingomyelin synthase, ceramidases, β-glucosylceramide synthase, dihydroceramide desaturase, and S1P lyase.
Other Structural Analogs of Ceramide with High Antiproliferative Activity
The bioactivity of many synthetic ceramide analogs that bear modifications in the long-chain base or in the fatty amide chain has been studied.
Modifications in the sphingenine backbone
(2S,3R,4E,6E)-N-Octanoylamidooctadecadiene-1,3-diol, a ceramide analog with an additional double bond (between C-6 and C-7), induced accumulation of endogenous ceramide in multidrug-resistant breast cancer cells and induced apoptosis by the mitochondrial pathway without inhibiting the growth of normal epithelial cells (76).
Modifications in the length of the N-acyl chain
In vitro and in vivo studies have shown that short-chain ceramides induce growth arrest (154). A fluorescent analog of C6-ceramide partitioned into caveolin-enriched microdomains of rat aorta vascular smooth muscle cells and led to growth arrest via activation of PKC-zeta, which is recruited to lipid microdomains and subsequently reduces the activity of Akt (155).
Additional polar groups
Ceramide analogs with an additional hydroxy group in the long-chain base, as in 6-hydroxyceramide (76) and phytoceramide (156), are more effective than ceramide in arresting proliferation of some tumor cell lines. The introduction of a uracil or thiouracil group at C-1 of ceramide provided an effective apoptotic agent (157).
Reduction of the carboxamido group of ceramide to a methylene group
An analog called ceramine induced apoptosis in leukemic cells, which indicates that the carbonyl group of the amide group of ceramide is not required for in vitro cytotoxicity (66, 158). Ceramide analogs with an arylsulfonamido group had higher cytoxicity activity than the corresponding alkylsulfonamide analogs (159).
N-acylated 2-amino-1,3-diols
Reaction of amino diols with a fatty acid afforded a series of simple ceramide analogs with pro-apoptotic activity in human cancer cell lines (160). An example is N -oleoylserinol, which induced apoptosis in cells expressing the pro-apoptotic protein PAR-4 and was used to purge transformed cells from embryonic stem cells before implantation into mouse brain (161).
Targeting of ceramide analogs to specific cell organelles
Short- and long-chain ceramides form channels in the outer mitochondrial membrane, which enables proteins in the intermembrane space to be released (162). An aromatic “ceramidoid” in which the N-acyl chain terminates in an N-alkylpyridinium group bears a net positive charge (163) (Fig. 13). This ceramide analog preferentially targets mitochondria, where it accumulates and induces apoptosis by triggering the release of cytochrome c into the cytosol, activating the apoptotic cascade, and blocking the growth of cell carcinomas in vitro and in vivo (164). LCL-204 (also called AD2646) accumulates in lysosomal membranes, inhibiting acid ceramidase, inducing the release of cathepsins, and triggering apoptosis of prostate cancer cells (165) and head and neck squamous cell cancer cells (166). LCL-204 also reduced resistance to FasL.

Figure 13. Structures of a cationic ceramide analog and N-(N'-phenethylthiocarbamoyl)-sphingosine.
Ceramide analogs that inhibit protein kinase C isoforms and induce downstream loss of extracellular signal-regulated kinase (ERK1/ERK2)
Inhibition of the PKC isozyme superfamily elicits apoptosis in tumor cells. Phenethyl isothiocyanate (PEITC) conjugates of sphingosine and sphinganine (Fig. 13) exert potent antineoplastic effects in human leukemia HL-60 cells by the inhibition of conventional PKC/novel PKC activity and ERK1/ERK2 activity (167). The activity of these derivatives surpassed that of safingol (L-threo-sphinganine, a non-natural analog of the natural lipid D-erythro-sphinganine), which is known to modulate the activity of PKC and induce apoptosis.
Sphingolipid Regulation of Signaling via Control of Raft Formation and Stability
Sphingomyelin and glycosylated sphingolipids (GSLs), together with sterols and glycerophospholipids, are key lipid building blocks of rafts. As rafts are considered to represent sites for the initiation of many receptor-mediated signaling events including the uptake of pathogens, sphingolipids play pivotal roles in a variety of dynamic cellular events such as membrane trafficking and the activity of membrane proteins.
Disruption of plasma membrane microdomains by sphingolipid analogs that cannot pack tightly with neighboring lipids
Fluorescent analogs of D-erythro-(or 2S,3R)-lactosylceramide and D-erythro-sphingomyelin undergo endocytosis from the plasma membrane via caveolae, whereas non-natural analogs such as L-threo-(or 2S,3S)-lactosylceramide and L-threo-sphingomyelin follow a predominantly clathrin-dependent route of endocytosis (168). These observations suggest that the stereochemistry at the C-3 position of the sphingosine backbone plays a role in the internalization pathway of the sphingolipid. Studies with a fluorescent, excimer-forming derivative of lactosylceramide (BODIPY-LacCer, Fig. 14) indicated that the D-erythro stereoisomer forms clusters in membranes, from which caveolar endocytosis is initiated. The corresponding L-threo stereoisomer was excluded from these domains; molecular modeling indicated that it does not pack as tightly with neighboring lipids as the natural stereoisomer.
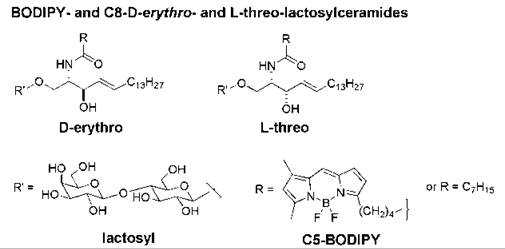
Figure 14. Structures of the D-erythro and L-threo stereoisomers of BODIPY- and C8-lactosylceramides.
A non-natural glycosphingolipid inhibited caveolar uptake, viral binding and infectivity, and β1-integrin signaling
The addition of D-erythro-N-octanoyl-lactosylceramide (C8-D-e-LacCer, Fig. 14) to human skin fibroblasts at low temperature promoted the formation of plasma membrane microdomains as shown by confocal fluorescence microscopic studies. The addition of C8-D-e-LacCer to cells also initiated the clustering of β1-integrins within these domains and the activation of β1-integrins. On warming to 37° C, β1-integrins were internalized rapidly via caveolar endocytosis in cells treated with C8-D-e-LacCer, whereas little β1-integrin underwent endocytosis in untreated fibroblasts. The incubation of cells with C8-D-e-LacCer followed by a brief warm-up also caused src activation and a reorganization of the actin cytoskeleton (169). Conversely, addition of C8-L-threo-LacCer (Fig. 14) inhibited the formation of microdomains in the plasma membrane, which resulted in the inhibition of both endocytosis and β1-integrin signaling (170). Thus, non-natural sphingolipids that can block the formation of microdomains in the plasma membrane may offer a novel means for interfering with the cell entry mechanism that is employed by various pathogens and viruses.
Synthetic Immunomodulatory Sphingolipids: Analogs of the Immunosuppressant FTY720 and the Immunostimulator α-Galactosylceramide
FTY720 analogs interfere with S1P signaling and modulate lymphocyte function
Sphingosine 1-phosphate (S1P) mediates numerous biological processes; therefore, potential new drug candidates may be specific agonists and antagonists of S1P receptors. Chemical manipulation of myriocin led to the development of a new immunosuppressive sphingosine analog known as FTY720 (2-amino-2-[2-(4-octylphenylethyl]-1,3-propanediol), which is phosphorylated in vivo to form the (S )-phosphate (FTY720-phosphate, Fig. 15) (171). This synthetic analog of S1P is a potent agonist of the S1P-type 1 receptor but does not activate the S1P-type 2 receptors on the surface of thymocytes and lymphocytes. FTY720 possesses more potent immunosuppressive activity than myriocin without inhibiting sphingolipid biosynthesis and host immune defense responses to many infectious agents. FTY720 inhibits lymphocyte trafficking in vivo, which promotes the sequestration of lymphocytes into lymph nodes and impairs the S1P-type 1 receptor-mediated migration of lymphocytes between secondary lymphoid tissues and the blood. Thus, the cells become unresponsive to S1P and external signals that direct these cells to sites of inflammation (172). In addition, FTY720 has other potent suppressive effects on T cells unrelated to migration, including the inhibition of the S1P-evoked generation of cytokines that promote autoimmune inflammation such as IL-17 (173). (S)-FTY720-phosphate and other FTY720 analogs that activate S1P receptors and stimulate various signaling pathways seem to be useful for the treatment of a variety of pathologic conditions, including angiogenesis, inflammation, respiratory distress syndrome, and autoimmune diseases.
Promotion of myelination by FTY720 and (S)-FTY720-phosphate
Although FTY720 failed to improve efficacy for preventing renal allograft rejection in Phase III clinical studies (174), a different purpose has been found for its potential use. Phase III clinical trials are underway to examine the use of FTY720 for potential treatment of systemic lupus erythematosus and autoimmune demyelinating diseases such as multiple sclerosis (175). (S)-FTY720-phosphate stimulated, via induction of ERK1/2 and Akt phosphorylation, the survival of progenitor cells that give rise to myelin-producing mature oligodendrocytes (176). Therefore, in addition to its immunosuppressive function, FTY720 shows promise as a therapeutic agent in the treatment of multiple sclerosis via the protection of oligodendrocytes and the replenishment of lost oligodendrocytes, thus promoting remyelination.
Other potential clinical applications of FTY720
The combination of FTY720 with a tyrosine kinase inhibitor induced apoptosis in melanoma cells (177). (R)-AAL, an ether analog of FTY720 (which also has a methyl group in place of a hydroxymethyl group) (Fig. 15), was phosphorylated in vascular cells and blocked vascular endothelial growth factor-(VEGF)-induced vascular permeability in vivo (178). (S)- FTY720-phosphate also may regulate calcium ion channels in smooth muscle cells in an S1P-receptor independent manner (179). FTY720 also enhances pulmonary endothelial cell barrier integrity by a mechanism that seems to be different than that used by S1P, which also augments endothelial cell vascular barrier integrity (180).
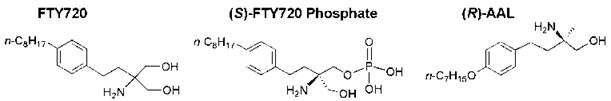
Figure 15. Structures of FTY720, (S)-FTY720-phosphate, and a deoxy ether analog of FTY720 [(ft)-AAL].
Glycosphingolipid-Based Immunotherapies: Presentation of Glycosphingolipids to a Subset of T Cells
A naturally occurring α-anomer of galactosylceramide known as KRN7000 (Fig. 16) was isolated from the marine sponge Agelas mauritianus in 1992 and was subsequently identified as an antitumor agent and a potent immunostimulant of invariant natural killer T (iNKT) cells. These cells are an innate subset of T lymphocytes that express a semi-invariant T-cell receptor (TCR). They rapidly produce immunoregulatory cytokines on recognition of glycolipid ligands bound to the CD1 d glycoprotein on the surface of antigen-presenting cells, which results in the activation of dendritic cells, NK cells, B cells, and T cells. The function of CD1 d is related to the nonclassical major histocompatibility complex (MHC) class I molecules. CD1 d presents glycolipid antigens to the TCR of NKT cells (181). A synthetic analog of KRN7000 known as α-galactosylceramide (α-GalCer, Fig. 16) is the most extensively studied lipid antigen that binds to CD1 d. Although α-GalCer is a highly potent glycolipid antigen for cells that express CD1 d, it is not a natural product of mammalian cells. The identity of the natural mammalian glycolipid antigen that binds to CD1 d for presentation to iNKT cells has remained elusive despite intense study. X-ray crystallographic studies showed that the phytosphingosine backbone and fatty amide chain of α-GalCer reside in the two CD1 d clefts, with hydrogen bonding between residues in CD1 d and the 2’-, 3’-, and 4’-hydroxy groups of galactose and the 3-hydroxy group of phytosphingosine (182). The 4'- and 6’-positions of the sugar head group are exposed for recognition of the αGalCer/Cd1 d complex by the TCR. CD1 d is recycled through the endosomal and/or lysosomal pathway, and this trafficking is essential for the loading of CD1 d with glycolipid antigens (183).
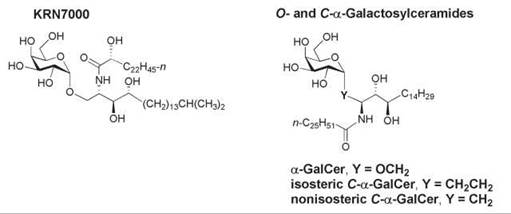
Figure 16. Structures of KRN7000 and the O- and C-glycosides of α-galactosylceramide.
Manipulation of the Th1 versus Th2 polarization with α-GalCer analogs
Although the therapeutic potential of α-GalCer as an immunomodulator for treating autoimmune and infectious diseases, cancer, hepatitis B, and malaria is well established, several limitations to its therapeutic use have been recognized. These include the production of both T-helper 1 (Th1) and T-helper 2 (Th2) cytokines, long-term iNKT cell unresponsiveness in mice on repeated administration, and a very low solubility of the glycolipid in aqueous media. Therefore, many α-GalCer analogs with modifications in the two lipid chains or in the galactose head group have been synthesized and studied.
The balance between the levels of Th1 and Th2 cytokine levels secreted may be crucial to attaining therapeutic efficacy of the immunostimulant because Th1 and Th2 have opposing actions (184). For example, a glycolipid agonist that stimulates NKT cells to produce Th1-type cytokines (such as interferon-γ and the interleukins IL-2 and IL-12) would be beneficial for the treatment of intracellular infections caused by viruses and some bacteria and for the stimulation of antitumor immunity. Several α-GalCer analogs have been synthesized that modulate the NKT cell responses by polarizing them to produce a desired cytokine profile. An N-acyl analog of α-GalCer that has a C20:2∆11∆14 chain with two cis-double bonds in place of the C26:0 amide chain of the natural agelasphins induced a Th1-type response (185) as did analogs that have an aromatic group at the ω-terminus of the N-acyl chain (186). However, an analog of α-GalCer with a truncated phytosphingosine chain stimulated NKT cells to selectively produce Th2-type cytokines, such as IL-4, and suppressed autoimmunity in animal models of multiple sclerosis (187), collagen-induced arthritis (188), and autoimmune diabetes (189). An α-GalCer analog with an elongated phytosphingosine chain induced NKT cells to produce a weaker cytokine response, which resulted in the suppression of an arthritis disease model in mice (190).
An increased production of Th1 cytokines has also been achieved in mice with a C-glycosidic analog of α-GalCer, in which the linker region between the sugar and the backbone was altered by replacing the α-anomeric oxygen atom of galactose with a methylene group (isosteric C-α-GalCer, Fig. 16) (191). A nonisosteric C-glycoside of α-GalCer also was prepared in which the anomeric carbon is bonded directly to C-1 of the phytosphingosine backbone (nonisosteric C-α-GalCer, Fig. 16) (192). This analog elicited a higher ratio of Th1-type cytokine/Th-2-type cytokine response in human NKT cells in culture than α-GalCer and the isosteric C-α-GalCer. The mechanism of the Th1 bias remains to be elucidated, but factors such as aqueous solubility, access to lipid transfer proteins, the stability of the CD1 d-glycosphingolipid-TCR ternary complex, and receptor-mediated uptake may be involved. Thus, new α-GalCer analogs may have therapeutic relevance.
Conclusion
Many natural compounds, dietary constituents, and chemotherapeutic agents alter the activities of enzymes in the de novo biosynthetic pathway of sphingolipids, which thereby modifies the relative concentrations of endogenous sphingolipid species present in cells. During the last two decades the importance of sphingolipids in cell signaling pathways has been recognized, and many bioactivities of sphingolipid metabolites have been studied. The mechanisms by which sphingolipids exert their biological activities on intra- and extracellular targets have been elucidated, but the roles of sphingolipid-metabolizing enzymes and their isozymes in disease processes are not yet understood. The attention that has been devoted to the enzymes that metabolize sphingolipids has resulted in an improved understanding of the pathways for interconversions among sphingolipid messengers with opposing bioactivities.
Manipulation of the levels of sphingolipid metabolites seems to be a means of determining cell fate. In vitro and in vivo studies have shown that several enzymes are potential targets for therapeutic applications, including sphingomyelinases, ceramidases, GlcCer synthase, sphingosine kinases, and S1P lyase. Other enzyme targets probably will be identified in future research, such as ceramide synthase, ceramide kinase, autotaxin, and dihydroceramide desaturase. In addition to enzymes involved in sphingolipid metabolism, lysophospholipid GPCRs hold promise as attractive pharmacologic targets. Agents that disrupt lipid microdomains may also have therapeutical applications.
Several human clinical trials have been undertaken to date with synthetic sphingolipids in the hope of assessing their efficacy and safety as chemotherapeutic agents for various diseases: with α-GalCer (193, 194), safingol (195), NB-DNJ (miglustat) (196-198), and FTY720 (175, 199, 200). It remains to be shown whether synthetic sphingolipids will be successful agents for the treatment of infectious and autoimmune diseases and hyperproliferative diseases such as cancer.
References
1. Carter HE, Hirschberg CB. Phytosphingosines and branched sphingosines in kidney. Biochemistry 1968; 7:2296-2300.
2. Karlsson KA, Martensson E. Studies on sphingosines. XIV. On the phytosphingosine content of the major human kidney glycolipids. Biochim. Biophys. Acta 1968; 152:230-233.
3. Sullards MC, Lynch DV, Merrill AH Jr, Adams J. Structure determination of soybean and wheat glucosylceramides by tandem mass spectrometry. J. Mass Spectrom. 2000; 35:347-353.
4. Karlsson KA, Leffler H, Samuelsson BE. Characterization of cerebroside (monoglycosylceramide) from the sea anemone, Metridium senile. Identification of the major long-chain base as an unusual dienic base with a methyl branch at a double bond. Biochim. Biophys. Acta 1979; 574:79-93.
5. Chitwood DJ, Lusby WR, Thompson MJ, Kochansky JP; Howarth OW. The glycosylceramides of the nematode Caenorhabditis elegans contain an unusual, branched-chain sphingoid base. Lipids 1995; 30:567-573.
6. Abeytunga DT, Glick JJ, Gibson NJ, Oland LA, Somogyi A, Wysocki VH, Polt R. Presence of unsaturated sphingomyelins and changes in their composition during the life cycle of the moth Manduca sexta. J. Lipid Res. 2004; 45:1221-1231.
7. Noda N, Tanaka R, Miyahara K, Kawasaki T. Isolation and characterization of a novel type of glycosphingolipid from Neanthes diversicolor. Biochim. Biophys. Acta 1993; 1169:30-38.
8. Ishii T, Okino T, Mino Y. A ceramide and cerebroside from the starfish asterias amurensis Lutken and their plant-growth promotion activities. J. Nat. Prod. 2006; 69:1080-1082.
9. Diaz de Vivar ME, Seldes AM, Maier MS. Two novel glucosylceramides from gonads and body walls of the Patagonian starfish Allostichaster inaequalis. Lipids 2002; 37:597-603.
10. Ohashi Y, Tanaka T, Akashi S, Morimoto S, Kishimoto Y, Nagai Y. Squid nerve sphingomyelin containing an unusual sphingoid base. J. Lipid Res. 2000; 41:1118-1124.
11. Nudelman ED, Levery SB, Igarashi Y, Hakomori S. Plasmalopsychosine, a novel plasmal (fatty aldehyde) conjugate of psychosine with cyclic acetal link. Isolation and characterization from human brain white matter. J. Biol. Chem. 1992; 267:11007-11016.
12. Jin ZQ, Karliner JS. Low dose N,N-dimethylsphingosine is cardioprotective and activates cytosolic sphingosine kinase by a PKCe-dependent mechanism. Cardiovasc. Res. 2006; 71:725-734.
13. Salcedo M. Cuevas C, Alonso JL, Otero G, Faircloth G, Fernandez-Sousa JM, Avila J, Wandosell F. The marine sphingolipid-derived compound ES 285 triggers an atypical cell death pathway. Apoptosis 2007; 12:395-409.
14. Den Brok MWJ, Nuijen B, Beijnen JH, Manada del Campo C. Antitumor pharmaceutical compositions comprising a spisulosine and a cyclodextrin. PCT Int. Appl. 2006, WO 2006034849; Chem. Abstr. 2006; 144:376489.
15. Nakao Y, Takada K, Matsunaga S, Fusetani N. Calyceramides A-C: neuraminidase inhibitory sulfated ceramides from the marine sponge Discodermia calyx. Tetrahedron 2001; 57:3013-3017.
16. Bittman R. The 2003 ASBMB-Avanti award in lipids address: applications of novel synthetic lipids to biological problems. Chem. Phys. Lipids 2004; 129:111-131.
17. Wertz PW, Hoogstraate AJ, Squier CA. Biochemical basis of the permeability barrier in skin and oral mucosa. Drugs Pharm. Sci. 1996; 74:27-49.
18. Norlen L, Gil IP, Simonsen A, Descouts P. Human stratum corneum lipid organization as observed by atomic force microscopy on Langmuir-Blodgett films. J. Struct. Biol. 2007; 158:386-400.
19. Stewart ME, Downing DT. Free sphingosines of human skin include 6-hydroxysphingosine and unusually long-chain dihydrosphingosines. J. Invest. Dermatol. 1995; 105:613-618.
20. Wertz PW, Madison KC, Downing DT. Covalently bound lipids of human stratum corneum. J. Invest. Dermatol. 1989; 92:109-111.
21. Farwanah H, Pierstorff B, Schmelzer CEH, Raith K, Neubert RHH, Kolter T, Sandhoff K. Separation and mass spectrometric characterization of covalently bound skin ceramides using LC/APCI-MS and nano-ESI-MS/MS. J. Chromatogr. B. 2007; 852:562-570.
22. van Meeteren LA, Ruurs P, Christodoulou E, Goding JW, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem. 2005; 280:21155-21161.
23. Karlsson AA, Michelsen P, Odham G. Molecular species of sphingomyelin: determination by high-performance liquid chromatography/mass spectrometry with electrospray and high- performance liquid chromatography/tandem mass spectrometry with atmospheric Pressure chemical ionization. J. Mass Spectrom. 1998; 33:1192-1198.
24. Yappert MC, Borchman D. Sphingolipids in human lens membranes: an update on their composition and possible biological implications. Chem. Phys. Lipids 2004; 129:1-20.
25. Malewicz B, Valiyaveettil JT, Jacob K, Byun HS, Mattjus P, Baumann WJ, Bittman R, Brown RE. The 3-hydroxy group and 4,5-trans double bond of sphingomyelin are essential for modulation of galactosylceramide transmembrane asymmetry. Biophys. J. 2005; 88:2670-2680.
26. Nylund M, Kjellberg MA, Molotkovsky JG, Byun HS, Bittman R, Mattjus P. Molecular features of phospholipids that affect glycolipid transfer protein-mediated galactosylceramide transfer between vesicles. Biochim. Biophys. Acta 2006; 1758:807-812.
27. Talbott CM, Vorobyov I, Borchman D, Taylor KG, DuPre DB, Yappert MC. Conformational studies of sphingolipids by NMR spectroscopy. II. Sphingomyelin. Biochim. Biophys. Acta 2000; 1467:326-337.
28. Epand RM. Cholesterol in bilayers of sphingomyelin or dihydrosphingomyelin at concentrations found in ocular lens membranes. Biophys J. 2003; 84:3102-3110.
29. Subbaiah PV, Sargis RM. Sphingomyelin: a natural modulator of membrane homeostasis and inflammation. Med. Hypotheses 2001; 572:135-138.
30. Stallberg-Stenhagen S, Svennerholm L. Fatty acid composition of human brain sphingomyelins. Normal variation with age and changes during myelin disorders. J. Lipid Res. 1965; 6:146-155.
31. Waarts BL, Bittman R, Wilschut J. Sphingolipid and cholesterol dependence of alphavirus membrane fusion. Lack of correlation with lipid raft formation in target liposomes. J. Biol. Chem. 2002; 277:38141-38147.
32. Epand RM, Epand RF. Non-raft forming sphingomyelin-cholesterol mixtures. Chem. Phys. Lipids 2004; 132:37-46.
33. Rao RP, Yuan C, Allegood JC, Rawat SS, Edwards MB, Wang X, Merrill AH, Acharya U, Acharya JK. Ceramide transfer protein function is essential for normal oxidative stress response and lifespan. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:11364-11369.
34. Karlander SG, Karlsson KA, Leffler H, Lilja A, Samuelsson BE, Steen GO. The structure of sphingomyelin of the honey bee (Apis mellifera). Biochim. Biophys. Acta 1972; 270:117-131.
35. Rietveld A, Neutz S, Simons K, Eaton S. Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J. Biol. Chem. 1999; 274:12049-12054.
36. Subbaiah PV, Subramanian VS, Wang K. Novel physiological function of sphingomyelin in plasma. Inhibition of lipid peroxidation in low density lipoproteins. J. Biol. Chem. 1999; 274:36409-36414.
37. Sargis RM, Subbaiah PV. Protection of membrane cholesterol by sphingomyelin against free radical-mediated oxidation. Free Radic. Biol. Med. 2006; 40:2091-2102.
38. Bittman R, Kasireddy CR, Mattjus P, Slotte JP. Interaction of cholesterol with sphingomyelin in monolayers and vesicles. Biochemistry 1994; 33:11776-11781.
39. Terova B, Heczko R, Slotte JP. On the importance of the phosphocholine methyl groups for sphingomyelin/ cholesterol interactions in membranes: a study with ceramide phospho- ethanolamine. Biophys. J. 2005; 88:2661-2669.
40. Watanabe Y, Nakajima M, Hoshino T, Jayasimhulu K, Brooks EE, Kaneshiro ES. A novel sphingophosphonolipid head group 1-hydroxy-2-aminoethylphosphonate in Bdellovibrio stolpii. Lipids 2001; 36:513-519.
41. Jayasimhulu K, Hunt SM, Kaneshiro ES, Watanabe Y, Giner JL. Detection and identification of Bacteriovorax stolpii UKi2 sphingophosphonolipid molecular species. J. Am. Soc. Mass Spectrom. 2007; 18:394-403.
42. Figueiredo JM, Dias WB, Mendonca-Previato L, Previato JO, Heise N. Characterization of the inositol phosphorylceramide synthase activity of Trypanosma cruzi. Biochem. J. 2005; 387: 519-529.
43. de Lederkremer RM, Lima CE, Ramirez MI, Goncalvez MF, Colli W. Hexadecylpalmitoylglycerol or ceramide is linked to similar glycophosphoinositol anchor-like structures in Trypanosma cruzi. Eur. J. Biochem. 1993; 218:929-936.
44. Fankhauser C, Homans SW, Thomas-Oates JE, McConville MJ, Desponds C, Conzelmann A, Ferguson MA. Structures of glycosylphosphatidylinositol membrane anchors from Saccharomyces cerevisiae. J. Biol. Chem. 1993; 268:26365-26374.
45. Nagiec MM, Nagiec EE, Baltisberger JA, Wells GB, Lester RL, Dickson RC. Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the Aur1 gene. J. Biol. Chem. 1997; 272:9809-9817.
46. Sugimoto Y, Sakoh H, Yamada K. IPC synthase as a useful target for antifungal drugs. Curr. Drug Targets Infect. Disord. 2004; 4:311-322.
47. Nakamura M, Mori Y, Okuyama K, Tanikawa K, Yasuda S, Hanada K, Kobayashi S. Chemistry and biology of khafrefungin. Large-scale synthesis, design, and structure-activity relationship of khafrefungin, an antifungal agent. Org. Biomol. Chem. 2003; 1:3362-3376.
48. Liliom K, Sun G, Bunemann M, Virag T, Nusser N, Baker DL, Wang DA, Fabian MJ, Brandts B, Bender K, Eickel A, Malik KU, Miller DD, Desiderio DM, Tigyi G, Pott L. Sphingosylphospho- choline is a naturally occurring lipid mediator in blood plasma: a possible role in regulating cardiac function via sphingolipid receptors. Biochem. J. 2001; 355:189-197.
49. Jeon E, Lee MJ, Sung SM, Kim JH. Sphingosylphosphorylcholine induces apoptosis of endothelial cells through reactive oxygen species-mediated activation of ERK. J. Cell. Biochem. 2007; 100:1536-1547.
50. Kleger A, Busch T, Liebau S, Prelle K, Paschke S, Beil M, Rolletschek A, Wobus A, Wolf E, Adler G, Seufferlein T. The bioactive lipid sphingosylphosphorylcholine induces differentiation of mouse embryonic stem cells and human promyelocytic leukaemia cells. Cell Signal. 2007; 19:367-377.
51. Peters SLM, Alewijnse AE. Sphingosine-1-phosphate signaling in the cardiovascular system. Curr. Opin. Pharmacol. 2007; 7:186-192.
52. Kihara A, Mitsutake S, Mizutani Y, Igarashi Y. Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2007; 46:126-144.
53. Marcu AC, Chalfant CE. Ceramide 1-phosphate, a new bioactive sphingolipid in regulating cell signaling. Future Lipidol. 2007; 2:75-84.
54. Harouse JM, Collman RG, Gonzalez-Scarano F. Human immunodeficiency virus type 1 infection of SK-N-MC cells: domains of gp120 involved in entry into a CD4-negative, galactosyl ceramide/3’-sulfogalactosyl ceramide-positive cell line. J. Virol. 1995; 69:7383-7390.
55. Sonnino S, Mauri L, Chigorno V, Prinetti A. Gangliosides as components of lipid membrane domains. Glycobiology 2007; 17:1-13.
56. Karlsson KA. Animal glycosphingolipids as membrane attachment sites for bacteria. Annu. Rev. Biochem. 1989; 58:309-350.
57. Merritt EA, Sarfaty S, van den Akker F, L’Hoir C, Martial JA, Hol WG. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci; 1994; 3:166-175.
58. Velter IA, Politi M, Podlipnik C, Nicotra F. Natural and synthetic cholera toxin antagonists. Mini-Rev. Med. Chem. 2007; 7:159-170.
59. Martinez Z, Zhu M, Han S, Fink AL. GM1 specifically interacts with α-synuclein and inhibits fibrillation. Biochemistry 2007; 46:1868-1877.
60. Kabayama K, Sato T, Saito K, Loberto N, Prinetti A, Sonnino S, Kinjo M, Igarashi Y, Inokuchi J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:13678-13683.
61. El Bawab S, Usta J, Roddy P, Szulc ZM, Bielawska A, Hannun YA. Substrate specificity of rat brain ceramidase. J. Lipid Res. 2002; 43:141-148.
62. Okino N, He X, Gatt S, Sandhoff K, Ito M, Schuchman EH. The reverse activity of human acid ceramidase. J. Biol. Chem. 2003; 278:29948-29953.
63. Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res. 2004; 45:496-506.
64. Moesby L, Corver J, Erukulla RK, Bittman R, Wilschut J. Sphingolipids activate membrane fusion of Semliki Forest virus in a stereospecific manner. Biochemistry 1995; 34:10319-10324.
65. Laulederkind SJF, Bielawska A, Raghow R, Hannun YA, Ballou LR. Ceramide induces interleukin 6 gene expression in human fibroblasts. J. Exp. Med. 1995; 182:599-604.
66. Karasavvas N, Erukulla RK, Bittman R, Lockshin R, Zakeri Z. Stereospecific induction of apoptosis in U937 cells by N-octanoyl-sphingosine stereoisomers and N-octyl-sphingosine. The ceramide amide group is not required for apoptosis. Eur. J. Biochem. 1996; 236:729-737.
67. Merrill AH Jr, Nimkar S, Menaldino D, Hannun YA, Loomis C, Bell RM, Tyagi SR, Lambeth JD, Stevens VL, et al. Structural requirements for long-chain (sphingoid) base inhibition of protein kinase C in vitro and for the cellular effects of these compounds. Biochemistry 1989; 28:3138-3145.
68. Bielawska A, Crane HM, Liotta D, Obeid LM, Hannun YA. Selectivity of ceramide-mediated biology. Lack of activity of erythro-dihydroceramide. J. Biol. Chem. 1993; 268:26226-26232.
69. Usta J, El Bawab S, Roddy P, Szulc ZM, Hannun YA, Bielawska A. Structural requirements of ceramide and sphingosine based inhibitors of mitochondrial ceramidase. Biochemistry 2001; 40: 9657-9668.
70. Chiantia S Kahya N, Schwille, P. Raft domain formation driven by short- and long-chain ceramide: a combined AFM and FCS study. Langmuir 2007; 23:7659-7665.
71. Megha, Sawatzki P, Kolter T, Bittman R, London E. Effect of ceramide N-acyl chain and polar head group structure on the properties of ordered lipid domains (lipid rafts). Biochim. Biophys. Acta 2007; 1768:2205-2212.
72. Brockman HL, Momsen MM, Brown RE, He L, Chun J, Byun HS, Bittman R. The 4,5-double bond of ceramide regulates its dipole potential, elastic properties, and packing behavior. Biophys. J. 2004; 87:1722-1731.
73. Kishida E, Kasahara M, Takagi Y, Matsumura M, Hayashi T, Kobayashi S, Masuzawa Y. Evaluation of a trans configuration for the apoptosis-inducing activity of ceramide. J. Lipid Mediat. Cell Signal. 1997; 16:127-137.
74. Wijesinghe DS, Massiello A, Subramanian P, Szulc Z, Bielawska A, Chalfant CE. Substrate specificity of human ceramide kinase. J. Lipid Res. 2005; 46:2706-2716.
75. Lu X, Arthur G, Bittman R. Synthesis of a novel ceramide analogue via Tebbe methylenation and evaluation of its antiproliferative activity. Org. Lett. 2005; 7:1645-1648.
76. Struckhoff AP, Bittman R, Burow ME, Clejan S, Eliot S, Hammond T, Scandurro AB, Tang Y, Beckman BS. Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. J. Pharmacol. Exp. Ther. 2004; 309:523-532.
77. Crawford KW, Bittman R, Chun J, Byun HS, Bowen WD. Novel ceramide analogues display selective cytotoxicity in drugresistant breast tumor cell lines compared to normal breast epithelial cells. Cell. Mol. Biol. 2003; 49:1017-1023.
78. Stiban J, Fistere D, Colombini M. Dihydroceramide hinders ceramide channel formation: Implications on apoptosis. Apoptosis 2006; 11:773-780.
79. Kowluru A, Metz SA. Ceramide-activated protein phosphatase-2A activity in insulin-secreting cells. FEBS Lett. 1997; 418:179-182.
80. Brodesser S, Sawatzki P, Kolter T. Bioorganic chemistry of ceramide. Eur. J. Org. Chem. 2003; 11:2021-2034.
81. van Echten-Deckert G, Giannis A, Schwarz A, Futerman AHK., 1-Methylthiodihydroceramide, a novel analog of dihydroceramide, stimulates sphinganine degradation resulting in decreased de novo sphingolipid biosynthesis. J. Biol. Chem. 1998; 273:1184-1191.
82. Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 2006; 29:449-456.
83. Hannun YA, Obeid LM. The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J. Biol. Chem. 2002; 277:25847-25850.
84. Goswami R, Singh D, Phillips G, Kilkus J, Dawson G. Ceramide regulation of the tumor suppressor phosphatase PTEN in rafts isolated from neurotumor cell lines. J. Neurosci. Res. 100. 2005; 1:541-550.
85. Yard BA, Carter LG, Johnson KA, Overton IM, Dorward M, Liu H, McMahon SA, Oke M, Puech D, Barton GJ, Nai- smith JH, Campopiano DJ. The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J. Mol. Biol. 2007; 370:870-886.
86. Hanada K, Nishijima M, Fujita T, Kobayashi S. Specificity of inhibitors of serine palmitoyltransferase (SPT), a key enzyme in sphingolipid biosynthesis, in intact cells. A novel evaluation system using an SPT-defective mammalian cell mutant. Biochem. Pharmacol. 2000; 59:1211-1216.
87. Park TS, Panek RL, Rekhter MD, Mueller SB, Rosebury WS, Robertson A, Hanselman JC, Kindt E, Homan R, Karathanasis SK. Modulation of lipoprotein metabolism by inhibition of sphingomyelin synthesis in ApoE1 knockout mice. Atherosclerosis 2006; 189:264-272.
88. Ikushiro H, Hayashi H, Kagamiyama H. Reactions of serine palmitoyltransferase with serine and molecular mechanisms of the actions of serine derivatives as inhibitors. Biochemistry 2004; 43:1082-1092.
89. Kobayashi S, Furuta T, Hayashi T, Nishijima M, Hanada K. Catalytic asymmetric syntheses of antifungal sphingofungins and their biological activity as potent inhibitors of serine palmitoyltransferase (SPT). J. Am. Chem. Soc. 1998; 120:908-919.
90. Merrill AH Jr, Gilchrist DG, Riley RT. Fumonisins and other inhibitors of de novo sphingolipid biosynthesis. Adv. Lipid Res. 1993; 26:215-234.
91. Desai K, Sullards MC, Allegood J, Wang E, Schmelz EM, Hartl M, Humpf HUl, Liotta DC, Peng Q, Merrill AH. Fumonisins and fumonisin analogs as inhibitors of ceramide synthase and inducers of apoptosis. Biochim. Biophys. Acta 2002; 1585:188-192.
92. Jiang Q, Wong J, Fyrst H, Saba JD, Ames BN. γ-Tocopherol or combinations of vitamin E forms induce cell death in human prostate cancer cells by interrupting sphingolipid synthesis. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:17825-17830.
93. Hanada K, Kumagai K, Tomishige N, Kawano M. CERT and intracellular trafficking of ceramide. Biochim. Biophys. Acta 2007; 1771:644-653.
94. Bibel DJ, Aly R, Shinefield HR. Antimicrobial activity of sph- ingosines. J. Invest. Dermatol. 1992; 98:269-273.
95. Kihara A, Mitsutake S, Mizutani Y, Igarashi Y. Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2007; 46:126-144.
96. van Veldhoven PP, Mannaerts GP. Sphingosine-phosphate lyase. Adv. Lipid Res. 1993; 26:69-98.
97. Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren. Trends Molec. Pharmacol. 2007; 66:1671-1678.
98. Giussani P, Maceyka M, Le Stunff H, Mikami A, Lepine S, Wang E, Kelly S, Merrill AH Jr, Milstien S, Spiegel S. Sphingosine-1-phosphate phosphohydrolase regulates endoplasmic reticulum-to-Golgi trafficking of ceramide. Mol. Cell. Biol. 2006; 26:5055-5069.
99. Mechtcheriakova D, Wlachos A, Sobanov J, Kopp T, Reuschel R, Bornancin F, Cai R, Zemann B, Urtz N, Stingl G, Zlabinger G, Woisetschlaeger M, Baumruker T, Billich A. Sphingosine 1-phosphate phosphatase 2 is induced during inflammatory responses. Cell Signal. 2007; 19:748-760.
100. Rotolo JA, Zhang J, Donepudi M, Lee H, Fuks Z, Kolesnick R. Caspase-dependent and -independent activation of acid sphingomyelinase signaling. J. Biol. Chem. 2005; 280:26425-26434.
101. Schenck M, Carpinteiro A, Grassme H, Lang F, Gulbins E. Ceramide: Physiological and pathophysiological aspects. Arch. Biochem. Biophys. 2007; 462:171-175.
102. Chen JK, Capdevila J, Harris RC. Cytochrome p450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Mol. Cell Biol. 2001; 21:6322-6331.
103. Nara F, Tanaka M, Masuda-Inoue S, Yamasato Y, Doi-Yoshioka H, Suzuki-Konagai K, Kumakura S, Ogita T. Biological activities of scyphostatin, a neutral sphingomyelinase inhibitor from a discomycete, Trichopeziza mollissima. J. Antibiot. (Tokyo) 1999; 52:531-535.
104. Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA. Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-a-induced cell death J. Biol. Chem. 1998; 273:11313-11320.
105. Martin SF, Sawai H, Villalba JM, HannunYA. Redox regulation of neutral sphingomyelinase-1 activity in HEK293 cells through a GSH-dependent mechanism. Arch. Biochem. Biophys. 2007; 459:295-300.
106. Arenz C, Thutewohl M, Block O, Waldmann H, Altenbach HJ, Giannis, A. Manumycin A and its analogues are irreversible inhibitors of neutral sphingomyelinase. ChemBioChem. 2001; 2:141-143.
107. Matera M, Bellinghieri G, Costantino G, Santoro D, Calvani M, Savica V. History of L-carnitine: implications for renal disease. J. Ren. Nutr. 2003; 13:2-14.
108. Andrieu-Abadie N, Jaffrezou JP, Hatem S, Laurent G, Levade T, Mercadier JJ. L-Carnitine prevents doxorubicin-induced apoptosis of cardiac myocytes: role of inhibition of ceramide generation. FASEB J. 1999; 13:1501-1510.
109. Koelzer M, Arenz C, Ferlinz K, Werth N, Schulze H, Klingenstein R, Sandhoff K. Phosphatidylinositol-3,5-bisphosphate is a potent and selective inhibitor of acid sphingomyelinase. Biol. Chem. 2003; 384:1293-1298.
110. Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005; 309:1735-1739.
111. Ogretmen B, Schady D, Usta J, Wood R, Kraveka JM, Luberto C, Birbes H, Hannun YA, Obeid LM. Role of ceramide in mediating the inhibition of telomerase activity in A549 human lung adenocarcinoma cells. J. Biol. Chem. 2001; 276:24901-24910.
112. Ogretmen B, Pettus BJ, Rossi MJ, Wood R, Usta J, Szulc Z, Bielawska A, Obeid LM, Hannun YA. Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. J. Biol. Chem. 2002; 277:12960-12969.
113. Shashidhar MS, Volwerk JJ, Keana JFW, Griffith OH. Inhibition of phosphatidylinositol-specific phospholipase C by phosphonate substrate analogs. Biochim. Biophys. Acta 1990; 1042:410-412.
114. Cui P, Tomsig JL, McCalmont WF, Lee S, Becker CJ, Lynch KR, Macdonald TL. Synthesis and biological evaluation of phosphonate derivatives as autotaxin (ATX) inhibitors. Bioorg. Med. Chem. Lett. 2007; 17:1634-1640.
115. Dinev Z, Gannon CT, Egan C, Watt JA, McConville MJ, Williams SJ. Galactose-derived phosphonate analogues as potential inhibitors of phosphatidylinositol biosynthesis in mycobacteria. Org. Biomol. Chem. 2007; 5:952-959.
116. MacDougall JM, Zhang XD, Polgar WE, Khroyan TV, Toll L, Cashman Jr. Synthesis and in vitro biological evaluation of a carbon glycoside analog of morphine-6-glucuronide. Bioorg. Med. Chem. Lett. 2005; 15:1583-1586.
117. Walker JR, Alshafie G, Nieves N, Ahrens J, Clagett-Dame M, Abou-Issa H, Curley RW Jr. Synthesis and preliminary chemotherapeutic evaluation of the fully C-linked glucuronide of N-(4-hydroxyphenyl)retinamide. Bioorg. Med. Chem. 2006; 14:3038-3048.
118. Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003; 426:803-809.
119. Yasuda S, Kitagawa H, Ueno M, Ishitani H, Fukasawa M, Nishijima M, Kobayashi S, Hanada K. A novel inhibitor of ceramide trafficking from the endoplasmic reticulum to the site of sphingomyelin synthesis. J. Biol. Chem. 2001; 276:43994-44002.
120. Lister MD, Ruan ZS, Bittman R. Interaction of sphingomyelinase with sphingomyelin analogs modified at the C-1 and C-3 positions of the sphingosine backbone. Biochim. Biophys. Acta 1995; 1256:25-30.
121. Taguchi M, Goda K, Sugimoto K, Akama T, Yamamoto K, et al. Biological evaluation of sphingomyelin analogues as inhibitors of sphingomyelinase. Bioorg. Med. Chem. Lett. 2003; 13: 3681-3684.
122. Darroch PI, Dagan A, Granot T, He X, Gatt S, Schuchman EH. A lipid analogue that inhibits sphingomyelin hydrolysis and synthesis, increases ceramide, and leads to cell death. J. Lipid Res. 2005; 46:2315-2324.
123. Soeda S, Tsuji Y, Ochiai T, Mishima K, Iwasaki K, et al. Inhibition of sphingomyelinase activity helps to prevent neuron death caused by ischemic stress. Neurochem. Int. 2004; 45:619-626.
124. Hakogi T, Taichi M, Katsumura S. Synthesis of a nitrogen analogue of sphingomyelin as a sphingomyelinase inhibitor. Org. Lett. 2003; 5:2801-2804.
125. Wascholowski V, Giannis A. Sphingolactones: selective and irreversible inhibitors of neutral sphingomyelinase. Angew. Chem. Int. Ed. 2006; 45:827-830.
126. Claus RA, Wustholz A, Muller S, Bockmeyer CL, Riedel NH, Kinscherf R, Deigner HP. Synthesis and antiapoptotic activity of a novel analogue of the neutral sphingomyelinase inhibitor scyphostatin. ChemBioChem 2005; 6:726-737.
127. Luberto C, Hannun YA. Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. Does sphingomyelin synthase account for the putative phosphatidylcholine-specific phospholipase C? J. Biol. Chem. 1998; 273:14550-14559.
128. Meng A, Luberto O, Meier P, Bai A, Yang X, Hannun YA, Zhou D. Sphingomyelin synthase as a potential target for D609-induced apoptosis in U937 human monocytic leukemia cells. Exp. Cell Res. 2004; 292:385-392.
129. Larsen EC, Hatcher JF, Adibhatla RM. Effect of tricyclodecan-9-yl potassium xanthate (D609) on phospholipid metabolism and cell death during oxygen-glucose deprivation in PC12 cells. Neuroscience 2007; 146:946-961.
130. Boucheron C, Desvergnes V, Compain P, Martin OR, Lavi A, Mackeen M, Wormald MR, Dwek RA, Butters TD. Design and synthesis of imino-sugar-based inhibitors of glucosylceramide synthase: the search for new therapeutic agents against Gaucher disease. Tetrahedron: Asymmetry 2005; 16:1747-1756.
131. Radin NS, Shayman JA, Inokuchi J. Metabolic effects of inhibiting glucosylceramide synthesis with PDMP and other substances. Adv. Lipid Res. 1993; 26:183-213.
132. Bieberich E, Freischutz B, Suzuki M, Yu RK. Differential effects of glycolipid biosynthesis inhibitors on ceramide-induced cell death in neuroblastoma cells. J. Neurochem. 1999; 72:1040-1049.
133. Makino A, Ishii K, Murate M, Hayakawa T, et al. D-threo-1-Phenyl-2-decanoylamino-3-morpholino-1-propanol alters cellular cholesterol homeostasis by modulating the endosome lipid domains. Biochemistry 2006; 45:4530-4541.
134. Schneider JS, Bradbury KA, Anada Y, Inokuchi JI, Anderson DW. The synthetic ceramide analog L-PDMP partially protects striatal dopamine levels but does not promote dopamine neuron survival in murine models of Parkinsonism. Brain Res. 2006; 1099:199-205.
135. Azuma N, O’Brien JS, Moser HW, Kishimoto Y. Stimulation of acid ceramidase activity by saposin D. Arch. Biochem. Biophys. 1994; 311:354-357.
136. Klein A, Henseler M, Klein C, Suzuki K, Harzer K, Sandhoff K. Sphingolipid activator protein D (sap-D) stimulates the lysosomal degradation of ceramide in vivo. Biochem. Biophys. Res. Commun. 1994; 200:1440-1448.
137. Quintans J, Kilkus J, McShan CL, Gottschalk AR, Dawson G. Ceramide mediates the apoptotic response of WEHI 231 cells to anti-immunoglobulin, corticosteroids and irradiation. Biochem. Biophys. Res. Commun. 1994; 202:710-714.
138. Grijalvo S, Bedia C, Triola G, Casas J, Llebaria A, Teixido J, Rabal O, Levade T, Delgado A, Fabrias G. Design, synthesis and activity as acid ceramidase inhibitors of 2-oxooctanoyl and N-oleoylethanolamine analogues. Chem. Phys. Lipids 2006; 144:69-84.
139. Park JH, Schuchman EH. Acid ceramidase and human disease. Biochim. Biophys. Acta 2006; 1758:2133-2138.
140. Kita K, Okino N, Ito M. Reverse hydrolysis reaction of a recombinant alkaline ceramidase of Pseudomonas aeruginosa. Biochim. Biophys. Acta 2000; 1485:111-120.
141. Bielawska A, Greenberg MS, Perry D, Jayadev S, Shayman JA, McKay C, Hannun YA. (1S,2R)-D-erythro-2-(N-Myristoyl amino)-1-phenyl-1-propanol as an inhibitor of ceramidase. J. Biol. Chem. 1996; 271:12646-12654.
142. Samsel L, Zaidel G, Drumgoole HM, Jelovac D, Drachenberg C, Rhee JG, Brodie AM, Bielawska A, Smyth MJ. The ceramide analog, B13, induces apoptosis in prostate cancer cell lines and inhibits tumor growth in prostate cancer xenografts. Prostate 2004; 58:382-393.
143. Selzner M, Bielawska A, Morse MA, Rudiger HA, Sindram D, Hannun YA, Clavier PA. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001; 61:1233-1240.
144. Bielawska A, Bielawski J, Szulc ZM, Mayroo N, Liu X, Bai AP, Elojeimy S, Rembiesa B, Pierce J, Norris JS, Hannun YA. Novel analogs of D-e-MAPP and B13. Part 2: Signature effects on bioactive sphingolipids. Bioorg. Med. Chem. 2007, in Press.
145. Triola G, Fabrias G, Casas J, Llebaria A. Synthesis of cyclo-ropene analogues of ceramide and their effect on dihydroceramide desaturase. J. Org. Chem. 2003; 68:9924-9932.
146. Schulz A, Mousallem T, Venkataramani M, Persaud-Sawin DA, Zucker A, Luberto C, Bielawska A, Bielawski J, Holthuis JCM, Jazwinski SM, Kozhaya L, Dbaibo GS, Boustany RMN. The CLN9 protein, a regulator of dihydroceramide synthase. J. Biol. Chem. 2005; 281:2784-2794.
147. Triola G, Fabrias G, Dragusin M, Niederhausen L, Broere R, Llebaria A, van Echten-Deckert G. Specificity of the dihydroceramide desaturase inhibitor N-((1R,2S)-2-hydroxy-1-hydroxy methyl-2-(2-tridecyl-1-cyclopropenyl)ethyl)octanamide (GT11) in primary cultured cerebellar neurons. Mol. Pharmacol. 2004; 66:1671-1678.
148. Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, Obeid LM, Bielawska A. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J. Biol. Chem. 2007; 282:16718-16728.
149. Morales MC, Perez-Yarza G, Rementeria NN, Boyano MD, Apraiz A, Gomez-Munoz A, Perez-Andres E, Asumendi A. 4-HPR-mediated leukemia cell cytotoxicity is triggered by ceramide-induced mitochondrial oxidative stress and is regulated downstream by Bcl-2. Free Radical Res. 2007; 41:591-601.
150. Wang H, Maurer BJ, Reynolds CP, Cabot MC. N-(4-Hydroxyphenyl)retinamide elevates ceramide in neuroblastoma cell lines by coordinate activation of serine palmitoyltransferase and ceramide synthase. Cancer Res. 2001; 61:5102-5105.
151. Bandhuvula P, Tam YY, Oskouian B, Saba JD. The immune modulator FTY720 inhibits sphingosine-1-phosphate lyase activity. J. Biol. Chem. 2005; 280:33697-33700.
152. Boumendjel A, Miller SPF. Synthesis of an inhibitor of sphingosine-1-phosphate lyase. Tetrahedron Lett. 1994; 35:819-822.
153. Reiss U, Oskouian B, Zhou J, Gupta V, Sooriyakumaran P, Kelly S, Wang E, Merrill AH Jr, Saba JD. Sphingosine-phosphate lyase enhances stress-induced ceramide generation and apoptosis. J. Biol. Chem. 2004; 279:1281-1290.
154. Charles R, Sandirasegarane L, Yun J, Bourbon N, Wilson R, Rothstein RP, Levison SW, Kester M. Ceramide-coated balloon catheters limit neointimal hyperplasia after stretch injury in carotid arteries. Circulation Res. 2000; 87:282-288.
155. Fox TE, Houck KL, O’Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M. Ceramide recruits and activates protein kinase C Z (PKC Z) within structured membrane microdomains. J. Biol. Chem. 2007; 282:12450-12457.
156. Hwang O, Kim G, Jang YJ, Kim SW, Choi G, Choi HJ, et al. Synthetic phytoceramides induce apoptosis with higher potency than ceramides. Mol. Pharmacol. 2001; 59:1249-1255.
157. Macchia M, Barontini S, Bartini S, DiBussolo V, Fogli S, Giovannetti E, et al. Design, synthesis, and characterization of the antitumor activity of novel ceramide analogues. J. Med. Chem. 2001; 44:3994-4000.
158. Dagan A, Wang C, Fibach E, Gatt S. Synthetic, non-natural sphingolipid analogs inhibit the biosynthesis of cellular sphingolipids, elevate ceramide and induce apoptotic cell death. Biochim. Biophys. Acta 2003; 1633:161-169.
159. Kim S, Kang J, Kim S, Choi S, Lim S, Im C, Yim C. Synthesis and cytotoxicity of new aromatic ceramide analogs with alkylsulfonamido chains. Arch. Pharm. Res. 2007; 30:570-580.
160. Bieberich E, Hu B, Silva J, MacKinnon S, Yu RK, Fillmore H, Broaddus WC, Ottenbrite RM. Synthesis and characterization of novel ceramide analogs for induction of apoptosis in human cancer cells. Cancer Lett. 2002; 181:55-64.
161. Bieberich E, Silva J, Wang G, Krishnamurthy K, Condie BG. Selective apoptosis of pluripotent mouse and human stem cells by novel ceramide analogues prevents teratoma formation and enriches for neural precursors in ES cell-derived neural transplants. J. Cell Biol. 2004; 167:723-734.
162. Siskind LJ, Kolesnick RN, Colombini M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion 2006; 6:118-125.
163. Szulc ZM, Bielawski J, Gracz H, Gustilo M, Mayroo N, Hannun YA, Obeid LM, Bielawska A. Tailoring structure-function and targeting properties of ceramides by site-specific cationization. Bioorg. Med. Chem. 2006; 14:7083-7104.
164. Novgorodov SA, Szulc ZM, Luberto C, Jones JA, Bielawski J, Bielawska A, Hannun YA, Obeid LM. Positively charged ceramide is a potent inducer of mitochondrial permeabilization. J. Biol. Chem. 2005; 280:16096-16105.
165. Holman DH, Turner LS, El-Zawahry A, Elojeimy S, Liu X, Bielawski J, Szulc ZM, Norris K, Zeidan YH, Hannun YA, Bielawska A, Norris JS. Lysosomotropic acid ceramidase inhibitor induces apoptosis in prostate cancer cells. Cancer Chemother. Pharmacol. 2008; 61:231-242.
166. Elojeimy S, Liu X, Mckillop JC, El-Zawahry AM, Holman DH, Cheng JY, Meacham WD, Mahdy AEM, Saad AF, Turner LS, Cheng J, Day TA, Dong JY, Bielawska A, Hannun YA, Norris JS. Role of acid ceramidase in resistance to FasL: therapeutic approaches based on acid ceramidase inhibitors and FasL gene therapy. Mol. Ther. 2007; 15:1259-1263.
167. Johnson CR, Chun J, Bittman R, Jarvis WD. Intrinsic cytotoxicity and chemomodulatory actions of novel phenethylisothio- cyanate sphingoid base derivatives in HL-60 human promyelocytic leukemia cells. J. Pharmacol. Exp. Ther. 2004; 309:452-461.
168. Singh RD, Liu Y, Wheatley CL, Holicky EL, Makino A, Marks DL, Kobayashi T, Subramaniam G, Bittman R, Pagano RE. Caveolar endocytosis and microdomain association of a glycosphingolipid analog is dependent on its sphingosine stereochemistry. J. Biol. Chem. 2006; 281:30660-30668.
169. Sharma DK, Brown JC, Cheng Z, Holicky EL, Marks DL, Pagano RE. The glycosphingolipid, lactosylceramide, regulates β1-integrin clustering and endocytosis. Cancer Res. 2005; 65:8233-8241.
170. Singh RD, Holicky EL, Cheng ZJ, Kim SY, Wheatley CL, Marks DL, Bittman R, Pagano RE. Inhibition of caveolar uptake, SV40 infection, and ^1-integrin signaling by a nonnatural glycosphin- golipid stereoisomer. J. Cell Biol. 2007; 176:895-901.
171. Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007; 115:84-105.
172. Chiba K, Matsuyuki H, Maeda Y, Sugahara K. Role of sphingosine 1-phosphate receptor type 1 in lymphocyte egress from secondary lymphoid tissues and thymus. Cell Mol. Immunol. 2006; 3:11-19.
173. Liao JJ, Huang MC, Goetzl EJ. Cutting edge: alternative signaling of Th 17 cell development by sphingosine 1-phosphate. J. Immunol. 2007; 178:5425-5428.
174. Budde K, Schutz M, Glander P, Peters H, Waiser J, Liefeldt L, Neumayer HH, Bohler T. FTY720 (fingolimod) in renal transplantation. Clin. Transplant. 2006; 20 (Suppl. 17): 17-24.
175. Baumruker T, Billich A, Brinkmann V. FTY720, an immunomodulator sphingolipid mimetic: translation of a novel mechanism into clinical benefit in multiple sclerosis. Expert Opin. Invest. Drugs 2007; 16:283-289.
176. Coelho RP, Payne SG, Bittman R, Spiegel S, Sato-Bigbee C. The immunomodulator FTY720 has a direct cytoprotective effect in oligodendrocyte progenitors: implications for the treatment of multiple sclerosis. J. Pharmacol. Exp. Ther. 2007; 323:626-635.
177. LaMontagne K, Littlewood-Evans A, Schnell C, O’Reilly T, Wyder L, Sanchez T, Probst B, Butler J, Wood A, Liau G, Billy E, Theuer A, Hla T, Wood J. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006; 66:221-231.
178. Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, Claffey K, Hla T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J. Biol. Chem. 2003; 278:47281-47290.
179. Watterson KR, Berg KM, Kapitonov D, Payne SG, Miner AS, Bittman R, Milstien S, Ratz PH, Spiegel S. Sphingosine 1-phosphate and the immunosuppresant FTY720-phosphate regulate detrusor muscle tone. FASEB J. 2007; 21:2818-2828.
180. Dudek SM, Camp SM, Chiang ET, Singleton PA, Usatyuk PV, Zhao Y, Natarajan V, Garcia JGN. Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell Signal. 2007; 19:1754-1764.
181. Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu. Rev. Immunol. 2005; 23:877-900.
182. Zajonc DM, Cantu C 3rd, Mattner J, Zhou D, Savage PB, Bendelac A, Wilson IA, Teyton L. Structure and function of a potent agonist for the semi-invariant natural killer T cell receptor. Nature Immunol. 2005; 6:810-818.
183. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu. Rev. Immunol. 2007; 25:297-336.
184. Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J. Clin. Invest. 2004; 114:1379-1388.
185. Chang YJ, Huang JR, Tsai YC, Hung JT, Wu D, Fujio M, Wong CH, Yu AL. Potent immune-modulating and anticancer effects of NKT cell stimulatory glycolipids. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:10299-10304.
186. Yu KOA, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, Fujiwara N, Arias I, Miyake S, Yamamura T, Chang YT, Besra GS, Porcelli SA. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of α-galactosylceramides. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:3383-3388.
187. Miyamoto K, Miyake S, Yamamura T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing Th2 bias of natural killer T cells. Nature 2001; 413:531-534.
188. Chiba A, Oki S, Miyamoto K, Hashimoto H, Yamamura T, Miyake S. Suppression of collagen-induced arthritis by natural killer T cell activation with OCH, a sphingosine-truncated analog of α-galactosylceramide. Arthritis Rheum. 2004; 50:305-313.
189. Miyake S, Yamamura T. Therapeutic potential of glycolipid ligands for natural killer (NK) T cells in the suppression of autoimmune diseases. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005; 5:315-322.
190. Kaieda S, Tomi C, Oki S, Yamamura T, Miyake S. Activation of invariant natural killer T cells by synthetic glycolipid ligands suppresses autoantibody-induced arthritis. Arthritis Rheum. 2007; 56:1836-1845.
191. Yang G, Schmieg J, Tsuji M, Franck RW. The C-glycoside analogue of the immunostimulant α-galactosylceramide (KRN7000): synthesis and striking enhancement of activity. Angew. Chem. Int. Ed. 2004; 43:3818-3822.
192. Lu X, Song L, Metelitsa LS, Bittman R. Synthesis and evaluation of an α-C-galactosylceramide analogue that induces Th1-biased responses in human natural killer T cells. ChemBioChem 2006; 7:1750-1756.
193. Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, Iizasa T, Nakayama T, Taniguchi M, Fujisawa T. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. 2005; 11:1910-1917.
194. Uchida T, Horiguchi S, Tanaka Y, Yamamoto H, Kunii N, Motohashi S, Taniguchi M, Nakayama T, Okamoto Y. Phase I study of alpha-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer. Cancer Immunol. Immunother. 2008; 57:337-345.
195. Morales PR, Dillehay DL, Moody SJ, Pallas DC, Pruett S, Allgood JC, Symolon H, Merrill AH Jr. Safingol toxicology after oral administration to TRAMP mice: demonstration of safingol uptake and metabolism by N-acylation and N-methylation. Drug Chem. Toxicol. 2007; 30:197-216.
196. Lachmann RH. Miglustat: substrate reduction therapy for glycosphingolipid lysosomal storage diseases. Drugs Today (Bare.) 2006; 42:29-38.
197. Elstein D, Dweck A, Attias D, Hadas-Halpern I, Zevin S, Altarescu G, Aerts JF, van Weely S, Zimran A. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood 2007; 110:2296-2301.
198. Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007; 6:765-772.
199. Oppenheimer F, Mulgaonkar S, Ferguson R, Grinyo J, Juarez F, Ostrowski M, Klinger M, Walker R, Torres A, Preiss R, Cremer M, Jardine A. Impact of long-term therapy with FTY720 or mycophenolate mofetil on cardiac conduction and rhythm in stable adult renal transplant patients. Transplantation 2007; 83:645-648.
200. Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphocytic leukemia. J. Clin. Invest. 2007; 117:2408-2421.
Further Reading
Biochim. Biophys. Acta, Special Issue: Sphingolipids, Apoptosis, and Disease 2006; 1758(12).
Brown HA, ed. Methods in Enzymology, Volume 434: Lipidomics and Bioactive Lipids: Lipids and Cell Signaling. 2007. Academic Press, San Diego.
Merrill AH Jr, Hannun YA, eds. Methods in Enzymology, Volumes 311 and 312: Sphingolipid Metabolism and Cell Signaling, Parts A and B. 2000. Academic Press, San Diego.
Merrill AH Jr, Schmelz EM, Dillehay DL, Spiegel S, Shayman JA, Schroeder JJ, Riley RT, Voss KA, Wang E. Sphingolipids - The enigmatic lipid class: biochemistry, physiology, and pathophysiology. Toxicol. Appl. Pharmacol. 1997; 142:208-225.
Ogretman B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004; 4:604-616.
Thevissen K, Francois IE, Winderickx J, Pannecouque C, Cammue BP. Ceramide involvement in apoptosis and apoptotic diseases. Mini-Rev. Med. Chem. 2006; 6:699-709.
Website: sphingolipid classifaction list: http://www.lipidmaps.org/data/classification/sp.html
See Also
Extracellular Lipid Signals
Glycolipids, Synthesis of
Lipidomics