CHEMICAL BIOLOGY
Transcription Factors
KE Hauschild, CD Carlson, LJ Donato, R Moretti and AZ Ansari, Department of Biochemistry and the Genome Center, University of Wisconsin-Madison, Madison, Wisconsin
doi: 10.1002/9780470048672.wecb603
The genome of an organism is deciphered by transcriptional processes to generate RNA and protein molecules that determine cellular fate and perform all cellular functions. The transcriptional machinery itself displays limited specificity and is involved in transcribing all genes in the genome. The exquisite specificity with which gene networks are expressed is mediated by regulatory proteins called transcription factors. Transcription factors bind to gene-specific regulatory sites in the genome and help assemble the complex multi-subunit machinery that transcribes the target gene(s). Spatio-temporal regulation of gene expression permits selective expression of a subset of genes within the genome and thus governs the diversity of cell types and cellular function in response to physiologic signals. The central role of transcription factors in regulation of specific genes and networks is underscored by the fact that their malfunction is linked to the onset of a wide array of diseases, including developmental disorders, cancer, and diabetes. Thus, a major goal at the interface of chemistry, biology, and molecular medicine is the ability to generate synthetic molecules that function as transcription factors. The key requirement in the creation of these artificial transcription factors is the ability to define the properties of natural transcription factors fully. Here we discuss the common properties of transcription factors, summarize the alluring value of targeting gene expression with small molecules, and summarize the current advances toward regulating the expression of desired genes and gene networks with artificial transcription factors.
Introduction
Multicellular organisms consist of a diverse array of cell types, yet this diversity belies the fact that they all contain an identical genomic complement. Specific gene regulatory programs are set into motion during cellular differentiation to confer a unique cellular identity (1, 2). Selective gene expression is directed by gene-specific transcription factors (TFs) that decipher various signals to regulate the expression of relevant target genes and gene regulatory networks. TFs target unique sites in the genome and nucleate the assembly of the transcriptional machinery (Fig. 1) (1). TFs achieve this action by interacting with several protein complexes, including nucleosome-remodeling and -modifying enzymes, the proteasome, coactivators, corepressors, general transcription factors (GTFs), and RNA polymerase II (3-14). Generally, the process of gene transcription by RNA polymerase II can be described by three stages: 1) initiation, which involves the assembly of the transcriptional machinery at the gene promoters, 2) elongation of the nascent transcript, and finally, (3) termination, with release of the full-length transcript that is processed to yield mature mRNA (15, 16). Initially, TFs were thought to act only at the first stage of transcription, but recently they have been implicated in other steps of this intricately choreographed process (1) (Fig. 1a). In addition to TFs that control common cellular functions, different cell types have a unique cell type-specific complement of TFs that regulates the expression of genes and transcriptional networks that are unique to a given cell type.

Figure 1. Function of transcription factors. (a) TF binding. Highly compact chromatin, which correlates with hypoacetylation and reduced transcription, limits TF binding. To overcome this inhibition many TFs recruit chromatin-remodeling and -modifying enzymes to promoters. TFs typically recruit the transcriptional machinery to a promoter through protein-protein interactions with the mediator complex. (b) TF transport. Many nuclear receptors are kept in the cytoplasm until they bind their ligand. Ligand binding induces transport into the nucleus. (c) TF modification. Posttranslational modification of TFs before, during, or after transcription initiation frequently regulates TF activity or even DNA binding.
Most TFs are composed minimally of two key functional protein domains: the regulatory domain (RD), which is used to activate or repress transcription, and the DNA binding domain (DBD), which targets the TFs to specific sites within the genome (1). Different TFs may contain additional domains that confer unique properties. For example, in the case of ligand-sensitive TFs, such as the nuclear receptor family, TFs also contain a ligand binding domain (LBD), which allows the TF to alter its functional state based on the presence or absence of small molecule signals (17-20) (Fig. 1b). The individual protein domains often are functionally self contained, and these functional modules have been shown to be interchangeable between TFs (1). This realization led to the development of the widely used two-hybrid assay where DBD and RD are noncovalently assembled by the dimerization of attached proteins or small molecule ligands (21-25). In an additional application of modular design, ligand binding domains of nuclear receptors have been used to confer small molecule control on other transcription factors (19, 26, 27). More recently, engineered DBDs have been assembled in a modular fashion with RD and LBD to generate a class of artificial transcription factors (ATFs) (28-30). Modular assembly that uses synthetic counterparts of DBDs and RDs have also yielded synthetic ATFs that are not limited to peptidic RD and DBD, which allows the use of a wide variety of synthetic compounds as functional modules (29, 31-34). The assembly of ATFs with synthetic modules that harness the cooperative DNA binding properties of eukaryotic TFs has led to a class of molecules that function as protein-DNA dimerizers (34-36).
A significant amount of research has been dedicated to identifying the numerous factors involved in specific gene transcription. However, a greater focus has been on identifying the gene targets of TFs. Understanding the patterns of gene expression that are produced by TFs, individually and subsequently in complex combinatorial assemblages, will provide an increased understanding of how the genome is deciphered to give rise to a functional organism (37, 38). Nearly 6% of the human genome is thought to encode TFs (39, 40). Only a handful of these ~2000 TFs have been characterized to the point that we can comprehensively define their gene targets (41-48). As careful analysis of TFs from other organisms is performed, it is becoming increasingly clear that complex transcriptional circuits regulate a network of genes, which leads to the final cellular response to a given stimulus (49-51). The gene regulatory networks also have led to the realization that key nodes may regulate a defined cascade of gene transcription (2, 52-55). To aid in understanding this major problem of the postgenomic era, it will be essential to have an arsenal of engineered modulators of transcription, including ATFs, to help elucidate these mechanisms fully (29, 33, 34). These factors also will be useful for targeting transcription to alter the gene expression profiles of individual cells or even entire organisms to suit a required need. A striking example of this use has been seen in the emerging field of synthetic biology that unites several scientific disciplines toward the goal of manipulating genetic networks (56) and generating molecular precursors for industrial use (57). The ability to use ATFs to modulate transcription selectively, without the need for direct manipulation of endogenous factors, holds promise to expand their application into several emerging technologies such as metabolic engineering (58).
These finely tuned transcriptional networks can go awry with devastating consequence (59). Although cells possess active systems to prevent and repair transcriptional errors, the disruption of transcription through mutations in TFs has been implicated in a wide variety of diseases (Table and references therein). As such, this situation makes transcriptional regulators a logical target for drug therapies. The vast majority of TF protein assemblies are driven by cooperative interactions that involve several protein-protein interfaces, yet enzymes involved in transcription are a more common target for drug design. Because of the inherent difficulty in disrupting such interfaces, TF protein-protein interactions often are an underexplored target (1, 60).
Below we discuss examples where chemical approaches have been used to alter transcriptional function by using small molecule inhibitors or activators. We also address strategies to overcome the limitation of targeting protein-protein interfaces. It is clear that the ability to manipulate transcription will be a powerful tool not only to elucidate gene networks or to generate industrial metabolites but also to treat a wide array of diseases. A glimpse into the incredible potential of targeting TFs with small molecules is provided by the success achieved through regulating nuclear receptors and histone deacetylases. Below we touch on the insights gleaned from studying the nuclear receptor class of TFs and on the impact of targeting histone-modifying enzymes. We then discuss exciting new developments in the creation of ATFs and the importance of additionally targeting transcriptional processes for chemical intervention.
Ligand-Dependent Transcription Factors: Nuclear Receptors
Nuclear hormone receptors are ligand-inducible TFs that modulate transcriptional rates in response to their cognate ligands (18). Based on homology, the receptors contain a variable amino terminal region that harbors a ligand-independent activation activity, a highly conserved zinc finger DNA binding domain, and a carboxy-terminal ligand-dependent activation domain (LBD) (Fig. 2a) (61, 62) (19). Ligands for nuclear receptors include a diverse array of lipophilic compounds such as steroid hormones, fatty acids, eicosenoids, bile acids, and oxysterols (18). However, some receptors identified by sequence homology have no known ligands and therefore are referred to as orphan receptors; a subset of these receptors may have no natural ligand. When ligand binding occurs, the LBD undergoes a conformational change in which the carboxy-terminal helix 12 is positioned to form a hydrophobic groove that is necessary for coactivator binding (Fig. 2b) (63). Whereas some receptors are bound to heat shock proteins in the absence of a ligand, the structure of helix 12 in other unliganded and antagonist bound receptors favors the binding of corepressors (proteins that maintain the surrounding chromatin in a transcriptionally inactive state) to a region of the receptor that overlaps the coactivator binding site. The change in helix 12 position and the resulting switch from corepressor to coactivator interaction when agonist binding occurs translates into a transcriptional response (17).

Figure 2. Nuclear hormone receptor structure and ligands. (a) The functional domains of nuclear hormone receptors. They act as either homodimers or heterodimers with a ligand binding domain and a DNA binding domain that are separated by a linker sequence. (b) The conformational change in helix 12 when ligand binding occurs (61, 62). All-trans retinoic acid is shown behind helix 12. (c) Examples of synthetic ligands for estrogen receptor (SERMs) and thyroid hormone receptor (Thyromimetics).
Synthetic ligands to modulate nuclear receptor activity
The biologic systems that are modulated by nuclear receptors include many metabolic responses, which makes them attractive targets for drug discovery. In fact, currently marketed drugs exist for many receptors including the vitamin D receptor (VDR), glucocorticoid receptor (GR), estrogen receptor (ER), and peroxisome proliferator activated receptors (PPARs) (64-67). Transcriptional activation by nuclear receptors is accomplished through ligand binding to the receptor; therefore, the most common mode of repressing the activity of this TF class is through the use of antagonist ligands that function by binding to the ligand pocket of the receptor and retaining the inactive, corepressor-bound state (17). Some of these ligands can act as agonists or antagonists depending on the target tissue (68). One of the most well-studied examples of non-natural ligands used in the clinical setting is the selective estrogen receptor modulators (SERMs) in the treatment of breast cancer (69). Two examples of SERMs, Tamoxifen and Raloxifene, bind to and act as antagonists of the estrogen receptor in breast tissue (Fig. 2c). Both drugs also have been shown to protect against osteoporosis. However, Tamoxifen treatment has shown an increased risk of uterine cancer because it acts as an agonist in the uterus, whereas Raloxifene does not (68, 70). The tissue-selective function of SERMs seems to be determined by the coregulatory proteins that are expressed in the target tissues. SERMs induce a conformational change in the receptor such that it selectively interacts with a subset of coregulators and correspondingly activates or represses the transcription of the target genes (70).
Similarly, synthetic thyromimetics such as CGS 23425 that target the thyroid hormone receptor beta are designed to lower plasma concentrations of cholesterol in patients with hypothyroidism without incurring the negative cardiac side effects that administering the natural hormone would induce (Fig. 2c) (71). Recently, a new thyromimetic compound, CO23, has been designed specifically to target the TRa isoform in vitro and in vivo and could be used to study the effects of the thyroid hormone receptor mediated transcription in the heart (72). SERMs and thyromimetics are only a small selection of the plethora of synthetic agonist and antagonist ligands that exist for nuclear receptors. Many of these synthetic ligands can target specific isotypes/isoforms of their target receptors and target these receptors in a tissue-specific manner. This class of small molecule regulators of transcription contains the largest and most diverse compilation of molecules that regulate transcription and has yielded numerous therapeutic agents.
As an alternative to using synthetic analogs for disease treatment, the natural ligand for the retinoic acid receptor, all-trans-retinoic acid (ATRA), is used effectively in the treatment of acute promyelocytic leukemia (APL) (73). Chromosomal translocations in APL patients are responsible for the cellular transformation in this disease. The transformation results in a fusion of the retinoic acid receptor alpha (RARa) with another protein (73). Treatment of APL patients with pharmacologic doses of ATRA induces complete remission by restoring normal granulocytic differentiation (74, 75).
Table 1. Transcription factors in disease
|
Transcription factor |
Event |
Disease |
Refs |
|
Gata4 |
G296S, E359del |
Congenital Heart Defects |
Nature, 2003. 424(6947): p. 443-7. |
|
MEF2a |
aa 440-446 del |
Coronary Artery disease |
Science, 2003. 302(5650): p. 1578-81. |
|
CRX |
Mutation R90W |
Leber congenital amaurosis |
Hum Mol Genet, 1999. 8(2): p. 299-305. |
|
MAF |
Mutation R288P |
Cataract |
Hum Mol Genet, 2002. 11(1): p. 33-42. |
|
PRAKG2 |
R531G |
Wolff-Parkinson-White |
Circulation, 2001. 104(25): p. 3030-3. |
|
FKHL7 |
11bp deletion from chromosome fusion |
Glaucoma |
Trends Endocrinol Metab, 2005. 16(4): p. 176-82. |
|
EGR2 |
Missense mutation |
Congenital hypomyelinating neuropathy |
Nat Genet, 1998. 18(4): p. 382-4. |
|
NR4A2 |
291Tdel and 245T → G |
Familial Parkinson disease |
Nat Genet, 2003. 33(1): p. 85-9. |
|
TRβ |
Mutations resulting in reduced or no T3-binding affinity and transcriptional capacity |
Thyroid hormone resistance syndrome |
Trends Endocrinol Metab, 2005. 16(4): p. 176-82. |
|
Mineralcorticoid receptor |
inactivating mutations |
PHA1 |
Trends Endocrinol Metab, 2004. 15(6): p. 264-70. |
|
GR |
Mutations causing abnormal interactions with the ligand, target DNA sequences, or aberrant nucleocytoplasmic trafficking |
Glucocorticoid resistance |
Mol Med, 2004. 10(7-12): p. 80-8. |
|
AR |
Mutations |
Prostate cancer |
Endocr Rev, 2004. 25(2): p. 276-308; Cancer Res, 2000. 60(4): p. 944-9; N Engl J Med, 1995. 332(21): p. 1393-8. |
|
E2F |
Loss of function mutation |
Retinoblastoma, osteosarcomas, breast cancer |
Nature, 1986. 323(6089): p. 643-6; Genes Dev, 2000. 14(19): p. 2393-409; Oncogene, 1993. 8(2): p. 279-88; Hum Mol Genet, 2001. 10(7): p. 699-703. |
|
ERα |
Increased expression |
Breast cancer |
Endocrine-Related Cancer, 1998. 5: p. 271-282; Endocr Relat Cancer, 2003. 10(2): p. 193-202. |
|
HTF-1α |
Stabilized in hypoxic conditions of solid tumors |
Breast, cervical, colon, prostate, clear cell renal carcinoma |
MolCancer Res, 2006. 4(9): p. 601-5; Cancer Res, 2000. 60(17): p. 4693-6; J Biol Chem, 2002. 277(33): p. 29936-44;Curr Pharm Des, 2005. 11(22): p. 2873-87; Cancer Res, 2001.61(13): p. 5215-22; Cancer Res, 1999. 59(22): p. 5830-5. |
|
c-Jun |
Increased phophorylation status due to ocogenic Ras signaling |
Chronic myelogenous leukemia, small cell lung cancer |
Mol Cell Biol,1996. 16(8): p. 4504-11; Oncogene, 2001. 20(19): p. 2365-77; Cancer Res, 2000. 60(2): p. 400-8; Proc Natl Acad Sci USA, 1995. 92(25): p. 11746-50. |
|
c-MYC |
Overexpression |
Burkitt’s lymphoma, pediatric acute lymphoblatic leukemia, medulloblastoma, breast cancer |
Endocrine-Related Cancer, 1998. 5: p. 271-282; Proc Natl Acad Sci USA, 1982. 79(24): p. 7824-7; Leukemia, 2006. 20(9): p. 1572-81; Endocr Relat Cancer, 2000. 7(3): p. 143-64; Mol Cell Biol, 2006. 26(5): p. 1666-78. |
|
NFKB |
Constitutively active due to decrease level of IK Ba |
Pediatric acute lymphoblastic leukemia, renal cell carcinoma, retinoblastoma, melanoma |
Cancer Res, 1997. 57(14): p. 3032-9; Am J Pathol, 2002. 161(6): p. 2229-40; Oncogene, 2001. 20(29): p. 3888-96; Leukemia, 2000. 14(3): p. 399-402. |
|
p53 |
Mutationsin p53 or overexpression of MDM2 |
Colon, breast, leukemias, lymphomas, lung, esophageal, sarcomas, Li-Fraumeni syndrome |
Oncogene, 1993. 8(2): p. 279-88; Endocrine-Related Cancer, 1998. 5: p. 271-282; Science, 1991. 253(5015): p. 49-53; Science, 1989. 244(4901): p. 217-21; Science, 1989. 246(4929): p. 491-4; Proc Natl Acad Sci USA, 1990. 87(15): p. 5863-7; Nucleic Acids Res, 1998. 26(15): p. 3453-9; Blood, 1994. 84(9): p. 3158-65. |
|
RARα |
Chromosomal translocation of RARa gene to either PML, PLZF, NPM, NuMA, or STAT5B |
Acute promyelocytic leukemia |
Oncogene, 2001. 20(24): p. 3116-27; Blood, 1999. 93(10): p. 3167-215; Mol Cell, 2000. 6(5): p. 1131-41. |
|
REST |
Overexpression |
Medulloblastoma |
Mol Cancer Ther, 2005. 4(3): p. 343-9. |
|
STAT3 |
Constututively activated |
Head and neck, multiple myeloma, leukemias/lymphomas, breast cancer |
Immunity, 1999. 10(1): p. 105-15; Oncogene, 2000. 19(21): p. 2474-88; Cell Growth Differ, 1997. 8(12): p. 1267-76; Leuk Lymphoma, 1997. 28(1-2): p. 83-8; Oncogene, 2000. 19(21): p. 2489-95. |
|
TWIST |
Haploinsufficiency |
Skull and linb anomolies, acrocephaly |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
MITF |
Haploinsufficiency |
Waardenburg syndrome type II |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
FOXC1 |
Haploinsufficiency |
Axenfeld-Rieger syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
FOXC2 |
Haploinsufficiency |
Lymphedema-distichisis syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
HFN1B |
Haploinsufficiency |
Kidney disease and diabetes |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
HFN1A |
Haploinsufficiency |
Maturity onset diabetes of the young |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
SOX10 |
Haploinsufficiency |
Waardenburg-Shah syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
SOX9 |
Haploinsufficiency |
Campomelic dysplasia |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
MSX2 |
Haploinsufficiency |
Enlarged parietal foramina |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
PITX1 |
Haploinsufficiency |
Rieger syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
PITX3 |
Haploinsufficiency |
Eye defects |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
LIMlxb |
Haploinsufficiency |
Abnormal skeletal patterning and renal dysplasia |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
NKX2.1 |
Haploinsufficiency |
Choreoathetosis and pulmonary dysfunction |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
NKX2.5 |
Haploinsufficiency |
Atrial sepral defect and atrioventricular conduction defects |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
SHOX |
Haploinsufficiency |
Lero-Weill dyschondrosteosis |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
ZFHX1B |
Haploinsufficiency |
Hirschprung disease and microcephaly, Mental retardation, hypertelorism, submucous cleft palate, short stature |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
IPF1 |
Haploinsufficiency |
MODY4 |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
PAX2 |
Haploinsufficiency |
Renal coloboma syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
PAX8 |
Haploinsufficiency |
Thyroid dysgenesis |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
PAX6 |
Haploinsufficiency |
Aniridia type II |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
PAX3 |
Haploinsufficiency |
Waardenburg syndrome type I |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
CREBBP |
Haploinsufficiency |
Rubinstein-Taybi syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
CBAF1 |
Haploinsufficiency |
Cleidocranial dysplasia |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
CBAF2 |
Haploinsufficiency |
Familial platelet disorder with associated myeloid malignancy |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
TBX1 |
Haploinsufficiency |
Velocardial facial syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
TBX5 |
Haploinsufficiency |
Holt-Oram syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
TBX3 |
Haploinsufficiency |
Ulnar-mammary syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
WT1 |
Haploinsufficiency |
WAGR syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
GATA3 |
Haploinsufficiency |
Hypoparathyroidism, sensorineural deafness, and renal dysplasia |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
SALL1 |
Haploinsufficiency |
Townes-Brocks syndrome |
J Clin Invest, 2002. 109(4): p. 451-5. |
|
TRPS1 |
Haploinsufficiency |
Trichorhinophalangeal syndrome types I and III |
J Clin Invest, 2002. 109(4): p. 451-5. |
Progress in chemically targeting the nuclear receptor class of TFs continues unabated. The development of assays that use time-resolved fluorescence energy transfer and fluorescence polarization techniques has enabled high-throughput screening (HTS) to detect equilibrium binding, nucleic acid hybridization, and enzymatic activity with such targets as kinases, phosphatases, proteases, G-protein coupled receptors, and nuclear receptors (70, 76, 77). These HTS assays also are being used to screen large libraries of natural or synthetic ligands in search of both endogenous ligands of orphan receptors and synthetic ligands for receptors of potential therapeutic use. As SERMs show unanticipated side effects, new criteria for small molecule regulators of nuclear receptors are being formulated. An ideal drug lead would be a ligand that retained the beneficial effects of the activated receptor in the target tissues without incurring negative side effects. With information from ERα and TRβ and the power of HTS assays, synthetic NR ligands can be pursued to maximize potential therapeutic efficacy.
Small Molecule Regulation of the TF Environment: HDAC Inhibitors
Another approach to regulating the activity of transcription factors is to modify the chromatin state and therefore the accessibility of the promoter and TF binding sites (78, 79). Chromatin states are modulated partially by posttranslational modifications of the histones in nucleosomes, which have significant effects on transcription levels (14, 80). These modifications include phosphorylation, methylation, acetylation, and ubiquitination (80, 81). An early indication of the importance of these covalent modifications on transcription emerged from the observation that many transcriptional coactivators contain histone acety- lase activity, whereas corepressors contain deacetylase activity (Fig. 1b) (82). Several clinically important transcription factors, such as ERa, recruit chromatin-modifying enzymes that constitute a critical step in the function of the TF (14, 83). In the active or agonist bound state, ERa interacts with histone acetyl transferases (HATs), and in the inactive or antagonist bound state the TF instead recruits histone deacetylases (HDACs) (84).
When a transcriptional repressor recruits a HDAC to a gene promoter, the HDAC de-acetylates proximal histones, a process which is thought to enhance chromosomal condensation and thereby reduce the ability of transcriptional activators to bind the promoter (14). The observation that chromatin condensation correlates with histone deacetylation was possible only by using small molecule HDAC inhibitors (85). The HDAC inhibitor activity of the short chain fatty acid sodium butyrate was identified and led to the suggestion that histone acetylation increases DNA accessibility for TFs and the transcriptional machinery (85). The hydroxamic acid containing natural product Trichostatin A (TSA) was a known antifungal agent for 14 years before it was determined to be a potent inhibitor of HDACs (86). Like many HDAC inhibitors, TSA acts by using a bulky hydrophobic group to block the HDAC pocket and a polar region to interact with the HDAC active site zinc atom (Fig. 3) (87).
HDAC inhibitors that were purified from natural sources and/or synthesized chemically have shown exciting biologic properties such as the induction of cellular differentiation, growth arrest, and apoptosis in several different cancer cell types in culture (88). Additionally, currently three classes of synthetic HDAC inhibitors are undergoing clinical trials (Fig. 3c) (89). These inhibitors include hydroxamic acid containing molecules like suberoylanilide hydroxamic acid (SAHA), PXD-101, and LBH-589 (90-92). The drug MS-275 is from a class of benzamide containing HDAC inhibitors that show efficacy in treating some tumors and lymphoma (93). Both the hydroxamic acid and benzamide-containing inhibitors are general HDAC inhibitors thought to affect most of the more than nine different classes of HDACs. In contrast, a sulfonamide anilide-containing inhibitor currently being tested in clinical trials, MGCD-0103, is thought to be isotype specific, which could reduce negative side effects (94). In contrast to all of these synthetically prepared inhibitors, depsipeptide is a natural product purified from Chromobacterium violaceum that functions as an HDAC inhibitor and is proving to be highly effective in clinical trials (95). The search for highly specific HDAC inhibitors with extremely low toxicity and other negative side effects continues.
ERα serves as an example of a TF that is dependent on an HDAC for its activity (84). The reduction in expression of this nuclear receptor is a key step in the carcinogenesis of breast cancer and correlates with poor prognosis. ERα has been shown to bind HDAC1 directly in vitro and in vivo, and the overexpression of HDAC1 in MCF-7 cells leads to a reduction in both ERα protein levels and ERα transcriptional activity. Overexpression of HDAC1 also causes increased cell proliferation of MCF-7 cells (96). Thus, it has been suggested that HDAC1 plays a critical role in breast cancer progression. Consistent with this hypothesis, HDAC inhibitors including TSA, SAHA, and others have been shown to induce a re-expression of ERα and a corresponding increase in the expression of ER target genes in breast cancer cells that lack ERα activity. Moreover, cells treated with HDAC inhibitors have reduced proliferation and, separately, because of the increase in ERα levels, these cells have an increased sensitivity to anticancer drugs (97). These studies have led to clinical trials that use HDAC inhibitors in the treatment of breast cancer and several other cancers (84).
HDAC inhibitors have revealed the complex relationship between chromatin and transcription. One limitation of using HDAC inhibitors as therapeutics is the effects on the deacetylation of other nonhistone cellular targets. HDAC inhibitors are known to have effects beyond chromatin remodeling, including microtubule and aggresome regulation (98). Despite these limitations, applying a chemical strategy has unveiled the importance of chromatin dynamics in the mechanism of transcriptional regulation. Next generation HDAC inhibitors that specifically inhibit targeted HDACs or HDAC-substrate interactions in desired cell types and at specific promoters would increase the use of HDAC inhibitors (99). However, engineering such specificity is nontrivial, and targeting protein-protein interactions presents a significant challenge for a small molecule. Some successes in addressing this challenge are described below and offer lessons in the design of molecules that use coupled equilibria to target protein interfaces.

Figure 3. Histone deacetylase inhibitors. (a) The mechanism by which many HDAC inhibitors function. A hydrophobic moiety blocks the active site of the HDAC. A flexible linker extends into the cleft, and a polar group interacts with the active zinc atom. (b) Crystal structure of TSA (left) and SAHA (right) in the active site of a HDAC-like protein HDLP (87). (c) Examples from four major classes of HDAC inhibitors.
TF Interfaces: Targeting Molecular Interactions
The rewards of targeting NRs and HDACs strongly imply that transcriptional regulation (TFs in particular) is an exciting yet underexplored target for chemical intervention. Despite their seemingly simple architecture, TFs have been fairly recalcitrant to chemical perturbation—this situation may well be because of the weak molecular interfaces between TFs, their myriad and perhaps redundant interactions with components of the transcriptional machinery, or the lack of our understanding of the functional properties of the targeted TF or its closely related isoforms that also may exist in the same cell (60). Even so, examples exist of TF inhibitors that bind TFs and prevent their association with cellular partners. These examples include small molecules that bind DNA to prevent TF-DNA interaction and include several TF-protein binding inhibitors. Moreover, new approaches have been developed to block (or enhance) competitively the function of TFs.
TF-DNA interactions
Several current cancer therapeutic agents, such as cisplatin, can inhibit cancer by conjugating to DNA and thereby preventing transcription, replication, and repair in actively reproducing cancer cells (100). Other successful therapeutic agents trap topoisomerases in a covalent complex with genomic DNA, and these nucleoprotein complexes act as physical barriers to RNA and DNA polymerases (101). Unfortunately, such approaches can lead to a wide range of side effects in several normal cell types that exhibit rapid growth. To overcome this significant limitation, new strategies are being developed to inhibit sequence-specific interactions between TFs and their binding sites. In a recent study, the Hypoxia-inducible factor-1 a (HIF-1α) was targeted because this TF has been implicated in tumor evolution and metastasis (102). An ELISA-based screen led to small molecules capable of blocking DNA binding by HIF-1a. One screened molecule, echinomycin (a quinoxaline antibiotic, Fig. 4a) (103), could disrupt HIF-1α DNA binding by specifically targeting the hypoxia-responsive element (HRE) in vitro and in U251 cells (102).
Polyamides, a class of sequence-specific, minor-groove DNA binders, also have been used to target HIF-1α and many other TF DNA interactions successfully (104-109). This targeting is achieved by steric occlusion, as when a polyamide occupies the site of a minor-groove binding TF (or a TF that has minor groove contacts), by steric interference of a major-groove binding TF with a tripeptide-conjugated polyamide (104), or by allosterically modifying the DNA groove width that inhibits binding in cases in which a TF alters DNA structure when binding (106). In addition, a polyamide conjugated to acridine, a DNA intercalator, was used to prevent DNA binding by a bZip TF (107). Conjugating the acridine to a sequence-specific polyamide conferred site specificity to the acridine moiety that intercalated between the base steps of the bZip binding site and prevented TF binding to DNA (107). Such strategies offer a rational approach to disrupting TF-DNA interactions at targeted sites.
An interesting approach to disrupting DNA binding by nuclear receptors relies on dislodging the zinc atom from its zinc finger DBD. The cysteine thiolates of zinc fingers are particularly labile, and recently disulfide benzamide (DIBA), an electrophile, was found to block both ligand-dependent and ligand-independent ERa-mediated cell growth. DIBA disrupts the ERα zinc finger, which releases the chelated Zn(II), and thereby inhibits ERα dimerization and DNA binding, which leads to the loss of both ERa-mediated transcription and estrogen-mediated breast cancer growth (Fig. 4a) (110, 111). The attractive feature of this alternative method for modulating nuclear receptor activity lies in the fact that it targets a completely different region of the TF. Breast cancer patients taking anti-estrogens or prostate cancer patients taking androgen receptor antagonists often develop resistance to the treatment after acquiring a mutation in the LBD (112). A small molecule inhibitor of the DNA binding domain would be a useful treatment to overcome such drug resistance.
DNA-binding small molecules have been isolated to enhance the DNA binding of a mutated C2H2 zinc finger protein (30). A structural variant of zif268, C7, was mutated at two critical residues involved in Zn(II) coordination and subsequently used in a screen to identify small molecules that would complement the mutation. A potent compound (2-(4'-quinoline)benzimidazole) was identified that significantly restored the DNA binding and transcriptional capability of the mutated protein (Fig. 4a). This method for developing a small molecule-dependent zinc finger TF may be a valuable tool in the study of this class of TFs.
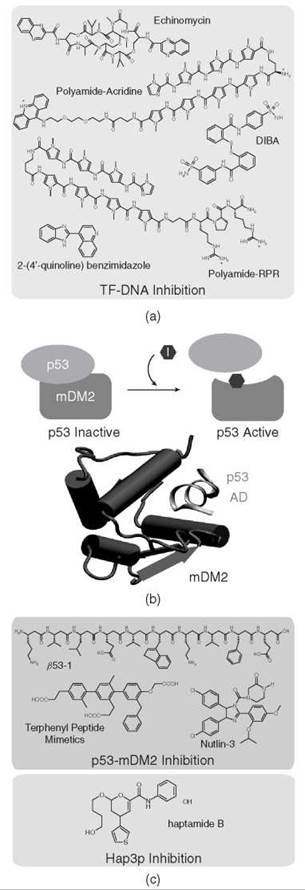
Figure 4. Protein-protein and protein-DNA inhibition. (a) Examples of inhibitors of protein-DNA interactions. (b) p53 is bound by MDM2 and inhibits p53-dependent transcription. In some cancers, MDM2 is overexpressed and p53 is bound continuously by MDM2 despite cellular signals. Inhibitors can be used to disrupt this interface and regain p53 function. The structure of p53 activation domain bound to MDM2 (103). (c) Inhibitors of the p53-MDM2 interaction and of the TF Hap3p.
TF-protein interactions
The transcription factor p53 is a tumor suppressor protein important in regulating genes that control the cell cycle and especially programmed cell death, or apoptosis (Fig. 4b) (113, 114). Therefore, it is not surprising that p53 protein mutations are involved in the genesis of several cancers (115-117). Other than the direct protein mutations in p53, several other routes to p53 inactivation in cancer exist that involve MDM2 (hDM2 in humans). In normal functioning cells, MDM2 binds to p53 and maintains the p53 in an inactive state until cellular stress or DNA damage activate cell signals to induce p53 phosphorylation (118). These signal cascades lead to the phosphorylation of p53 at several key residues important for the p53-MDM2 interaction, which disrupts the interface with MDM2 and releases p53 to perform its regulatory function. In several cancers, the expression of MDM2 is highly upregulated and prevents p53 dissociation despite cellular signals that should trigger the p53 release (115). The p53-MDM2 interface is a well defined deep hydrophobic pocket and has become an important target for small molecule inhibitors (Fig. 4b) (115-117, 119, 120). Several studies have discovered small molecule inhibitors of the p53-MDM2 interface (Fig. 4c) (116, 121-124). These compounds fall into distinct categories. The first category consists of the compounds that mimic the naturally occurring interface by using peptidomimetic drugs. Several examples of this exist including those based on P-peptides, such as β53-1 and β53-3, which were identified as inhibitors of the p53-hDM2 interface (Fig. 4c) (122). β53-1 not only disrupted the p53-hDM2 interaction but also, more importantly, did not disrupt other protein interactions tested, which implies strong specificity for the p53-hDM2 complex. Another class of compounds based on a terphenyl backbone was used to mimic the secondary structure of the p53 N-terminal peptide (121, 124). Here, several terphenyl compounds were identified that disrupted the p53-MDM2 interaction and, importantly, also were shown to be membrane-permeable and able to activate p53 when tested in vivo.
The second category comprises small molecule inhibitors, including chalcones (123), sulfonamides (125), and a promising new class of inhibitors that were identified from a chemical screen, the cis-imidazoline analogs called nutlins (Fig. 4c) (116, 120). The nutlin compounds not only increased p53 activity in vivo but also showed a 90% inhibition of tumor growth when added to a cancer cell line. Other successful examples of using small molecules to disrupt the interactions between TFs and protein targets, such as the CBP/CREB and Myc/Max interfaces, are discussed in recent reviews (126-128).
In many cases, small molecules are found for a TF of interest through assays developed with known protein partners. Structures of the interacting protein domains also have been invaluable for drug discovery. For several important TFs, however, the specific cellular partners with which they interact are unknown. In this case, using small molecule screens on solid supports is an extremely useful tool for drug discovery. In one such screen, diversity-oriented synthesis (DOS (129) was used to identify a small molecule binder of Hap3p, a yeast protein of the Hap2/3/4/5p TF complex that is involved with aerobic respiration (130). For this assay, 12,396 DOS compounds were attached covalently to a solid surface and subsequently probed with Hap3p to yield a specific small molecule (Fig. 4c). HTS approaches have been limited by the fact that chemical libraries often are designed to interact in deep hydrophobic binding pockets. Future studies need to target the relatively shallow protein-protein interactions that are found at TF interfaces.
Transcription Factor Mimics: ATFs
Although in some cases small molecules can be effective at influencing protein-protein or protein-DNA associations, not all interactions can be so targeted, nor do these molecules allow for gain-of-function effects. To surmount these difficulties, researchers are working to create artificial molecules with functional properties akin to those of natural transcription factors.
Modular design
The construction of ATFs has been aided by the modular nature of natural transcription factors (Fig. 5a) (1, 29). TFs tend to have their DNA recognition and regulatory functions located on separable domains, as discussed earlier (19,131). ATFs have been constructed mirroring this modularity. Early work on ATFs focused on combining the DNA binding module from one protein with the regulatory domain of another, which results in a TF with the DNA recognition of the first and the regulatory activity of the second (29). In addition to RD and DBD, the modularity of nuclear receptors also has been exploited in such a fashion for nearly 2 decades with fusion of the ligand binding domains of an NR conferring ligand dependence on the chimeric protein (26, 132-134).
DNA binding domains
Zinc fingers commonly are used as DBDs in the construction of ATFs (Fig. 5a). The number of sequences that can be recognized by zinc finger proteins has been extended through structure-based engineering, phage-display, and other selection techniques (135, 136). Engineered zinc finger proteins have been expressed as fusions with several regulatory domains, which yield engineered regulators with diverse DNA recognition properties (137-139).
Although zinc finger-based ATFs have been invaluable in the creation of the engineered TFs, the use of proteins as exogenously applied ATFs is limited somewhat by the requirement for cell entry, nuclear localization, and potential immune response (140, 141). Although cell uptake peptides and nuclear localization signals can be attached to proteins (142), these proteins usually are introduced into cells with gene therapy techniques. Thus, synthetic “drug-like” molecules may be more useful for therapeutics and in vivo studies.
Most effort thus far on artificial transcription factors has focused on the DNA binding domain (Fig. 5c). Several synthetic DNA binding molecules have been used. One approach is to recognize the double-stranded DNA, either with triplex forming oligonucleotides (143, 144) or with peptide nucleic acids (145). Although triplex-forming polymers can display activation domains and increase expression (142), they are limited to targeting purine-rich DNA strands. A more versatile DNA binding molecule is the pyrrole-imidazole polyamide. Polyamides attached to natural activation peptides can promote transcription over 30-fold (147-152).

Figure 5. Artificial transcription factors. (a) Protein-based ATFs can consist of a DNA binding domain (DBD) that recognizes DNA and an activation domain (AD) that interacts with and recruits the transcriptional machinery. (b) Protein-DNA dimerizers do not interact directly with the transcriptional machinery but can bind and recruit other DNA binding molecules, which in turn interact with the transcriptional machinery. (c) Examples of molecules that act as functional domains used in ATFs. A DNA binding domain is connected, via a linker domain, to either an activation domain or a repression domain.
Regulatory domains
Efforts in ATF design have used naturally occurring regulatory modules, which typically include multikilodalton protein domains. Many regulatory modules can be reduced to short peptides that contain most of the regulatory activity (Fig. 5c) (29, 32). In addition to these naturally occurring peptides, genetic screens have found new activating peptides, many that have similarities with natural activator peptides (33, 153). Peptoids with regulatory properties that can function in intact cells also have been identified (154) (Fig. 5c).
Comparatively few nonpeptidic activation domains exist. Some of the first were found in genetic screens, which identify RNA molecules that can promote transcription in yeast (155-157). This finding provided the first evidence that activation modules did not necessarily have to be peptides. More recently, grafting functional groups (similar to those in natural peptidic activation domains) onto an isoxazolidine skeleton resulted in a class of small molecules that are capable of activating transcription to the same extent as a much larger peptide (158). Library screening and structure-based design studies have identified larger synthetic compounds that can function as activators (32). For example, screening for inhibitors of the endothelial specific transcription factor (ESX)/Sur2 interaction revealed compounds that bound to Sur2, part of the mediator complex in general transcription (159). More structure-based engineering to improve Sur2 binding produced wrenchnolol, which, when conjugated to a polyamide DNA binding molecule, produced a synthetic ATF that activated transcription 3.5-fold in vitro (160, 161). Unfortunately, this synthetic artificial transcription factor is not cell permeable (160).
In comparison with activation, much less has been done with transcriptional repression. The work done thus far has relied exclusively on naturally occurring repression domains and a few synthetic peptides with modest activity (29, 31, 162). The strategy employed to reduce gene expression with small molecules has focused on the inhibition of TF-DNA binding with a competing DNA binding molecule rather than on the active repression of transcription (29). One possible application toward direct repression effects is targeting histone-modifying complexes to gene promoters (29, 163). Active repression has great potential, as demonstrated by recent reports of the reduction of HIV virus production by zinc finger-repression domain fusions (164, 165).
Protein-DNA dimerizers
Although direct interaction with the transcriptional machinery is a common property of TFs, certain TFs do not interact with the machinery directly but instead interact with other TFs, which leads to the assembly of regulatory complexes (34). Synthetic molecules based on this principle were successful in nucleating the cooperative assembly of an ATF-TF complex on a target DNA binding site. The first generation of such “protein-DNA dimerizers” uses polyamides to display dipeptides that can interact with a specific TF. This class of bifunctional molecules is sufficient to change the distribution of a TF on DNA (35, 36, 166). This interaction also has been shown to be modulated by the linker that is used to connect the two domains (36). Protein-DNA dimerizers represent another advance in mimicking the characteristics of natural TFs.
Ligand-responsive artificial transcription factors
Although in the past few years much success with creating artificial molecules with functions similar to TFs has been achieved, these synthetic molecules still do not exhibit the versatility of TFs. Most notably, ATFs lag in their ability to be regulated by external signals. The activity of TFs is controlled finely by signals from the external environment and by internal cellular conditions. Some success has been found in the use of nuclear receptor LBDs in conferring small molecule control of zinc finger-based ATFs (28, 58, 167, 168) and found with small molecule binding RNA aptamers (169). Other techniques have used chemical inducers of dimerization (170) or small molecule-dependent DNA binding elements, such as engineered zinc fingers and the Tet regulatory system (30, 171, 172), for ligand responsive gene control. In the Tet regulatory system, tetracycline is used to regulate transcription through repression (Tet-Off) or activation (Tet-On). In the Tet-Off system, tetracycline transactivator (tTA) binds to the tet promoter (Ptet) and activates a gene of interest. The addition of doxycycline (Dox, a tetracycline derivative) prevents tTA binding to Ptet and effectively inhibits gene expression. For the Tet-On system, a reverse tetracycline transactivator protein (rtTA) requires Dox for binding to Ptet; therefore, dosing with Dox will activate the transcription of a gene of interest 173). A few reports of ligand-responsive small molecule-based ATFs used for gene control have been made.
Targeting TF-Modifying Enzymes: Indirect Regulators
A parallel approach to modulating gene expression is to target enzymes that modify and thereby control the function of TFs (Fig. 1c). Targeting the active sites of enzymes or “druggable” pockets that permit the interaction between enzymes and their substrates has been highly successful in yielding several important chemical tools and even therapeutic agents (59, 96, 174, 175). As more cell signaling pathways are investigated, it is becoming increasingly clear that the signal transduction culminates in the posttranslational modification of a specific set of TFs (176, 177). These modifications define the function of the TF: In some cases, the modification leads to the ability of the TF to translocate to the nucleus and to regulate target genes; in other cases, modifications can lead to proteolysis and degradation of the TF. A wide array of posttranslational modifications of TFs have been reported, including phosphorylation, glycosylation, acetylation, methylation, ubiquitination, sumoylation, and ribosylation (178-184). This wide array provides numerous opportunities to target enzymes that act on TFs and thus indirectly regulate the expression of desired genes and networks. Although promising examples of this approach already exist, the rational targeting of TFs by chemically perturbing the modifying enzyme is not practiced widely yet. An interesting recent example is that of the Myocyte enhancer factor 2 (MEF2) that is specifically deacetylated by HDAC3 (185). Like many other HDACs, HDAC3 has biologic targets beyond histones and MEF2 is one of its known substrates (186). In cells, hyperacetylated MEF2 is a potent activator and deacetylation by HDAC3 greatly attenuates MEF2 activation potency. Chemical inhibition of HDAC3 prolongs the acetylated state of MEF2, which leads to increased expression of its target genes (185). As a key regulator of the gene networks that trigger cells to differentiate into skeletal muscle cells, prolonged MEF2 function because of HDAC3 inhibition increases myogenesis of C3H10T1/2 cells (185). MEF2 activity also is regulated by other modifications, for example a phosphorylation-dependent switch between an acetylation and a sumoylation on lysine 403 that changes MEF2 from a potent transcriptional activator to a transcriptional repressor (187). The complex interplay of posttranslational modifications on the transcription factor MEF2 shows the rich promise of exogenously regulating transcription by targeting indirect regulators.
Several other examples of indirect targeting of TF activity have been reported, and a prominent example is Nuclear Factor kappa B (NF-KB), a TF that plays a central role in rapid cellular response to stress signals (188). NF-KB regulates the immune response to infection and is involved, therefore, in several inflammation pathways that contribute to diverse ailments such as arthritis, asthma, and cancer (189-191). The activity of NF-KB is modulated by Inhibitor of kappaB (IKB), which binds and inhibits the function of NF-KB (192). The inhibitory protein is in turn regulated by the IKB Kinases (IKKs). When cellular signaling occurs, the IKK enzyme is activated and it phosphorylates IKB. This modification marks IKB for degradation and frees NF-KB to translocate to the nucleus and to activate transcription of its target genes (193). The aberrant overactivity of NF-KB in several diseases has prompted a concerted effort to identify antagonists of IKK activity (111). Many IKK inhibitors have been identified and exploited to modulate the activity of NF-KB and ameliorate disease symptoms (111). As we develop a better understanding of the relationship between cellular signals and the enzymes that regulate downstream TF function, we can anticipate the use of indirect targeting approaches to decouple signaling from the undesirable expression of genes and transcriptional circuits.
Future Directions
The genomic revolution is dramatically changing our perspective on gene regulatory networks that govern cell fate and function. The rapid pace of discovery at this scientific frontier will provide unprecedented insight into the networks that define cellular differentiation, the response to various internal and external signals, and a broad array of devastating diseases. In addition, the potential for regulating gene networks in non-natural ways for metabolic engineering offers an unimaginable boon to the biotechnology industry (194). Similarly, the ability to regulate gene networks that determine cell fate is crucial for regenerative medicine. The allure of the rational programming of gene networks increasingly will propel the creation of powerful tools and strategies to achieve this goal. In the coming years we will see the development of sophisticated molecules that mimic the function of natural TFs and molecules that counteract the effect of malfunctioning TFs that are responsible for numerous diseases. A preview to such control of transcriptional networks is the rapid progress in the application of RNAi strategies to downregulate the expression of any desired gene (195). However, it still is not possible to upregulate endogenous genes at will, and complicated methods are employed to express genes and determine their functional role in cellular physiology. ATFs provide the promise of positive regulation of any desired gene or gene regulatory network (33). As such, they serve as a complementary tool to negative regulation by RNAi. A recent series of reports suggests that in addition to ATFs, duplex RNA molecules in some contexts may stimulate the expression of target genes (196).
As Niels Bohr famously noted, “It is very difficult to make an accurate prediction, especially about the future.” Although we cannot anticipate the new and creative approaches that will be developed to regulate desired genes and networks, no doubt exists that transcriptional regulatory methods will be developed to explore how information encoded in genomes is deciphered in the genesis of organisms. Once that understanding is at hand, these same molecules, be they RNAi, ATFs, or tools that remain to be invented, will be used to correct the malfunctioning TFs that participate in the onset of disease or to program the cells to perform new metabolic tasks. This exciting challenge faces the chemical biologists, synthetic biologists, genomicists, and future practitioners of personalized molecular medicine.
References
1. Ptashne M, Gann A. Genes & Signals. 2001. Cold Spring Harbor Laboratory, New York.
2. Levine M, Davidson EH. Gene regulatory networks for development. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:4936-4942.
3. Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes Dev. 1996; 10:2657-2683.
4. Verrijzer CP, Tjian R. TAFs mediate transcriptional activation and promoter selectivity. Trends Biochem. Sci. 1996; 21:338-342.
5. Kadonaga JT. Eukaryotic transcription: an interlaced review network of transcription factors and chromatin-modifying machines. Cell 1998; 92:307-313.
6. Koh SS, et al. An activator target in the RNA polymerase II holoenzyme. Mol. Cell 1998; 1:895-904.
7. Struhl K. Histone acetylation and transcriptional regulatory mechanisms. 1998. Cold Spring Harbor Laboratory, New York.
8. Vignali M, et al. ATP-Dependent Chromatin-Remodeling Complexes. 2000. American Society for Microbiology, Washington, D.C.
9. Krebs JE, et al. Global role for chromatin remodeling enzymes in mitotic gene expression. Cell 2000; 102:587-598.
10. Myers LC, Kornberg RD. Mediator of transcriptional regulation. Ann. Rev. Biochem. 2000; 69:729-749.
11. Tan Q, et al. Activation mutants in yeast RNA polymerase II subunit RPB3 provide evidence for a structurally conserved surface required for activation in eukaryotes and bacteria. Genes Dev. 2000; 14:339-348.
12. Fry CJ, Peterson CL. Chromatin remodeling enzymes: who’s on first? Curr. Biol. 2001; 11:185-197.
13. Naar AM, Lemon BD, Tjian R. Transcriptional coactivator complexes. Annu. Rev. Biochem. 2001; 70:475-501.
14. Berger SL. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002; 12:142-148.
15. Luse DS, Samkurashvili I. The transition from initiation to elongation by RNA polymerase II. Cold Spring Harb Symp Quant Biol, 1998; 63:289-300.
16. Lee TI, Young RA. Transcription of Eukaryotic Protein-Coding Genes. Ann. Rev. Genet. 2000; 34:77-137.
17. Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Devel. 2000; 14:121-141.
18. Mangelsdorf DJ, et al. The nuclear receptor superfamily: the second decade. Cell 1995; 83:835-839.
19. Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996; 10:940-954.
20. McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002; 108: p.465-474.
21. Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature 1989; 340:245-246.
22. Chien C, et al. The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. 1991; 88:9578-958.
23. Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:12817-12821.
24. Huang J, Schreiber SL. A yeast genetic system for selecting small molecule inhibitors of protein-protein interactions in nanodroplets. 1997; 94:13396-13401.
25. Althoff EA, Cornish VW. A bacterial small-molecule three-hybrid system. Angew Chem. Int. Ed. Engl. 2002; 41:2327-2330.
26. Picard D, Salser SJ, Yamamoto KR. A movable and regulable inactivation function within the steroid binding domain of the glucocorticoid receptor. Cell 1988; 54:1073-1080.
27. Eilers M, et al. Chimaeras of Myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature 1989; 340:66-68.
28. Beerli RR, et al. Chemically regulated zinc finger transcription factors. J. Biol. Chem. 2000; 275:32617-32627.
29. Ansari AZ, Mapp AK. Modular design of artificial transcription factors. Curr. Opin. Chem. Biol. 2002; 6:765-772.
30. Lin Q, Barbas CF 3rd, Schultz PG. Small-molecule switches for zinc finger transcription factors. J. Am. Chem. Soc. 2003; 125:612-613.
31. Mapp AK. Regulating transcription: a chemical perspective. Org. Biomol. Chem. 2003; 1:2217-2220.
32. Lum JK, Mapp AK. Artificial transcriptional activation domains. ChemBioChem. 2005; 6:1311-1315.
33. Mapp AK, Ansari AZ. A TAD further: exogenous control of gene activation. ACS Chem. Biol. 2007; 2:62-75.
34. Ansari AZ. Chemical crosshairs on the central dogma. Nat. Chem. Biol. 2007; 3:2-7.
35. Arndt HD, et al. Toward artificial developmental regulators. J. Am. Chem. Soc. 2003; 125:13322-13323.
36. Hauschild KE, et al. Temperature-sensitive protein-DNA dimerizers. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:5008-5013.
37. Levine M, Davidson EH. Gene regulatory networks for development. 2005; 102:4936-4942.
38. Carroll SB, Weatherbee S. From DNA to diversity: molecular genetics and the evolution of animal design. Heredity 2002; 89:411.
39. Venter JC, et al. The Sequence of the Human Genome. 2001.
40. Lander ES, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409:860-921.
41. Wells J, et al. The identification of E2F1-specific target genes. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3890-3895.
42. Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005; 122:947-956.
43. Horak CE, et al. GATA-1 binding sites mapped in the beta-globin locus by using mammalian chip-chip analysis. Proc. Natl. Acad. Sci. 2002; 99:2924.
44. Carroll JS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005; 122:33-43.
45. Wei CL, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006; 124:207-219.
46. Loh YH, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006; 38:431-440.
47. Martone R, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:12247-12252.
48. Odom DT, et al. Control of pancreas and liver gene expression by hnf transcription factors. Science 2004; 303:1378-1381.
49. Ren B, et al. Genome-wide location and function of DNA binding proteins. Science 2000; 290:2306-2309.
50. Giaever G, et al. Functional profiling of the saccharomyces cerevisiae genome. Nature 2002; 418:387-391.
51. Harbison CT, et al. Transcriptional regulatory code of a eukaryotic genome. Nature 2004; 431:99-104.
52. Tavazoie S, et al. Systematic determination of genetic network architecture. Nature Genet. 1999; 22:281-285.
53. Lee TI, et al. Transcriptional regulatory networks in saccharomyces cerevisiae. Science 2002; 298:799-804.
54. Milo R, et al. Network motifs: simple building blocks of complex networks. 2002; 298:824-827.
55. Basso K, et al. Reverse engineering of regulatory networks in human B cells. Nature Genet. 2005; 37:382-390.
56. McDaniel R, Weiss R. Advances in synthetic biology: on the path from prototypes to applications. Curr. Opin. Biotechnol. 2005; 16:476-483.
57. Weikert S, et al. Engineering Chinese hamster ovary cells to maximize sialic acid content of recombinant glycoproteins. Nature Biotechnol. 1999; 17:1116-1121.
58. Fussenegger M. The impact of mammalian gene regulation concepts on functional genomic research, metabolic engineering, and advanced gene therapies. Biotechnol. Prog. 2001; 17:1-51.
59. Darnell JE. Transcription factors as targets for cancer therapy. Nature Rev. Cancer 2002; 2:740-749.
60. Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov, 2004; 3:301-317.
61. Renaud JP, et al. Crystal structure of the RAR-big gamma ligand-binding domain bound to all-trans retinoic acid. Nature 1995; 378:681-689.
62. Bourguet W, et al. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-a. Nature 1995; 375:377-382.
63. Feng W, et al. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science 1998; 280:1747-1749.
64. Howell A, Howell SJ, Evans DG. New approaches to the endocrine prevention and treatment of breast cancer. Cancer Chemother. Pharmacol. 2003; 52(suppl 1):S39-S44.
65. Adcock I. M. Glucocorticoids: new mechanisms and future agents. Curr. Allergy Asthma Rep. 2003; 3:249-257.
66. Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu. Rev. Biochem. 2001; 70:341-367.
67. Takahashi T, Morikawa K. Vitamin D receptor agonists: opportunities and challenges in drug discovery. Curr. Top.{\nobreak} Med. Chem. 2006; 6:1303-1316.
68. Bryant HU. Selective estrogen receptor modulators. Rev. Endocr. Metab. Disord. 2002; 3:231-241.
69. Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science 2002; 295:2465-2468.
70. Zhou G, et al. Nuclear receptors have distinct affinities for coactivators: characterization by fluorescence resonance energy transfer. Mol. Endocrinol. 1998; 12:1594-1604.
71. Taylor AH, et al. Beneficial effects of a novel thyromimetic on lipoprotein metabolism. Mol. Pharmacol. 1997; 52:542-547.
72. Ocasio CA, Scanlan TS. Design and characterization of a thyroid hormone receptor alpha (TRalpha)-specific agonist. ACS Chem. Biol. 2006; 1:585-593.
73. Zelent A, et al. Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 2001; 20:7186-7203.
74. Kalantry S, et al. Gene rearrangements in the molecular pathogenesis of acute promyelocytic leukemia. J. Cell Physiol. 1997; 173:288-296.
75. Licht JD. Reconstructing a disease: What essential features of the retinoic acid receptor fusion oncoproteins generate acute promyelocytic leukemia? Cancer Cell. 2006; 9:73-74.
76. Liu J, et al. A homogeneous in vitro functional assay for estrogen receptors: coactivator recruitment. Mol. Endocrinol. 2003; 17:346-355.
77. Parker GJ, et al. Development of high throughput screening assays using fluorescence polarization: nuclear receptor-ligandbinding and kinase/phosphatase assays. J. Biomol. Screen. 2000; 5:77-88.
78. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997; 389:349-352.
79. Luger K, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997; 389:231-233.
80. Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074-1080.
81. Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41-45.
82. Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002; 108:475-487.
83. Kurtev V, et al. Transcriptional regulation by the repressor of estrogen receptor activity via recruitment of histone deacetylases. J. Biol. Chem. 2004; 279:24834-24843.
84. Margueron R, et al. Histone deacetylase inhibition and estrogen signalling in human breast cancer cells. Biochem. Pharmacol. 2004; 68:1239-1246.
85. Vidali G, et al. Butyrate Suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:2239-2243.
86. Yoshida M. et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990; 265:17174-17179.
87. Finnin MS, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999; 401:188-193.
88. Marks PA, et al. Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol. 2001; 13:477-483.
89. Garcia-Manero G, Issa JP. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer Invest. 2005; 23:635-642.
90. George P, et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005; 105:1768-1776.
91. Kelly WK, et al. Phase I clinical trial of histone deacetylase inhibitorsuberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003; 9:3578-3588.
92. Plumb JA, et al. Pharmacodynamic Response and inhibition of growth of human tumor xenografts by the novel histone deacety- lase inhibitor PXD101. Molec. Cancer Therapeut. 2003; 2:721-728.
93. Ryan QC, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J. Clin. Oncol. 2005; 23: 3912-3922.
94. Gelmon K, et al. Phase I trials of the oral histone deacetylase (HDAC) inhibitor MGCD0103 given either daily or 3x weekly for 14 days every 3 weeks in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2005; 23:3147.
95. Sandor V, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin. Cancer Res. 2002; 8:718-728.
96. Kawai H, et al. Overexpression of histone deacetylase HDAC 1 modulates breast cancer progression by negative regulation of estrogen receptor a. Internat. J. Cancer 2003; 107:353-358.
97. Yang X, et al. Transcriptional activation of estrogen receptor a in human breast cancer cells by histone deacetylase inhibition 1. Biochem. Pharmacol. 2000; 68:1239-1246.
98. Catley L, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006; 108:3441.
99. Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 2002; 9:3-16.
100. Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 1999; 99:2467-2498.
101. Liu LF. DNA Topoisomerase poisons as antitumor drugs. Ann. Rev. Biochem. 1989; 58:351-375.
102. Kong D, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005; 65:9047-9055.
103. Kussie PH, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996; 274:948.
104. Bremer RE, Baird EE, Dervan PB. Inhibition of major-groovebinding proteins by pyrrole-imidazole polyamides with an Arg-Pro-Arg positive patch. Chem. Biol. 1998; 5:119-133.
105. Dickinson LA, et al. Inhibition of RNA polymerase II transcription in human cells by synthetic DNA-binding ligands. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12890-12895.
106. Nguyen-Hackley DH, et al. Allosteric inhibition of zinc-finger binding in the major groove of DNA by minor-groove binding ligands. Biochemistry 2004; 43:3880-3890.
107. Fechter EJ, Dervan PB. Allosteric inhibition of protein-DNA complexes by polyamide-intercalator conjugates. J. Am. Chem. Soc. 2003; 125:8476-8485.
108. Olenyuk BZ, et al. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:16768-16773.
109. Burnett R, et al. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA.TTC repeats in Friedreich’s ataxia. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:11497.
110. Wang LH, et al. Disruption of estrogen receptor DNA-binding domain and related intramolecular communication restores tamoxifen sensitivity in resistant breast cancer. Cancer Cell. 2006; 10:487-499.
111. Karin M, Yamamoto Y, Wang QM. The IKK NF-KB system: a treasure trove for drug development. Nature Rev. Drug Disc. 2004; 3:17-26.
112. Feldman BJ, Feldman D. The development of androgen- independent prostate cancer. Nature Rev. Cancer 2001; 1:34-45.
113. Kastan MB. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991; 51:6304-6311.
114. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88:323-331.
115. Chene P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat. Rev. Cancer 2003; 3:102-109.
116. Klein C, Vassilev LT. Targeting the p53-MDM2 interaction to treat cancer. Br. J. Cancer 2004; 91:1415-1419.
117. Lane DP, Hupp TR. Drug discovery and p53. Drug Discov. Today 2003; 8:347-355.
118. Shieh SY, et al. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. DNA 1997; 91:325-334.
119. Foster BA, et al. Pharmacological rescue of mutant p53 conformation and function. Science 1999; 286:2507-2510.
120. Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle 2004; 3:419- 421.
121. Chen L, et al. p53 alpha-Helix mimetics antagonize p53/MDM2 interaction and activate p53. Mol. Cancer Ther. 2005; 4:1019-1025.
122. Kritzer JA, et al. Helical beta-peptide inhibitors of the p53-hDM2 interaction. J. Am. Chem. Soc. 2004; 126:9468-9469.
123. Stoll R, et al. Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry 2001; 40:336-344.
124. Yin H, et al. Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew Chem. Int. Ed. Engl. 2005; 44:2704-2707.
125. Galatin PS, Abraham DJ. A nonpeptidic sulfonamide inhibits the p53-mdm2 interaction and activates p53-dependent transcription in mdm2-overexpressing cells. J. Med. Chem. 2004; 47:4163-4165.
126. Arndt HD. Small molecule modulators of transcription. Angew. Chem. Int. Ed. 2006; 45:4552-4560.
127. Berg T, et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3830-3835.
128. Majmudar CY, Mapp AK. Chemical approaches to transcriptional regulation. Curr. Opin. Chem. Biol. 2005; 9:467-474.
129. Schreiber SL. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000; 287:1964.
130. Koehler AN, Shamji AF, Schreiber SL. Discovery of an inhibitor of a transcription factor using small molecule microarrays and diversity-oriented synthesis. J. Am. Chem. Soc. 2003; 125:8420-8421.
131. van der Saag PT. Nuclear retinoid receptors: mediators of retinoid effects. Eur. J. Clin. Nutr. 1996; 50(suppl 3):S24-S28.
132. Eilers M, et al. Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature 1989; 340:66-68.
133. Godowski PJ, Picard D, Yamamoto KR. Signal transduction and transcriptional regulation by glucocorticoid receptor-LexA fusion proteins. Science 1988; 241:812-816.
134. Webster NJ, et al. The hormone-binding domains of the estrogen and glucocorticoid receptors contain an inducible transcription activation function. Cell 1988; 54:199-207.
135. Segal DJ, Barbas CF 3rd. Custom DNA-binding proteins come of age: polydactyl zinc-finger proteins. Curr. Opin. Biotechnol. 2001; 12:632-637.
136. Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2 His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 2000; 29:183-212.
137. Blancafort P, Segal DJ, Barbas C.F 3rd. Designing transcription factor architectures for drug discovery. Mol. Pharmacol. 2004; 66:1361-1371.
138. Jamieson AC, Miller JC, Pabo CO. Drug discovery with engineered zinc-finger proteins. Nat Rev. Drug Discov. 2003; 2:361-368.
139. Choo Y, Klug A. Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:11163-11167.
140. Nagahara H, et al. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998; 4:1449-1452.
141. Wender PA, et al. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:13003.
142. Prive GG, Melnick A. Specific peptides for the therapeutic targeting of oncogenes. Curr. Opin. Genet. Dev. 2006; 16:71-77.
143. Moser HE, Dervan PB. Sequence-specific cleavage of double helical DNA by triple helix formation. Science 1987; 238:645.
144. Thuong NT, Helene C. Sequence-specific recognition and modification of double-helical DNA by oligonucleotides. Angew. Chem. Internat. Ed. Eng. 1993; 32:666-690.
145. Egholm M, et al. Peptide nucleic acids (PNA). Oligonucleotide analogues with an achiral peptide backbone. J. Am. Chem. Soc. 1992; 114:1895-1897.
146. Kuznetsova S, et al. Gene activation by triplex-forming oligonucleotide coupled to the activating domain of protein VP16. Nucleic Acids Res. 1999; 27:3995-4000.
147. Schmitz K, Schepers U. Polyamides as artificial transcription factors: novel tools for molecular medicine? Angew. Chem. Int. Ed. Engl. 2004; 43:2472-2475.
148. Dervan PB, et al. Regulation of gene expression by synthetic DNA-binding ligands. Top.{\nobreak} Curr. Chem, 2005; 253:1-31.
149. Ansari AZ, et al. Towards a minimal motif for artificial transcriptional activators. Chem. Biol. 2001; 8:583-592.
150. Arora PS, et al. Design of artificial transcriptional activators with rigid poly-L-proline linkers. J. Am. Chem. Soc. 2002; 124:13067-13071.
151. Mapp AK, et al. Activation of gene expression by small molecule transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:3930-3935.
152. Melander C, Burnett R, Gottesfeld JM. Regulation of gene expression with pyrrole-imidazole polyamides. J. Biotechnol. 2004; 112:195-220.
153. Lu Z, et al. A target essential for the activity of a nonacidic yeast transcriptional activator. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:8591-8596.
154. Liu B, et al. A potent transactivation domain mimic with activity in living cells. J. Am. Chem. Soc. 2005; 127:8254-8255.
155. Sengupta DJ, Wickens M, Fields S. Identification of RNAs that bind to a specific protein using the yeast three-hybrid system. Rna 1999; 5:596-601.
156. Saha S, et al. RNA sequences that work as transcriptional activating regions. Nucleic Acids Res. 2003; 31:1565-1570.
157. Buskirk AR, et al. In vivo evolution of an RNA-based transcriptional activator. Chem. Biol. 2003; 10:533-540.
158. Minter AR, Brennan BB, Mapp AK. A small molecule transcriptional activation domain. J. Am. Chem. Soc. 2004; 126:10504- 10505.
159. Asada S, Choi Y, Uesugi M. A gene-expression inhibitor that targets an alpha-helix-mediated protein interaction. J. Am. Chem. Soc. 2003; 125:4992-4993.
160. Kwon Y, et al. Small molecule transcription factor mimic. J. Am. Chem. Soc. 2004; 126:15940-15941.
161. Shimogawa H, et al. A wrench-shaped synthetic molecule that modulates a transcription factor-coactivator interaction. J. Am. Chem. Soc. 2004; 126:3461-3471.
162. Denison C, Kodadek T. Small-molecule-based strategies for controlling gene expression. Chem. Biol. 1998; 5.
163. Fisher AL, Caudy M. Groucho proteins: transcriptional corepressors for specific subsets of DNA-binding transcription factors in vertebrates and invertebrates. Genes Dev. 1998; 12:1931-1940.
164. Eberhardy SR, et al. Inhibition of human immunodeficiency virus type 1 replication with artificial transcription factors targeting the highly conserved primer-binding site. J. Virol. 2006; 80:2873-2883.
165. Segal DJ, et al. Attenuation of HIV-1 replication in primary human cells with a designed zinc finger transcription factor. J. Biol. Chem. 2004; 279:14509-14519.
166. Warren CL, et al. Defining the sequence-recognition profile of DNA-binding molecules. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:867-872.
167. Buskirk AR, Liu DR. Creating small-molecule-dependent switches to modulate biological functions. Chem. Biol. 2005; 12:151-161.
168. Xu L, et al. A versatile framework for the design of ligand-dependent, transgene-specific transcription factors. Mol. Ther. 2001; 3:262-273.
169. Buskirk AR, Landrigan A, Liu DR. Engineering a ligand-dependent RNA transcriptional activator. Chem. Biol. 2004; 11:1157-1163.
170. Pollock R, Clackson T. Dimerizer-regulated gene expression. Curr. Opin. Biotechnol. 2002; 13:459-467.
171. Cuenoud B, Schepartz A. Altered specificity of DNA-binding proteins with transition metal dimerization domains. Science 1993; 259:510-513.
172. Gossen M, et al. Transcriptional activation by tetracyclines in mammalian cells. Science 1995; 268:1766-1769.
173. Gossen M, Bujard H. Studying gene function in eukaryotes by conditional gene inactivation. Ann. Rev. Genet. 2002; 36:153-173.
174. Chen, H., M. Tini, and R.M. Evans, HATs on and beyond chromatin. Curr. Opin. Cell Biol. 2001; 13:218-224.
175. Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science 2004; 303:1800-1805.
176. Karin M, Hunter T. Transcriptional control by protein phosphorylation: signal transmission from the cell surface to the nucleus. Curr. Biol. 1995; 5:747-757.
177. Verger A, Perdomo J, Crossley M. Modification with SUMO. A role in transcriptional regulation. EMBO Rep. 2003; 4:137-142.
178. Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 1996; 272:1347-1349.
179. Reason AJ, et al. Localization of O-GlcNAc modification on the serum response transcription factor. J. Biol. Chem. 1992; 267:16911-16921.
180. Boyes J, et al. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 1998; 396:594-598.
181. Mowen KA, et al. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell 2001; 104:731-741.
182. Kuras L, et al. Dual regulation of the met4 transcription factor by ubiquitin-dependent degradation and inhibition of promoter recruitment. Mol. Cell 2002; 10:69-80.
183. Eloranta JJ, Hurst HC. Transcription factor AP-2 interacts with the SUMO-conjugating enzyme UBC9 and is sumolated in vivo. J. Biol. Chem. 2002; 277:30798-30804.
184. Mendoza-Alvarez H, Alvarez-Gonzalez R. Regulation of p53 sequence-specific DNA-binding by covalent poly(ADP-ribosyl)ation. J. Biol. Chem. 2001; 276:36425-36430.
185. Gregoire S, et al. Histone deacetylase 3 interacts with and deacetylates MEF2 transcription. Mol. Cell Biol. 2006; 27:1280-1295.
186. Glozak MA, et al. Acetylation and deacetylation of non-histone proteins. Gene 2005; 363:15-23.
187. Shalizi A, et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 2006; 311:1012-1017.
188. Karin M, Lin A. NF-KB at the crossroads of life and death. Nature Immunol. 2002; 3:221-227.
189. Barnes PJ, Karin M. Nuclear factor-KB-A pivotal transcription factor in chronic inflammatory diseases. New Eng. J. Med. 1997; 336:1066-1071.
190. Bharti AC, Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Biochem. Pharmacol. 2002; 64:883-888.
191. Orlowski RZ, Baldwin AS Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002; 8:385-389.
192. Zandi E, et al. The IKB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IKB phosphorylation and NF-KB activation. Cell 1997; 91:243-252.
193. Yaron A, et al. Inhibition of NF-kappa B cellular function via specific targeting of the I kappa B-ubiquitin ligase. EMBO J. 1997; 16:6486-6494.
194. Schubert C. Can biofuels finally take center stage? Nat. Biotechnol. 2006; 24:777-784.
195. Matzke MA, Birchler JA. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 2005; 6:24-35.
196. Janowski BA, et al. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol. 2007; 3:166-173.