CHEMICAL BIOLOGY
Click Peptides, Design and Applications of
Yoshiaki Kiso and Atsuhiko Taniguchi Department of Medicinal Chemistry, Kyoto Pharmaceutical University, Yamashina-ku, Kyoto, Japan
Youhei Sohma Department of Physical Chemistry, Kyoto Pharmaceutical University, Yamashina-ku, Kyoto, Japan
doi: 10.1002/9780470048672.wecb632
A ''click peptide'' is a chemically modified peptide analog used to understand the biologic function of peptides. A click peptide does not exhibit the inherent biologic activity of the original peptide because of a simple chemical modification of the peptide backbone and; by adding an exogenous action (''click'') such as pH-change or photo-irradiation, easily affords the native peptide in situ with a quick and easy one-way conversion via a native amide bond-forming reaction. For example, the designed click peptides of Alzheimer's amyloid fi peptide (Aβ) 1 -42, which did not exhibit a self-assembling nature because of a backbone isomerization from a native Gly25-Ser26 bond to a fi-ester bond, could migrate to the intact Aβ1-42 under physiologic conditions by pH- or photo-triggered ''click'' via an O→N intramolecular acyl migration reaction. This click peptide overcomes the difficulties in handling Aβ1 -42 in syntheses and biologic experiments, which clarifies the currently unexplained processes of Alzheimer's disease.
The use of intact peptides in an in vitro experiment sometimes causes considerable discrepancies in the biologic data because of the difficulties in handling and controlling the properties of peptides such as low water-solubility and highly aggregative nature. A “click peptide” is a chemically modified peptide analog used to understand the biologic function of peptides while overcoming the problems associated with the peptide properties (Fig. 1) (1-3).
A click peptide has the ability to:
• Control the following natures of the original peptide.
• Physicochemical property (e.g., water-solubility, self-assembly, aggregation, or folding)
• Biologic activity (e.g., ligand-receptor binding affinity, or enzyme-substrate binding affinity)
• Convert to the native peptide in situ by an exogenous action (“click”).
• Examples of such an action include pH-change and photo-irradiation.
• Because a rapid amide bond formation is required, an intramolecular reaction is desired.
• An atom-economical reaction should be selected as the conversion reaction to avoid a side effect derived from coreleased products in biologic experimental conditions.
Additionally, in the design of the click peptide, a simple modification of the peptide backbone is desired to minimize difficulties in chemical synthesis of the click peptide and the conversion process to the native peptide.
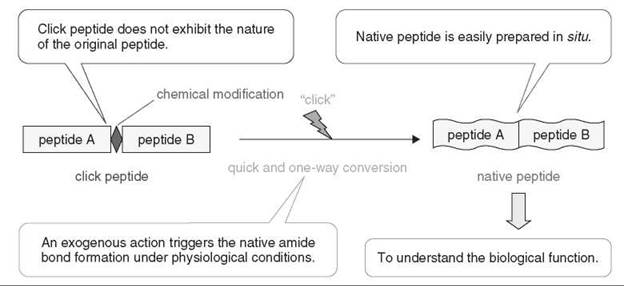
Figure 1. A concept of chemical biology-oriented click peptide.
Biologic Background
The highly aggregative feature of peptides is a significant obstacle against establishing a reliable in vitro biologic experimental system to investigate the major causative agents of diseases.
Recent amyloid fi peptide (Aβ)-related studies have encountered such problems because of their highly aggregative nature. A more clear understanding of the pathologic mechanism of Aβ would be of great value to discover novel drug targets against Alzheimer’s disease (AD). A is the main proteinaceous component of amyloid plaques found in the brain as a pathognomonic feature of AD (4), and it has have been found to be neurotoxic in vitro and in vivo (5). Several studies have supported the hypothesis that neurotoxicity and the kinetics of Aβ1-42 aggregation are related directly to the assembly state in solution. However, the pathologic self-assembly of Aβ1-42 in amyloid plaque formation, a process currently unexplained, is very difficult to demonstrate in vitro because of its uncontrolled self-assembly. The synthesized Aβ1-42 in itself contains various oligomeric forms (6), and Aβ1-42 undergoes time- and concentration-dependent aggregation in an aqueous TFA (trifluoroacetic acid)-acetonitrile solution used in high performance liquid chromatography (HPLC) purification (7). Moreover, the Aβ1-42 monomer forms aggregates easily even in a standard storage solution such as dimethylsulfoxide (8). This uncontrolled self-assembly in an in vitro experiment causes considerable discrepancies in the biologic data. As a result of its highly aggregative nature, difficulties in handling Aβ1-42 have hampered the progress of Aβ1-42-related AD research such that constructs have been used instead of the native peptide.
Recently, it has been ascertained that the pathologic self-assembly nature of inherent peptides or proteins is one major event that leads to the development of many diseases such as prion protein in prion disease, α-synuclein in Parkinson’s disease, and islet amyloid polypeptide in type 2 diabetes, as well as Aβ1-42 in AD (9).
Chemistry
We designed and synthesized the click peptide based on the O-acyl isopeptide (Fig. 2) (1-3). The click peptide has an O-acyl isopeptide structure of the corresponding native peptide. Hydrogen bond interactions between peptide chains often play a crucial role in the onset of physicochemical and biologic actions through their contributions to the conformational stability of the higher order structure. In the click peptide, a newly formed ester bond results in the inhibition of the ordered hydrogen bond interactions, which lead to conformational changes, and mask the activity of the native peptide. Additionally, the target peptide was generated via an O→N intramolecular acyl migration reaction. The O→N intramolecular acyl migration is a very attractive chemical reaction for the click peptide. The O→N intramolecular acyl migration is capable of rapid amide bond formation under physiologic conditions (pH 7.4) because of the energetically favorable intramolecular five-membered ring intermediate. It is an atom-economical reaction; thus, no byproduct is released during conversion to the parent peptide, which is a great advantage in toxicology in biologic experimental systems.

Figure 2. A click peptide based on the O-acyl isopeptide method.
O-Acyl isopeptide method
In 2003, we discovered that the presence of an O-acyl instead of N-acyl residue within the peptide backbone changed the secondary structure of the native peptide significantly (10). The coupling and deprotection efficacies improved during solid-phase peptide synthesis of peptide derivatives that possess “difficult sequences,” most likely because the O-acyl isopeptide prevented the formation of secondary structures that disfavor coupling. Generally, the difficult sequences are hydrophobic and are prone to aggregate in solvent during synthesis and purification. This aggregation is attributed to inter-/intra-molecular hydrophobic interactions and the hydrogen bond network among resin-bound peptide chains, which results in the formation of extended secondary structures such as β-sheets (11). In addition, the target peptide was generated subsequently by an O→N intramolecular acyl migration reaction. This finding marked the beginning of the development of the “O-acyl isopeptide method” for peptide synthesis. The method has begun to be used by several other groups, especially Mimna et al. (12), Coin et al. (13) and Borner et al. (14), which indicates that the O-acyl isopeptide method is widely advantageous for peptide preparation.
Synthesis of O-acyl isopeptide
The synthetic procedure of O-acyl isopeptides has been well established (10). O-Acyl isopeptides can be prepared easily using Fmoc-based solid-phase peptide synthesis. After Boc-Ser/Thr-OH is coupled to a peptide-resin to obtain Boc-Ser/Thr-peptide-resin, Fmoc-Xaa-OH is esterified to the β-hydroxy group of Ser/Thr residue using a DIC (1,3-diisopropylcarbodiimide)-DMAP (4-dimethylaminopyridine) method. Although esterification on the resin might induce epimerization at the esterified amino acid residue, the use of an O-acyl isodipeptide unit, which can be synthesized easily in solution, could avoid this problem (15). The protected O-acyl isopeptide-resin is obtained through coupling of additional amino acid residues using conventional techniques. During Fmoc removal of the second amino acid residue at the N-terminal side next to the ester bond, caution should be used to prevent the formation of diketopiperazines. Finally, the desired O-acyl isopeptide is obtained by TFA treatment followed by HPLC purification. Because O-acyl isopeptides generally exhibit high solubility in various media and are eluted as a sharp single peak in HPLC purification, they are purified easily to give pure isopeptides (1, 3, 10).
pH-Triggered click peptide of Aβ1-42
An application of the method to Alzheimer’s Aβ1-42 revealed that the O-acyl isopeptide of Aβ1-42 (in which the Gly25-Ser26 sequence of Aβ1-42 was isomerized to a β-ester bond) could be synthesized effectively and stored without spontaneous self-assembly (1-3). Intact monomer Aβ1-42 could then be obtained from the isopeptide under physiologic experimental conditions. The water solubility of the TFA salt of the isopeptide was 100-fold higher than that of Aβ1-42 (0.14 mg mL-1). Purified isopeptides could be converted quantitatively to Aβ1-42 via O→N intramolecular acyl migration in phosphate buffered saline (pH 7.4) at 37 °C with a half-life of 1 minute (Fig. 3), whereas the TFA salt of the isopeptide was stable at 4 °C in both solid state and dimethylsulfoxide solution. The Aβ1-42 isopeptide proved to be easier to synthesize and purify than Aβ1-42; moreover, it provided an opportunity to prepare Aβ1-42 in situ rapidly under physiologic conditions. The spontaneous self-assembly of Aβ1-42 could thus be more studied effectively to elucidate the inherent pathologic functions of the aggregative Aβ1-42 in AD. Moreover, slower migration (t1/2 = 3 hours) was observed at pH 4.9, and no migration at pH 3.5 after incubating for 3 hours. These results suggest that this pH-dependent O→N intramolecular acyl migration enables pH-triggered click for controlled in situ production of an intact Aβ1-42 from the click peptide.

Figure 3. The production of Aβ1-42 from the pH-triggered click peptide via a pH-dependent O→N intramolecular acyl migration reaction.
Photo-triggered click peptide of Aβ1-42
Furthermore, we have synthesized a photo-triggered click peptide of Aβ1-42, in which a photocleavable 6-nitroveratryloxy-carbonyl group (16) was introduced at the α-amino group of Ser26 in the Aβ1-42 isopeptide (Fig. 4) (2, 3). The photo-triggered system had been employed to provide well-defined, time-controllable, and position-selective activation by light irradiation when studying biologic systems, because light is the fastest medium and can be focused to a restricted area. In size-exclusion chromatography, oligomers of Aβ1-42 increased in quantity with incubation time (pH 7.4, 37 °C) at the expense of the monomer. On the other hand, the photo-triggered click peptide remained in the monomeric form after 24 hours of incubation. Similarly, thioflavin-T fluorescence intensity (17), which corresponds to the extent of fibril formation, increased with incubation time in the case of Aβ1-42, yet unchanged in the case of the photo-triggered click peptide after 24 hours of incubation. Our results indicated that the click peptide was nonaggregative. The isopeptide structure resulted in complete inhibition of the aggregative nature of Aβ1-42. Photo-irradiation of the click peptide and subsequent O→N intramolecular acyl migration afforded intact Aβ1-42 rapidly in situ. In the absence of light, the click peptide was stable during storage. This method may provide a useful system to investigate the biologic dynamics of Aβ1-42 in AD with high spatial and temporal resolution by photochemically inducible activation of peptide self-assembly.

Figure 4. A photo-triggered click peptide. The production of Aβ1-42 by photo-triggered click followed by O→N intramolecular acyl migration reaction.
Chemical Tools and Techniques
An experimental system that can prepare a bioactive peptide easily in situ is advantageous to investigate biologic functions. The click peptide would be a useful tool to establish such an experimental system. As a tool with a similar concept, a “caged” compound is a synthetic molecule whose biologic activity is masked by a covalently attached photocleavable protecting group, and affords the bioactive molecule by photo-irradiation. Generally, such a photo-triggered system is considered to be advantageous for studying the dynamic processes of peptides and proteins, because upon photoactivation, only a short duration of time is required to control the spatiotemporal dynamics of the native compounds (18, 19). A fundamental drawback of the caged strategy on large peptides and proteins is that a small photocleavable group does not always mask biologic activity, because high potency may be attributed to large sections of the peptide structure (19, 20). This drawback can be overcome by the “click peptide” strategy because the native properties are masked in the isopeptide and released by O-acyl to N-acyl migration. This advantage should open new doors for the development of novel and useful photo-triggered tools to probe systems in chemical biology and medical science.
Because difficulties in handling Aβ1-42 in syntheses and biologic experiments would hamper the progress of Aβ1-42-related AD research, we expect that the “click peptide” method will help to clarify the currently unexplained processes of AD. Moreover, many amyloidogenic diseases, as well as Aβ1-42 in AD, have recently attracted much attention and are being studied to discover novel drug targets (9). Thus, we hope that the “click peptide” strategy would be applied widely to these amyloid-related peptides or proteins. Click peptide can serve as a tool to study peptide folding and aggregation in chemical biology-oriented research, because acyl migration can be induced chemically or photochemically to release the native peptide.
References
1. Sohma Y, Sasaki M, Hayashi Y, Kimura T, Kiso Y. Design and synthesis of a novel water-soluble Aβ1-42 isopeptide: an efficient strategy for the preparation of Alzheimer’s disease-related peptide, Ap1-42, via O-N intramolecular acyl migration reaction. Tetrahedron Lett. 2004; 45:5965-5968.
2. Taniguchi A, Sohma Y, Kimura M, Okada T, Ikeda K, Hayashi Y, Kimura T, Hirota S, Matsuzaki K, Kiso Y. “Click peptide” based on the “O-acyl isopeptide method”: control of Aβ1-42 production from a photo-triggered Aβ1-42 analogue. J. Am. Chem. Soc. 2006; 128:696-697.
3. Sohma Y, Kiso Y. “Click peptides”-chemical biology-oriented synthesis of Alzheimer’s disease-related amyloid β peptide (Aβ) analogues based on the “O-acyl isopeptide method”. ChemBioChem. 2006; 7:1549-1557.
4. Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999; 399:A23-31.
5. Geula C, Wu CK, Saroff D, Lorenzo A, Yuan ML, Yankner BA. Aging renders the brain vulnerable to amyloid p-protein neurotoxicity. Nature Med. 1998; 4:827-831.
6. Soto C, Castano EM, Kumar RA, Beavis RC, Frangione B. Fibrillogenesis of synthetic amyloid-β peptides is dependent on their initial secondary structure. Neurosci. Lett. 1995; 200:105-108.
7. Shen C-L, Murphy RM. Solvent effects on self-assembly of β-amyloid peptide. Biophys. J. 1995; 69:640-651.
8. Stine WB Jr, Dahlgren KN, Krafft GA, Ladu MJ. In vitro characterization of conditions for amyloid-p peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003; 278:11612-11622.
9. Gazit E. The role of prefibrillar assemblies in the pathogenesis of amyloid diseases. Drugs Fut. 2004; 29:613-619.
10. Sohma Y, Yoshiya T, Taniguchi A, Kimura T, Hayashi Y, Kiso Y. Development of “O-acyl isopeptide method”. Biopolymers 2007; 88:253-262.
11. Sheppard R. The fluorenylmethoxycarbonyl group in solid phase synthesis. J. Pept. Sci. 2003; 9:545-552.
12. Mimna R, Camus MS, Schmid A, Tuchscherer G, Lashuel HA, Mutter M. Disruption of amyloid-derived peptide assemblies through the controlled induction of a p-sheet to a-helix transformation: application of the switch concept. Angew. Chem. Int. Ed. Engl. 2007; 46:2681-2684.
13. Coin I, Dolling R, Krause E, Bienert M, Beyermann M, Sferdean CD, Carpino LA. Depsipeptide methodology for solid-phase peptide synthesis: circumventing side reactions and development of an automated technique via depsidipeptide units. J. Org. Chem. 2006; 71:6171-6177.
14. Borner HG. Functional polymer-bioconjugates as molecular LEGO® bricks. Macromol. Chem. Phys. 2007; 208:124-130.
15. Sohma Y, Taniguchi A, Skwarczynski M, Yoshiya T, Fukao F, Kimura T, Hayashi Y, Kiso Y. “O-Acyl isopeptide method” for the efficient synthesis of difficult sequence-containing peptides: use of “O-acyl isodipeptide unit”. Tetrahedron Lett. 2006; 47:3013-3017.
16. Bochet CG. Wavelength-selective cleavage of photolabile protecting groups. Tetrahedron Lett. 2000; 41:6341-6346.
17. Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Interactions of amyloid p-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry. 2002; 41:7385-7390.
18. Adams SR, Tsien RY. Controlling cell chemistry with caged compounds. Annu. Rev. Physiol. 1993; 55:755-784.
19. Shimizu M, Yumoto N, Tatsu Y. Preparation of caged compounds using an antibody against the photocleavable protecting group. Anal. Biochem. 2006; 348:318-320.
20. Bosques CJ, Imperiali B. Photolytic control of peptide self-assembly. J. Am. Chem. Soc. 2003; 125:7530-7531.
See Also
Peptides, Chemistry of;
Peptide Synthesis;
Proteins, Chemical Modifications of;
Protein Misfolding;
Synthetic Proteins to Elucidate Biologic Function