CHEMICAL BIOLOGY
Synthetic Chemistry: Formation of the Amide Bond
Christian A. G. N. Montalbetti and Virginie M. Falque, Evotec, United Kingdom
doi: 10.1002/9780470048672.wecb635
Amide bonds are found ubiquitously in natural or synthetic molecules of biologic interest. Since the early days of synthetic organic chemistry, methods for the formation of amides have been described. More recently, with the development of solid-phase chemistry and automated peptide synthesis, new strategies and reagents have been devised to overcome typical problems such as low conversion and racemization. This article provides an overview of the methodology that is available today. Depending on the nature of the synthetic target and the associated synthetic challenges, different approaches can be envisaged. Methods range from the rather straightforward use of acyl halides, anhydrides, and carbodiimides, to the more elaborate, low-racemization inducing methods that use phosphonium/uronium-based reagents. New amide bond-mediated ligation methodologies now offer new convergent strategies for the synthesis of highly functionalized molecules of biologic interest.
Amide bonds are found commonly in small or complex synthetic or natural molecules of biologic interest. Amide bonds are, for example, the structural backbone of proteins that play a crucial role in almost every biologic process. In nature, amides are formed via complex enzymatic pathways that ensure selectivity and specificity of the formed molecule. An analysis of the Comprehensive Medicinal Chemistry database revealed that more than 25% of known drugs bear amide functionality (1). For example, Taxol (pacitaxel; Bristol Myers Squibb, New York) 1 is a highly functionalized diterpenoid, which is extracted originally from the bark of the pacific yew tree. It is used to treat ovarian and breast cancers (2). Fuzeon (enfuvirtide; Roche Pharmaceuticals, Nutley, NJ) 2 is a synthetic biomimetic peptide and is the first of a novel class of fusion inhibitor antiretroviral drugs used to treat HIV-1 infection (3). Lipitor (atorvastatin hemicalcium salt; Pfizer, Inc., New York) 3 is the best-selling drug in the world, and it is used to treat high cholesterol (4). Sprycel (dasatinib; Bristol Myers Squibb) 4 is an oral dual BCR/ABL and Src family tyrosine kinase inhibitor, which is approved for use in patients with chronic myelogenous leukemia (5) (See Fig. 1).
Since the early 1840s (6), amide bond formation has been a very active field of research in organic chemistry. A multitude of synthetic methods have been elaborated and optimized (7). Relevant examples of these methods available to the bioorganic, organic, medicinal, or combinatorial chemist are reported in this article.

Figure 1. Examples of marketed drugs that contain amide bonds.
Amide Bond Formation: Overview
Amide bond formation by direct condensation between an acid and an amine is not obvious and must overcome adverse thermodynamics (8). This dehydrative process can be achieved under forcing conditions such as high temperatures (160-180° C), which are usually incompatible with the presence of other functionalities. Contrary to the ester formation between an acid and an alcohol, which is an equilibrium, the acid and the amine undergo first an acido-basic reaction that yields the stable salt. Therefore, the acid must be activated by the attachment of a leaving group before being reacted with the amine (see Fig. 2).
These two steps can be carried out separately with intermediate isolation of the activated species or by a one-pot synthesis with late introduction of the amine. More recent conditions allow coupling to occur with both the acid and the amine present in the reaction mixture.
Often, the choice of the methodology for one specific amide formation is not only governed by the yield. Avoiding racemization at neighboring chiral centers, improving difficult isolation from reaction by-products or reducing the costs of the reagents, might be the key elements for decision.
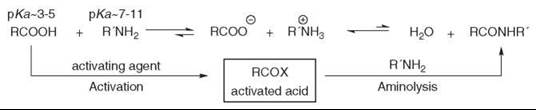
Figure 2. Activation and aminolysis process.
Amide Bond Formations: Methods and Strategies
Acyl or acid chlorides are used frequently in amide formation as activated forms of the corresponding carboxylic acid. A wide selection of acyl chlorides is available commercially. Otherwise, they can be prepared readily from the corresponding carboxylic acid in the presence of reagents such as thionyl chloride (9), oxalyl chloride 5 (10), phosphorus trichloride (11), and phosphorus pentachloride (12). Reactions that use oxalyl chloride or thionyl chloride are promoted by the addition of a catalytic amount of DMF (13) (see Fig. 3).

Figure 3. Acyl chloride-mediated amide coupling and potential side reactions.
Despite the high reactivity and low cost, these chlorinating reagents generate hydrogen chloride in situ and are not suitable for amide formation in the presence of acid labile groups. Hence, alternative basic conditions have been studied. For example, cyanuric chloride is used commonly to generate acyl chloride in nonacidic conditions (14). The presence of an organic or inorganic base maintains the basic pH conditions throughout the reaction. The cyanuric chloride method is also used in large-scale synthesis (15).
Neutral conditions (i.e., non-acid generating) that use triphenylphosphine and a source of chloride as carbon tetrachloride (16), hexachloroacetone (17), trichloroacetonitrile (18), or trichloroisocyanuric acid (19) have been developed. Alternatively, 1-chloro-N,N,2-trimethylpropenylamine (20) converts acids into acyl chlorides readily without HCl formation.
The coupling reaction with the amine usually requires an additional acid scavenger (often a base like triethylamine, DIEA, or NMM) to trap the formed HCl. The reaction can also be accelerated in the presence of a catalytic amount of DMAP (21), pyridine, or metallic zinc (22).
Nevertheless, acyl chlorides have some limitations. Acyl chlorides that bear α-hydrogens can undergo ketene 6 formation under basic conditions (23). The subsequent amine addition occurs with potential loss of chirality and side reactions (24). Oxazolone-mediated racemization is encountered in peptide chemistry. N-protected peptidoyl chlorides yield the corresponding oxazolones 7 spontaneously. These transient species react readily with nucleophiles; but, under the standard aminolysis conditions, the α-proton is acidic enough to enable acido-basic equilibrium, which compromises the chirality of the α-center. Peptides are, therefore, grown usually at their N-terminus, thus avoiding the oxazolone formation. Furthermore, the acyl chloride activation of N-urethane-protected amino acid (e.g., Boc, Fmoc, or Cbz) is unadvisable, as they react with the carbonyl of the neighboring urethane to yield the corresponding NCA 8 (25). NCA functionalities are notoriously reactive and promote many side reactions (see Fig. 3). Similar problems can also be observed with other activation methods.
Alternatively, acid fluorides are used to activate the acid. Acyl fluorides are less sensitive to moisture and are more reactive toward primary and secondary amines than the corresponding acyl chloride. Furthermore, they are compatible with basic- (Fmoc and Cbz) or even acid- (Boc) labile amine protecting groups and less prone to promote racemization than their chlorinated homologs (26). Thus, the acid fluoride method is often used in peptide synthesis (27). Cyanuric fluoride 9 (28), TFFH (29), DAST (30), and deoxofluor (31) are used commonly as fluorinating reagents (see Fig. 4).
The acyl azide strategy was developed for peptide synthesis in the early 1900s. The original preparation of the acyl azide 10 from the corresponding methyl ester is a two-step synthesis. Displacement of the methyl ester with the hydrazine, which generates the acyl hydrazide, is followed by the nitrosation reaction to yield the acyl azide 10. Isocyanate formation, also called the Curtius rearrangement, is a possible side reaction, and ureas 11 are often observed as side products (see Fig. 4). An improvement of this method is the one-pot synthesis of the acyl azide from the carboxylic acid using DPPA 12 (32). Acyl azides are, however, potential explosives and the leaving group (free azide) is toxic, which provides some limitation to this method.
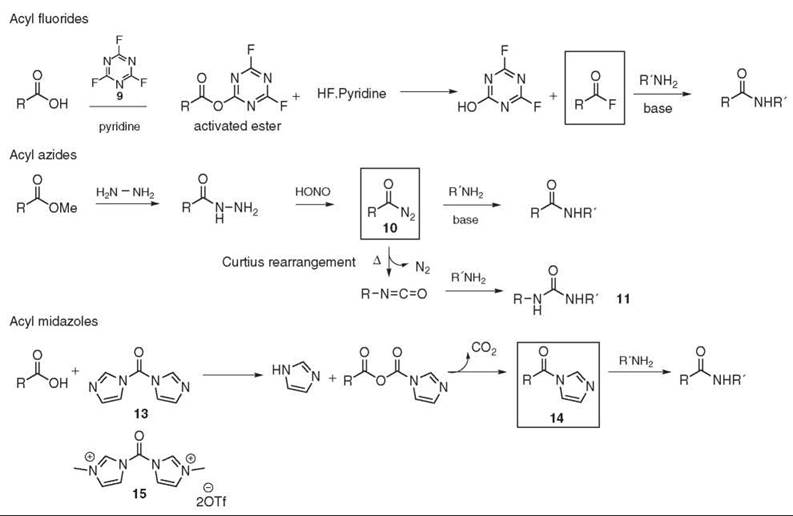
Figure 4. Acyl fluorides, acyl azides, and acyl imidazoles.
Acyl imidazoles
CDI 13 (33) allows one-pot amide bond formation and is also used for large-scale peptide chemistry (34). Initially, the mechanism may involve the formation of acyl carboxy imidazole and imidazole. Both intermediates react together to lead to the activated species as the acyl imidazole 14. Then the amine is added to undergo aminolysis (see Fig. 4). As imidazole is generated in situ, the reaction does not need an additional base and it is usually compatible with amine HCl salts (35). Incidentally, the acyl imidazole intermediate can also be isolated and stored. Some simple acyl imidazoles are even available commercially.
Similarly, carbonylimidazolium salts have been introduced. For example, CBMIT 15 is described as an efficient amino acylating reagent for peptide synthesis with sterically hindered amino acids (36).
Recently, acyl benzotriazoles have been described as O-, N-, and S-acylating agents (37). These agents are prepared easily by reacting benzotriazole with acyl chlorides. N-protected α-aminoacyl- or peptidoyl-benzotriazoles are coupled in aqueous acetonitrile solution with free amino acids or dipeptides (38). Under some specific conditions, hydroxyl carboxamides can be prepared directly from the corresponding hydroxyl acids without protection of the alcohol (39).
Anhydrides and mixed anhydrides
Anhydrides react readily with diverse nucleophiles such as amines. For example, the use of acetic anhydride was reported in the 1850s to produce acetamides (40). Symmetric anhydrides 16 can be prepared by dehydrating the corresponding acid under strong acidic conditions or at high temperatures. A more practical approach, however, consists of treating the carboxylic acid with DCC 17 (41). The anhydride is then subjected to aminolysis with the desired amine (see Fig. 5). This method is usually applicable to peptide synthesis, and, in theory, no additional base is required, as the carboxylate anion is produced in situ. Unfortunately, half of the acid is wasted during the process, which practically limits the application of this method to cheap and commercially available symmetric anhydrides.
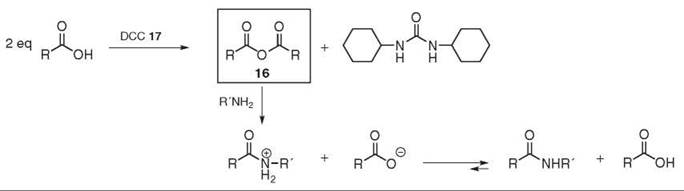
Figure 5. Symmetric anhydride preparation and coupling reaction.
Several mixed anhydride methods have, therefore, been developed. The acid is coupled to a second acid moiety that is considered to be disposable. This strategy relies fully on the regioselectivity of the aminolysis where the reactive center a is more reactive than b (see Fig. 6). The concept of mixed anhydrides has been extended to carbonic, boric, iso-ureas, phosphoric, and phosphinic acid-derived species. Iso-ureas and phosphorous-containing reagents will be discussed in separate articles.
Mixed pivalic anhydride 18 (42) is an example of selective mixed carboxylic anhydride. Selective aminolysis could be caused by the steric hindrance of the tert-butyl group. A mixed carbonic anhydride strategy has also been studied to overcome the aminolysis selectivity problem. This method exhibited excellent selectivity when ethoxycarbonyl 19 (43) or tert-butyloxycarbonyl 20 (44) anhydrides were used. The selectivity is mainly caused by the higher electrophilicity of one of the carboxylic centers toward the amine. Ethoxycarbonyl anhydrides can be prepared conveniently using ethyl chloroformate or EDDQ 21 (45).
Acyl aryl boron species also react with amines to yield amides with mixed results. For example, catecholborane was used to generate lactams successfully (46). Aryl boronic acids with electron-withdrawing groups, such as 3,4,5-trifluorobenzeneboronic acid and 3,5-bis-(trifluoromethyl)benzeneboronic acid, can act efficiently as an amidation catalyst when added to a mixture of acid and amine (47).

Figure 6. Different types of mixed anhydrides used for amid bond formation.
O-acyl iso-ureas
O-acylisourea species 22 are generated easily by reacting car- bodiimides with a mixture of the desired carboxylic acid and amine. Then, they undergo aminolysis readily to form the amide and the urea by-product (48). Often, formation of unreactive N-acylurea 23 by acetyl transfer and racemization are observed (see Fig. 7). This side reaction can be minimized substantially by reacting the acid with the carbodiimides at 0°C before adding the amine or by using DMAP or HOBt 24 as adjuvants (49).
DCC 17 (50), DIC 25, EDC 26, and polymer supported PS-CC (51) 27 are available commercially. Elimination of the resulting ureas can be achieved easily by filtration or solvent wash depending on their solubility. In the case of PS-CC, the urea by product is formed advantageously on the solid support while the amide is released in solution.

Figure 7. Carbodiimides.
Esters
Alkyl esters
Alkyl esters (e.g., methyl, ethyl, benzyl esters) are usually stable toward amines and, thus, are used as protecting groups. Some anecdotic examples of amide bond formation with alkyl esters, however, are reported in the literature. High reaction temperature, addition of a Lewis acid (52), or use of organoaluminium species generated from DIBAL-H-H2NR (53) can enable these reactions. Saturated ammonia in methanol can also react with methyl esters at room temperature to form the primary amides (54).
Recently, amidation of methyl esters with unprotected amino alcohols in the presence of a catalytic amount of IMes, a readily available carbene, was achieved in good yields (see Fig. 8). Initially, the carbene was proposed to react with the methyl ester to generate the activated C2-acylimidazolium intermediate (55). X-ray evidence, however, suggests a more complex mechanism (56).
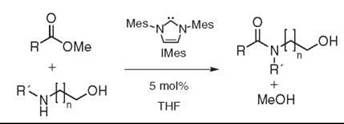
Figure 8. Hetereocyclic carbene catalyzed amidation.
Activated esters
Usually, esters of phenols are easier to hydrolyze than alkyl esters and they also react with a wide range of nucleophiles such as amines. The reactivity is increased when electron withdrawing groups are present on the phenyl ring. Activated esters can be obtained from the acid using DCC-17 mediated coupling or via the acid chloride. They can be used immediately without additional purification or they can be stored. For example, PNP 28 esters are purified easily by recrystallization in alcoholic solvents with which they do not react. Then, aminolysis is performed at room temperature (57). Other examples of alcohols used to activate acids are represented in Fig. 9.

Figure 9. Activated esters.
PFP 29 esters have been recommended for the synthesis of heterocyclic amides, when DCC 17 or DIC 25 alone had failed (58). HOSu 30 is coupled to acids easily using N,N’-disuccinimidyl carbonate (see chapter on DIC) and, being water soluble, it can be removed readily at the purification stage (59).
PFP and OSu esters are of significant importance, especially in the preparation of biomaterials and in bioconjugation chemistry. An increasing number of these activated esters are available now commercially.
HOBt 24 is one of the most common reagents in peptide chemistry. Good yields and low racemization are observed with O-Bt esters (60). Furthermore, yields and enantiomeric purities are increased when used in conjunction with DCC 17 (61) or even HOBt-based phosphonium- or uronium-coupling reagents (see chapter on Onium salts). In 2005, HOBt monohydrate, the standard form for this reagent, was reclassified by the United Nations as a desensitized explosive. This measure limited its availability drastically (UN3380). Other efficient racemization suppressants are HOAt 31, HODhbt 32, HOCt 33, and N-hydroxytetrazole 34 (62). The lack of popularity of HODhbt 32 in peptide coupling, despite very low racemization, can be explained by the high degradation levels of the activated ester, which are observed during the aminolysis step (63). HOAt 31 has been reported to be more efficient than HOBt 24 in some difficult cases. Additional chelation or neighboring effect caused by the pyridine ring could explain this increased reactivity (see Fig. 10) (64). Racemization during DIC-25 / HOCt-33 mediated coupling is negligible with all amino acids except histidine (65).
Another field of application for active esters is solid-phase synthesis. Some polymer-supported reagents are available commercially (see Fig. 9). The acid is first immobilized on a polymer support as an active ester and the excess reagents are washed away conveniently. Finally, the amide is released by amine treatment. During the cleavage, a limited amount of amine can be used to avoid the presence of excess amine in the final mixture. The acid is loaded onto the resin using classic ester condensation methods for TFP resin 35 (66), HOBt resin 36 (67), and oxime resin 37 (68). In the case of the triazine resin 38, the acid is loaded via an aromatic nucleophilic substitution in the presence of a base (69).
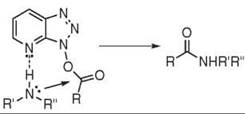
Figure 10. Additional chelation with HOAt 31.
Onium salts
An elegant one-pot coupling method based on both mixed anhydride and activated ester strategy was introduced by peptide chemists in the early 1970s. These coupling reagents were produced by combining reactive onium salts, such as phosphonium, uronium, iminium, or quaternized nitrogen species, with racemization suppressants, such as HOBt 24 or HOAt 31. Most of these coupling reagents are now available commercially and are compatible with both Boc- or Fmoc-strategy peptide synthesis (70, 71), using solid-phase parallel and combinatorial techniques (72). Usually, the coupling is performed by reacting the desired amine and acid in the presence of the onium-coupling reagent and an organic base such as triethylamine, DIEA, NMM, 2,6-lutidine, or collidine. In most cases, good yields and low racemizations are reported.
Phosphonium-based coupling reagents
BOP 39, also known as Castro’s reagent, was the first of the HOBt-based onium reagents (73) (see Fig. 11). The deprotonated acid reacts first with BOP 39 to produce both the highly reactive (acyloxy)phosphonium 40 species and the OBt anion. The initial mechanism postulated the direct attack of the OBt anion on the (acyloxy)phosphonium 40, which generates the aminolysable-activated O-Bt ester 41. This process is driven by the formation of the stable phosphoric triamide 42 (74). More recent studies suggested the existence of an additional step. The (acyloxy)phosphonium intermediate 40 first reacts with the carboxylate to form the symmetric anhydride 43. The subsequent activation with HOBt 24 is relatively slow and yields the anticipated O-Bt ester while regenerating the carboxylate. The additional step can be accelerated by the addition of HOBt 24 to the reaction mixture (75). Whether it is really necessary to use HOBt 24 in conjunction with HOBt-based uronium or phosphonium reagent is still debatable and it must be investigated on a case-by-case basis. PyBop (G.L. Biochem (Shanghia) Ltd., Shanghai, China) 44 and PyAOP 45 (76) where the dimethyl moiety was replaced were introduced to avoid the generation of toxic HMPA 42 (77). Other examples of phosphonium-coupling reagents are shown in Fig. 12.
Halogenophosphonium reagents often give better yields and lower racemization than HOBt-based ones for the coupling of N-methylated amino acids. The Bt ester is believed to be too stable to react with these hindered amines, which enables degradation or racemization to occur. Some effective reagents that eliminate the need for HOBt 24 have therefore been developed (see Fig. 12). For example, PyBrop 48 is an efficient peptide-coupling reagent for N-methylated amino esters (78). It is interesting to notice that reagents such as CloP 46 (79) and BroP 47 (80) had been considered initially as poor peptide-coupling reagents as they lead to noticeable racemization with primary amino acids.
Other organo-phosphorous reagents are based on the mixed carboxylic-phosphoric or phosphinic anhydrides. Initially used to convert carboxylic acids into acyl azides, DPPA 12 has been introduced as a one-pot coupling reagent for peptide chemistry (32), and it was adapted later to solid-phase chemistry (81). The driving force of these reactions is the formation of the phosphoric or phosphinic acids and their salts. Later DPP-Cl 49 (82) and FDPP 50 were introduced. FDPP 50 has been used successfully in macro cyclizations (83). Examples of phosphoric- and phosphinic-based coupling reagents are shown in Fig. 12 [DEPBT 51 (84), BDP 52 (85), BOP-Cl 53 (86)].
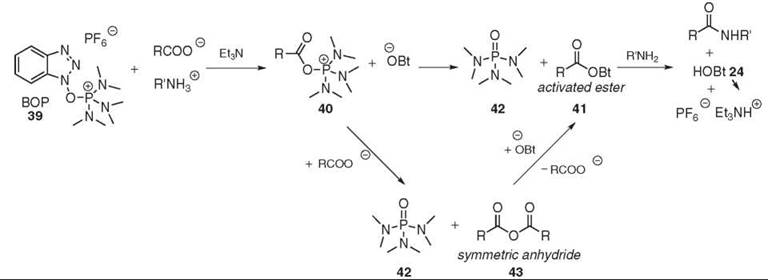
Figure 11. BOP 39-mediated coupling.

Figure 12. Organophosphorous coupling reagents.
Uronium/guanidium-based coupling reagents
Another popular family of reagents is based on uronium species such as HBTU 54 and TBTU 55 (87, 88). These species are the hexafluorophosphate and tetrafluoroborate salts of the same molecule, respectively. The coupling is performed the same as the phosphonium reagents, and the nature of the non-nucleophilic counter ion has no influence on the outcome. The driving force is, in this case, the formation of the stable tetra-methylurea 56 (see Fig. 13). In some cases, the amine can react directly with the coupling reagent to form the undesired guanidine 57. This side reaction can be suppressed by adding HOBt 24 to the reaction. Usually, these reagents are found in their more stable guanidinium form (N-form) (89, 90). The uronium species of HBTU 54 and HATU 58, however, have been isolated and are more active than the guanidinium species. Unfortunately, under standard coupling conditions, the O-form is converted quickly to the N -form (91) (see Fig. 14).
Some examples of uronium-based coupling reagents are represented in Fig. 15 [e.g., BCC 59 (92), TDBTU 60, TNTU 61, TPTU 62, TSTU 63 (88), HAPyU 64 (93), TAPipU 65 (94), CIP 67 (95), BTFFH 68 (96), HOTT / TOTT 69 (97)]. As for phosphonium-based coupling reagents, poor results are observed with sterically hindered amines when HOBt 24 is present. Therefore, some alternative reagents have been designed. For example, HATU 58 has proved to be very efficient in some sterically hindered couplings (64, 98). The superior reactivity of the At-activated ester toward amines is discussed in the Activated Esters section. TOTU 70 enables the formation of an activated acyl oxime intermediate, and low racemization in peptide couplings has been reported (99).
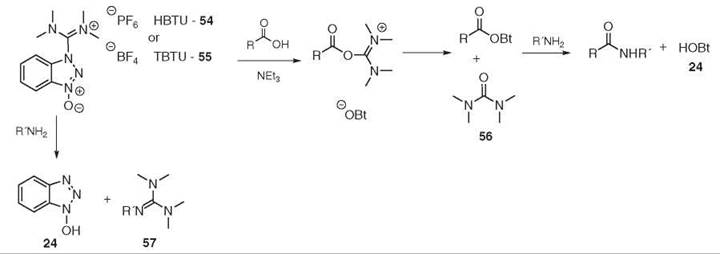
Figure 13. HBTU 54/TBTU 55-mediated coupling.

Figure 14. Uronium and guanidinium forms of HBTU (X = C) 54/HATU (X = N) 58.
Iminium-based coupling reagents
Iminium reagents are inspired directly from the uronium family. They differ structurally by the replacement of the amino groups with a hydrogen, an alkyl, or an aryl group. For example, BOMI 71 was reported to be more reactive than the correspondent uronium reagents (100). The increased reactivity could stem from the reduced number of mesomeric forms observed in the iminium species compared with the uronium forms. Representative examples of iminium reagents are shown in Fig. 15 [BOMI 71, BDMP 72 (101), BPMP 73, FOMP 74 (100)].

Figure 15. Uronium/guanidinium and iminium-based coupling reagents.
Quaternized nitrogen-based coupling reagents
Triazinyl esters
Recently, DMTMM 75 has been described as an efficient, one-pot coupling reagent for ester and amide bond formation (102). This reagent first undergoes an SNAr reaction as in the case of cyanuric fluoride 9. The activated ester then undergoes aminolysis. The in situ liberation of N-methyl morpholine avoids the use of an additional base conveniently. The triazinone 76 by-product is eliminated easily by an aqueous wash (see Fig. 16).
An inexpensive substitute to DMTMM is 2-chloro-4, 6-dimethoxy-1,3,5-triazine in the presence of an additional organic base (103).
Mukaiyama's reagent
2-Chloro-1-methylpyridinium iodide 77, also called Mukaiyama’s reagent, in the presence of a carboxylic acid and a tertiary amine yields an activated 2-acyloxy-pyridinium species 78. This intermediate reacts with a range of nucleophiles. The driving force for this reaction is the generation of a stable tautomeric pyridone. Mukaiyama’s reagent has been reported in the preparation of activated esters, the formation of amides, and the conversion of β-amino acids to the corresponding β-lactams (104, 105). The poor solubility of pyridinium iodides in conventional solvents indicates that the reaction requires refluxing in methylene chloride. To alleviate this limitation, novel reagents have been suggested: BEMT 79 (106), BEP 80, FEP 81, BEPH 82, and FEPH 83 (107) (see Fig. 16). Tetrafluoroborate and hexachloroantimonate have been adopted as counter ions. These reagents were used successfully in the synthesis of oligopeptides and in solid-phase peptide synthesis.
Isoxazolinium Salts
Woodward’s reagent K or NEPIS 84 is a zwitterionic isoxazolinium that reacts with N-protected amino acids in presence of triethylamine to form the activated enol ester 85 (108) (see Fig. 16). This intermediate can be reacted without additional purification with an amine to yield the desired amide and the sulfonate by-product that can be easily removed by aqueous extraction.

Figure 16. Quaternized nitrogen-based reagents.
Amide bonds by chemical ligation
New ligation strategies allow the selective formation of an amide bond between two highly functionalized fragments such as unprotected peptides, glycopeptides, or other molecules of biologic interest. The convergent assembling of complex, preformed peptidic sequences overcomes the inevitable contamination issues observed during extended linear peptide synthesis (109).
Native thioligation
This original methodology (110) requires the presence of a cysteine at the N-terminal position of fragment B and the introduction of a thioester at the C-terminal position of fragment A. Regiospecific coupling of the two unprotected fragments can be achieved in a two-step process described in Fig. 17. The first step is a chemoselective transthioesterification between the thiol functionality of the terminal cysteine and the terminal thioester to form the thioester linked intermediate. This step is called the “capture reaction.” The second step is the rapid, intramolecular acyl transfer from the thio- to the amino-position of the cysteine to yield the desired amide bond. Usually, no racemization is observed (111).
Different Boc (112) or Fmoc (113) compatible solid-phase strategies have been devised to allow the preparation of peptides bearing a C-terminal thioester. Recently the introduction of a 2-(ethyldisulfanyl)phenol ester at the C-terminal position of fragment A has been used in an elegant solution phase approach (114) (see Fig. 18). The first amino acid of fragment A is coupled to 2-(ethyldisulfanyl)phenol 86. The resulting phenol ester is sufficiently stable to be used as a protecting group and to allow the growing fragment A to use standard Boc strategy peptide synthesis. After final deprotection, fragment A can undergo native thioligation. First, the disulfide bond is cleaved under reductive conditions or in the presence of an excess of thiol reagent. The resulting 2-mercaptophenyl ester might be in an unfavorable, yet dynamic, equilibrium with the corresponding S-2-hydroxyphenyl thioester via intramolecular O- to S-acyl transfer, which generates in situ the appropriate setup for native thioligation.
A similar type of native chemical ligation has been extended to B fragments that contain homocysteine (115), selenocysteine (116), and histidine (117) at their A-terminal positions.
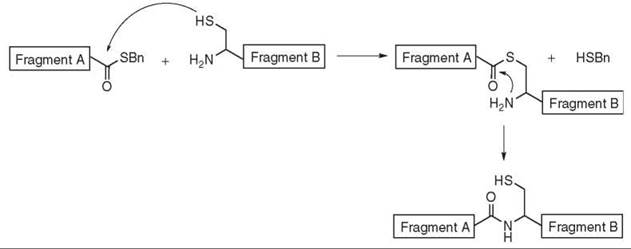
Figure 17. Native thioligation.
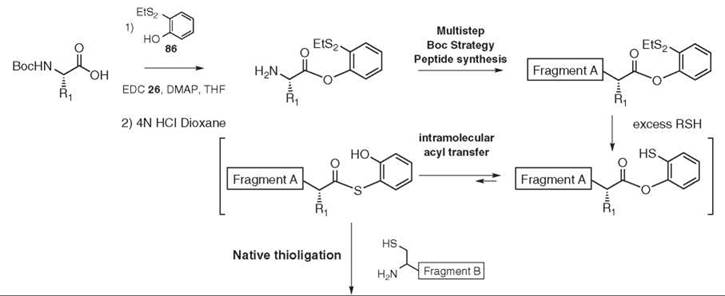
Figure 18. Convenient fragment A preparation.
Staudinger ligation
The Staudinger ligation (118) is a less restrictive approach that can be applied to couple unprotected peptides. First, a C-terminal phosphinomethylthioester is introduced on Fragment A 87, and the A-terminal amine of fragment B is converted to the corresponding azide 88. The two fragments are then reacted together via a Staudinger reaction (see Fig. 19), which yields the iminophosphorane 89. This intermediate undergoes intramolecular S- to A-acyl transfer readily to produce the corresponding hydrolysable amidophosphonium salt 90.
In principle, this methodology is independent of the nature of amino acids present at the ligation point and can be even extended to all types of acids and amines. For example, α-glycosylated amides (119) and peptides have been prepared via stereoselective Staudinger ligation (120). Challenging, medium-sized (7- to 10-membered) lactams have been ring-closed in good yield using the Staudinger ligation sequence described in Fig. 19 (121). In this example, the phosphine reagent has been protected judiciously as a borane complex. The Staudinger ligation is then triggered by the deprotection of the phosphine under basic conditions.

Figure 19. Staudinger ligation.
Enzymes such as proteases (122), subtilisin (123), acylases, peptidases, amidases, and lipases (124) are reported to catalyze amide bond formation with, in some cases, enantiospecificity of over 99%. Despite limited enzyme-substrate compatibility, specific conditions have been developed to reverse their natural reactivity, which is in favor of the hydrolysis. For example, Kyotorphin (Tyr-Arg) (125), a potent analgesic, was produced on an industrial scale using α-chymotrypsin, a peptidase isolated from bovine pancreas.
Conclusions
This article presents an overview of the different amide bond formation methodologies that are available to the organic and biochemist. For nearly two centuries, the methods have evolved from the original symmetric anhydrides and acyl chlorides. When Fischer started to study peptidic couplings in the early 1900s, it became obvious that poor yields and racemization would present a major challenge. During the last three decades, the design and synthesis of new coupling reagents have been an area of intense investigation. The predominance of carbodiimide and active ester procedures have been replaced gradually by onium salts approaches. The introduction of racemization suppressants combined with the development of solid-phase chemistry and urethane-based protecting group have allowed high-throughput chemistry and automated peptide synthesis to be a reality. Emerging technologies such as ligation allow convergent synthesis by amide coupling between two highly functionalized molecules.
Abbreviations
|
AOP At |
(1H-7-azabenzotriazol-1-yloxy)tris (dimethylamino)phosphonium hexafluorophosphate azabenzotriazole |
|
BCC BDMP BDP BEMT BEP BEPH Boc BOMI |
benzotriazolyloxy-bis (pyrrolidino)carbonium hexafluorophosphate 5-(1H-benzotriazol-1-yl)-3,4-dihydro-1-methyl 2H-pyrrolium hexachloroantimonate N-oxide benzotriazol-1-yl diethyl phosphate 2-bromo-3-ethyl-4-methylthiazolium tetrafluoroborate 2-bromo-1-ethylpyridinium tetrafluoroborate 2-bromo-1-ethylpyridinium hexachloroantimonate ferf-butyloxycarbonyl N-(1H-benzotriazol-1-ylmethylene)-N-methylmethanaminium hexachloroantimonate N-oxide |
|
BOP BPMP BOP-Cl BroP Bt BTFFH CBMIT Cbz CIP CDI CloP DAST DCC DEPBT DCU DIC DIEA DMAP DMF DMTMM DPPA DPP-Cl EDC or EDCI EDDQ FDPP FEP FEPH Fmoc FOMP HATU HAPyU HBTU |
(benzotriazol-1-yloxy)-tris(dimethylamino)phosphonium hexafluorophosphate or Castro’s reagent 1-(1H-benzotriazol-1-yloxy)phenylmethylene pyrrolidinium hexachloroantimonate bis(2-oxo-3-oxazolidinyl)phosphinic chloride bromo-tris-(dimethylamino)phosphonium hexafluorophosphate benzotriazole 1,1,3,3-bis(tetramethylene)fluorouronium hexafluorophosphate N,N’-carbonylbis(3-methylimidazolium) triflate benzyloxycarbonyl 2-chloro-1,3-dimethylimidazolidinium hexafluorophosphate carbonyl diimidazole chloro-tris-(dimethylamino)phosphonium hexafluorophosphate diethylaminosulfur trifluoride dicyclohexylcarbodiimide 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one dicyclohexylurea diisopropylcarbodiimide N,N-diisopropylethylamine also known as Hunig’s base N,N-dimethylaminopyridine N,N-dimethylformamide 4-(4,6-dimethoxy-(1,3,5)triazin-2-yl)-4-methyl-morpholiniumchloride diphenylphosphoryl azide diphenylphosphinic chloride 1-ethyl-3-(3’-dimethylamino)carbodiimide HCl salt, also known as Water Soluble Carbodiimide, HCl (WSC-HCl) 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline pentafluorophenyl diphenyl phosphinate 2-fluoro-1-ethylpyridinium tetrafluoroborate 2-fluoro-1-ethylpyridinium hexachloroantimonate 9-fluorenylmethoxycarbonyl 5-(pentafluorophenyloxy)-3,4-dihydro-1-methyl 2H-pyrrolium hexachloroantimonate O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate 1-(1-pyrrolidinyl-1H-1,2,3-triazolo[4,5-b]pyridine-1-ylmethylene)pyrrolidinium hexafluorophosphate N-oxide O-(1 H -benzotriazol-1-yl)-1,1,3,3’-tetramethyluronium hexafluorophosphate |
|
HMPA HOAt HOBt HOCt HODhbt HONB HOSu HOTT IMes NCA NEPIS NMM PFP PNP PS-CC PyAOP PyBop PyBrop TAPipU TBTU TDBTU TFFH TFP TNTU TPTU TOTT TOTU TSTU |
hexamethylphosphoric triamide 1-hydroxy-7-azabenzotriazole 1-hydroxybenzotriazole ethyl-1-hydroxy-1H-1,2,3-benzotriazine 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine N-hydroxy-5-norbornene-2,3-dicarboximide N-hydroxysuccinimide S-(1-oxido-2-pyridinyl)-1,1,3,3-tetramethylthiouronium hexafluorophosphate N,N-bismesitylimidazolylidene N-carboxyanhydride N-ethyl-phenylisoxazolium-3’-sulphonate, also known as Woodward’s reagent K N-methylmorpholine pentafluorophenyl para-nitrophenyl N-cyclohexylcarbodiimide-N’-methyl polystyrene 7-azabenzotriazol-1-yloxy-trispyrrolidinophosphonium hexafluorophosphate benzotriazol-1-yloxy-trispyrrolidinophosphonium hexafluorophosphate bromotri(pyrrolidino)phosphonium hexafluorophosphate O-(7-azabenzotriazol-1-yl)-1,1,3,3- bis(pentamethylene)uronium tetrafluoroborate O-(1H -benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate N,N,N’,N’-tetramethyl-O-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)uronium tetrafluoroborate N,N-tetramethylfluoroformamidinium hexafluorophosphate tetrafluorophenol 2-(5-norbornene-2,3-dicarboximido)-1,1,3,3-tetramethyluronium tetrafluoroborate O-(1,2-dihydro-2-oxo-pyridyl)-1,1,3,3-tetramethyluronium tetrafluoroborate S-(1-oxido-2-pyridinyl)-1,1,3,3-tetramethylthiouronium tetrafluoroborate O-((ethoxycarbonyl)cyanomethylene amino)-N,N,N’,N’-tetramethyluronium hexafluorophosphate N,N,N’,N’-tetramethyl-O-(N- succinimidyl)-uronium tetrafluoroborate |
References
1. Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999; 1:55-68.
2. Wani M, Taylor H, Wall M, Coggon P, McPhail A. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971; 93:2325-2327.
3. Greenberg ML, Cammack N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004; 54:333-340.
4. Krishan M. Best-selling human medicines 2002-2004. Drug Disc. Today 2005; 10:739-742.
5. Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, et al. 2-Aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-Chloro-6-methylphenyl)-2- [6-[4-(2-hydroxyethyl)-1-piperazinyl).-2-methyl-4-pyrimidinyl. amino).-1,3-thiazole-5-carboxamide (Dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J. Med. Chem. 2006; 49:6819-6832.
6. Gerhardt MC. Sur une nouvelle classe de composes organiques. Ann. Chim. Phys. 1845; 3:117-125.
7. Han S-Y, Kin Y-A. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron 2004; 60:2447-2467.
8. Ulijn RV, Moore BD, Janssen AEM, Halling PJ. A single aqueous reference equilibrium constant for amide synthesis- hydrolysis. J. Chem. Soc., Perkin Trans. 2002; 2:1024-1028.
9. Pearson, AJ, Roush, WR, eds. Handbook of Reagents for Organic Synthesis: Activating Agents and Protecting Groups. 1999. John Wiley & Sons, New York. pp. 370-373.
10. Knapp S, Gibson FS. Iodolactamization: 8-exo-Iodo-2-azabicyclo [3.3.0.octan-3-one. Org. Synth. 1998; 9:516-521.
11. Pearson, AJ, Roush WR, eds. Handbook of Reagents for Organic Synthesis: Activating Agents and Protecting Groups. 1999. John Wiley & Sons, New York. pp. 333-335.
12. Pearson AJ, Roush WR, eds. Handbook of Reagents for Organic Synthesis: Activating Agents and Protecting Groups. 1999. John Wiley & Sons, New York. pp. 335-338.
13. Bruckner R. Advanced Organic Chemistry, Reaction Mechanisms. 2002. Harcourt/Academic Press, San Diego, CA. pp. 239.
14. Venkataraman K, Wagle DR. Cyanuric chloride: a useful reagent for converting carboxylic acids into chlorides, esters, amides and peptides. Tetrahedron Lett. 1979; 20:3037-3040.
15. Rayle HL, Fellmeth L. Development of a process for triazine-promoted amidation of carboxylic acids. Org. Process Res. Dev. 1999; 3:172-176.
16. Lee JB. Preparation of acyl halides under very mild conditions. J. Am. Chem. Soc. 1966; 88:3440-3441.
17. Villeneuve GB, Chan TH. A rapid, mild and free procedure for the preparation of acyl chlorides including formyl chloride. Tetrahedron Lett. 1997; 38:6489-6492.
18. Jang DO, Park DJ, Kim J. A mild and efficient procedure for the preparation of acid chlorides from carboxylic acids. Tetrahedron Lett. 1999; 40:5323-5326.
19. Rodrigues RC, Barros IMA, Lima LS. Mild one-pot conversion of carboxylic acids to amides or esters with Ph3P/trichloroiso-cyanuric acid. Tetrahedron Lett. 2005; 46:5945-5947.
20. Devos A, Remion J, Frisque-Hesbain A.-M, Colens A, Ghosez L. Synthesis of acyl halides under very mild conditions. J. Chem. Soc. 1979; 1180-1181.
21. Ragnarsson U, Grehn L. Novel amine chemistry based on DMAP-catalysed acylation. Acc. Chem. Res. 1998; 31:494-501.
22. Meshram HM, Reddy GS, Reddy MM, Yadav JS. Zinc mediated facile amide formation: application to alkyl, aryl, heterocycle, carbohydrate and amino acids. Tetrahedron Lett. 1998; 39: 4103-4106.
23. Lynch JE, Riseman SM, Laswell WL, Volante RP, Smith GB, Shinkai I, Tschae DM. Mechanism of an acid chloride-imine reaction by low-temperature FT-IR: beta-lactam formation occurs exclusively through a ketene intermediate. J. Org. Chem. 1989; 54:3792-3796.
24. Seikaly HR, Tidwell TT. Addition reactions of ketenesTetrahedron. 1986; 42:2587-2613.
25. Poduska K, Gross H. Uber-Halogenather, VII: Reaktionen von Dichlormethyl-alkyl-athern mit Aminosaurederivaten. Chem. Ber. 1961; 94:527-537.
26. Carpino LA, Beyermann M, Wenschuh H, Bienert M. Peptide synthesis via amino acid halides. Acc. Chem. Res. 1996; 29: 268-274.
27. Lippert JW. Amide bond formation by using amino acid fluorides. Arkivoc 2005; xiv:87-95.
28. Olah GA, Nojima M, Kerekes I. Synthetic methods and reactions; IV.1 Fluorination of carboxylic acids with cyanuric fluoride. Synthesis 1973; 8:487.
29. Carpino LA, El-Faham A. Tetramethylfluoroformamidinium hexafluorophosphate: a rapid-acting peptide coupling reagent for solution and solid phase peptide synthesis. J. Am. Chem. Soc. 1995; 117:5401-5402.
30. Lal GS, Pez GP, Pesaresi RJ, Prozonic FM, Cheng H. Bis(2-methoxyethyl)aminosulfur trifluoride: a new broad-spectrum de-oxofluorinating agent with enhanced thermal stability. J. Org. Chem. 1999; 64:7048-7054.
31. LaI GS, Pez GP, Pesaresi RJ, Prozonic FM. Bis(2-methoxyethyl) aminosulfur trifluoride: a new broad-spectrum deoxofluorinating agent with enhanced thermal stability. Chem. Commun. 1999; 215-216.
32. Shioiri T, Ninomiya K, Yamada SY. Diphenylphosphoryl azide. New convenient reagent for a modified Curtius reaction and for peptide synthesis. J. Am. Chem. Soc. 1972; 94:6203-6205.
33. Paul R, Anderson WJ. N,N’-Carbonyldiimidazole, a new peptide forming reagent. J. Am. Chem. Soc. 1960; 82:4596-4600.
34. Dale DJ, Draper J, Dunn PJ, Hughes ML, Hussain F, Levett PC, Ward GB, Wood AS. The process development of a scaleable route to the PDE5 inhibitor UK-357,903. Org. Process Res. Dev. 2002;6:767-772.
35. Staab HA, Liiking M, Durr FH. Darstellung von imidazoliden. Synthese von amiden, hydraziden und hydroxamsauren nach der imidazolidmethode. Chem. Ber. 1962; 95:1275-1283.
36. Saha AK, Schultz P, Rapoport H. 1,1’-Carbonylbis(3-methyli-midazolium) triflate: an efficient reagent for aminoacylations. J. Am. Chem. Soc. 1989; 111:4856-4859.
37. Katritzky AR, Suzuki K, Wang Z. Acylbenzotriazoles as advantageous N-, C-, S-, and O-acylating agents. Synlett 2005; 11:1656-1665.
38. Katritzky AR, Angrish P, Hiir D, Suzuki K. N-(Cbz- and Fmoc-α-aminoacyl)benzotriazoles: stable derivatives enabling peptide coupling of Tyr, Trp, Cys, Met, and Gln with free amino acids in aqueous media with complete retention of chirality. Synthesis 2005; 3:397-402.
39. Katritzky AR, Singh SK, Cai C, Bobrov S. Direct synthesis of esters and amides from unprotected hydroxyaromatic and -aliphatic carboxylic acids. J. Org. Chem. 2006; 71:3364-3374.
40. Gerhardt C. Utersuchungen liber die wasserfreien organischen sauren. Jstus Liebigs, Ann. Chem. 1853; 149-179.
41. Mikolajczyk M, Kielbasinski P. Recent developments in the carbodiimide chemistry. Tetrahedron 1981; 37:233-284.
42. Wittenberger SJ, McLaughlin MA. Preparation of endothelin antagonist ABT-627. Tetrahedron Lett. 1999; 40:7175-7178.
43. Pelter A, Levitt TE. Investigations of the mechanisms of interaction of acyloxyalkoxyboranes with amines. Tetrahedron 1970; 26:1545-1553.
44. Jakopin Z, RoSkar R, Sollner Dolenc M. Synthesis of 3,5-disubstituted 1,2,4-oxadiazoles as peptidomimetic building blocks. Tetrahedron Lett. 2007; 48:1465-1468.
45. Belleau B, Malek G. A new convenient reagent for peptide syntheses. J. Am. Chem. Soc. 1968; 90:1651-1652.
46. Collum DB, Chen S-C, Ganem B. A new synthesis of amides and macrocyclic lactams. J. Org. Chem. 1978; 43:4393-4394.
47. Ishihara K, Ohara S, Yamamoto H. 3,4,5-trrifluorobenzeneboronic acid as an extremely active amidation catalyst. J. Org. Chem. 1996; 61:4196-4197.
48. Sheehan J, Cruickshank PA. Notes- a convenient synthesis of water-soluble carbodiimides. J. Org. Chem. 1961; 26:2525-2528.
49. Windridge GC, Jorgensen EC. 1-hydroxybenzotriazole as a racemisation-suppressing reagent for the incorporation of im-Benzyl-histidine into peptides. J. Am. Chem. Soc. 1971; 17:6318-6319.
50. Sheehan JC, Hess GP. A new method of forming peptide bonds. J. Am. Chem. Soc. 1955; 77:1067-1068.
51. Senten K, Daniels L, Van der Veken P, De Meester I, Lambeir A-M, Scharpe S, Haemers A, Augustyns K. Rapid parallel synthesis of dipeptide diphenyl phosphonate esters as inhibitors of dipeptidyl peptidases. J. Comb. Chem. 2003; 5:336-344.
52. Chakrabarti JK, Hotten TM, Pullar IA, Tye NC. Heteroarenoben- zodiazepines. Part 7. Synthesis and pharmacological evaluation of a series of 4-Piperazinylpyrazolo[3,4.- and -[4,3-b.[1,5.benzo-diazepines as potential anxiolytics. J. Med. Chem. 1989; 32:2573-2582.
53. Huang P-Q, Zheng X, Deng X-M. DIBAL-H-H2NR and DIBAL-H-HNR1R2-HCl complexes for efficient conversion of lactones and esters to amides. Tetrahedron Lett. 2001; 42:9039-9041.
54. Corey EJ, Ohtani M. Enantiospecific synthesis of a rigid, C2-symmetric, chiral guanidine by a new and direct method. Tetrahedron Lett. 1989; 30:5227-5230.
55. Connor EF, Nyce GW, Myers M, Mock A, Hedrick JL. First example of A-heterocyclic carbenes as catalysts for living polymerization: organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002; 124:914-915.
56. Movassaghi M, Schmidt MA. A-heterocyclic carbene-catalysed amidation of unactivated esters with amino alcohols. Org. Lett. 2005; 7:2453-2456.
57. Bodanszky M, du Vigneaud V. A method of synthesis of long peptide chains using a synthesis of oxytocin as an example. J. Am. Chem. Soc. 1959; 81:5688-5691.
58. Rajappan VP, Hosmane RS. Pentafluorophenol: a superior reagent for condensations in heterocyclic chemistry. Synth. Comm. 1998; 28:753-764.
59. Ogura H, Kobayashi T, Shimizu K, Kawabe K, Takeda K. A novel active ester synthesis reagent (N,N’-disuccinimidyl carbonate). Tetrahedron Lett. 1979; 20:4745-4746.
60. Koenig W, Geiger R. A-Hydroxy compounds as catalysts for the aminolysis of activated esters. Chem. Ber. 1973; 106:3626-3635.
61. Koenig W, Geiger R. New method for the synthesis of peptides: activation of the carboxyl group with dicyclohexylcarbodiimide by using 1-hydroxybenzotriazoles as additives. Chem. Ber. 1970; 103:788-798.
62. Spetzler JC, Meldal M, Felding J, Veds0 P, Begtrup M. Novel acylation catalysts in peptide synthesis: derivatives of A-hydroxytriazoles and A-hydroxytetrazoles. J. Chem. Soc. PerkinTrans. 1998; 10:1727-1732.
63. Knorr R, Trzeciak A, Bannwarth W, Gillessen D. New coupling reagents in peptide chemistry. Tetrahedron Lett. 1989;3 0:1927-1930.
64. Carpino LA. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 1993; 115:4397-4398.
65. Roberston N, Ramage R. Racemisation studies of a novel coupling reagent for solid phase peptide synthesis. Tetrahedron 1999; 55:2713-2720.
66. Salvino JM, Kumar NV, Orton E, Airey J, Kiesow T, Crawford K., Mathew R, Krolikowski P, Drew M, Engers D, Krolikowski D, Herpin T, Gardyan M, McGeehan G, Labaudiniere R. Polymer-supported tetrafluorophenol: a new activated resin for chemical library synthesis. J. Comb. Chem. 2000; 2:691-697.
67. Baxendale IR, Ley SV. Polymer-supported reagents for multi-step organic synthesis: application to the synthesis of sildenafil. Bioorg. Med. Chem. Lett. 2000; 10:1983-1986.
68. Scialdone MA. Diisocyanates as scaffolds for combinatorial libraries. The solid-phase synthesis of bis-ureas from polymer- supported diisocyanates. Tetrahedron Lett. 1996; 37:8141-8144.
69. Masala S, Taddei M. Solid-supported Chloro[1,3,5.triazine. A versatile new synthetic auxiliary for the synthesis of amide libraries. Org. Lett. 1999; 1:1355-1357.
70. Llyod-Williams P, Albericio F, Giralt E. Chemical Approaches to the Synthesis of Peptides and Proteins. 1997. CRC Press, New York. pp. 48-53.
71. Chan WC, White PD, eds. Fmoc Solid-Phase Peptide Synthesis: A Practical Approach. 2000. Oxford University Press, Oxford. pp. 53-54.
72. Bunin BA. The combinatorial index. In: Combinatorial Solid-Phase Synthesis. 1998. Academic Press. pp. 77-82.
73. Castro B, Dormoy JR, Evin G, Selve C. Reactifs de couplage peptidique IV (1) - L’hexafluorophosphate de benzotriazolyl A-oxytrisdimethylamino phosphonium (B.O.P.). TetrahedronLett. 1975; 16:1219-1222.
74. Castro B, Dormoy J-R, Dourtoglou B, Evin G, Selve C, Ziegler J-C. A convenient synthesis of 3-Imino-2-phenylindazolines. Synthesis 1976; 11:751.
75. Hudson D. Methodological implications of simultaneous solid-phase peptide synthesis. 1. Comparison of different coupling procedures. J. Org. Chem. 1988; 53:617-624.
76. Chen S, Xu J. Pentafluorophenyl diphenylphosphinate anew efficient coupling reagent in pepetide. Tetrahedron Lett. 1991; 32:6711-6714.
77. Coste J, Le-Nguyen D, Castro B. PyBOP®: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron. Lett. 1990; 31:205-208.
78. Coste J, Frerot E, Jouin P, Castro B. Oxybenzotriazole free peptide coupling reagents for A-methylated amino acids. Tetrahedron Lett. 1991; 32:1967-1970.
79. Castro B, Dormoy JR. Le perchlorate de chlorotrisdimethy-laminophosphonium: un nouveau reactif pour le couplage peptidique. Tetrahedron Lett. 1972; 13:4747-4750.
80. Coste J, Dufour M-N, Pantaloni A, Castro B. BROP: a new reagent for coupling A-methylated amino acids. Tetrahedron Lett. 1990; 31:669-672.
81. Yamada S, Ikota N, Shioiri T. Diphenyl phosphorazidate (DPPA) and diethyl phosphorocyanidate (DEPC). Two new reagents for solid-phase peptide synthesis and their application to the synthesis of porcine motilin. J. Am. Chem. Soc. 1975; 97:7174-7175.
82. Yang T, Lin C, Fu H, Jiang Y, Zhao Y. Synthesis of sterically hindered peptide analogs using diphenyl phosphite as the coupling reagent. Biorg. Chem. 2005; 33:386-392.
83. Chen S, Xu J. Pentafluorophenyl diphenylphosphinate a new efficient coupling reagent in peptide chemistry. Tetrahedron Lett. 1991; 32:6711-6714.
84. Li H, Jiang X, Ye Y-H, Fan C, Romoff T, Goodman M. 3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT): a new coupling reagent with remarkable resistance to racemization. Org. Lett. 1999; 1:91-93.
85. Kim S, Chang H, Ko YK. Benzotriazol-1-yl diethyl phosphate. Anew convenient coupling reagent for the synthesis of amides and peptides. Tetrahedron Lett. 1985; 26:1341-1342.
86. Diago-Meseguer J, Palomo-Coll AL, Fernandez-Lizarde JR, Zugaza-Bilbao A. A new reagent for activating carboxyl groups; preparation and reactions of N,N-Bis[2-oxo-3-ox-azolidinyl. phosphorodiamidic chloride. Synthesis 1980; 7:547.
87. Dourtoglou V, Ziegler J-C, Gross B. L’hexafluorophosphate de)-benzotriazolyl-A,A-tetramethyluronium:un reactif de couplage peptidique nouveau et efficace. Tetrahedron Lett. 1978; 19: 1269-1272.
88. Knorr R, Trzeciak A, Bannwarth W, Gillessen D. New coupling reagents in peptide chemistry. Tetrahedron Lett. 1989; 30:1927-1930.
89. Abdelmoty I, Albericio F, Carpino LA, Foxman BM, Kates SA. Structural studies of reagents for peptides bond formation: crystal and molecular structures of HBTU and HATU. Lett. Peptide Sci. 1994; 1:57-67.
90. Carpino LA, Henklein P, Foxman BM, Abdelmoty I, Costisella B, Wray V, Domke T, El-Faham A, Miigge C. The solid state and solution structure of HAPyU. J. Org. Chem. 2001; 66:5245-5247.
91. Carpino LA, Imazumi H, El-Faham A, Ferrer FJ, Zhang C, Lee Y, Foxman BM, Henklein P, Hanay C, Miigge C, Wenschuh H, Klose J, Beyermann M, Bienert M. The uronium/guanidinium peptide coupling reagents: finally the true uronium salts. Angew. Chem. Int. Ed. 2002; 41:441-445.
92. Chen S, Xu J. A new coupling reagent for peptide synthesis. Benzotriazolyloxy-bis (pyrrolidino)-carbonium hexaflouorophosphate (BBC). Tetrahedron Lett. 1992; 33:647-650.
93. Albericio F, Cases M, Alsina J, Triolo SA, Carpino LA, Kates SA. On the use of PyAOP, a phosphonium salt derived from HOAt, in solid-phase peptide synthesis. Tetrahedron Lett. 1997; 38:4853-4856.
94. Ehrlich A, Rothemund S, Brudel M, Beyermann M, Carpino LA, Bienert M. Synthesis of cyclic peptides via efficient new coupling reagents. Tetrahedron Lett. 1993; 34:4781-4784.
95. Akaji K, Kuriyama N, Kiso Y. Convergent synthesis of (-)-Mirabazole C using a chloroimidazolidium coupling reagent, CIP. J. Org. Chem. 1996; 61:3350-3357.
96. El-Faham A. Bis(tetramethylene)fluoroformamidinium Hexaflu-orophosphate (BTFFH): a convenient coupling reagent for solid phase peptide synthesis. Chem. Lett. 1998; 27:671-672.
97. Albericio F, Bailen MA, Chinchilla R, Dodsworth DJ, Najera C. 2-Mercaptopyridine-1-oxide-based peptide coupling reagents. Tetrahedron 2001; 57:9607-9613.
98. Carpino LA, El-Faham A, Albericio F. Racemization studies during solid-phase peptide synthesis using azabenzotriazole-based coupling reagents. Tetrahedron Lett. 1994; 35:2279-2282.
99. Stowasser B, Budt K-H, Jian-Qi L, Peyman A, Ruppert D. New hybrid transition state analog inhibitors of HIV protease with peripheric C2-symmetry. Tetrahedron Lett. 1992; 33:6625-6628.
100. Li P, Xu J-C. New and highly efficient immonium type peptide coupling reagents: synthesis, mechanism and application. Tetrahedron 2000; 56:4437-4445.
101. Li P, Xu J-C. 5-(1H-Benzotriazol-1-yloxy)-3,4-dihydro-1-methyl 2H-Pyrrolium Hexachloroantimonate (BDMP®): a highly efficient immonium type peptide coupling reagent. Chem. Lett. 1999; 28:1163-1164.
102. Kunishima C, Kawashi C, Morita J, Terao K, Iwasaki F, Tani S. 4(4,6-Dimethyl-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride: an efficient condensing agent leading to the formation of amides and esters. Tetrahedron 1999; 55:13159-13170.
103. Taylor EC, Schrader TH, Walensky LD. Synthesis of 10-substitued open-chain analogues of 5,10-Dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF, Lometrexol). Tetrahedron 1992; 48:19-32.
104. Bald E, Saigo K, Mukaiyama T. A facile synthesis of carboxamides by using 1-methyl-2-halopyridimium iodides as coupling reagents. Chem. Lett. 1975; 4:1163-1164.
105. Huang H, Iwasawa N, Mukaiyama T. A convenient method for the construction of β-lactam compounds from β-amino acids using 2-chloro-1-methylpyridinium iodide as condensing reagent. Chem. Lett. 1984; 13:1465-1466.
106. Xu JC, Li P. A novel thiazolium type peptide coupling reagent for hindered amino acids. Tetrahedron Lett. 1999; 40:8301-8304.
107. Li P, Xu J-C. 1-Ethyl 2-Halopyridinium salts, highly efficient coupling reagents for hindered peptide synthesis both in solution and the solid-phase. Tetrahedron 2000; 56:8119-8131.
108. Woodward R, Olofson RA, Mayer M. A new synthesis of peptides. J. Am. Chem. Soc. 1961; 83:1010-1012.
109. Kimmerlin T, Seebach D. ‘100 years of peptide synthesis’: ligation methods for peptide and protein synthesis with applications to β-peptide assemblies. J. Peptide Res. 2005; 65:229-260.
110. Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science 1994; 266:776-779.
111. Lu W, Qasim MA, Kent SBH. Comparative total syntheses of turkey ovomucoid third domain by both stepwise solid phase peptide synthesis and native chemical ligation. J. Am. Chem. Soc. 1996; 118:8518-8523.
112. Canne LE, Walker SM, Kent SBH. A general method for the synthesis of thioester resin linkers for use in the solid phase synthesis of peptide-α-thioacids. Tetrahedron Lett. 1996; 36:1217-1220.
113. Sewing A, Hilvert D. Fmoc-compatible solid-phase peptide synthesis of long C-terminal peptide thioesters. Angew. Chem. Int. Ed. 2001; 40:3395-3396.
114. Chen G, Warren JD, Chen J, Wu B, Wan Q, Danishefsky SJ. Studies related to the relative thermodynamic stability of C-terminal peptidyl esters of O-hydroxy thiophenol: emergence of a doable strategy for non-cysteine ligation applicable to the chemical synthesis of glycopeptides. J. Am. Chem. Soc. 2006; 128:7460-7462.
115. Tam JP, Yu Q. Methionine ligation strategy in the biomimetic synthesis of parathyroid hormones. Biopolymers 1998; 46:319-327.
116. Roelfes G, Hilvert D. Incorporation of selenomethionine into proteins through selenohomocysteine-mediated ligation. Angew. Chem. Int. Ed. 2003; 42:2275-2277.
117. Lianshan Z, Tam JP. Orthogonal coupling of unprotected peptide segments through histidyl amino terminus. Tetrahedron Lett. 1997; 38:3-6.
118. Soellner MB, Nilsson BL, Raines RT. Staudinger ligation of α-azido acids retains stereochemistry. Staudinger ligation of α-azido acids retains stereochemistry. J. Org. Chem. 2002; 67:4993-4996.
119. Temelkoff DP, Smith CR, Kibler DA, McKee S, Duncan SJ, Zeller M, Hunsen M, Norris P. Application of bis(diphenylphosphino)ethane (DPPE) in Staudinger-type N-glycopyranosyl amide synthesis. Carbohydrate Res. 2006; 341:1645-1656.
120. He Y, Hinklin RJ, Chang J, Kiessling LL. Stereoselective N-glycosylation by staudinger ligation. Org. Lett. 2004; 6:4479-4482.
121. David O, Meester WJN, Bieraugel H, Schoemaker HE, Hiemstra H, van Maarseveen JH. Intramolecular staudinger ligation: a powerful ring-closure method to form medium-sized lactams. Angew. Chem. Int. Ed. 2003; 42:4373-4375.
122. Schellenberger V, Jakubke H-D. Protease-catalysed kinetically controlled peptide synthesis. Angew. Chem. Int. Ed. Engl. 1991; 30:1437-1449.
123. Moree WJ, Sears P, Kawashiro K, Witte K, Wong CH. Exploitation of Subtilisin BPN’ as catalyst for the synthesis of peptides containing noncoded amino acids, peptide mimetics and peptide conjuguates. J. Am. Chem. 1997; 119:3942-3947.
124. Gotor V. Non-conventional hydrolase chemistry: amide and carbamate bond formation catalysed by lipases. Biorg. Med. Chem. 1999; 7:2189-2197.
125. Fischer A, Bommarius AS, Drauz K, Wandrey C. A novel approach to enzymatic peptide synthesis using highly solubilizing N[α]-protecting groups of amino acids. Biocatalysis 1994; 8:289-307.
Han S-Y, Kim Y-A. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron. 2004; 60:2447-2467.
Montalbetti CAGN, Falque V. Amide bond formation and peptide coupling. Tetrahedron 2005; 61:10827-10852.
Pearson, AJ, Roush, WR, eds. Handbook of Reagents for Organic Synthesis: Activating Agents and Protecting Groups. 1999. John Wiley & Sons, New York.
Zabicky, J, ed. Synthesis of Amides. 1970. Interscience, London.
Biomolecules, Asymmetric Synthesis of
Biomolecules, Enzymatic Synthesis of
Chemical Ligation: Peptide Synthesis
Organic Chemistry in Biology
Peptide Combinatorial Synthesis
Peptide Synthesis
Small Molecules Synthesis, Key Reaction of