CHEMICAL BIOLOGY
Protein and DNA Thermostability, Physics and Evolution of
Igor N. Berezovsky, Computational Biology Unit, Bergen Center for Computational Science, University of Bergen, Bergen, Norway
doi: 10.1002/9780470048672.wecb638
Adaptation to different environmental temperatures establishes specific requirements on the stability of DNA and protein macromolecules. Organismal strategies of thermophilic adaptation, structure- and sequence-based, and their physical origins provide a consistent picture of the evolution of protein thermostability. A strong correlation between the optimal growth temperature (OGT) and the frequency of ApG dinucleotides in both sense and antisense strands of genomic DNA along with the absence of any ''thermophilic'' bias in the nucleotide composition highlights a key role of base stacking in the thermostabilization of the DNA double helix. The codon bias provides an excess of ApG pairs, which ensures the thermophilic adaptation of genomic DNA. The concentration of seven amino acids, Ile, Val, Tyr, Trp, Arg, Glu, Leu (IVYWREL), serves as a universal proteomic predictor of the OGT prokaryotes. The IVYWREL combination manifests a generic ''thermophilic'' trend in amino acid composition: the increase of hydrophobic and charged residues at the expense of polar ones. This so-called ''from both ends of the hydrophobicity scale'' trend is a result of the positive (stabilizing the native state) and the negative (destabilizing misfolded conformations) components of protein design. The pressure to preserve energies of important native and non-native contacts results in a correlation in mutations of amino acid residues involved into these contacts. A comparison of energy (Myiazawa-Jernigan potential) and substitution (BLOSUM62) matrices reveals a high rate of substitutions between amino acids that strongly attract each other (native contacts) and between residues that strongly repel each other (non-native contacts).
What makes thermophilic adaptation so attractive for researchers from the very beginning of protein and DNA molecular studies (1,2)? Although life exists in different extreme conditions, such as temperature, pressure, salinity, radioactivity (3), the adaptation to extreme temperatures is an outstanding phenomenon. Indeed, organisms belonging to one level of organization, prokaryotes, thrive under environmental temperatures that cover the entire range from -10 to +110 °C, one third of the absolute temperature interval. A significant difference in the optimal growth temperature of prokaryotes results in a distinct stability of their proteins and DNA, which makes them a central subject in the studies of mechanisms of molecular adaptation.
The thermostabilization of biomolecules is a result of the mutual contribution from fundamental interactions [e.g., hydrophobic forces (4, 5) or ionic interactions (3, 6, 7)] that stabilize individual molecules and prevent their aggregation (6), structure modifications [such as DNA superhelicity (8, 9) and posttranslational modification of proteins], interactions with an environment (10), intermolecular interactions (11), and oligomerization (12). The possible dependence of fundamental interactions, for example, hydrophobic forces, on temperature may also affect stability. However, it remains a subject of controversy as to how and to what extent the dependence of the interaction strength on temperature should be taken into account (13-16). This article reviews the very basic level of protein and DNA thermostability, fundamental interactions, and their sequence/structure determinants.
Basics of Protein and DNA Thermostability
Various factors that contribute to protein thermostability, such as van der Waals interactions (17), core hydrophobicity (18-20), networks of hydrogen bonds (4, 5, 21), amount of secondary structure (4, 22), ionic interactions (6, 7, 23), packing density (24), and decreased length of surface loops (25), have been a subject of intense study for several decades. The major challenge, however, is to find out how the above factors are chosen and their combinations are formed by natural selection responding to the environmental temperature and depending on the evolutionary history of the organism (26).
Thermostabilization of double-stranded DNA is provided by base pairing (1) and base stacking (see Reference 27 and references therein) complemented by positive supercoiling by reverse gyrase [in hyperthermophiles (8, 9, 28)] and by stabilization via interactions with histone-like proteins (29, 30). The relative contribution of base paring and base stacking into the thermostability of double-stranded DNA has been a subject of extensive studies for more than four decades (1, 27, 31). We will consider here this question, based on the results of recent experimental and computational works (31, 32).
Sequence/Structure Signals of Thermophilic Adaptation
Major “recipes” for increasing the thermostability observed in previous computational sequence/structure analysis and confirmed in experiment vary from the optimization of hydrophobic core interactions (18-20) to the introduction of additional ion pairs (7, 33). Respectively, thermophilic trends known so far include a large difference between the proportion of charged (DEKR) versus polar (noncharged, NQST) residues (34, 35), an increase of long and branched side chain hydrophobic residues (36), an excess of some aromatic amino acid (35), and an over-representation of Pro (37). Recently, Sslensminde et al. (37) illuminated the importance of structure dependence in the relationship between amino acid composition and optimal growth temperature (OGT) of the organism. In particular, the difference in amino acid frequencies between core and surface residues is getting more pronounced under higher temperatures (38), but not during adaptation to a cold environment (37). It also was demonstrated that amino acid biases in thermophilic adaptation are independent of the (G + C) content of coding nucleotide sequences, and the (G + C) content itself is not a determinant of the thermophilic adaptation of the double-stranded DNA (39-41).
Experimental (Re)Design of Thermostable Proteins
All experimental techniques of protein thermostabilization can be related to one of three major directions (42). First, the rational design concept is based on using previously known stabilizing factors. The limited predictive power of the rational design concept prompts one to test all potentially thermostabilizing mutations by using site-directed mutagenesis. The second approach is a directed evolution approach. Selective pressure or screening for a desired trait applied after random mutagenesis and/or DNA shuffling provides another possibility for engineering protein stability. Limited sequence space, amenable to testing in directed evolution, makes it necessary to eliminate more effectively the neutral and deleterious mutations, to increase the number of recombination events, and to improve the selection tools. Third, the “consensus concept” is based on the assumption that consensus amino acid contributes more to the stability of the protein chain than the nonconsensus amino acid at a given position in the alignment of the amino acid sequences.
Physics and Evolution of Thermophilic Adaptation
The tight connection between the “recipes” for thermostability immediately raises a question about the common evolutionary and/or physical basis for the variety of mechanisms of thermophilic adaptation. To address this question, one has to go beyond the analysis of specific stabilizing interactions and their various combinations. Conceptually, then, two major directions lead to the selection of proteins with high thermal stability. First, thermostable proteins have a structural bias such as enhanced packing. Second, stabilization is achieved by a few particularly strong, strategically placed interactions. The choice between these directions is affected by several evolutionary and environmental factors, and thermostabilization is a result of the intimate interaction between the physics of protein stability and the phylogenetics of the host organism.
Physical Basics of Protein Thermostability
Given a structural similarity between meso- and thermophilic orthologs, the variation of the stability across different proteins stems from the differences in the physical mechanisms of thermostability (3). Sequence/structure analysis and the unfolding simulations of hyperthermophilic proteins and their mesophilic homologues (43) reveal two major mechanisms of thermostabilization (Table 1). The first mechanism is “structure-based.” Some hyperthrmophilic proteins are significantly more stable than their mesophilic homologues because of their high compactness (44). In this case, no single type of interaction is extremely strong and dominates stabilization, but the sheer number of interactions provides enhanced stability. Structure-based thermostability is nonspecific in the sense that no or minimal special features of sequences are needed to achieve thermostability, which makes it robust under a wide range of environmental conditions. A possible evolutionary disadvantage of such a robust stabilization mechanism is that it makes the protein less adaptable to rapid and specific changes in environmental conditions. The second mechanism is “sequence-based.” This way, several substitutions made in sequences of mesophilic proteins provide the formation of “staples,” that is, specific and strong interactions that do not significantly alter the protein structure. Therefore, just a few strategic substitutions in the sequence can lead to a significant stabilization of the existing structure through the formation of several strong interactions specific to certain demands of the environment. These “staples” work locally, which leaves the bulk of the structure and its compactness unchanged. A possible disadvantage to this mechanism, however, also exists. Sequence-based stabilization may not be robust because it is tailored typically to a specific and narrow range of environmental conditions.
Table 1. Important features of structure-based and sequence-based strategies of thermophilic adaptation
|
Strategy of adaptation |
Structure-based |
Sequence-based |
|
Important features |
Enhanced packing of the structure |
Small number of strong strategically placed interactions |
|
|
||
|
|
Compactness |
Bulk of the structure is not changed |
|
Advantage(s) |
No or minimal demands on sequence specificity Robust under a wide range of conditions |
Provides fast adaptation |
|
Disadvantage(s) |
Less adaptable to changes of environment |
Tailored to narrow range of conditions |
|
|
Role of Evolutionary History in Establishing Organismal Strategies of Thermophilic Adaptation
Structure- and sequence-based mechanisms of stabilization were sequestered en route of protein evolution into distinct organ- ismal strategies of thermophilic adaptation. Some ancient organisms, for example, hyperthermophilic archaebacteria, started in hot conditions (45) and developed adaptation mechanisms “from scratch.” Thermostable proteins in these organisms were designed de novo by the concomitant selection of sequences and structures. This selection introduces evolutionary pressure toward a more designable structure. Designability is a property of protein structure that indicates how many sequences can fold into that structure at various levels of stability (46, 47). Designability of structure is reflected in the certain properties of the contact matrix of a structure, C (44, 46). In particular, designability correlates with the second order of C (Tr2C), that is, the compactness of a structure (number of contacts per residue). It was demonstrated (44) that more designable structures provide initial advantage because a greater number of sequences can fold into them with low energy. Therefore, a sequence search in the design of a thermostable protein will be less severe given a highly designable structure. A high contact density of LUCA domains (48) is suggestive, which shows that nature used high designability in the creation of the first thermostable proteins for ancient species. The role of designability in the design of ancient thermostable proteins is corroborated additionally by the high-throughput analysis of major folds (43). The Van der Waals contact density in hyperthermophilic archaea Pyrococcus is higher than in hyperthermophilic (T. maritima and A. aeolicus) or mesophilic (E. coli) bacteria. It indicates that on the organismal level, archaea used a structure-based mechanism and developed a respective strategy of thermophilic adaptation. What evolutionary scenario can one imagine for the emergence of another, sequence-based, strategy of adaptation? When mesophilic organisms recolonized in a hot environment, it was necessary to find a fast and effective way of tuning protein stability. To increase the stability of the protein without a redesign of the whole structure is possible via making sequence substitutions that introduce “staples,” a restricted set of a strong specific interaction (e.g., ion pairs). Hyperthermophilic bacteria (T. maritima and A. aeolicus), which recolonized in hot conditions, exemplify a sequence-based strategy. A high-throughput comparison of T. maritima and A. aeolicus proteomes with those of hyperthermophilic archaea shows the crucial role of sequence-based strategy in achieving the thermostability of proteins in hyperthermophilic bacteria (43). An analysis of the phylogenetic relationships between hyperythermophilic archaea and bacteria provides additional evidence for different organismal strategies of adaptation. 24% and 16% of the genes of T. maritima and A. aeolicus, respectively, were transferred to bacteria via lateral gene transfer (LGT) from archaea (49, 50), and corresponding bacterial proteins are the most similar to those of archaea. The importance of LGT in specific biochemical and environmental adaptations was demonstrated undoubtedly by the comparison of complete genomes, codon analysis within genomes, and phylogenetic trees based on single gene families (see Reference 51 and references therein). Alternatively, it may be problematic to assess the relative contributions of LGT and vertical inheritance. For example, T. maritima and A. aeolicus belong to two lineages (Thermotogales and Aquificales) believed to have diverged earliest from the rest of bacteria. Therefore, it is possible that T. maritima and A. aeolicus retained ancestral genes and share some primitive features with archaea, whereas these genes were lost in the rest of the bacterial species. However, regardless of the scenario working in Thermotoga and Aquifex (genes are received via LGT or, alternatively, are descendants of retained ancestral ones), the so-called “archaeal” parts of their genomes are reflective of the hyperthermophilic lifestyle and the distant evolutionary past (51). In particular, the archaeal parts of the above bacterial proteomes (extracted according to the listing in the taxonomic distributions of the homolog TaxMap, available athttp://www.ncbi.nlm.nih.gov) exhibit compositional features typical for structure-based strategy, whereas the bacterial parts follow a sequence-based strategy of thermophilic adaptation (43). Later events in protein evolution affected structures/sequences of both archaeal and bacterial species which combine strategies of adaptation (52) or use complementary mechanisms of stability (53).
Genomics/Proteomics of Thermophilic Adaptation
A better understanding of how nature adapts life to elevated temperatures of the environment helps us to get a deeper insight into the basic physical laws that govern protein design. In particular, the explosion of information on crystallized proteins and complete genomes/proteomes makes it realistic to perform high-throughput analysis of sequences and structures. In this part, we will show how 1) alignments of proteomic sequences reveal a signal of a new entropic mechanism of thermostability, 2) exhaustive enumeration of all possible combinations of amino acid residues identifies a particular combination of them, which can serve as the best predictor of the optimal growth temperature (OGT) prokaryotes, and 3) correlation analysis of coding nucleotide sequences illuminates the major role of stacking in the thermostabilization of double-stranded DNA and shows that stabilization by stacking is provided by the codon bias.
Entropic Mechanism of Protein Thermostability
The compositional bias toward increasing charged residues in (hyper)thermophilic proteomes compared with mesophilic ones is well documented. However, the enrichment in positively charged residues is almost entirely because of lysines (34) (see Table 2). If only the total content of arginine (Arg) plus lysine (Lys) residues would matter in determining the stability of hyperthermophilic proteins, then no preference for the Lys over the Arg should exist. Arg and Lys are similar residues by their physical and chemical features; both residues are charged and have the same maximal number [81] of possible rotamers. An examination of the substitutions of types Arg/Lys versus Lys/Arg in the alignments of mesophilic sequences versus hyperthermophilic ones (Fig. 1) sheds light on the relationship between Arg and Lys content. The number before the slash (Table 3) is the percentage of amino acid residues in the mesophilic sequence, for example, Arg that was replaced by the other amino acid in the hyperthermophilic sequence, for example, Lys. The number after the slash reflects the same data for the opposite replacement, for example, Lys in the mesophilic sequence replaced by Arg in the hyperthermophilic sequence. Numbers in parenthesis show the ratio of forward to backward substitutions. The control groups are pairs Leu/Ile and Ser/Thr. Residues in each pair possess similar physical and chemical features (Leu/Ile are hydrophobic; Ser/Thr-are polar), and both have the same maximal number of possible rotamers (9 and 3, respectively). In nine hyperthermophilic organisms, the pairs RK/KR demonstrate a remarkable bias toward the replacement of arginine in the mesophilic sequence with lysine in the hyperthermophilic sequence (up to almost four times in N. equitans). In all alignments of E. coli sequences against those from a hyperthermophilic genome, numbers of residues substitutions in control pairs Leu/Ile and Ser/Thr are equal or very similar. The exceptions are pairs LI/IL and RK/KR in A. pernix and M. kandleri, which show bias in the opposite (hyperthermo-to-meso) direction, perhaps, as a consequence of high GC content (53). The above observation challenges the idea that arginine and lysine play the same role in thermostability (34) and hints to the specific role of lysine in protein stabilization. The complementary all-atom unfolding simulations show that lysines have a much greater number of accessible rotamers than arginines of similar degree of burial in folded states of proteins (53). Significant residual dynamics of lysine in folded states of proteins make the entropic cost to fold lysine-rich proteins less favorable compared with arginine-rich ones. The arginine-to-lysine replacement stabilizes the folded state, preserving, however, the charged nature of the substitution position. Positively charged residues, therefore, are the choice of nature for the evolutionary optimization of hyperthermostable proteins via entropic mechanism.
Table 2. Percentage of charged amino acids and (G + C) content of 10 hyperthermophilic archaea (A), 2 hyperthermophilic bacteria (B), and mesophilic bacteria E. coli. A strong prevalence of lysine over arginine in proteomes of hyperthermophiles is obtained for nine organisms. A bold font marks the exception from the general trend
|
|
EC |
AA |
AF |
MJ |
NE |
PA |
PH |
PF |
ST |
TM |
AP |
MK |
|
Lys (K) |
4.4 |
9.4 |
6.9 |
10.3 |
10.8 |
7.8 |
8.0 |
8.1 |
8.0 |
7.6 |
4.0 |
4.0 |
|
Arg (R) |
5.5 |
4.9 |
5.8 |
3.9 |
3.9 |
5.7 |
5.6 |
5.3 |
4.2 |
5.5 |
7.8 |
8.3 |
|
Gln (E) |
5.8 |
9.6 |
8.9 |
8.6 |
7.9 |
8.9 |
8.7 |
8.9 |
7.0 |
8.9 |
7.3 |
10.0 |
|
Asp (D) |
5.1 |
4.3 |
5.8 |
5.5 |
5.0 |
4.6 |
4.4 |
4.4 |
4.6 |
5.0 |
4.2 |
5.8 |
|
G + C content |
50.8 |
43.5 |
48.6 |
31.4 |
31.6 |
44.7 |
41.9 |
40.8 |
32.8 |
46.2 |
56.3 |
62.1 |
|
Life kingdom |
B |
B |
A |
A |
A |
A |
A |
A |
A |
B |
A |
A |
A, archaea; AA, A. aeolicus; AF, A. fulgidus; AP, A. pernix; B, bacteria; EC, E. coli; MJ, M. jannaschii; MK, M. kandleri; NE, N. equitans; PA, P. abyssi; PH, P. horikoschii; PF, P. furiosis; ST, S. tokodaii; TM, T. maritima.
Table 3. Percentage of the forward/backward replacements in alignments of hyperthermophilic genomes against mesophilic E. coli. A bold font shows two hyperthrmophilic organisms without the general trend of arginine-to-lysine replacement
|
Hyperthermophilic genome |
RK/KR |
LI/IL |
TS/ST |
|
A. aeolicus |
20.0/8.1 (2.47) |
14.2/19.3 (0.74) |
7.5/6.8 (1.10) |
|
A. fulgidus |
14.5/10.6 (1.37) |
14.4/17.5 (0.83) |
8.4/6.6 (1.27) |
|
M. jannaschii |
22.4/6.0 (3.73) |
20/16.7 (1.2) |
7.0/6.5 (1.08) |
|
N. equitans |
23.7/6 (3.95) |
19.5/19 (1.03) |
6.8/6.8 (1.00) |
|
P. abyssi |
16.3/10.0 (1.63) |
16.1/18.3 (0.88) |
7.7/7.0 (1.10) |
|
P. horikoschii |
16.7/9.6 (1.74) |
16.7/18.3 (0.92) |
8.1/7.2 (1.13) |
|
P. furiosis |
16.5/9.9 (1.67) |
16.3/18.2 (0.90) |
7.8/7.5 (1.04) |
|
S. tokodaii |
18.2/7.4 (2.46) |
18.5/17.8 (1.04) |
9.8/7.3 (1.34) |
|
T. maritima |
16.3/9.5 (1.72) |
14.2/18.3 (0.78) |
8.6/7.7 (1.17) |
|
A. pernix |
8.1/15.6 (0.52) |
10.7/20.9 (0.52) |
9.8/7.2 (1.36) |
|
M. kandleri |
8.1/15.8 (0.51) |
9. 8/19.1 (0.51) |
6.6/7.0 (0.95) |
A, archaea; AA, A. aeolicus; AF, A. fulgidus; AP, A. pernix; B, bacteria; EC, E. coli; MJ, M. jannaschii; MK, M. kandleri; NE, N. equitans; PA, P. abyssi; PH, P. horikoschii; PF, P.furiosis; ST, S. tokodaii; TM, T. maritima.
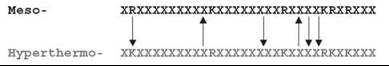
Figure 1. Scheme of the pair - wise alignment of mesophilic versus hyperthermophilic coding sequences. Only extended segments of alignments were considered (length 45 residues or larger) with gaps less than 3 residues and high sequence similarity (e = 0.05).
Proteomic Sequence Determinant of Thermophilic Adaptation
The availability of complete genomes/proteomes makes it possible to search systematically for the combination of amino acid residues, which is most important for protein themostability (41, 54). An exhaustive enumeration of all possible subsets of 20 amino acids is performed by representing sets of amino acids by vectors, where each component ai of the vector takes the value of 1 if the amino acid of type i is presented in the set and 0 otherwise. Thus, 219 — 2 = 524,286 linearly independent nontrivial combinations exist. Given fi(j), the fraction of amino acid i in proteome j, total fraction F(j)of the amino acids from a particular subset, ![]() Linear regression between the values of F(j) and the optimal growth temperature (OGT) of the organism allows us to determine the best predictor of OGT. For 86 complete proteomes of prokaryotes thriving under temperatures from —10 to +110 °C, the combination of Ile, Val, Tyr, Trp, Arg, Glu, and Leu (IVYWREL) gives the highest correlation coefficient between the fraction (FIVYWREL) in the proteome and OGT of the organism. The correlation coefficient R is 0.930, and the quantitative relationship between the OGT (in degrees Celsius) and fraction F of IVYWREL amino acids reads Topt = 937F-335. The accuracy of Topt prediction (root-mean-square deviation) is 8.9 °C. Additional analysis of thermostability predictors of major protein folds shows that they are very similar to universal combination IVYWREL. The correlation coefficient between IVYWREL content in sequences of the most abundant protein folds and OGT is very high, for example, α/β barrel (R = 0.87), β barrel (0.87), Rossman fold (0.86), and bundle (0.82). However, if complementary mechanisms of stabilization are invoked, such as heme and metal binding (globin, cytochrome C, ferredoxin) or S-S bridges (lysozyme), then the correlation coefficient is significantly lower: 0.53, 0.44, 0.45, and 0.5, respectively. Thermostability predictors for two major types of membrane proteins, α-helical bundle and β-barrel, reveal the low slope of the correlation FIVYWREL with OGT in the former and the CVYP predictor in the latter. This result suggests that thermal adaptation in membrane proteins is governed by different rules than in globular ones, in particular, the stability and folding of membrane proteins are affected by the interactions with the lipid bilayer (55). Various control tests show the statistical significance and robustness of the IVYWREL predictor (32). The IVYWREL fraction is a better predictor of thermostability than fractions of charged Asp, Glu, Lys, and Arg (DEKR) or hydrophobic Ile, Val, Trp, and Leu (IVWL) amino acids, which predict OGT with the accuracy 21 and 16.8 °C, respectively. Thus, both hydrophobic and charged residues are important for achieving thermostabilization, contrary to earlier beliefs that only hydrophobic or charged residues are major determinants of thermostability. In addition to IVYWREL amino acids, several residues exist that are favorable for thermostabilization. For example, the addition of Met (M) or a combination of Phe and Pro (F,P) results in the correlation coefficient 0.921 and 0.917 for the predictor, and the substitution of Trp (W) into His (H) or the substitution of pair Trp, Arg (W,R) into Gly, Pro (G,P) gives the correlation coefficient 0.914 and 0.902, respectively. Importantly, Ala (A) and Gln (Q) are extremely disadvantageous for thermostabilization. If Ala (A) or the combination Ala, Gln (A,Q) are added to IVYWREL it practically destroys a predictor (R = 0.47 and 0.24). The same situation is observed when Ala (A) or a combination Ala, Gln (A,Q) replaces Glu (E) or a combination Val, Glu (V,E): The correlation coefficient R is 0.18 and 0.23, respectively.
Linear regression between the values of F(j) and the optimal growth temperature (OGT) of the organism allows us to determine the best predictor of OGT. For 86 complete proteomes of prokaryotes thriving under temperatures from —10 to +110 °C, the combination of Ile, Val, Tyr, Trp, Arg, Glu, and Leu (IVYWREL) gives the highest correlation coefficient between the fraction (FIVYWREL) in the proteome and OGT of the organism. The correlation coefficient R is 0.930, and the quantitative relationship between the OGT (in degrees Celsius) and fraction F of IVYWREL amino acids reads Topt = 937F-335. The accuracy of Topt prediction (root-mean-square deviation) is 8.9 °C. Additional analysis of thermostability predictors of major protein folds shows that they are very similar to universal combination IVYWREL. The correlation coefficient between IVYWREL content in sequences of the most abundant protein folds and OGT is very high, for example, α/β barrel (R = 0.87), β barrel (0.87), Rossman fold (0.86), and bundle (0.82). However, if complementary mechanisms of stabilization are invoked, such as heme and metal binding (globin, cytochrome C, ferredoxin) or S-S bridges (lysozyme), then the correlation coefficient is significantly lower: 0.53, 0.44, 0.45, and 0.5, respectively. Thermostability predictors for two major types of membrane proteins, α-helical bundle and β-barrel, reveal the low slope of the correlation FIVYWREL with OGT in the former and the CVYP predictor in the latter. This result suggests that thermal adaptation in membrane proteins is governed by different rules than in globular ones, in particular, the stability and folding of membrane proteins are affected by the interactions with the lipid bilayer (55). Various control tests show the statistical significance and robustness of the IVYWREL predictor (32). The IVYWREL fraction is a better predictor of thermostability than fractions of charged Asp, Glu, Lys, and Arg (DEKR) or hydrophobic Ile, Val, Trp, and Leu (IVWL) amino acids, which predict OGT with the accuracy 21 and 16.8 °C, respectively. Thus, both hydrophobic and charged residues are important for achieving thermostabilization, contrary to earlier beliefs that only hydrophobic or charged residues are major determinants of thermostability. In addition to IVYWREL amino acids, several residues exist that are favorable for thermostabilization. For example, the addition of Met (M) or a combination of Phe and Pro (F,P) results in the correlation coefficient 0.921 and 0.917 for the predictor, and the substitution of Trp (W) into His (H) or the substitution of pair Trp, Arg (W,R) into Gly, Pro (G,P) gives the correlation coefficient 0.914 and 0.902, respectively. Importantly, Ala (A) and Gln (Q) are extremely disadvantageous for thermostabilization. If Ala (A) or the combination Ala, Gln (A,Q) are added to IVYWREL it practically destroys a predictor (R = 0.47 and 0.24). The same situation is observed when Ala (A) or a combination Ala, Gln (A,Q) replaces Glu (E) or a combination Val, Glu (V,E): The correlation coefficient R is 0.18 and 0.23, respectively.
Finally, the fundamental question in thermophilic adaptation is a relationship between the amino acid composition of proteins and the nucleotide composition of coding DNA sequences (39-41). The availability of prokaryotic complete genomes, which consist mostly of coding DNA (on average ~85% of the total genome size), clarifies a relationship between the thermophilic adaptation of protein and DNA. If the IVYWREL predictor depends on nucleotide composition only, it must remain the same after the reshuffling of coding sequences given a nucleotide composition. However, the reshuffling of nucleotide sequences results in a non IVYWREL thermostability predictor (32). Therefore, amino acid composition and thermal adaptation of proteins are not affected by the nucleotide composition of DNA sequences. The amino acid composition of the proteome, on the contrary, introduces a bias in the purine loading (A + G content) of nucleotide sequences. Indeed, purine loading of coding sequences reversely translated from protein sequences without codon bias, for example, by using synonymous codons with equal probabilities, is very close to a natural nucleotide sequence. The correlation coefficient between the purine loading and OGT is 0.48 and 0.6 in sequences without codon bias and natural ones, respectively (32).
Major Role of Stacking in DNA Thermostability
Pairing and stacking are two major factors of DNA stability. In a base pairing, the G●C pair contains three hydrogen bonds compared with the A●T pair that has two hydrogen bonds. The classical Marmur and Doty work (1) gives a linear relationship between the (G + C) content of the double-stranded DNA and its melting temperature, which strongly suggests that stability of the G●C and A●T pairs is different independently of their neighbors. This result originated a belief that DNA thermostability is provided mainly by pairing interactions and is achieved via (G + C) loading. Futhermore, the role of (G + C) content in establishing specific “thermophilic” biases in the amino acid composition of corresponding proteins was discussed extensively. A high-throughput analysis of genomic sequences conclusively demonstrates the absence of any connection between the (G + C) content and the OGT of the organism (39-41). The only bias in the nucleotide composition that correlates with the OGT is the (A + G) content (purine loading). It was shown, however, that purine loading is determined chiefly by the amino acid composition of proteins (32). Thus, thermostabilization of DNA does not work on the level of nucleotide composition.
The next step in the description of the DNA sequence is the analysis of the pair-wise nearest-neighbor correlations cij, for example, the normalized probabilities to find successive pairs of the nucleotides i and j. For all 16 possible successive dinucleotides in the coding strand of DNA, only the functions cAG and cCT correlate with OGT. The excess probabilities to find ApG and CpT pairs in the coding DNA are increasing significantly with OGT, correlation coefficient R = 0.68 and 0.601 (Fig. 2, upper row). Remarkably, the codon bias explains the observed sequence correlations in the coding parts of DNA. First, correlation in the nucleotide sequences does not depend on sequence correlations in amino acid sequences because removing an effect of the codon interface does not destroy a correlation between cAG/cCT and OGT, R = 0.736/0.574 (Fig. 2, middle row). Second, the correlation in DNA sequences stems from the neighboring nucleotides within a codon. Indeed, removing the natural codon bias results in eliminating the correlation between cAG/cCT and OGT, R = 0.177/0.216 (Fig. 2, bottom row). Thus, the codon bias establishes an excessive use of codons that contain successive ApG ans CpT pairs, which is manifested in the correlation of cAG/cCT with the optimal growth temperature. The above sequence correlations in the coding strand of DNA sequences point to base stacking as a major factor of DNA thermostabilization. ApG dinucleotides have a low energy characteristic for a purine-purine stacking. The cCT correlation also shows, although indirectly, the role of stacking in themostabilization. Indeed, the abundance of CpT pairs in the sense strand points to the equal enrichment of the antisense strand with ApG pairs because of the opposite directionality of sense and antisense strands of DNA. Therefore, the thermostabilization of double-stranded DNA is based on the stacking interactions provided by ApG pairs that are spread in different locations of both sense and antisense strands. This picture holds also for the whole DNA of prokaryotes, including its coding and noncoding parts. Therefore, in the scenario of thermophilic adaptation of double-stranded DNA, the stacking interactions play a major role. The codon bias provides an increase in the number of ApG dinucleotides with OGT in both sense and antisense strands of the DNA double helix. The necessity for ApG pairs can be explained by their low free energies of stacking obtained both theoretically (56) and experimentally (57). First, the study of the free energy contribution to the nucleic base stacking in aqueous solution shows that the free energy of stacking in order of decreasing stability follows the order purine-purine >> purine-pyrimidine>pyrimidine-purine>pyrimidine-pyrimidine in general, and the free energy of ApG stacking is one of the lowest in particular (56). Second, the experimental study on the coaxial stacking contribution to the stabilization of gel-immobilized duplexes reveals that adenine stacking with other bases is significantly stronger than the stacking of other bases (57). The reasons for the discrepancy between the latter and the parameters of duplex stability obtained in the nearest neighbor approximation are yet to be explored (see Reference 27 and References 16-19 therein).
Recent experimental efforts also corroborate a major role of the base stacking (31) in DNA thermostability and the independence of the latter on G●C base pairing (1). In particular, DNA stacking parameters are determined directly (31) for the temperatures from below room temperature to close to melting temperature and for the salt concentrations from 15 to 100 mM Na+. It seems that base stacking is the main stabilizing factor in the double-stranded DNA that determines the temperature and the salt dependence of DNA stability parameters. The A●T pairing is always destabilizing, and G●C pairing contributes almost no stabilization (31). It is important to note that base stacking interactions always are stabilizing for both A●T- and G●C-containing contacts in double-stranded DNA. Bioinformatics studies display the importance of stacking by showing the independence of the DNA thermostability on (G + C) content (32, 39-41) and by illuminating a specific role of ApG stacking in the thermostability of the DNA double helix via a consideration of pair-wise nearest-neighbor correlations (32) or a regression analysis of the dinucleotide composition of genomic DNA (41).
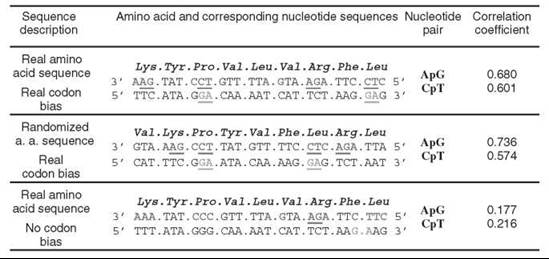
Figure 2. Base stacking provided by the correlations in nucleotide sequences is the major mechanism of DNA thermostability. Upper row. Real amino acid sequence and original codon bias. Middle row. The effect of codon interface is removed through the reshuffling of protein sequences while retaining the actual codons used for each amino acid. Bottom row. Codon bias in natural protein sequences is removed by using synonymous codons with equal probabilities. ApG and CpT pairs in the sense strand and ApG pairs in the antisense strand of DNA are underlined if they are located inside one codon. For example (upper row), the first ApG pair in the sense strand is in the Lys codon, whereas the second ApG pair is on the border between the codons of Leu and Val.
Minimalist Physical Model of Protein Thermostability
It is of a great importance for protein design to elucidate how physical principles work in the evolution of natural proteins and how they provide viability and adaptation to different environments. An analysis of individual prokaryotic and eukaryotic proteins reveals a direct connection between their stability (expressed in melting temperature, Tmelt) and the average living temperature of the organism, Tenv (4); hence, environmental temperature should be incorporated in the model of protein thermophilic design. In terms of statistical physics, the stability of the native state of a protein is determined by the Boltzmann factor exp(-∆E/kBT), where ∆E is the energy gap between the native state and the lowest energy completely misfolded structure (58, 59). This factor imposes a requirement on the energy gap: It must increase with the temperature (Fig. 3), and, as a result, the unique lowest energy native state will be preserved from the destruction by thermal fluctuations. The widening of the energy gap can be achieved by lowering the energy of the native state (positive design) and by increasing the energy of the misfolded structures (negative design) or by both processes working simultaneously.

Figure 3. The widening of the energy gap between the native state and the misfolded structures during an increase in the environmental temperature. Positive design provides the lowering of the native state energy, whereas negative design contributes to the increase of the energy of misfolded structures.
Design of Model Proteins with Selected Thermal Stability
The first approach to simulation of protein thermophilic adaptation is to start from a purely statistical-mechanical analysis of protein thermostability. A specific Monte-Carlo procedure [the so-called P-design (60, 61)] exists that maximizes the Boltzmann probability Pnat of being in the lowest energy (native state) conformation, ![]() where E0 is the lowest energy among all conformations and Tenv is the environmental temperature. It takes the environmental temperature Tenv as an input physical parameter, introduces mutations in the amino acid sequence, and accepts or rejects them according to the Metropolis criterion. As a result, this procedure designs proteins stable at given Tenv. The stability of designed proteins is characterized by their melting temperature Tmeltthat can be found numerically from the condition Pnat(Tmelt) = 0.5.
where E0 is the lowest energy among all conformations and Tenv is the environmental temperature. It takes the environmental temperature Tenv as an input physical parameter, introduces mutations in the amino acid sequence, and accepts or rejects them according to the Metropolis criterion. As a result, this procedure designs proteins stable at given Tenv. The stability of designed proteins is characterized by their melting temperature Tmeltthat can be found numerically from the condition Pnat(Tmelt) = 0.5.
"From Both Ends of the Hydrophobicity Scale'' Trend in Thermophilic Adaptation
The design of model protein for thermostability by using an exactly solvable lattice model [103346 compact conformations of 3 x 3 x 3 lattice proteins (62)] discovers the fundamental rules of thermophilic adaptation. First, the amino acid composition of designed proteins reveals a specific “thermophilic” trend: Thermostabilization is accompanied by an increase of the amount charged (DEKR) and the hydrophobic amino acids (MPCLVWIF) at the expense of weak hydrophobic and polar (AGNQSTHY) ones. Importantly, the amino acid composition of 83 proteomes of psycho-, meso-, thermo-, and hyperthermophilic prokaryotes reveals similar trend. Thus, the “from both ends of the hydriohibicty scale” trend, that is combining amino acids with the maximum variance in their hydrophobicity, observed in simulations is, indeed, crucial for the thermostabilization of proteins.
The “from both ends” trend is related to the positive and negative components of the design. The positive design is a major contributor to the temperature-dependent energy decrease of the native state, and the negative design ensures an increase of the average energy of misfolded structures (when an increase occurs in the Tenv). Interactions between strongly hydrophobic residues in the protein core and ion pairs formed by amino acids of the opposite charge on the protein surface are responsible for the positive design. The repulsion between charged residues of the similar sign contributes to the negative design by raising the average energy of misfolded conformations (see Fig. 3). Importantly, both positive and negative components of the design are based on the conservative native and non-native contacts between residues that play an especially important role in the stabilization of the native state and the destabilization of the misfolded conformations (62). Whereas identities of amino acids that form such a contact may vary from sequence to sequence, the strength (or interaction energy) of the key native and non-native contacts is preserved: These contacts are either strongly repulsive or strongly attractive for all sequences that fold into a given structure (see Fig. 6 in Reference 62).
Design simulations confirm an existence of the energy-conserved strongly attractive (native) and most repulsive (nonnative) contacts (62). The standard deviation of the contact energy is the lowest for these contacts. When the design is performed under hyperthermophilic temperatures, it results in stronger and more conserved (lower dispersion of the energy) native and non-native interactions compared with the design under the mesophilic temperatures (see Fig. 7 in Reference 62). As a result, the gap between the energies of native and misfolded structures is widening and the thermostability of the structure is increasing in response to the elevated environmental temperature Tenv.
Positive and Negative Design in Evolution and Thermal Adaptation of Natural Proteins
The requirement to preserve energy of key contacts in multiples sequences that fold into the same structure implies that amino acids forming such contacts should mutate in a correlated way. For example, correlated mutations may occur as swaps to keep specific attractive native and repulsive non-native interactions (see Fig. 8 in Reference 62). This scenario invokes a peculiar dependence between the amino acid substitution rates [e.g., BLOSUM matrices (63)] and the interaction energy between corresponding amino acid residues [e.g., the Miyazawa-Jernigan quasi-chemical potential (64)]. Frequent substitutions are expected between amino acids that strongly attract each other (to preserve specific stabilizing native contacts) and between amino acids that strongly repel each other (to preserve specific non-native repulsive contacts). The dependence of elements of substitution matrix BLOSUM (62, 63) for 190 pairs of amino acids (synonymous substitutions are excluded) versus their interaction energy as approximated by the knowledge-based Miyazawa-Jernigan potential (64) has a nonmonotonic nature (Fig. 4, top chart; the dependence is highlighted by the parabolic fit). The most frequent substitutions are observed between the most attractive and most repulsive amino acids. A blow-up of the right top part of Fig. 4 (bottom chart) shows that along with conserved substitutions that reflect a positive design (arginine to lysine and glutamic acid to aspartic acid substitutions), frequent substitutions exist between mutually repulsive amino acids with vastly different physical-chemical properties and encoded by very dissimilar amino codons, such as glutamine to arginine, serine to asparagine, and so forth (Fig. 4; bottom chart). The high frequencies of substitutions between residues that strongly repeal each other explain the correlated mutations observed between the residues that are distant in the native structure (62). These residues may form important repulsive contacts, which increase the energies of the misfolded conformations (see Fig. 10 in Reference 62).
Whereas a positive design is used widely in experiments (65), the big challenge in using the negative design originates from the difficulties in the modeling of relevant misfolded conformations (66). Nevertheless, charged residues were used effectively in negative design (65, 66). Site-directed mutagenesis provides other, although indirect, evidence of the contribution of charged residues to negative design: The mutation of polar groups to charged ones on the protein surface leads to structure stabilization even in the absence of the salt-bridge partners of the mutated group. It also has been shown (67-70) that surface electrostatic interactions provide a marginal contribution to stability of the native structure; hence, the possible importance of charged amino acids is in making unfavorable high-energy contacts in misfolded structures. In the case of thermophilic adaptation, positive and negative components of design work concomitantly and provide stabilization of the structure via an “opening” of the energy gap from both sides: A decreasing energy of the native state and, at the same time, an increasing energy of misfolded conformations can both exist.

Figure 4. The dependence of the elements of the BLOSUM62 substitution matrix on the interaction energies between amino acid residues (approximated by the Miyazawa-Jernigan parameter). Top chart. Only nonsynonymous substitutions are presented. The curved line represents the parabolic fit to highlight the nonmonotonic nature of the plot. Bottom chart. Blowup of the right upper corner of the top chart. Amino acid pairs are labeled, and pairs of amino acids that can contribute to positive and negative components of design are shown.
Conclusions
A deep understanding of the physical mechanisms and the evolution of thermophilic adaptation is crucial for the engineering and design of biologic catalysts with desired stability (20). This knowledge also is important for establishing a trade-off between the stability and flexibility in a directed evolution of protein function (66, 71). Current predictors of the stability effects of protein mutations are based on empirical potentials that are calibrated to fit experimentally observed ∆∆G values (20, 72, 73). Although predictions of ∆∆Gs during mutation in the native state are in a good agreement with experimentally observed ones, they lack the effect of mutations on misfolded conformations, the structure-dependence of mutation effects (37, 38), and the dependence of mutations on the evolutionary strategy of thermophilic adaptation (43).
Recent computational studies of thermophilic adaptation described in this article make use of genomic/proteomic data (32, 43, 53, 62), simulations of model lattice proteins (62), and off-lattice all-atom simulations of natural proteins (43, 53). High-throughput analysis reveals signals of novel mechanisms of protein [entropic mechanism (53)] and DNA [purine-purine base stacking (32)] thermostability and urges us to consider what evolutionary strategy was followed in the process of thermal adaptation (43). Proteomic analysis and simulations of thermophilic adaptation also demonstrate that negative design necessarily should be taken into account to properly predict the effect of protein mutations (62).
References
1. Marmur J, Doty P. Determination of the base composition of deoxyribonucleic acid from its thermal denaturation temperature. J. Mol. Biol. 1962; 5:109-118.
2. Perutz MF, Raidt H. Stereochemical basis of heat stability in bacterial ferredoxins and in haemoglobin A2. Nature 1975; 255:256- 259.
3. Jaenicke R, Bohm G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998; 8:738-748.
4. Gromiha MM, Oobatake M, Sarai A. Important amino acid properties for enhanced thermostability from mesophilic to thermophilic proteins. Biophys. Chem. 1999; 82:51-67.
5. Szilagyi A, Zavodszky P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure 2000; 8:493-504.
6. Greaves RB, Warwicker J. Mechanisms for stabilisation and the maintenance of solubility in proteins from thermophiles. BMC Struct. Biol. 2007; 7:18.
7. Xiao L, Honig B. Electrostatic contributions to the stability of hyperthermophilic proteins. J. Mol. Biol. 1999; 289:1435-1444.
8. Forterre P, Elie C. The Biochemistry of Archaea (Archaebacteria). Kates M, Kushner, D., and Matheson, A., eds. 1993. pp. 325-361.
9. Kikuchi A. Cozarelli NR, Wang JC, editors; 1990. pp. 285-298.
10. Marguet E, Forterre P. DNA stability at temperatures typical for hyperthermophiles. Nucleic Acids Res. 1994; 22:1681-1686.
11. Kirino H, Aoki M, Aoshima M, Hayashi Y, Ohba M, Yamagishi A, Wakagi T, Oshima T. Hydrophobic interaction at the subunit interface contributes to the thermostability of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur. J. Biochem. 1994; 220:275-281.
12. Tanaka Y, Tsumoto K, Yasutake Y, Umetsu M, Yao M, Fukada H, Tanaka I, Kumagai I. How oligomerization contributes to the thermostability of an archaeon protein. Protein L-isoaspartyl- O-methyltransferase from Sulfolobus tokodaii. J. Biol. Chem. 2004; 279:32957-32967.
13. Makhatadze GI, Privalov PL. Heat capacity of proteins. I. Partial molar heat capacity of individual amino acid residues in aqueous solution: hydration effect. J. Mol. Biol. 1990; 213:375-384.
14. Makhatadze GI, Privalov PL. Contribution of hydration to protein folding thermodynamics. I. The enthalpy of hydration. J. Mol. Biol. 1993; 232:639-659.
15. Prabhu NV, Sharp KA. Heat capacity in proteins. Annu. Rev. Phys. Chem. 2005; 56:521-548.
16. Privalov PL, Makhatadze GI. Contribution of hydration to protein folding thermodynamics. II. The entropy and Gibbs energy of hydration. J. Mol. Biol. 1993; 232:660-679.
17. Berezovsky IN, Tumanyan VG, Esipova NG. Representation of amino acid sequences in terms of interaction energy in protein globules. FEBS Lett. 1997; 418:43-46.
18. Chen J, Stites WE. Replacement of staphylococcal nuclease hydrophobic core residues with those from thermophilic ho- mologues indicates packing is improved in some thermostable proteins. J. Mol. Biol. 2004; 344:271-280.
19. Holder JB, Bennett AF, Chen J, Spencer DS, Byrne MP, Stites WE. Energetics of side chain packing in staphylococcal nuclease assessed by exchange of valines, isoleucines, and leucines. Biochemistry 2001; 40:13998-14003.
20. Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science 2005; 308:857-860.
21. Jaenicke R. Stability and folding of domain proteins. Prog. Biophys. Mol. Biol. 1999; 71:155-241.
22. Querol E, Perez-Pons JA, Mozo-Villarias A. Analysis of protein conformational characteristics related to thermostability. Protein Eng. 1996; 9:265-271.
23. Vetriani C, Maeder DL, Tolliday N, Yip KS, Stillman TJ, Britton KL, Rice DW, Klump HH, Robb FT. Protein thermostability above 100 degrees C: a key role for ionic interactions. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12300-12305.
24. Hurley JH, Baase WA, Matthews BW. Design and structural analysis of alternative hydrophobic core packing arrangements in bacteriophage T4 lysozyme. J. Mol. Biol. 1992; 224:1143-1159.
25. Thompson MJ, Eisenberg D. Transproteomic evidence of a loop-deletion mechanism for enhancing protein thermostability. J. Mol. Biol. 1999; 290:595-604.
26. Pe’er I, Felder CE, Man O, Silman I, Sussman JL, Beckmann JS. Proteomic signatures: amino acid and oligopeptide compositions differentiate among phyla. Proteins 2004; 54:20-40.
27. Protozanova E, Yakovchuk P, Frank-Kamenetskii MD. Stacked-unstacked equilibrium at the nick site of DNA. J. Mol. Biol. 2004; 342:775-785.
28. Bouthier de la Tour C, Portemer C, Huber R, Forterre P, Duguet M. Reverse gyrase in thermophilic eubacteria. J. Bacteriol. 1991; 173:3921-3923.
29. Sandman K, Krzycki JA, Dobrinski B, Lurz R, Reeve JN, HMf, a DNA-binding protein isolated from the hyperthermophilic archaeon Methanothermus fervidus, is most closely related to histones. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:5788-5791.
30. Stein DB, Searcy DG. Physiologically important stabilization of DNA by a prokaryotic histone-like protein. Science 1978; 202:219-221.
31. Yakovchuk P, Protozanova E, Frank-Kamenetskii MD. Basestacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006; 34:564-574.
32. Zeldovich KB, Berezovsky IN, Shakhnovich EI. Protein and DNA sequence determinants of thermophilic adaptation. PLoS. Comput. Biol. 2007; 3:e5.
33. Loladze VV, Ibarra-Molero B, Sanchez-Ruiz JM, Makhatadze GI. Engineering a thermostable protein via optimization of charge-charge interactions on the protein surface. Biochemistry 1999; 38:16419-16423.
34. Cambillau C, Claverie JM. Structural and genomic correlates of hyperthermostability. J. Biol. Chem. 2000; 275:32383-32386.
35. Chakravarty S, Varadarajan R. Elucidation of factors responsible for enhanced thermal stability of proteins: a structural genomics based study. Biochemistry 2002; 41:8152-8161.
36. Pack SP Yoo YJ. Protein thermostability: structure-based difference of amino acid between thermophilic and mesophilic proteins. J. Biotechnol. 2004; 11:269-277.
37. Saelensminde G Halskau O, Jr., Helland R, Willassen NP, Jonassen I. Structure-dependent relationships between growth temperature of prokaryotes and the amino acid frequency in their proteins. Extremophiles 2007; 11:585-596.
38. Glyakina AV, Garbuzynskiy SO, Lobanov MY, Galzitskaya OV. Different packing of external residues can explain differences in the thermostability of proteins from thermophililic and mesophilic organisms. Bioinformatics 2007; 23:2231-2238.
39. Galtier N, Lobry Jr, Relationships between genomic G + C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J. Mol. Evol. 1997; 44:632-636.
40. Hurst LD, Merchant AR. High guanine-cytosine content is not an adaptation to high temperature: a comparative analysis amongst prokaryotes. Proc. Biol. Sci. 2001; 268:493-497.
41. Nakashima H, Fukuchi S, Nishikawa K. Compositional changes in RNA, DNA and proteins for bacterial adaptation to higher and lower temperatures. J. Biochem. 2003; 133:507-513.
42. Lehmann M, Wyss M. Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Curr. Opin. Biotechnol. 2001; 12:371-375.
43. Berezovsky IN, Shakhnovich EI. Physics and evolution of thermophilic adaptation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:12742-12747.
44. England JL, Shakhnovich BE, Shakhnovich EI. Natural selection of more designable folds: a mechanism for thermophilic adaptation. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:8727-8731.
45. Ogata Y, Imai E, Honda H, Hatori K, Matsuno K. Hydrothermal circulation of seawater through hot vents and contribution of interface chemistry to prebiotic synthesis. Orig. Life Evol. Biosph. 2000; 30:527-537.
46. Li H, Helling R, Tang C, Wingreen N. Emergence of preferred structures in a simple model of protein folding. Science 1996; 273:666-669.
47. Zeldovich KB, Berezovsky IN, Shakhnovich EI. Physical origins of protein superfamilies. J. Mol. Biol. 2006; 357:1335-1343.
48. Shakhnovich BE, Deeds E, Delisi C, Shakhnovich E. Protein structure and evolutionary history determine sequence space topology. Genome Res. 2005; 15:385-392.
49. Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, Huber R, Feldman RA, Short JM, Olsen GJ, Swanson RV. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature 1998; 392:353-358.
50. Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, Fraser CM, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 1999; 399:323-329.
51. Nesbo CL, L’Haridon S, Stetter KO, Doolittle WF. Phylogenetic analyses of two “archaeal” genes in thermotoga maritima reveal multiple transfers between archaea and bacteria. Mol. Biol. Evol. 2001; 18:362-375.
52. Ausili A, Cobucci-Ponzano B, Di Lauro B, D’Avino R, Perugino G, Bertoli E, Scire A, Rossi M, Tanfani F, Moracci M. A comparative infrared spectroscopic study of glycoside hydrolases from extremophilic archaea revealed different molecular mechanisms of adaptation to high temperatures. Proteins 2007; 67:991-1001.
53. Berezovsky IN, Chen WW, Choi PJ, Shakhnovich EI. Entropic stabilization of proteins and its proteomic consequences. PLoS. Comput. Biol. 2005; 1:e47.
54. Ponnuswamy P Muthusamy, R Manavalan, P. Amino acid composition and thermal stability of proteins. Internat. J. Biol. Macromol. 1982; 4:186-190.
55. Bowie JU. Solving the membrane protein folding problem. Nature 2005; 438:581-589.
56. Friedman RA, Honig B. A free energy analysis of nucleic acid base stacking in aqueous solution. Biophys. J. 1995; 69:1528-1535.
57. Vasiliskov VA, Prokopenko DV, Mirzabekov AD. Parallel multiplex thermodynamic analysis of coaxial base stacking in DNA duplexes by oligodeoxyribonucleotide microchips. Nucleic Acids Res. 2001; 29:2303-2313.
58. Goldstein RA, Luthey-Schulten ZA, Wolynes PG. Optimal protein-folding codes from spin-glass theory. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:4918-4922.
59. Shakhnovich EI. Protein Folding thermodynamics and dynamics: where physics, chemistry, and biology meet. Chem. Rev. 2006; 106:1559-1588.
60. Morrissey MP, Shakhnovich EI. Design of proteins with selected thermal properties. Fold. Des. 1996; 1:391-405.
61. Seno F, Vendruscolo M, Maritan A, Banavar JR, Optimal protein design procedure. Phys. Rev. Lett. 1996; 77:1901-1904.
62. Berezovsky IN, Zeldovich KB, Shakhnovich EI. Positive and negative design in stability and thermal adaptation of natural proteins. PLoS. Comput. Biol. 2007; 3:e52.
63. Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:10915-10919.
64. Miyazawa S, Jernigan RL. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 1996; 256:623-644.
65. Butterfoss GL, Kuhlman B. Computer-based design of novel protein structures. Annu. Rev. Biophys. Biomol. Struct. 2006; 35:49-65.
66. Bolon DN, Grant RA, Baker TA, Sauer RT. Specificity versus stability in computational protein design. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:12724-12729.
67. Perez-Jimenez R, Godoy-Ruiz R, Ibarra-Molero B, Sanchez-Ruiz JM. The effect of charge-introduction mutations on E. coli thioredoxin stability. Biophys. Chem. 2005; 115:105-107.
68. Pjura P, Matsumura M, Baase WA, Matthews BW. Development of an in vivo method to identify mutants of phage T4 lysozyme of enhanced thermostability. Protein Sci. 1993; 2:2217-2225.
69. Sali D, Bycroft M, Fersht AR. Surface electrostatic interactions contribute little of stability of barnase. J. Mol. Biol. 1991; 220:779-788.
70. Zhang XJ, Baase WA, Shoichet BK, Wilson KP, Matthews BW. Enhancement of protein stability by the combination of point mutations in T4 lysozyme is additive. Protein Eng. 1995; 8:1017-1022.
71. Bloom JD, Meyer MM, Meinhold P, Otey CR, MacMillan D, Arnold FH. Evolving strategies for enzyme engineering. Curr. Opin. Struct. Biol. 2005; 15:447-452.
72. Gilis D, Rooman M, PoPMuSiC, an algorithm for predicting protein mutant stability changes: application to prion proteins. Protein Eng. 2000; 13:849-856.
73. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005; 33:W382-388.
Further Reading
Branden C, Tooze J. Introduction to Protein Structure. 1999. Garland Publishing Inc.
Cantor CR, Schimmel PR. Biophysical Chemistry. Part I: The conformation of biological macromolecules. Part III: The behavior of biological macromolecules. 1980. W.H. Freeman.
Hochachka P, Somero G. Biochemical Adaptation. Mechanism and Process in Physiological Evolution. 2002. Oxford University Press, Oxford.
Saenger W. Principles of Nucleic Acid Structure. 1984. Springer-Verlag, New York.
See Also
Amino Acids, Chemical Properties of
Physico-chemical Properties of Nucleic Acids: Character and Recognition of Watson-Crick Base Pairs
Protein Folding, Computation and Modeling of
Protein Folding, Energetics of
Proteins: Computational Analysis of Structure, Function and Stability
Thermophiles, Topics in Chemical Biology