CHEMICAL BIOLOGY
Catalytic Modes in Natural Ribozymes
Michael E. Harris, Center for RNA Molecular Biology, and Vernon E. Anderson, Department of Biochemistry, Case Western Reserve University School of Medicine, Cleveland, Ohio
doi: 10.1002/9780470048672.wecb640
In nature, RNA transmits genetic information, and it catalyzes reactions that are universal in biology. The reactions of phosphorus and carbon esters that are catalyzed by natural ribozymes occur with specific geometries and are susceptible to both acid and base catalysis as well as catalysis that involves metal ions. High-resolution structures of a few ribozymes implicate the involvement of individual nucleotides in performing these catalytic strategies. Detailed biochemical tests of the mechanisms of these ribozymes provide evidence for the involvement of nucleobase functional groups in acid/base catalysis, yet debate continues concerning their precise roles in proton transfer. Complementary structural and biochemical support has been gained for the involvement of specific RNA functional groups in positioning metal ions for catalysis. Still, only a few systems have been probed in sufficient depth to infer mechanistic detail and to achieve a complete chemical description of how ribozyme active sites enact specific catalytic strategies remains a key goal.
RNA Catalysis Is Ubiquitous in Biology
RNA holds a unique position in living systems because it can both convey genetic information as well as act as an enzyme to accomplish catalysis (1-3). RNAs that promote catalysis are called ribozymes, and the examples discovered thus far in nature catalyze addition/displacement reactions that involve phosphodiesters and carbon esters with rate enhancements approaching their protein counterparts. Despite the current understanding of its central role in biology, catalysis by RNA was initially considered initially heretical (4). Now appreciated as a fundamental aspect of terrestrial biology, the existence of ribozymes was a milestone recognized by the Nobel Prize in Chemistry to Cech and Altman in 1986 (5, 6). The discovery of RNA catalysis was important not only because it broadened our understanding of biologic catalysis, but also because it suggests a central role for RNA in the development of self-replicating systems that are believed to underlie the evolution of cells and ultimately all of biology (7).
The known ribozymes in biology may be divided into three different classes defined by differences in structural complexity as well as by the chemical reactions they catalyze (Fig. 1). Small ribozymes cleave an RNA phosphodiester backbone by catalyzing the intramolecular attack of a ribose 2'-OH on the adjacent 3',5' phosphodiester, which generates characteristic 5' hydroxyl and 2',3' cyclic phosphate products. In biology, small ribozymes are involved in replication of RNA viruses and are found in the 3' untranslated regions of genes where they are involved in gene regulation (8-11). Large ribozymes also catalyze addition/displacement reactions of phosphodiesters, but activate oxygen nucleophiles that include 2' and 3' ribose hydroxyls, or water for intermolecular nucleophilic attack displacing the 3’ O. A new 3',5' or 2',5' phosphodiester is generated or, if the nucleophile is in water (or rather its lyate ion hydroxide), the RNA chain is cleaved producing 5' monophospate and 2', 3' cis-diol termini. Large ribozymes include two classes of self-splicing introns (termed Group I and Group II), which catalyze two successive, site-specific transesterification reactions, and P RNA, the catalytic subunit of the tRNA processing endonuclease, ribonuclease P (12-14). The peptidyl transferase active site of ribosomal RNA, which is responsible for the synthesis of all proteins in the cell, comprises a third class (15). The RNA active site within the large ribosomal subunit RNA catalyzes attack of a primary amine on the carbon ester linkage between amino acid and 3’-OH of tRNA, which results in formation of a peptide bond and free 3’-OH.
By analogy with protein enzymes and precepts of transition state theory, ribozyme catalysis should involve an array of interactions between RNA functional groups and the reacting groups of the substrate that lower the free energy of the transition state relative to the ground state. As with any enzyme class, the specific mechanisms that ribozymes use to achieve catalysis are dependent necessarily on the intrinsic chemistry of phosphoryl and carbonyl transfer reactions in solution for which a well-developed understanding exists. This framework establishes that these reactions occur with specific geometries, and thus positioning of reactants provides rate enhancement. In addition, phosphoryl and carbonyl transfer are acid- and base-catalyzed, and interactions with metal ions and their complexes also provides rate enhancement. Substantial structural and biochemical evidence exists for use of substrate positioning, acid/base catalysis, and metal ion catalysis by ribozyme active sites. Here, we will describe the chemistry of the reactions catalyzed by ribozymes found in biology and review the current evidence for the existence of substrate positioning, metal ion catalysis, and acid/base catalysis for representative RNA catalysts.

Figure 1. General chemical mechanism of addition/displacement reactions at RNA phosphodiesters and amino acid esters catalyzed by ribozymes in biology. (a) Intramolecular hydrolysis of RNA via attack of an adjacent 2’-OH catalyzed by small ribozymes. The action of acid and base in leaving group protonation and nucleophile deprotonation are indicated in gray. Catalytic interactions with the nonbridging oxygens that result in protonation of a phosphorane intermediate or by stabilization of a negatively charged phosphorane-like transition state is also indicated. (b) Intermolecular phosphoryl transfer catalyzed by large ribozymes. In the reactions of large ribozymes, the nucleophile (R1) is a ribose 2’ or 3’ hydroxyl or a water molecule. Interactions that result in acid/base or electrostatic catalysis are indicated as in part A. (c) Amidolysis of an amino acid ester that results in peptide bond formation catalyzed by the peptidyl transferase active site of the ribosome. The R1 and R2 groups represent the 2’-OH groups of the aminoacyl-tRNAs in the ribosomal A-site and P-site, respectively. Similar catalytic interactions involved in facilitating proton transfer and charge stabilization are indicated. Note that the net proton transfer from the nucleophile to the leaving group could be mediated potentially by a single functional group.
Reactions Catalyzed by Natural Ribozymes
RNA phosphodiester bonds are cleaved under both basic and acidic conditions in reactions that involve intramolecular displacement of the 5’-O by attack of the adjacent the 2’-O, giving 2’,3’-cyclic phosphates (Fig. 1). Under acidic conditions, the 2’,3’-cyclic phosphates can isomerize to form 2’,5’-cyclic phosphates (See Reference 16 and references therein). Attack of nucleophiles on a tetrahedral phosphate ester gives a pentacoordinated species, the structure of which is a trigonal bipyramid that has two apical and three equatorial ligands. Nucleophiles may enter and leave the intermediate/transition state only through apical positions, but the pentacoordinated phosphorane may be sufficiently stable to allow ligand reorganization by a pseudorotation. In acid-catalyzed hydrolysis, a phosphorane intermediate forms, which is sufficiently stable to pseudorotate while the base-catalyzed reaction is thought to be concerted.
Rates of phosphodiester addition/displacement reactions in solution are sensitive to both nucleophile and leaving group pKa. Bronsted analyses support a concerted mechanism for the base-catalyzed reaction in which significant charge on both positions exists in the transition state. For attack of an adjacent ribose hydroxyl, a change in the degree of sensitivity occurs with leaving groups with pK > 12, which supports a change in the mechanism that involves formation of a phosphorane intermediate as the leaving group becomes less reactive (17). Kinetic 18O isotope effects on the nonbridging and leaving group oxygen atoms, as well as solvent deuterium isotope effects, have been measured for the hydrolysis of phosphate diesters with nitrophenol and nitrobenzyl leaving groups (18, 19). The observation of measurable nucleophile and leaving group KIEs for base-catalyzed nitrophenol ester hydrolysis demonstrate a concerted mechanism and indicate equilibrium deprotonation of the nucleophile prior to nucleophilic attack. For the acid-catalyzed reaction of less reactive nitrobenzyl esters, leaving group KIE analysis are consistent with a pre-equilibrium proton transfer to the ester oxygen atom, followed by rate-limiting P-O bond fission (20). Thus, enzyme active site interactions will facilitate proton transfer to and from the leaving group and nucleophile and will influence their reactivity provide powerful catalytic strategies.
In addition to acids and bases, metal ions and their complexes also catalyze phosphoryl transfer reactions in solution, which provides a model for possible mechanisms in enzyme catalysis (e.g., References 21-24). Metal ion coordination lowers the pKa of the interacting alcohol, which reduces its nucleophilicity but increases correspondingly the concentration of the lyate ion at neutral pH. Because both phosphodiester reactions are sensitive to the pKa of the nucleophile and leaving group, metal coordination at these positions can accelerate the reaction. Sensitivities of phosphoryl transfer reactions to ionic strength indicate that significant electrostatic repulsion occurs between anionic nucleophiles and the negatively charged phosphoryl center. Thus, metal ion interactions that offset this repulsion or interact more favorably with the negatively charged transition state relative to the ground state can provide catalysis as well. Additionally, as outlined above, these reactions occur with specific inline geometries for the nucleophile and leaving group. Because they can coordinate multiple electronegative ligands, divalent metal ions can also promote catalysis by simultaneous interactions with the nonbridging phosphate oxygens and the nucleophile to offset the free energy costs of decrease in entropy in the transition state. Such additional catalysis beyond effects on pKa and ionic strength have been referred to as induced intramolecularity (23).
Transpeptidation-aminolysis of a carbon ester
The transpeptidation reaction catalyzed by the ribosome involves nucleophilic attack of an amine on a carbon ester with subsequent displacement of the alcohol (Fig. 1c). Isotope exchange from water into the substrate ester during aqueous ester hydrolysis provided evidence for exchange between the carbonyl oxygen and water, which implicates the formation of a tetrahedral intermediate (e.g., Reference 25). Aminolyses of esters in solution can be general base- or general acid-catalyzed; however, a break in the pH-rate profile at lower pH exists, which suggests a change in the rate-limiting step. Extensive analysis of pH and substituent effects show that with most esters, nucleophilic attack to form the intermediate is rapid and reversible, and the rate-determining step at high pH is proton transfer within the intermediate or its breakdown. For very fast reactions, evidence exists for a change to rate-determining formation of the intermediate (e.g., References 26 and 27).
Nucleophilic attack on carbon esters also shows dependence on both nucleophile and leaving group pKa, and structure reactivity studies support a stepwise mechanism in which a tetrahedral intermediate forms and is more stable for esters with poor leaving groups like the 3' ribose OH of aminoacyl-tRNA esters used by the ribosome (28). The transition state for formation of the intermediate is believed to be neutral with a zwitteri- onic character. The transfer of a proton from nucleophile to carbonyl oxygen or the leaving group oxygen is proposed to be the rate-determining step. Isotope effects on the nucleophile, esterified carbon, and ester oxygen provide strong support for a stepwise mechanism for attack of oxygen and nitrogen nucleophiles on nitrophenyl esters (29). Thus, like phosphodiester reactions, specific reaction geometry is dictated and facilitation of this geometry, as well as of proton transfer from the nucleophile to carbonyl oxygen, or the leaving group oxygen can provide catalysis.
The Potential for Catalysis by Ribozymes
The potential for RNA to act as a catalyst is dictated by its structure as a linear polymer of the four common ribonucleotides. Like DNA, RNA can form double stranded, antiparallel helices via traditional Watson-Crick base pairing. However, the backbone of nucleic acid is highly flexible and RNA can form complex tertiary structures that often involve non-Watson-Crick base pairing to create active site crevices for catalysis. The phosphodiester backbone is charged negatively and interacts electrostatically as well as by direct coordination with solution divalent cations. Ribose, purines, and pyrimidine bases contain both H-bond donors and acceptors that help stabilize higher-order structure and provide for substrate positioning, as well as participate in active site interactions.
The potential for general acid/base catalysis is constrained by the available functional groups that can participate in proton transfer. The pKas of RNA nucleobases are 3.5 and 4.2 for A and C, and 9.2 for G and U and the ribose 2'OH has a pKa of ~12 (Fig. 2a). As outlined below, metal ions can be bound tightly by RNA and such hydrated Mg(2+) ions have pKa of ~10. Thus, at neutral pH, A and C would be good acids, but at a low concentration relative to the unprotonated form. Although G and U could serve as bases, they exist predominantly in their protonated forms. Conversely, A and C could act as bases because they are predominantly unprotonated, but their low pKa provide less driving force for proton transfer at neutral pH. Similarly, G and U have protons to donate, but have high pKa. However, large pKa shifts to increase the concentration of the active protonation state of amino acid functional groups often are observed in protein enzymes (30), and no a priori reason seems to exist why RNA would not be prone to similar pKa perturbations. Indeed, nucleobases with altered pKa have been observed in model RNAs (31), and coupling structure formation has been highlighted as a means to provide the driving force for pKa shifting in RNA (32, 33). Furthermore, the sensitivity to pKa will depend on the Bronsted p value for that reaction, which can lessen the impact of nonoptimal pKas on reaction rate (34). Similarly, general acid/base catalysis will always occur when a large change occurs in the pKa of the reacting group, and when the pKa of the catalyst is intermediate between the initial and the final pKa values of the substrate group (35). For example, the pKa of the 2-OH nucleophile undergoes a very large change in pKa, from a value of 12 for the substrate 2-OH to a value of 0 for protonation of the product ester. Thus, nucleobase functional groups with pKas considered nonoptimal (pKa 3-4 or 9.2) are between these values and can nonetheless provide significant general acid/base catalysis.
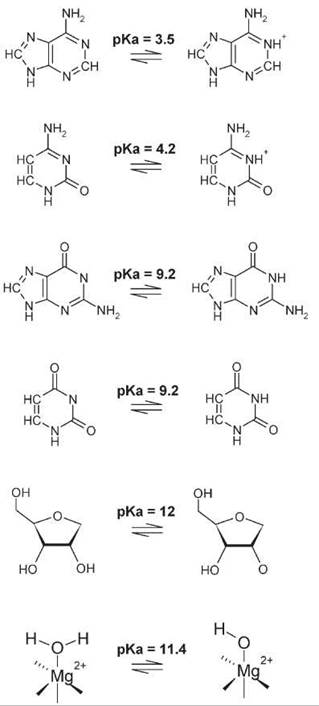
Figure 2. lonizable functional groups in RNA. The base and acid forms of the nucleobase and ribose pKa values that are nearest to neutral are shown. The pKa of a water that forms an inner sphere coordination interaction with Mg(2+) is also shown.
Like protein enzymes, an understanding of the involvement of ionized functional groups can be gained by analyzing the effects of changes in pH on rate. Much of the detailed analysis of RNA catalysis has involved analysis of mechanistic detail from such dependencies. Typically, functional groups that are active in their protonated state (acids) as well as in their unprotonated states (bases) are invoked. However, the same pH-rate profile can be observed for different mechanisms (Fig. 3). For example, a reaction that is catalyzed by both acid and base displays a bell-shaped rate dependence on pH with two apparent pKas for the acid and base. However, switching the pKas of these species does not change the observed data. This kinetic ambiguity (36) has a very large impact on mechanistic interpretations of structure-function relationships in catalytic RNAs (34, 36). Indeed, the presence of multiple titratable groups and/or the presence of specific acid/base catalysis adds even more ambiguity with respect to identification of mechanisms that can develop observed pH-rate behavior. Nonetheless, kinetic data provide a direct functional read-out and necessarily must be accommodated by models of ribozyme mechanism. An ongoing challenge, as described below, is in defining specific experimental tests to assign catalytic roles to individual active site interactions.

Figure 3. Idealized pH dependence of a ribozyme reaction. Ideal pH-species plots and pH-kobs profiles according to a kinetic model for general acid/base catalysis. The solid lines depict a mechanism in which the species with the lower pKa (pKa,1) acts as the general base (shown by blue lines), and the species with the higher pKa (pKa,2) acts as the general acid (shown by red lines). The black line indicates the observed pH dependence of the reaction rate. The dotted lines simulate a mechanism in which the catalytic roles of the species with pKa,1 and pKa,2 have been switched. Adapted from References 34 and 35.
Metal ions can promote phosphoryl and carbonyl reactions in solution in several ways and are a common feature of protein enzyme active sites. Like acid/base catalysis, information on the importance of metal ions to catalysis often has been gained by analyzing the dependence of activity on metal ion concentration and identity. However, the dependence of RNA catalysts on Mg(2+) concentrations can be complex. Importantly, metal ions make large contributions to RNA folding and promote activity because of stabilization of the active RNA conformation (37, 38). Thus, the observed concentration dependence of the ribozyme reaction may (or may not) represent a direct role in catalysis (Fig. 4). For example, metal ions may bind to the folded RNA conformation at the active site with high affinity. At concentrations of ions that are saturated for folding, the observed metal ion dependence will reflect ion binding to the active site. In contrast, if ion binding at a site or sites that stabilizes folding occurs at a lower affinity, then the observed dependence of the reaction on metal ion concentration will represent necessarily the binding to the structural, but nonetheless activating, site.
Additionally, RNA-metal ion interactions take two thermodynamically important forms: 1) diffuse binding in which the ions interact electrostatically with the negatively charged phosphodiester backbone and 2) site-specific binding in which metal hydrate makes inner sphere or direct H-bond contact with an RNA functional group (39). Although diffuse ion interactions make weak contact, they are numerous and overall have a high thermodynamic contribution to folding and to apparent catalytic activity. Also, site-bound interactions are fundamentally important, but may only occur at certain steps in the ribozyme mechanism, or be linked indirectly to catalysis by stabilization of structure. The difficulty in characterizing these interactions biochemically is described picturesquely as equivalent to searching for them amid the “sea” of other RNA-metal ion interactions. Accordingly, titration experiments are difficult to interpret in terms of specific metal ion binding sites, and development of structural and biochemical probes for functional metal ion interactions has been a considerable focus of ribozyme research.

Figure 4. Direct and indirect models of metal ion activation of ribozyme catalysis. Model of classes of metal sites that influence ribozyme activity. The scheme on top depicts the binding of a metal ion important for ribozyme folding that binds distant from the active site but promotes a structural transition that permits catalysis or the binding of catalytic metal ions. Such binding interactions may result directly in overall folding or may merely foster small structural changes near the active site that are critical to ribozyme chemistry. The scheme below depicts the direct activation of ribozyme activity by binding of metal ions that interact with the reactive phosphate and are involved in metal ion catalysis. Adapted from Reference 38.
Evidence for Acid/Base Catalysis in the Hepatitis Delta Virus Ribozyme Active Site
Evidence for involvement of a cytosine nucleobase in proton transfer
The hepatitis delta virus (HDV) ribozyme is a member of the class of small ribozymes and functions as a self-cleaving RNA sequence critical to the replication of the virus’ RNA genome (1, 8, 40). HDV ribozymes are proposed to employ several catalytic strategies that include an important example of general acid/base catalysis that involves a specific cytosine residue in the active site. Indeed, a milestone in our understanding of RNA catalysis was the observation that HDV and other small ribozymes could function in the absence of divalent metal ion cofactors, provided that high (molar) concentrations of monovalent ions are present (41, 42). These high monovalent ion concentrations are believed to stabilize the active RNA conformation, which implies that the primary role of divalent metal ions is in structural stabilization (42).
An additional insight into HDV ribozyme active site interactions came from the determination of the structure of the ribozyme including the 3' cleavage product. The 5'-OH terminus of this product is located in a cleft formed by the intersection of conserved helical elements. Within this crevice, the 5'-OH is in sufficient proximity to form a hydrogen bond with the N3 of C75 (C76 in the antigenomic version of the ribozyme). If C75 becomes protonated, it could act as a general acid to affect leaving group stabilization (43) (Fig. 5). Participation of titrat-able groups in the HDV reaction is indicated by a bell-shaped pH-rate profile that reveals two groups with apparent pKa values. ~6.5 and 9 (44). However, in the absence of divalent metal ions, the pH dependence reversed, with the rate of cleavage increasing with lowering of pH (42, 45, 46), which indicates that a metal ion hydrate could perform as a general base. Although deletion of C75/C76, or mutation to G or U, reduces cleavage activity dramatically (45, 47) a C76A mutation retains significant activity but displays an altered apparent pKa for the pH dependence of the self-cleavage reaction (44, 45). Functional group modification experiments with C analogs with altered N3 pKas demonstrated the importance of C75 ionization in the HDV cis-cleavage reaction. Inhibition of ribozyme function also occurs at C75 with a C analog that lacks the N4 amino group, which implicates the exocyclic amine in critical interactions in the active site (48). These data develop a model of general acid/base catalysis, with C75 potentially acting as the acid as indicated by the ribozyme-product structure and a hydrated divalent metal ion as the general base.

Figure 5. Proposed acid/base catalytic interactions in the HDV ribozyme active site. The cleavage site for the HDV ribozyme is shown with the nucleotide 5' to the site of bond cleavage shown in red and the nucleotide 3' to that site shown in blue. Two proposed mechanisms for the function of the C76 cytisine nucleobase and a hydrated active site metal ion are shown in which C76 acts as either an acid (left) or base (right) as described in the text.
Remarkably, addition of exogenous cytosine or certain nucleobase and imidazole analogs can rescue partially the activity of the nearly inactive C76U mutant ribozyme (44, 49). Structure-function studies of the rescuing analogs provided additional insight into mechanism. The pH-rate profiles for the rescued reactions were bell shaped with one pKa attributed to ionization of the exogenous base. When a second potential ionizable nucleobase (C41) was removed, one leg of the bell-shaped curve was eliminated (50). The purported ionizable base, C41, is distant from the active site but may enhance cleavage rates through structural stabilization. The rescue of cleavage activity in the C75 deletion mutant, and by analogy the native ribozyme, can be explained by a model in which three ionizable groups exist (8, 34). These include: 1) the 2'OH group nucleophile (or a general-base catalyst with a relatively high pKa that acts at the 2'OH group, 2) a C75 or an analogous rescuing buffer that acts as a proton donor, and 3) a nucleobase (C41) associated with structure formation.
Thus, the role of C75 seems to act as a general acid in its protonated form. The most likely candidate based on the product complex and on the chemistry of phosphodiesters would be protonation of the 5'-OH leaving group. Recently, Das and Piccirilli (51) provided strong evidence for this model using a chemically modified “activated” RNA substrate in which the 5'O leaving group is replaced by sulfur, which has a lower pKa and thus is a better leaving group. The activated 5'S substrate specifically suppressed the effects of C75 mutation and modifications that alter its pKa, consistent with it providing general acid catalysis, which mediates proton transfer to the leaving group through a protonated N3-imino nitrogen. However, this model for C75 function is distinct from that derived from crystallographic studies of precursor forms of the genomic ribozyme in which catalysis is blocked by the C75U mutation, chelation of divalent metal ions, or 2’-deoxy substitution of the 2'-OH nucleophile (52). Importantly, these structures reveal electron density for a divalent cation coordinated to the 5’-O, whereas the position of the N3 of U75 is equidistant from the 2'O and the 5'O. Comparison of the precursor and product structures reveals differences in the active site structure after cleavage. Interpretation of these differences in terms of the catalytic mechanism suggests that a conformational change occurs and results in dissociation of the catalytic metal ion. Thus, although strong evidence exists that the C75 nucleobase functions in acid/base catalysis, likely in its protonated form, the specific transition state interaction is still of considerable interest as is the interplay of catalysis and metal ion binding.
Other small ribozymes
The HDV ribozyme, however, is only one example of the class of small ribozymes that catalyze self-cleavage, which includes the hammerhead, hairpin, VS, and glmS riboswitch ribozymes (1, 2). Like the mechanistic detail described above, crystal structures have been solved for several modified versions of these ribozymes, and the structures thus derived provide candidate nucleobases that could act as acid/base catalysis. Biochemical tests of these interactions in the transition state have been made in several cases to support the involvement of nucleobases in proton transfer. Nonetheless, the same limitations with respect to kinetic ambiguity limit the detail with which specific active site mechanisms can be articulated. Riboswitches are a newly described class of self-cleaving RNA in which the catalytic activity is linked directly to regulation of gene expression (53, 54). The glmS ribozyme is a self-cleaving RNA that occurs in the 5' untranslated region of the mRNA of the gene that encodes glucosamine-6phosphate (GlcN6P) synthase in some Gram-positive bacteria and also acts to regulate gene expression in an analogous fashion. Self-cleavage represses gene expression when this ribozyme is activated by binding of GlcN6P (55). Recent structural and biochemical studies result in a model for this RNA in which it binds its activator such that the amine group of GlcN6P is in position to interact with the reactive phosphate potentially to provide nucleophilc activation. Such coenzyme usage raises the potential for an expanded catalytic repertoire for biologic ribozymes. Yet, obtaining a complete description of the transition state interactions for the glmS and other small ribozymes continues to present structural biologists, theoreticians, and experimentalists with significant challenges.
Analysis of Divalent Metal Ion Interactions in the Group I Self-Splicing Intron Active Site
Structural and biochemical evidence for catalytic metal ion interactions
Group I (GI) introns are large ribozymes that function as self-splicing intron sequences that catalyze two successive phosphotransesterification reactions using a single conserved active site (4, 12). In the first reaction, the 3'-O of a G cofactor attacks the phosphodiester bond at the 5' splicing site; the 3'-OH of the last exon nucleotide is the leaving group. In the second reaction, the 3'-O product from the first reaction attacks the 3' splice site that generates the spliced exons; the 3'-O of the last intron nucleotide (termed ®G) is the leaving group. Note that the 3'-O leaving group in the first step is the nucleophile for the second step, and the interactions with the leaving group in the second step will replicate interactions involved in nucleophilic activation in the first step (Fig. 6) (56). Recent crystal structures of a ribozyme intermediate formed after the first splicing step show that the substrate is in a highly constrained conformation with complete reversal of strand direction at the 3' splice site (57, 58). A network of hydrogen bonding interactions positions the 3’-OH of the 5’ exon proximal to the scissile phosphate, and the geometry is that expected for inline nucleophilic attack. With respect to active site interactions with the nucleophile/leaving group oxygens, most attention has focused on divalent metal ion interactions, and now strong evidence exists that nucleophilic activation at both 3’-O nucleophiles involves direct coordination to active site Mg(2+) ions.

Figure 6. Proposed divalent metal ion coordination interactions involved in metal ion catalysis by the GI intron ribozyme active site. The coordination interactions determined by substrate PS and 2' amino modification and quantitative metal rescue are depicted on the left. The individual interacting functional groups are shown in red. The observed metal ions (green spheres) in the crystal structure of the GI ribozyme active site is shown on the right and the metal ligands identified by functional studies are shown as red spheres. Adapted from Reference 56.
The GI ribozyme reaction depends on the presence of divalent metal ions; but as indicated above, the binding of these ions plays multiple roles that include folding and enhancing substrate binding affinities (59). The rate of the chemical step is Mg(2+) dependent, but these data do not distinguish between direct or indirect roles, or a combination of both. As indicated above, distinguishing active site metal ions from what has been referred to as the “sea” of other functionally important metal ion interactions presents a considerable challenge. For the GI ribozyme and other catalytic RNAs, site-specific evidence for active site metal interactions comes primarily from analyses of thiophilic metal ion rescue of phosphorothioate and other substrate modifications (e.g., References 60 and 61). These analyses rely on the fact that substitution of a substrate phosphate by a phosphorothioate weakens the affinity of coordinated Mg(2+) ions substantially such that catalysis is inhibited by several orders of magnitude. Because softer metal ions such as Mn(2+) and Cd(2+) coordinate more readily to sulfur, if inclusion of these ions in the reaction rescues activity, then a direct coordination interaction at the substituted position is likely. By comparing quantitative analyses of the concentration dependence of the metal ion rescue for different substrate modifications, a network of different metal ion coordination reactions has been revealed. The model that developed from a series of elegant biochemical studies of this kind combined with a wealth of additional structure-function and kinetic data can be summarized as follows: Metal ions coordinate to both the 3'-oxygen leaving group and to the 3'-oxygen on the G nucleophile in the first step (MA and MB, respectively), and a third metal ion interacts with the 2'-hydroxyl of the G nucleophile (MC). Two metal ions (MA and MC) also contact the pro-SP oxygen of the scissile phosphate. Extension of this analysis to encompass phosphate oxygens within the intron itself provide evidence that these metal ions are positioned by coordination interactions with nonbridging oxygens that constitute, in part, the active site of the ribozyme (12, 61, 62).
Recently, high-resolution structures of the ribozyme, which includes one that contains the 2'-OH of the substrate G, a key metal ion ligand, have been reported that provide the most consistent model to date of active site interactions that include metal ion coordination to the functional groups identified biochemically. In the most complete structure, two metals coordinate to nonbridging oxygens in the intron and substrate that include all biochemically identified interactions (56, 63). One Mg(2+) ion makes inner sphere contacts with both the 2'-O and the 3'-O nucleophile/leaving group of the G cofactor. One obvious difference in this model is that one metal ion makes the contacts attributed to metal ions MB and MC in the model derived biochemically. Although differences in the electrostatic environments of the active site because of different functional group modifications may complicate interpretation of the biochemical analyses, the remarkably high degree of correspondence between the structural and functional analyses provide the most detailed account to date for ribozyme active site metal ion interactions.
Other large ribozymes
The presence of active site metal ion interactions seems to be a general feature of the class of large ribozymes that catalyze phosphoryl transfer. Both Group II intron and RNase P RNA ribozymes that catalyze transesterification and phosphodiester bond hydrolysis, respectively, are thought to use metal ion catalysis as well. Although ample evidence exists for higher order structure that provides specificity via positioning the reactive phosphate, high-resolution structure of other examples of this class are limiting currently. Information that concerns active site interactions comes largely from kinetic analysis of substrate modifications including the kinds of PS rescue experiments described above. Both GII and P RNA are modeled to have two metal ions coordinated to the pro-Rp oxygen of the reactive phosphate and to the nucleophile and leaving group oxygens. However, the number and the position of coordinated metals and their ribozyme contacts are yet to be described fully. Under reaction conditions that favor the chemical step, both ribozymes react with rates consistent with general base catalysis but whether these ribozymes, like their smaller counterparts, employ functional groups in proton transfer remains unknown.
Substrate Positioning and Substrate Assisted Catalysis in the Peptidyl Transferase Center of the Ribosome
A landmark achievement in our understanding of biology has been the elucidation of the structure of ribosomes and their complexes with substrates, cofactors, and inhibitors (15). In addition to providing a wealth of functional insights, the structures showed that the peptidyl transferase active site involves RNA functional groups exclusively (64). Comparisons with model reactions indicate that the ribosome active site provides ~107-fold catalysis that is promoted significantly by positioning the reactive groups for catalysis. However, mechanistic studies have been providing evidence both for and against the involvement of active site functional groups in additional catalytic interactions (65, 66). Indeed, the extent to which the ribozyme employs catalytic interactions with the amine nucleophile and the oxyanion of the tetrahydral intermediate has framed several important studies that concern ribozyme mechanism (67).
Structures of the large subunit with low-molecular weight substrates and products bound are highly suggestive regarding peptidyl transferase active site interactions (64, 67-69). Three groups exist in the neighborhood of the reactive amino group that could conceivably interact to assist in positioning and deprotonation of the nucleophile. These include the 2'-OH of A76 at the 3' end of the P-site bound peptidyl tRNA itself, the N3, and the 2'-OH of A2451 (Escherichia coli) of the large subunit rRNA. Replacement of the 2’-OH group of A76 in the P site-bound tRNA with 2'-H or 2'-F resulted in a dramatic decrease in the rate of peptide bond formation but did not affect ground state substrate binding (70, 71). Combined with the structural perspective, a role in nucleophilic activation is proposed; however, the precise role of this substrate residue in assisting catalysis is not resolved entirely. Site-specific incorporation of nucleoside analogs into 23 S rRNA of thermus aquaticus 50 S subunits (72) provided evidence that the ribose 2'-OH of A2451 is also important for catalytic function. Also, mutation of A2451 reduces the rate of the chemical step of peptide-bond formation by ~100-fold (73, 74).
Using puromycin as an A-site substrate, the rate of the peptidyl transferase reaction is strongly pH-dependent, and pH-rate profiles suggest two ionizing groups with apparent pKa values of 7.5 and 6.9 (75). The second pKa of 6.9 has been attributed to the amine nucleophile of the puromycin substrate. In the single-protonated state in which the ribosomal group with pKa 7.5 is protonated whereas that with pKa 6.9 is deprotonated, the reaction is 105-fold faster compared with the uncatalyzed reaction. Protonation of a ribosomal group with pKa 7.5 thus seems to contribute a factor of ~100 to the observed rate that is likely to be caused by additional catalytic interactions. Kinetic analysis of a A2451U mutant showed that the pH dependence caused by the ribosomal ionizing group was eliminated, which is consistent with a direct role of A2451 in catalysis (75). Thus, the peptidyl transferase active site clearly makes contacts that position the aminoacyl and peptidyl tRNA termini in the appropriate geometry for nucleophilic attack. In addition to substrate-assisted catalysis by the adjacent tRNA 2'OH, the ribosome active site also provides a network of noncovalent interactions that assists in providing additional rate enhancement (Fig. 7) (76).
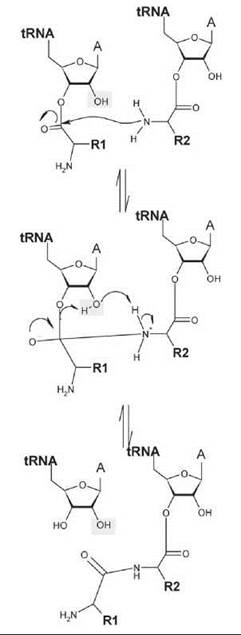
Figure 7. General chemical scheme for peptidyl transfer by the ribosome. The scheme shows nucleophilic attack by the amine group of the amino acid (with side chain R2) esterified to the tRNA in the ribosomal A site (right) on the ester linkage of the aminoacyl tRNA (with amino acid chain R1). The 2’OH of the peptidyl tRNA participates in the reaction and seems to transfer a proton to the 3’O leaving group, either directly or potentially via a solvent bridge. Adapted from Reference 76.
All enzymes are constrained necessarily by the intrinsic chemistry of the reactions that they catalyze and the availability of functional groups for interfacing with this chemistry. As transesterification of phosphodiesters and carbon esters involves proton transfer, acid/base catalysis is a fundamental strategy for which strong support now exists for ribozymes. Yet, a continuing challenge is “following the protons” to and from active site functional groups and the substrate. Similarly, it is accepted widely that metal ions are involved directly in phosphotransfer reactions catalyzed by large ribozymes, but the site and number of active site metal ion interactions and their precise contribution to rate enhancement is still the subject of intense interest. Confronting these challenges and developing new technical and intellectual tools for understanding RNA catalysis represents a significant focus of the current investigations of ribozyme chemistry.
Acknowledgments
We thank Drs. Eric Christian, Michael Been, and Scott Silverman for helpful discussions regarding to the concepts discussed in this article. We are grateful particularly to Drs. Scott Strobel, Donald Burke, and Phil Bevilacqua for review and insightful comments on the manuscript. We are indebted to the community of enzymologists who investigate RNA catalysis and whose foundational work is alluded to, but not referenced explicitly, because of limitations of space and scope of this article.
References
1. Fedor MJ, Williamson JR. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell. Biol. 2005; 6:399-412.
2. Doudna JA, Lorsch JR. Ribozyme catalysis: not different, just worse. Nat. Struct. Mol. Biol. 2005; 12:395-402.
3. Lilley DM. Structure, folding and mechanisms of ribozymes. Curr. Opin. Struct. Biol. 2005; 15:313-323.
4. Cech TR. Biologic catalysis by RNA. Harvey Lect. 1986; 82:123-144.
5. Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982; 31:147-157.
6. Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA16 moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983; 35:849-857.
7. Orgel LE. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004; 39:99-123.
8. Been MD. HDV ribozymes. Curr. Top. Microbiol. Immunol. 2006; 307:47-65.
9. Collins RA. The Neurospora Varkud satellite ribozyme. Biochem Soc Trans., 2002; 30:1122-1126.
10. Blount KF, Uhlenbeck OC. The hammerhead ribozyme. Biochem. Soc. Trans. 2002; 30:1119-1122.
11. Wilson TJ, Nahas M, Ha T, Lilley DM. Folding and catalysis of the hairpin ribozyme. Biochem. Soc. Trans. 2005; 33:461-465.
12. Hougland J, Piccirilli J, Forconi M, Lee J, Herschlag D. How the group intron works: a case study of RNA structure and function. In: RNA World. 3rd edition. Gesteland RF, Cech TR, Atkins JF, ed. 2005. Cold Spring Harbor Laboratory Press, New York. pp. 133-205.
13. Lambowitz AM, Zimmerly S. Mobile group II introns. Annu. Rev. Genet. 2004; 38:1-35.
14. Hsieh J, Andrews AJ, Fierke CA. Roles of protein subunits in RNA-protein complexes: lessons from ribonuclease P. Biopolymers 2004; 73:79-89.
15. Moore PB, Steitz TA. The structural basis of large ribosomal subunit function. Annu. Rev. Biochem. 2003; 72:813-850.
16. Oivanen M, Kuusela S, Lonnberg H. Kinetics and mechanism for the cleavage and isomerization of phosphodiester bonds of RNA by Bronsted acids and bases. Chem. Rev. 1998; 98:961-990.
17. Lonnberg H, Stromberg R, Williams A. Compelling evidence for a stepwise mechanism of the alkaline cyclisation of uridine 3'-phosphate esters. Org. Biomol. Chem. 2004; 2:2165-2167.
18. Hengge AC. Isotope effects in the study of phosphoryl and sulfuryl transfer reactions. Acc. Chem. Res. 2002; 35:105-112.
19. Cassano AG, Anderson VE, Harris ME. Understanding the transition states of phosphodiester bond cleavage: insights from heavy atom isotope effects. Biopolymers 2004; 73:110-129.
20. Grzyska PK, Czyryca PG, Purcell J, Hengge AC. Transition state differences in hydrolysis reactions of alkyl versus aryl phosphate monoester monoanions. J. Am. Chem. Soc. 2003; 125:13106- 13111.
21. Hendry P, Sargeson AM. Metal ion promoted phosphate ester hydrolysis. Intramolecular attack of coordinated hydroxide ion. J. Am. Chem. Soc. 1989; 111:2521-2527.
22. Cassano AG, Anderson VE, Harris ME. Analysis of solvent nucleophile isotope effects: evidence for concerted mechanisms and nucleophilic activation by metal coordination in nonenzymatic and ribozyme-catalyzed phosphodiester hydrolysis. Biochemistry 2004; 4:10547-10559.
23. Herschlag D, Jencks WP. Catalysis of the hydrolysis of phosphorylated pyridines by Mg(OH)+: a possible model for enzymatic phosphoryl transfer. Biochemistry 1990; 29:5172-5179.
24. Cowan JA. Metal-mediated hydrolysis of biological phosphate esters. J. Biol. Inorg. Chem. 1997; 2:168-176.
25. Bender ML. and H. H., Carbon oxygen exchange in general base catalyzed ester hydrolysis. J Am Chem Soc, 1967. 89(5):p. 1211-1218.
26. Cunningham BA, Schmir GL. Hydroxyl group participation in amide hydrolysis. The influence of catalysts on the partitioning of a tetrahedral intermediate. J. Am. Chem. Soc. 1967; 89:917-922.
27. Blackburn GM, Jencks WP. The mechanism of the aminolysis of methylformate. J. Am. Chem. Soc. 1968; 90:2638-2645.
28. Jencks WP, Gilchrist M. Nonlinear structure-reactivity correlations. The reactivity of nucleophilic reagents toward esters. J. Am. Chem. Soc. 1968; 90:2622-2637.
29. Marlier JF. Multiple isotope effects on the acyl group transfer reactions of amides and esters. Acc. Chem. Res. 2001; 34:283-290.
30. Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002; 53:85-98.
31. Ravindranathan S, Butcher SE, Feigon J. Adenine protonation in domain B of the hairpin ribozyme. Biochemistry 2000; 39:16026-16032.
32. Moody EM, Lecomte JT, Bevilacqua PC. Linkage between proton binding and folding in RNA: a thermodynamic framework and its experimental application for investigating pKa shifting. RNA 2005; 11:157-172.
33. Bevilacqua PC, Brown TS, Chadalavada D, Lecomte J, Moody E, Nakano SI. Linkage between proton binding and folding in RNA: implications for RNA catalysis. Biochem. Soc. Trans. 2005; 33:466-470.
34. Bevilacqua PC. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry 2003; 42:2259-2265.
35. Jencks WP. Catalysis in Chemistry and Enzymology. 1987. Dover Publications, New York.
36. Bevilacqua PC, Brown TS, Nakano S, Yajima R. Catalytic roles for proton transfer and protonation in ribozymes. Biopolymers 2004; 73:90-109.
37. Woodson SA. Metal ions and RNA folding: a highly charged topic with a dynamic future. Curr. Opin. Chem. Biol. 2005; 9:104-109.
38. DeRose VJ. Metal ion binding to catalytic RNA molecules. Curr. Opin. Struct. Biol. 2003; 13:317-324.
39. Draper DE, Grilley D, Soto AM. Ions and RNA folding. Annu. Rev. Biophys. Biomol. Struct. 2005; 34:221-243.
40. Shih IH, Been MD. Catalytic strategies of the hepatitis delta virus ribozymes. Annu. Rev. Biochem. 2002; 71:887-917.
41. Murray JB, Seyhan AA, Walter NG, Burke JM, Scott WG. The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol. 1998; 5:587-595.
42. Nakano S, Cerrone AL, Bevilacqua PC. Mechanistic characterization of the HDV genomic ribozyme: classifying the catalytic and structural metal ion sites within a multichannel reaction mechanism. Biochemistry 2001; 40:12022-12038.
43. Ferre-D’Amare, A.R., K. Zhou, and J.A. Doudna, Crystal structure of a hepatitis delta virus ribozyme. Nature, 1998; 395(6702): p. 567-74.
44. Perrotta AT, Shih I, Been MD. Imidazole rescue of a cytosine mutation in a self-cleaving ribozyme. Science 1999; 286:123-126.
45. Nakano S, Chadalavada DM, Bevilacqua PC. General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science 2000; 287:1493-1497.
46. Wadkins TS, Shih I, Perrotta AT, Been MD. A pH-sensitive RNA tertiary interaction affects self-cleavage activity of the HDV ribozymes in the absence of added divalent metal ion. J. Mol. Biol. 2001; 305:1045-1055.
47. Perrotta AT, Been MD. Core sequences and a cleavage site wobble pair required for HDV antigenomic ribozyme self-cleavage. Nucleic Acids Res. 1996; 24:1314-1321.
48. Oyelere AK, Kardon JR, Strobel SA. pK(a) perturbation in genomic Hepatitis Delta Virus ribozyme catalysis evidenced by nucleotide analogue interference mapping. Biochemistry 2002; 41:3667-3675.
49. Shih IH, Been MD. Involvement of a cytosine side chain in proton transfer in the rate-determining step of ribozyme self-cleavage. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:1489-1494.
50. Perrotta AT, Wadkins TS, Been MD. Chemical rescue, multiple ionizable groups, and general acid-base catalysis in the HDV genomic ribozyme. RNA 2006; 12:1282-1291.
51. Das SR, Piccirilli JA. General acid catalysis by the hepatitis delta virus ribozyme. Nat. Chem. Biol. 2005; 1:45-52.
52. Ke A, Zhou K, Ding F, Cate JH, Doudna JA. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 2004; 429:201-205.
53. Winkler WC. Riboswitches and the role of noncoding RNAs in bacterial metabolic control. Curr. Opin. Chem. Biol. 2005; 9:594-602.
54. Tucker BJ, Breaker RR. Riboswitches as versatile gene control elements. Curr. Opin. Struct. Biol. 2005; 15:342-348.
55. Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 2004; 428:281-286.
56. Stahley MR, Strobel SA. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science 2005; 309:1587-1590.
57. Stahley MR, Strobel SA. RNA splicing: group I intron crystal structures reveal the basis of splice site selection and metal ion catalysis. Curr. Opin. Struct. Biol. 2006; 16:319-326.
58. Adams PL, Stahley MR, Gill ML, Kosek AB, Wang J, Strobel SA. Crystal structure of a group I intron splicing intermediate. RNA 2004; 10:1867-1887.
59. McConnell TS, Herschlag D, Cech TR. Effects of divalent metal ions on individual steps of the Tetrahymena ribozyme reaction. Biochemistry 1997; 36:8293-8303.
60. Shan S, Yoshida A, Sun S, Piccirilli JA, Herschlag D. Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:12299-12304.
61. Hougland JL, Kravchuk AV, Herschlag D, Piccirilli JA. Functional identification of catalytic metal ion binding sites within RNA. PLoS Biol. 2005; 3:e277.
62. Szewczak AA, Kosek AB, Piccirilli JA, Strobel SA, Identification of an active site ligand for a group I ribozyme catalytic metal ion. Biochemistry 2002; 41:2516-2525.
63. Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:6498-6502.
64. Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science 2000; 289:920-930.
65. Sievers A, Beringer M, Rodnina MV, Wolfenden R. The ribosome as an entropy trap. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7897-7901.
66. Seila AC, Okuda K, Nunez S, Seila AF, Strobel SA. Kinetic isotope effect analysis of the ribosomal peptidyl transferase reaction. Biochemistry 2005; 44:4018-4027.
67. Moore PB, Steitz TA. After the ribosome structures: how does peptidyl transferase work? RNA 2003; 9:155-159.
68. Page MI, Jencks WP. Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. U.S.A. 1971; 68:1678-1683.
69. Schmeing TM, Seila AC, Hansen JL, Freeborn B, Soukup JK, Scaringe SA, Strobel SA, Moore PB, Steitz TA. A pre-translocational intermediate in protein synthesis observed in crystals of enzymatically active 50S subunits. Nat. Struct. Biol. 2002; 9:225-230.
70. Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate40 assisted catalysis of peptide bond formation by the ribosome. Nat. Struct. Mol. Biol. 2004; 11:1101-1106.
71. Weinger JS, Strobel SA. Participation of the tRNA A76 hydroxyl group throughout translation. Biochemistry 2006; 45:5939-5948.
72. Erlacher MD, Lang K, Wotzel B, Rieder R, Micura R, Polacek N. Efficient ribosomal peptidyl transfer critically relies on the presence of the ribose 2'-OH at A2451 of 23S rRNA. J. Am. Chem. Soc. 2006; 128:4453-4459.
73. Polacek N, Gaynor M, Yassin A, Mankin AS. Ribosomal peptidyl transferase can withstand mutations at the putative catalytic nucleotide. Nature 2001; 411:498-501.
74. Thompson J, Kim DF, O’Connor M, Lieberman KR, Bayfield MA, Gregory ST, Green R, Noller HF, Dahlberg AE. Analysis of mutations at residues A2451 and G2447 of 23S rRNA in the peptidyltransferase active site of the 50S ribosomal subunit. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:9002-9007.
75. Katunin VI, Muth GW, Strobel SA, Wintermeyer W, Rodnina MV. Important contribution to catalysis of peptide bond formation by a single ionizing group within the ribosome. Mol. Cell. 2002; 10:339-346.
76. Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural insights into the roles of water and the 2’ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol. Cell. 2005; 20:437-448.
Further Reading
Altman S. The road to RNase P. Nat. Struct. Biol. 2000; 7:827-828.
Bevilacqua PC. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry 2003; 42:2259-2265.
Cech TR. Nobel lecture. Self-splicing and enzymatic activity of an intervening sequence RNA 7 from Tetrahymena. Biosci. Rep. 1990; 10:239-261.
Draper DE. A guide to ions and RNA structure. RNA 2004; 10:335-343.
Fedor MJ, Williamson JR. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell Biol. 2005; 6:399-412.
Hougland J, Piccirilli J, Forconi M, Lee J, Herschlag D. How the Group I intron works: a case study of RNA structure and function. In: RNA World. 3rd edition. Gesteland RF, Cech TR, Atkins JF, eds. 2005. Cold Spring Harbor Laboratory Press, New York. pp. 133-205. Moore PB, Steitz TA. The ribosome revealed. Trends Biochem. Sci. 2005; 30:281-283.
Oivanen M, Kuusela S, Lonnberg H. Kinetics and mechanism for the cleavage and isomerization of phosphodiester bonds of RNA by Bronsted acids and bases. Chem. Rev. 1998; 98:961-990.
Pyle AM. Metal ions in the structure and function of RNA. J. Biol. Inorg. Chem. 2002; 7:679-690.
Weinger JS, Strobel SA. Participation of the tRNA A76 hydroxyl groups throughout 38 translation. Biochemistry 2006; 45:5939-5948.