CHEMICAL BIOLOGY
Quorum Sensing
Pratibha Singh, Research Center for Advanced Science and Technology, The University of Tokyo, Tokyo, Japan; Department of Advanced Interdisciplinary Studies, Graduate School of Engineering, The University of Tokyo, Tokyo, Japan
Jun Igarashi, The Otsuka Protech Laboratory, Komaba Open Laboratory, The University of Tokyo, Tokyo, Japan
Hiroaki Suga, Research Center for Advanced Science and Technology, The University of Tokyo, Tokyo, Japan; Department of Advanced Interdisciplinary Studies, Graduate School of Engineering, The University of Tokyo, Tokyo, Japan; The Otsuka Protech Laboratory, Komaba Open Laboratory; The University of Tokyo, Tokyo, Japan
doi: 10.1002/9780470048672.wecb642
Bacterial pathogenesis can cost in terms of loss of man-hours, loss of livestock, and damage to cash crops. Quorum sensing (QS) is a mechanism by which bacteria regulate the expression of their virulence factors and the resultant pathogenicity. It presents itself as an attractive target for the design of small-molecule inhibitors. An added advantage of targeting QS is that it does not threaten the viability of the bacteria as opposed to the traditional antibacterial agents. Thus, when compared with the traditional antibacterial agents, these small-molecule inhibitors promise to maintain their efficacy over a longer period of time. In this article, the mechanism of bacterial QS systems will be discussed along with a survey of the small molecules/strategies designed to combat them.
Introduction
Bacteria, long viewed as single unicellular organisms existing in a free-floating manner, exist in sessile colonies. This mode of existence is made possible by their ability to communicate among themselves via cell-to-cell signaling mechanisms. Not only can bacteria sense the presence of their own kind and coordinate the expression of their genes, but also they can recognize other bacteria as friend or foe and respond accordingly. Quorum sensing (QS) is one such cell-to-cell signaling mechanism (1, 2). Thus, QS enables bacteria to coordinate their behavior if they need to respond quickly to survive a rapid change of the environmental conditions. Such responses include adaptation to availability of nutrients, defense against other microorganisms that may compete for the same nutrients, and avoidance of toxic compounds that are potentially dangerous for the bacteria. It is very important during infection of a host (e.g., humans, other animals, or plants) for pathogenic bacteria to coordinate their virulence to escape the immune response of the host and to establish a successful infection.
The quorum-sensing signal is a self-generated small molecule (or molecules) called autoinducer (AI). Each bacterial cell secretes AI, but when the cell density is low, the AI concentration is too low to be sensed. However, when bacteria reach a critical mass, the secreted AI concentration also reaches a threshold that enables the bacteria to sense it. This event regulates (activates or represses) the transcription of genes that lie under the control of the QS system. Most bacteria that have the QS system exist in association with a plant or animal host, and this association can be symbiotic or pathogenic. Particularly, in the latter scenario, it would be imperative to understand the mechanism of the bacterial QS system to control their pathogenicity. Although several types of QS systems exist in bacteria, this article focuses on discussions of the mechanism of one major type of QS, the so-called luxIR system and its importance as a drug target. Other systems like luxS are referenced in the Further Reading section.
Mechanism of the luxIR System in Gram-Negative Bacteria
In the luxIR system (Fig. 1a), AI is an acylated homoserine lactone (AHL), and the family of AHL signal molecules is generally called AI-1 (Fig. 1a and Fig. 1b) (3). This name refers to the chronology of QS signal(s) discovery. LuxI is the synthase of AI-1, which is synthesized from S - adenosylmethionine (SAM) and acyl-acyl carrier protein (acyl-ACP) (Fig. 1a). Activation of the QS circuit(s) relies on the synthesis, accumulation, and subsequent sensing of AI-1. The minimal concentration of AI-1 is constitutively synthesized by its cognate synthase. In Vidua fischeri, for instance, the chemical structure of AI-1 is 3-oxo-C6-HSL (Fig. 1c), which is synthesized by LuxI (4, 5). As the cell density increases (i.e., more bacteria are present) AI-1 accumulates in the area surrounding the bacteria. When the concentration of AI-1 reaches a critical threshold, AI-1 reenters the cells and is specifically sensed by its cognate transcription factor called LuxR. The LuxR-AI-1 complex binds to a region of DNA (lux box) causing the activation of downstream genes (6). In addition, the LuxR-AI-1 complex also causes an increase in the expression levels of LuxI, which results in more production of AI-1. Thus, the AI (autoinducer) is named after the function of “auto-induction” of its own synthesis. In V. fischeri, activation of QS circuit results in activation of luminescence genes; thus, when V. fischeri exists at high cell densities, its colony becomes luminescent.

Figure 1. P. aeruginosa QS system. Its mechanism. (a) Biosynthesis of acyl-homoserine lactone (AHL). Abbreviations: SAM, 5-adenosyl methionine; ACP, acyl carrier protein. (b) General chemical structure of AHL molecules, generally called autoinducer-1 (AI-1). (c) Chemical structure of V. fischeri AI-1. (d) Chemical structure of P. aeruginosa 3-oxo-C12-HSL and (e) C4-HSL. (f) Pseuodomonas quinolone signal, PQS.
Why can bacteria only sense exogenous the AI signal, not the endogenously existing AI signal? The most recent biochemical studies have suggested that LuxR associates with the cell membrane, and its AI receptor site is not likely exposed to the interior of the cell when it is in an unbound form. Thus, LuxR would not see the AI signal inside of the cell. Moreover, some bacteria actively transport AI signals via an efflux pump. Such mechanisms generally operate to sense the exogenous AI signal, and thus bacteria can monitor the surrounding environment for its own species or for other species of bacteria.
The QS system of Pseudomonas aeruginosa has been studied extensively because of its importance as a human pathogen. P. aeruginosa has a unique system of QS that consists of las, rhl, and Pseudomonas quinolone signal (PQS) circuits (Fig. 2a). The first two circuits, las and rhl, have a set of proteins (synthase and receptor) and AI-1 like the typical luxIR circuit. The las circuit is composed of LasI and LasR along with 3-oxo-C12-HSL (Fig. 2b) as its cognate AI, whereas in the rhl circuit RhlI and RhlR exist with C4-HSL (Fig. 2c) as their cognate AI. The fundamental mechanism of the P. aeruginosa QS system is similar to that of V. fischeri, but activation of the rhlI circuit relies on activation of an upstream lasI circuit (7); for example, accumulation of LasR-3-oxo-C12-HSL activates the expression of the rhlI gene, which results in the activation of the rhl circuit. Significantly, activation of the rhl circuit is connected to the transcription of genes that express secondary metabolites and virulence factors, such as pyocyanin and elastase (8). Recently, more attention has been focused on the fact that the QS system controls biofilm formation/maturation (9).
AIs isolated from different Gram-negative bacteria differ in the N -acyl side-chain length (from C4 to C14) or degree of substitution (3-oxo, 3-hydroxy, saturated, or unsaturated). Generally, assumed to be freely diffusible in bacterial cells, radiolabeled V. fischeri AI (3-oxo-C6-HSL) (10) has been shown to be freely diffusible into and out of V. fischeri and Escherichia coli cells. However, in P. aeruginosa, whereas C4-HSL can diffuse freely into and out of P. aeruginosa cells, 3-oxo-C12-HSL is actively transported by an efflux pump (MexAB-OprM) (11) outside the cell, which results in three times higher levels of 3-oxo-C12-HSL inside the cell, which suggests that the length and/or degree of substitution of the N-acyl chain determines whether it diffuses freely or is actively pumped out from the cells.
In the PQS circuit, it has been shown that PQS (12) is involved in controlling genes required for virulence factor expression and biofilm formation. PQS regulates the expression of lasB, encoding for elastase, an important virulence factor, and both the las and rhl QS systems of P. aeruginosa affect the synthesis and bioactivity of PQS. LasR positively regulates the synthesis of PQS, whereas RhlR represses it. Both 3-oxo-C12-HSL and C4-HSL compete for pqsABCDE (PQS operon) regulation, and the levels of expression are thus dependent on the ratio of the two autoinducers (13). Furthermore, PQS also induces the rhl circuit (14). PQS is hydrophobic and is conveyed between P. aeruginosa cells via a specialized vesicular transport mechanism (15). The vesicles package the PQS and other quinolones/quinolines and traffic them between P. aeruginosa cells that exist in biofilms in cystic fibrosis sputum. Mutants that do not produce these vesicles also do not show PQS-mediated QS. Interestingly, PQS behaves as an antibiotic against Gram-positive cells (16, 17), which suggests that mechanisms similar to those of other known quinolones/quinolines antibiotics are likely to be in place.

Figure 2. Ribbon representation of dimeric LasR-LBD found in the crystal structure. Each protein monomer possesses a single 3-oxo-C12-HSL molecule buried deep in its pocket. The picture was taken from the paper reported by Bottomley et al. (43).
Cross-Species Communication between Bacteria Using LUXR Signals
P. aeruginosa is a soil bacterium and shares its habitat with antibiotic-producing bacteria such as Streptomyces tenebarius (the source of tobramycin, an antibiotic used commonly against P. aeruginosa) (18) and the pathogenic filamentous yeast Candida albicans (19). It thus may use its AI molecules to restrain antipseudomonal compound(s) that produce environmental pathogens. Recently, it has been shown that the degradation product of 3-oxo-C12-HSL resembles a class of antibacterial compounds called tetramic acids (Fig. 3) (20). Such a tetramic acid analog is produced through the enol form of the 3-oxo-carbonyl group on the fatty acid side chain undergoing intramolecular alkylation of the lactone ring of HSL, which gives rise to a HSL open-ring structure. This rearranged molecule turns out to resemble tetramic acid structurally. It has been shown that this tetramic acid-like molecule acts like an antibiotic toward several Gram-positive bacteria, while leaving the Gram-negative bacteria unaffected. Although the mechanism of bactericidal activity of this tetramic acid-like molecule derived from 3-oxo-C12-HSL is as yet unclear, it has been postulated that it chelates metal ions such as Fe3+ and that this complex somehow may increase its bactericidal potency (Fig. 3) (20). Alternatively, it simply may exhibit the antibiotic activity via a mechanism similar to that of reutericyclin, which is known to act as a proton ionophore that dissipates the transmembrane change in pH and leads to the cell lysis of Gram-positive bacteria (21).
4-hydroxy-2-heptylquinilone-N-oxide (HQNO), a quinolone family member, similarly has antistaphylococcal activity, which suppresses the growth of many Gram-positive bacteria. Paradoxically, it also allows some Gram-positive bacteria to grow, albeit slowly, in the presence of aminoglycoside antibiotics like streptomycin. This growth limitation exerted by HQNO causes Streptomyces aureus to form smaller colonies that revert back to the wild-type phenotype in the absence of HQNO (22). P. aeruginosa coexists with Burkholderia cepacia in the lungs of cystic fibrosis patients. A unidirectional communication (23) exists between the two pathogens where B. cepacia can perceive the autoinducer molecules produced by P. aeruginosa but not vice versa.
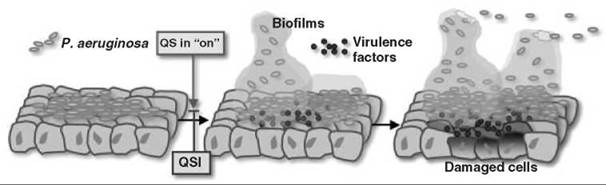
Figure 3. Schematic presentation of P. aeruginosa-mediated cellular damage in the host during infection.
Why is Quorum Sensing Important as a Therapeutic Target?
Traditional antibacterial agents target processes crucial to the survival of a bacterium. These processes include cell wall biosynthesis, replication, and protein synthesis, which endanger its survival and thus lead to the development of drug resistance in bacteria. Bacteria eliminate drugs by exporting them out via multidrug pumps or by circumventing/degrading the drug molecule. QS inhibitors (QSI(s)), however, do not threaten bacterial survival, and therefore, in principle, bacteria would not be forced to evolve resistant strains if QSI(s) are used. Because QS regulates the production of virulence factors, QSI(s) potentially can attenuate the virulence of pathogenic bacteria (Fig. 3). Thus, QSIs may offer us a new class of antimicrobial agents that would not suffer from the emergence of resistant strains.
P. aeruginosa is an example of one such attractive target for therapeutic drug design. It is the leading cause of mortality among cystic fibrosis patients. It also affects immunocompromised individuals with HIV infections, cancer, burns, or organ transplants. Through knockout studies, it has been firmly established that P. aeruginosa, incapable of QS, also is incapable of virulence. For instance, analysis of a lasI mutant, rhlI mutant, and lasI-rhlI double mutant in a neonatal mouse model revealed markedly decreased virulence (24), with the most notable reduction in the double mutant. Other infection models studied (25, 26), firmly establish that it is the QS mechanism that enables P. aeruginosa to form biofilms and cause sustained infections in the host system (Fig. 3). Thus, strategies geared toward elimination of QS can aid in clearing the infection because they retard the formation of biofilm and the resulting sustained virulence, which makes the QS system an attractive therapeutic target.
Small Molecular QS Modulators
The discovery of AI antagonists is challenging and therapeutically significant. A potent antagonist was recently derived by modification of natural AI antagonists, halogenated furanones (Fig. 4a), produced by a marine alga to prevent colonization by QS bacteria (27-31). These natural products disrupt the Serratia liquefaciens SwrR-C4-HSL (32) interaction and also inhibit the LuxR-3-oxo-C6-HSL (33) and CarR-3-oxo-C6-HSL (27) interactions but have little activity against the LasR-3-oxo-C12-HSL interaction. Givskov et al. synthesized an analog that lacks the alkyl side chain of the natural furanones and found that this compound had considerable inhibitory activity against the P. aeruginosa QS system (Fig. 4b). They showed that the molecule inhibited QS-controlled reporter genes and virulence factors in a QS mutant with exogenous AIs but not in wild-type P. aeruginosa with natural levels of AI. This compound did not inhibit biofilm formation, but it affected biofilm architecture and enhanced the process of bacterial detachment. Interestingly, the same group recently reported that the QS inhibition by the synthetic furanone was not because of the interaction with LuxR demonstrated in V. cholera; more likely, it accelerates the turnover, for example, degradation, of LuxR via an unknown mechanism(s). Although this finding might not be totally surprising because the synthetic furanone and natural furanones are structurally quite different from natural Als, it is significant that the molecules somehow associating with QS inhibition can attenuate bacterial virulence.
Natural furanones are not alone in having anti-AHL activity. They are joined in nature by garlic, vanilla, and phytochemicals present in dietary fruits and spices, and so forth. Garlic extracts have been shown to inhibit QS sensing in P. aeruginosa and also render it sensitive to tobramycin and phagocytosis by polymorphormonuclear leaucocytes (PMN) in a mice model of P. aeruginosa infection (34). However, the garlic-extract dose used to treat the mice equates to approximately 50 bulbs of garlic per day for an average human being. Thus, more work to identify and isolate the pure compounds responsible for inhibition of QS is awaited. Vanilla extract, however, mainly contains vanillin (85%). Vanillin, despite having no structural similarity to furanone derivatives or AIs, shows significant inhibition of QS in Chromobacterium violaceum (35). QS in Chromobacterium violaceum also is inhibited by phytochemicals in dietary fruits, herbs, and spices (36). These phytochemicals also affect the swarming motility of P. aeruginosa through a possible impact on synthesis and activity of AI molecules. Macrolides like azithromycin also repress the las and rhl in P. aeruginosa albeit at sublethal concentrations (37).
Molecular insights into the LasR-3-oxo-C12-HSL interactions, available from crystal studies, may provide an important tool into the process of antagonist design. Over the past decade, tremendous efforts have been made to generate a soluble form of various LuxR family proteins and their complex with their cognate AI. Three LuxR proteins, Erwinia chrysanthemi ExpR (38), Agrobacterium tumefaciens TraR (39), and Erwinia carotovora CarR (40) have been expressed successfully as a complex with the cognate AI and purified for in vitro characterization. Particularly, the recent success in solving the crystal structure of the dimer of TraR-AI (3-oxo-C8-HSL) complex interacting with the target DNA (41, 42) has provided the first visual information regarding the molecular interactions between a LuxR family protein and its cognate AI. It should be noted that the dimerization of TraR (as well as ExpR and CarR) has been predicted by the in vitro biochemical studies, and thus the X-ray structure most likely represents the active form in vivo.

Figure 4. Structure of QS antagonists. (a) Naturally occurring furanones and its derivative. X = H or Br. (b) Antagonists found from an AI analog library that contains HSL substituents, (c) those found from chemical library that contains fatty acid chains, and (d) those containing fatty acid substituents.
Despite the fact that many attempts to overexpress the full-length LasR failed, Bottomley et al. (43) have recently succeeded in crystallizing a construct of LasR consisting of only the predicted ligand binding domain (LasR-LBD, which consists of the amino acids from Met-1 to lys-173) in a complex with 3-oxo-Ci2-HSL. The complex was observed as a symmetrical dimer of LasR-LBD, and 3-oxo-C12-HSL was buried deeply inside the binding site. The monomer exhibits structural features similar to TraR, which consists of an α-β-α sandwich structure with three a-helices flanking a five-stranded antiparallel β-sheet. 3-oxo-C12-HSL lies parallel to the β-sheet and lies buried in a pocket formed between the β-sheet and a3-a5 helices. Also, all the polar groups of 3-oxo-Q2-HSL, except the oxygen of the lactone ring, make hydrogen bonds with the amino acid residues of LasR-LBD. The acyl chain extends into a hydrophobic residue-lined cavity. Some of these residues are observed only in LasR, thus providing high specificity and a minimum crosstalk between bacteria of different species. In silico, modeling of the interaction of available QS inhibitors with the LasR-LBD provides a clue to the mechanism of action of the inhibitors. This modeling also provides a scaffold for the de novo design of better inhibitor molecules.
Suga et al. (44, 45) have reported that the screening of a library of synthetic AI analogs with substituted HSL has yielded a novel class of QS inhibitors. Interestingly, two of these antagonists found in the screening, 3-oxo-C12-(S)-2 and 3-oxo-C12-3 (Fig. 4c), are structurally related to the synthetic agonist 3-oxo-C12-(S,S)-1 (Fig. 4b). Small structural changes of the agonist HSL substitute (1) altered activity dramatically from agonist to antagonist. In light of the strong agonist activity of 1, these antagonists most likely maintain binding to LasR but fail to activate it and hence act as potent inhibitors. These QS inhibitors inhibit expression of virulence factors, such as elastase B and pyocyanin. Moreover, 3-oxo-C12-(S )-2 inhibited biofilm formation in both QS mutant (in the presence of exogenous Als) and wild-type P. aeruginosa under static conditions. The latter compound 3-oxo-C12-3 did not inhibit biofilm formation but significantly altered biofilm architecture. Thus, these antagonists are promising leads to derive more potent antagonists by the screening of focused libraries.
In fact, a resurvey of focused libraries based on the antagonists found in the initial screening has given several new strong antagonists. 3-oxo-C12-4 and its related molecule 3-oxo-Q2-5 (which also is related to the 3-oxo-C12-3) have shown a strong inhibitory effect on las circuit and also show inhibition on the downstream rhl circuit (Fig. 4c). Likewise, 3-oxo-Q2-6 exhibited a strong inhibitory effect on the QS circuits. Significantly, 3-oxo-C12-5 and 3-oxo-C12-6 showed remarkable abilities in the inhibition of biofilm formation of wild-type P. aeruginosa in flow-cell experiments (Hiroaki Suga et al., unpublished data). Moreover, adding these compounds to wild-type P. aeruginosa that had formed mature biofilms resulted in the detachment of biofilms and the removal of bacteria from the glass surface in a flow cell. Along similar lines, Greenberg et al. (46) reported two new substituents of HSL that can inhibit P. aeruginosa QS circuits (Fig. 4d). These QS inhibitors are promising candidates for drug development, and more results from in-depth studies in various models of infection are awaited.
Alteration of the fatty acid side chain based on the structure of AHL is an obvious alternative approach to antagonist(s) discovery. Earlier works devoted to changing the length of the fatty acid or 3-functional group unfortunately did not yield notable antagonists. However, more recent investigations independently reported by Doutheau (47, 48) and Blackwell (49) successfully showed that more drastic derivatizations of the fatty acid side chain generated potent antagonists. Doutheau et al. has shown that AI analogs with Cn-acyl, Cn-sulfonyl, or Cn-ureido chains (Fig. 4e, 7-10) bearing aromatic groups such as phenyl at the end of an alkyl chain antagonize the QS circuit of V. fischeri. Blackwell et al. later observed that substitution of the aromatic ring with an indole ring on the C4-acyl chain gave approximately two orders of magnitude stronger inhibitory effect on P. aeruginosa QS system than the parental compounds reported by Doutheau. This molecule also could block the biofilm production under static conditions. It must be noted, however, that these HSL-based analogs likely suffer from an instability problem because of the hydrolysis of the lactone ring when the analogs are subjected to an in vivo environment (vide infra).
In conclusion, it is to be noted that although the current QSI candidates can disrupt the interaction of the AHL molecule with its cognate receptor, they are not potent enough to halt the QS process and the resultant pathogenesis. Thus more studies geared toward the discovery of molecules that antagonize the QS system are awaited, in particular, those that target not only the receptors but also the AHL synthases.
Modulating the QS System Outside of Bacterial Cells
Because the QS system is triggered by the exogenous AI signal entering the cell, if the QS signals were to be degraded (50) so that the signaling function is lost, then the QS system could be modulated. In fact, some prokaryotes and eukaryotes do have such defense systems.
The aiiA gene (51) encoding an AHL lactonase, recently discovered in a Gram-positive bacillus bacterium isolated from soil, enables it to compete against Gram-negative bacteria in the soil. Expression of aiiA in a transformed Erwin caratovora strain reduced the release of AI significantly and attenuated the pathogenic effects on important crops like potato, eggplant, Chinese cabbage, carrot, celery, cauliflower, and tobacco. Such genetically engineered crops would be expected to fare better against bacterial infections.
Paraxonases (PONs) (52) are a family of mammalian lactone hydrolases, expressed in liver and various tissues, and they can deactivate 3-oxo-C12-HSL by the hydrolysis of the lactone ring. Similarly, airway epithelial cells can inactivate 3-oxo-C12-HSL by an enzymatic mechanism. (53). The inactivation is selective for AHLs with certain carbon chain lengths, and C4-HSL is reportedly immune to such inactivation. This inactivation activity is cell associated and not mediated by a secreted factor. Also, the inactivation is shown to occur in cell-free lysates, and the ability of the cell-free lysate to inactivate 3-oxo-C12-HSL depends on the amount of lysate used.
An alternative approach to the inhibition of QS is the use of antibodies directed toward the AI molecule. Recently, Kaufmann et al. (54) has reported the generation of anti-AHL monoclonal antibodies (mAbs). Their hapten designed initially focused on the synthesis of AHL analogs with a carboxylic acid functionality to facilitate the binding of BSA or KLH. Because of the instability of the haptens, a lactam moiety replaced the lactone ring. Three haptens thus were synthesized, and after conjugation to KLH (18-23 hapten molecules per carrier protein), Balb/c mice were immunized for generation of hybridoma(s). The monoclonal antibodies generated against the 3-oxo-hapten had a good affinity (Kd = 150 nM to 5 μM) for 3-oxo-C12-AHL and the lactam analog and high specificity because short-chain 3-oxo-AHLs were not recognized. Also, these antibodies, in particular RS2-1G9, demonstrated strong inhibition of QS signaling in both wild-type and mutant P. aeruginosa PAO cells concomitant with inhibition of pyocyanin production. The crystal structure of FabRS2-1G9 in complex with the lactam analog has revealed that the polar lactam moiety is encapsulated completely in the antibody-combining site (55). This study provides insight into the immune recognition of a quorum-sensing molecule by an antibody. Furthermore, this structure can be used for protein engineering that leads to an enhanced interaction of the antibody with the AHL molecule. Lactonase activity could be added into the antibody through site-directed mutagenesis of the antibody. Lending additional credence to the antibody approach is the study on 3-oxo-C12-HSL-BSA conjugates (56). Mice immunized with 3-oxo-C12-HSL-BSA conjugate showed significant amounts of antibody in the serum. When challenged intranasally with P. aeruginosa, 36% of the mice survived for 4 days post challenge as compared with the control mice that died in 2 days. Interestingly, the bacterial numbers in the lungs were similar in the two groups. Thus, specific antibodies to 3-oxo-C12-HSL confer a protective advantage against acute P. aeruginosa infections.
Conclusion and Perspective
In this article we have seen how QS presents an alternate route to combating bacterial pathogenesis. Although more studies are awaited to develop efficient drug molecules to inhibit QS system, it nonetheless proves to be an attractive target for drug development. Also, the attenuation of virulence observed with the use of QSIs is not accompanied with a loss in viability of the bacterial pathogens. In this regard, QSIs are different from traditional antibacterial agents that act through affecting the viability of the bacteria in various ways. It is hoped that QSIs will not suffer from the drawback of resistance development, which traditional antibacterials have to battle against. Thus, more studies geared toward the design of QSIs, including both synthetic analogs of AI molecules and inhibitors of AI synthases, may lead to the availability of drug molecules capable of maintaining their efficacy over longer periods of time compared with the traditional antibacterial agents.
References
1. Fuqua C, Parsek MR, Greenberg EP. Regulation of gene expression by cell-to cell communication: acyl-homoserine lactone quorum sensing. Annu. Rev. Genet. 2001; 35:439-468.
2. Fuqua C, Greenberg EP. Listening in on bacteria: acyl-homoserine lactone signalling. Nat. Rev. Mol. Cell Biol. 2002; 3:685-695.
3. Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol. 1994; 176:269-275.
4. Eberhard A, Burlingame AL, Eberhard C, Kenyon GL, Nealson KH, Oppenheimer NJ. Structural identification of autoinducer of Photobacterium fischeri luciferase. Biochemistry 1981; 20:2444- 2449.
5. Engebrecht J, Silverman M. Identification of genes and gene products necessary for bacterial bioluminescence. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:4154-4158.
6. Hanzelka BL, Greenberg EP. Evidence that the N-terminal region of the Vibrio fischeri LuxR protein constitutes an autoinducerbinding domain. J. Bacteriol. 1995; 177:815-817.
7. Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol. Microbiol. 1996; 21:1137-1146.
8. Winson MK, Camara M, Latifi A, Foglino M, Chhabra SR, Daykin M, Bally M, Chapon V, Salmond GP, Bycroft BW, et al. Multiple N-acyl-L-homoserine lactone signal molecules regulate production of virulence determinants and secondary metabolites in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:9427-9431.
9. Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 1998; 280:295-298.
10. Kaplan HB, Greenberg EP. Diffusion of autoinducer is involved in regulation of the Vibrio fischeri luminescence system. J. Bacteriol. 1985; 163:1210-1214.
11. Pearson JP, Van Delden C, Iglewski BH. Active efflux and diffusion are involved in transport of Pseudomonas aeruginosa cell-to-cell signals. J. Bacteriol. 1999; 181:1203-1210.
12. Camilli A, Bassler BL. Bacterial small-molecule signaling pathways. Science 2006; 311:1113-1116.
13. McGrath S, Wade DS, Pesci EC. Dueling quorum sensing systems in Pseudomonas aeruginosa control the production of the Pseudomonas quinolone signal (PQS). FEMS Microbiol. Lett. 2004; 230:27-34.
14. McKnight SL, Iglewski BH, Pesci EC. The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 2000; 182:2702-2708.
15. Mashburn LM, Whiteley M. Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature 2005; 437:422-425.
16. Deziel E, Lepine F, Milot S, He J, Mindrinos MN, Tompkins RG, Rahme LG. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:1339-1344.
17. Kadurugamuwa JL, Beveridge TJ. Bacteriolytic effect of membrane vesicles from Pseudomonas aeruginosa on other bacteria including pathogens: conceptually new antibiotics. J. Bacteriol. 1996; 178:2767-2774.
18. Kharel MK, Basnet DB, Lee HC, Liou K, Woo JS, Kim BG, Sohng JK. Isolation and characterization of the tobramycin biosynthetic gene cluster from Streptomyces tenebrarius. FEMS Microbiol. Lett. 2004; 230:185-190.
19. Hogan DA, Vik A, Kolter R. A Pseudomonas aeruginosa quorumsensing molecule influences Candida albicans morphology. Mol. Microbiol. 2004; 54:1212-1223.
20. Kaufmann GF, Sartorio R, Lee SH, Rogers CJ, Meijler MM, Moss JA, Clapham B, Brogan AP, Dickerson TJ, Janda KD. Revisiting quorum sensing: Discovery of additional chemical and biological functions for 3-oxo-N-acylhomoserine lactones. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:309-314.
21. Ganzle MG, Vogel RF. Studies on the mode of action of reutericyclin. Appl. Environ. Microbiol. 2003; 69:1305-1307.
22. Hoffman LR, Deziel E, D’Argenio DA, Lepine F, Emerson J, McNamara S, Gibson RL, Ramsey BW, Miller SI. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:19890-19895.
23. Riedel K, Hentzer M, Geisenberger O, Huber B, Steidle A, Wu H, Hoiby N, Givskov M, Molin S, Eberl L. N-acylhomoserine-lactone-mediated communication between Pseudomonas aeruginosa and Burkholderia cepacia in mixed biofilms. Microbiology 2001; 147:3249-3262.
24. Wu H, Song Z, Givskov M, Doring G, Worlitzsch D, Mathee K, Rygaard J, Hoiby N. Pseudomonas aeruginosa mutations in lasI and rhlI quorum sensing systems result in milder chronic lung infection. Microbiology 2001; 147:1105-1113.
25. Rumbaugh KP, Griswold JA, Iglewski BH, Hamood AN. Contribution of quorum sensing to the virulence of Pseudomonas aeruginosa in burn wound infections. Infect. Immun. 1999; 67:5854- 5862.
26. Wu H, Song Z, Givskov M, Hoiby N. Effects of quorum-sensing on immunoglobulin G responses in a rat model of chronic lung infection with Pseudomonas aeruginosa. Microbes. Infect. 2004; 6:34-37.
27. Manefield M, Welch M, Givskov M, Salmond GP, Kjelleberg S. Halogenated furanones from the red alga, Delisea pulchra, inhibit carbapenem antibiotic synthesis and exoenzyme virulence factor production in the phytopathogen Erwinia carotovora. FEMS Microbiol. Lett. 2001; 205:131-138.
28. Olsen JA, Severinsen R, Rasmussen TB, Hentzer M, Givskov M, Nielsen J. Synthesis of new 3- and 4-substituted analogues of acyl homoserine lactone quorum sensing autoinducers. Bioorg. Med. Chem. Lett. 2002; 12:325-328.
29. Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Hoiby N, et al. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology 2002; 148:87-102.
30. Welch M, Dutton JM, Glansdorp FG, Thomas GL, Smith DS, Coulthurst SJ, Barnard AM, Salmond GP, Spring DR. Structure- activity relationships of Erwinia carotovora quorum sensing signaling molecules. Bioorg. Med. Chem. Lett. 2005; 15:4235-4238.
31. Glansdorp FG, Thomas GL, Lee JK, Dutton JM, Salmond GP, Welch M, Spring DR. Synthesis and stability of small molecule probes for Pseudomonas aeruginosa quorum sensing modulation. Org. Biomol. Chem. 2004; 2:3329-3336.
32. Rasmussen TB, Manefield M, Andersen JB, Eberl L, Anthoni U, Christophersen C, Steinberg P, Kjelleberg S, Givskov M. How Delisea pulchra furanones affect quorum sensing and swarming motility in Serratia liquefaciens MG1. Microbiology 2000; 146:3237-3244.
33. Manefield M, Rasmussen TB, Henzter M, Andersen JB, Steinberg P, Kjelleberg S, Givskov M. Halogenated furanones inhibit quorum sensing through accelerated LuxR turnover. Microbiology 2002; 148:in9-1127.
34. Bjarnsholt T, Jensen PO, Rasmussen TB, Christophersen L, Calum H, Hentzer M, Hougen HP, Rygaard J, Moser C, Eberl L, et al. Garlic blocks quorum sensing and promotes rapid clearing of pulmonary Pseudomonas aeruginosa infections. Microbiology 2005; 151:3873-3880.
35. Choo JH, Rukayadi Y, Hwang JK: Inhibition of bacterial quorum sensing by vanilla extract. Lett. Appl. Microbiol. 2006; 42:637- 641.
36. Vattem DA, Mihalik K, Crixell SH, McLean RJ. Dietary phytochemicals as quorum sensing inhibitors. Fitoterapia 2007; 78:302- 310.
37. Nalca Y, Jansch L, Bredenbruch F, Geffers R, Buer J, Haussler S. Quorum-sensing antagonistic activities of azithromycin in Pseudomonas aeruginosa PAO1: a global approach. Antimicrob. Agents Chemother. 2006; 50:1680-1688.
38. Nasser W, Bouillant ML, Salmond G, Reverchon S. Characterization of the Erwinia chrysanthemi expI-expR locus directing the synthesis of two N-acyl-homoserine lactone signal molecules. Mol. Microbiol. 1998; 29:1391-1405.
39. Zhu J, Winans SC. Autoinducer binding by the quorum-sensing regulator TraR increases affinity for target promoters in vitro and decreases TraR turnover rates in whole cells. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:4832-4837.
40. Welch M, Todd DE, Whitehead NA, McGowan SJ, Bycroft BW, Salmond GP. N-acyl homoserine lactone binding to the CarR receptor determines quorum-sensing specificity in Erwinia. EMBO J. 2000; 19:631-641.
41. Zhang RG, Pappas T, Brace JL, Miller PC, Oulmassov T, Molyneaux JM, Anderson JC, Bashkin JK, Winans SC, Joachimiak A. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature 2002; 417:971-974.
42. Vannini A, Volpari C, Gargioli C, Muraglia E, Cortese R, De Francesco R, Neddermann P, Marco SD. The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. EMBO J. 2002; 21:4393-4401.
43. Bottomley MJ, Muraglia E, Bazzo R, Carfi A. Molecular insights into quorum sensing in the human pathogen Pseudomonas aeruginosa from the structure of the virulence regulator LasR bound to its autoinducer. J. Biol. Chem. 2007; 282:13592-13600.
44. Smith KM, Bu Y, Suga H. Induction and inhibition of Pseudomonas aeruginosa quorum sensing by synthetic autoinducer analogs. Chem. Biol. 2003; 10:81-89.
45. Smith KM, Bu Y, Suga H. Library screening for synthetic agonists and antagonists of a Pseudomonas aeruginosa autoinducer. Chem. Biol. 2003; 10:563-571.
46. Muh U, Schuster M, Heim R, Singh A, Olson ER, Greenberg EP. Novel Pseudomonas aeruginosa quorum-sensing inhibitors identified in an ultra-high-throughput screen. Antimicrob. Agents Chemother. 2006; 50:3674-3679.
47. Reverchon S, Chantegrel B, Deshayes C, Doutheau A, Cotte-Pattat N. New synthetic analogues of N-acyl homoserine lactones as agonists or antagonists of transcriptional regulators involved in bacterial quorum sensing. Bioorg. Med. Chem. Lett. 2002; 12:1153-1157.
48. Castang S, Chantegrel B, Deshayes C, Dolmazon R, Gouet P, Haser R, Reverchon S, Nasser W, Hugouvieux-Cotte-Pattat N, Doutheau A. N-Sulfonyl homoserine lactones as antagonists of bacterial quorum sensing. Bioorg. Med. Chem. Lett. 2004; 14:5145-5149.
49. Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. Small molecule inhibitors of bacterial quorum sensing and biofilm formation. J. Am. Chem. Soc. 2005; 127:12762-12763.
50. Dong YH, Zhang LH: Quorum sensing and quorum-quenching enzymes. J. Microbiol. 2005; 43:101-109.
51. Dong YH, Xu JL, Li XZ, Zhang LH: AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:3526-3531.
52. Ozer EA, Pezzulo A, Shih DM, Chun C, Furlong C, Lusis AJ, Greenberg EP, Zabner J. Human and murine paraoxonase 1 are host modulators of Pseudomonas aeruginosa quorum-sensing. FEMS Microbiol. Lett. 2005; 253:29-37.
53. Chun CK, Ozer EA, Welsh MJ, Zabner J, Greenberg EP. Inactivation of a Pseudomonas aeruginosa quorum-sensing signal by human airway epithelia. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:3587-3590.
54. Kaufmann GF, Sartorio R, Lee SH, Mee JM, Altobell LJ, 3rd, Ku- jawa DP, Jeffries E, Clapham B, Meijler MM, Janda KD. Antibody interference with N-acyl homoserine lactone-mediated bacterial quorum sensing. J. Am. Chem. Soc. 2006; 128:2802-2803.
55. Debler EW, Kaufmann GF, Kirchdoerfer RN, Mee JM, Janda KD, Wilson IA. Crystal structures of a quorum-quenching antibody. J. Mol. Biol. 2007; 368:1392-1402.
56. Miyairi S, Tateda K, Fuse ET, Ueda C, Saito H, Takabatake T, Ishii Y, Horikawa M, Ishiguro M, Standiford TJ, et al. Immunization with 3-oxododecanoyl-L-homoserine lactone-protein conjugate protects mice from lethal Pseudomonas aeruginosa lung infection. J. Med. Microbiol. 2006; 55:1381-1387.
Further Reading
Dunny GM, Winans SC. Cell-Cell Signaling in Bacteria. 1999. ASM Press, Washington, DC.
Demuth DR, Lamont RJ. Bacterial Cell-to-Cell Communication: Role in Virulence and Pathogenesis. 2006. Cambridge University Press, Cambridge, UK.
England R. Microbial Signalling and Communication. 1999. Cambridge University Press, Cambridge, UK.
Hodgson DA Thomas CM Signals, Switches, Regulons, and Cascades: Control of Bacterial Gene ExpressionSociety for General Microbiology. 2002 Cambridge University Press, Cambridge, UK.
Vendeville A, Winzer K, Heurlier K, Tang CM, Hardie KR. Making ‘sense’ of metabolism: autoinducer-2, LuxS and pathogenic bacteria. Nat. Rev. Microbiol. 2005; 3:383-396.
Karina B, Bassler X, Bassler BL, LuxS quorum sensing: more than just a numbers game. Curr. Opin. Microbiol. 2003; 6:191-197.
McNab R, Lamont RJ. Microbial dinner-party conversations: the role of LuxS in interspecies communication. J. Med. Microbiol. 2003; 52:541-545.
Yoon SS, Hassett DJ. Chronic Pseudomonas aeruginosa infection in cystic fibrosis airway disease: metabolic changes that unravel novel drug targets. Expert Rev. Anti. Infect. Ther. 2004; 2:611-623.
Pritchard DI. Immune modulation by Pseudomonas aeruginosa quorumsensing signal molecules. Int. J. Med. Microbiol. 2006; 296:111-116.
See Also
Antibacterial Drugs, Design of
Chemical Signals in Sensing
Quorum Sensing