CHEMICAL BIOLOGY
Metabolomics: Topics in Chemical Biology
Julian L. Griffin, Helen J. Atherton and Michael R. Pears, Department of Biochemistry, Tennis Court Road, Cambridge, United Kingdom
doi: 10.1002/9780470048672.wecb655
With the development of systems biology, several approaches have been developed to profile a tier of organization in a cell, tissue, or organism globally. Metabolomics, also referred to as metabonomics, is an approach that attempts to profile all the small-molecule metabolites in a biological matrix. One major challenge of this approach, as with other "-omic" technologies, is that the metabolome is context dependent and varies with developmental stage/age, genetic modification, disease, and environment. Thus, by definition, the metabolome of an organism must take into consideration all the effects of these manipulations. Despite these challenges, the approach has already been applied to understand metabolism in a range of animal models and has more recently started to be applied to clinical studies. In this article, we will discuss some common approaches currently used in metabolomics and the results that they have produced. In particular, we will focus on the two most common analytical approaches, nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry, and some biological problems they have been used to address.
The postgenomic era seeks to define the biological function of genes and how these translate to define an organism’s phenotype. Unlike genome sequencing projects, the effective study of functional genomics necessitates a plethora of multidisciplinary techniques (1). The development of analytical tools that enable the high-throughput measurement of gene products has provided a comprehensive signature of the physiological state of the cell at several levels (Fig. 1). Metabolomics describes the large-scale analysis of endogenous metabolites that comprise the entire collection of small molecules in a cell, tissue, organism, or biofluid, and includes sugars, organic acids, amino acids, and nucleotides (2, 3).

Figure 1. The physiological state of the cell is measured at several levels in a functional genomic approach, all of which interact with one another to form a highly interconnected network. In particular, differences in the transcriptome, proteome, and metabolome are monitored as a means to study gene function and cellular responses to external stimuli.
Benefits of a Metabolomic Approach
A metabolomic approach has several advantages for use in functional genomics. Firstly, as with transcriptional and proteomic analyses, the technique is context-dependent, such that the metabolite complement varies according to the physiological, developmental, or pathological state of the cell, tissue, organ, or organism, which makes it a highly potent tool for measuring changes in phenotype.
Secondly, the importance of metabolites in biological control and communication, as building blocks for complex macromolecules and energy transporters, makes them effective markers of cellular function. Indeed, metabolites constitute molecular endpoints farther down the line from gene to function. Thirdly, metabolic control analysis suggests that, although changes in the quantities of individual enzymes are expected to exert little influence on metabolic fluxes, they can and do have a significant impact on metabolite concentrations, even when changes in flux are negligible. Fourthly, metabolites span the species barrier because they have the same chemical structure irrespective of the organism. Metabolomics is therefore a universal “omic” technology in contrast to transcriptomics and proteomics in which a priori knowledge of DNA and protein sequences from each organism is required. Little time is required for reoptimizing protocols for a new species. Finally, metabolomics presents several practical benefits, including cheap cost on a per sample basis, high throughput, and full automation. For example, after the initial purchase of a nuclear magnetic resonance (NMR) spectrometer or mass spectrometer (MS), samples can be analyzed at a cost in the region of £ ~1.00 per sample, in terms of consumables, with analytical acquisition times typically taking 10 min (NMR) to 70 min (GC-MS). This cost compares very favorably with transcriptional and proteomic analyses.
The terms metabolomics (2) and the related term metabonomics (3) were coined in the late 1990s to describe the use of global profiling tools combined with pattern recognition to define a metabolic phenotype of a cell, tissue, or organism. The confusion over what to call the field is still a persistent problem, and new researchers to the field should be aware of both terms. Many people use the terms metabolomics and metabonomics interchangeably (4, 5). However, Nicholson and coworkers state that metabonomics is the “quantitative measurement of the time-related multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification,” whereas metabolomics is the “measurement of metabolite concentrations and fluxes and secretion in cells and tissues in which there is a direct connection between the genetic activity, protein activity and the metabolic activity itself” (6). To add more confusion, additional terms have been put forward dependent on the analytical techniques adopted. “Metabolic profiling” has been proposed to refer to the detailed analysis of metabolites by hyphenated techniques such as gas chromatography-(GC)-MS and liquid chromatography-(LC)-MS. In contrast, “metabolic fingerprinting” is thought to measure a subset of metabolites and uses techniques such as NMR and direct infusion electrospray-MS to create a barcode of metabolism (7). The term “metabolomics” will be employed in this article throughout.
Analytical techniques
Metabolomics is challenged by the remarkable heterogeneity and complexity exhibited by metabolites. Metabolites span concentration ranges of the order 109, polarity ranges of ~1020, and mass ranges of the order of 1500 amu. Moreover, most extraction procedures do not provide complete representations, which results in only a portion of the metabolome being analyzed. A truly comprehensive coverage of the metabolome, therefore, necessitates multiple techniques and different sample preparation strategies to encompass such diversity. The principal analytical platforms for metabolomics include 1H NMR spectroscopy and GC- and LC-MS (Fig. 2), although others have also used other techniques including Fourier transform infrared spectroscopy, thin layer chromatography, high-pressure liquid chromatography, and Raman spectroscopy. The major techniques as judged by current number of publications will be discussed below.

Figure 2. The two most frequently used approaches for metabolomics are NMR spectroscopy and mass spectrometry. To maximize the coverage of the metabolome, these techniques can be combined. This figure shows the analysis of the aqueous fraction of a section of heart tissue that uses NMR spectroscopy, gas chromatography mass spectrometry and liquid chromatography mass spectrometry. Although mass spectrometry approaches are inherently more sensitive than NMR spectroscopy, metabolite identification becomes more difficult.
Nuclear magnetic resonance spectroscopy
High-resolution 1H nuclear magnetic resonance (NMR) spectroscopy is a relatively rapid technique and is highly robust in terms of reproducibility of results. The ubiquity of protons in cellular metabolites and the fact that other nuclei are observable by NMR (e.g., 31P and 13C) mean that a relatively large number of different metabolites can be detected. Furthermore, the technique requires minimal sample preparation, and its nondestructive nature allows for more analyses to be conducted. However, 1H NMR is an inherently insensitive analytical tool that only measures high-concentration metabolites. Typically, using a simple one-dimensional pulse sequence, 1H NMR can measure 30-100 metabolites in urine, 20-30 metabolites in blood plasma, and 10-30 metabolites in tissue extracts (8). Despite this, however, 1H NMR has proved highly discriminatory for liver toxins in rats (9, 10), mouse models of cardiac and neurological diseases (11, 12), and silent phenotypes in yeast (13). It has been suggested that this finding may be because of the high-concentration metabolites that represent central hubs of metabolism, whereby a perturbation at one point in a metabolic network would be transferred to other pathways through these highly connected hubs (14, 15). However, restricting the coverage of the metabolome to 30 or so metabolites may hinder the isolation of metabolites as unique biomarkers for disease processes and confound the deduction of which pathways are perturbed during a given modification. Strategies are being developed to make NMR a more sensitive approach, including the use of cryoprobes and hyphenated techniques. Cryoprobes use liquid helium to cool down the NMR coil assembly and preamplifier to ~4 K, which, by reducing thermal noise, results in a three- to four-fold increase in the signal-to-noise ratio. Hyphenated techniques involve first separating out high and low concentration metabolites using liquid chromatography. This separation improves sensitivity by reducing the likelihood of coresonant peaks and improves the dynamic range of the NMR experiment (16).
Other NMR-based techniques allow the quantification of metabolite concentrations in intact tissue, either in vivo or ex vivo. For example, magnetic resonance spectroscopy (MRS) allows the noninvasive interrogation of metabolite concentrations directly in vivo within a specific localized region. The technique has proved invaluable in the investigation of Alzheimer’s disease (17, 18), traumatic brain injury (19), multiple sclerosis (20), and many brain tumors (21). However, one disadvantage of MRS is that it is compromised by low spectral resolution, partly as a result of the lower fields used in vivo but also confounded by dipolar couplings, chemical shift anisotropy, and bulk magnetic field inhomogeneity effects, which act to broaden spectral resonances. These effects can be reduced dramatically by spinning the sample at the “magic angle” (54.7°). Indeed, using high-resolution magic angle spinning (HRMAS) 1H NMR spectroscopy, it is possible to produce high-resolution spectra comparable to those from tissue extracts, specifically from intact tissue ex vivo (22-24). This method circumvents the need for tissue extraction procedures and effectively eliminates a step that introduces more variation into metabolite quantification. Another advantage of HRMAS 1H NMR is the prospect of investigating metabolic compartmentation. A recent study demonstrated subcompartments of acetoacetate and glutamine in rat heart mitochondria that were not NMR-visible (22).
Mass spectrometry
Mass spectrometry (MS) allows the analysis of lower-concent ration metabolites compared with 1H NMR spectroscopy. However, a comprehensive metabolic analysis by MS often requires sample prefractionation, the most common being gas chromatography (GC) and liquid chromatography (LC). Although this prefractionation improves the resolution of the technique and reduces ion suppression, adding a chromatographic step can introduce variability into a data set.
In GC-MS, volatile and thermally stable compounds are first separated by GC and the eluted compounds detected by MS. To maximize the range of compounds that are volatile and thermally stable, samples are usually derivatized before analysis for metabolomic experiments. Methoximation in combination with trimethylsilylation is the most predominant protocol for examining aqueously soluble metabolites (25). Methoximation reduces the number of isomers present for sugars, with the number of isomeric forms being reduced from 5 to 2, which thereby dramatically simplifies chromatograms. Trimethylsilylation replaces exchangeable protons with silyl groups to make compounds less polar and therefore more volatile and to improve chromatography. The derivatized samples are vaporized within an inlet port and injected onto the column where they are carried by an inert gas. Compounds are separated according to their partition between the mobile gas phase and the column-bound stationary phase, and the eluted compounds are subsequently ionized within the ion source. The most frequently used ionization method is electron impact in which a beam of electrons ionizes the sample molecules, which concomitantly fragment. Because the energy of these electrons is predefined, the fragmentation pattern is reproducible and effectively provides a near-unique metabolite fingerprint. Metabolites, therefore, can be identified easily by comparing spectra with those in commercially available mass spectral databases (e.g., http://www.nist.gov). Although this approach generally works well, distinction between sugar diastereomers is confounded by both species having identical fragmentation patterns. In such cases, retention times of standards are imperative for accurate assignments. Furthermore, spectral databases are not exhaustive, and so not all peaks can be assigned. Currently, efforts are being made to create metabolomic-specific mass spectral libraries (26). More detailed characterization of unknown peaks can be conducted by running the same samples using chemical ionization as opposed to electron impact ionization or using ion trap mass spectrometers. Chemical ionization is a less energetic method of ionization and so produces less fragmentation. This method increases the probability of observing the molecular ion and so can facilitate compound identification.
The sensitivity of the GC-MS approach is impressive. Using a simple single quadrupole GC-MS, Fiehn and coworkers working on Arabidopsis thaliana reported the detection of 326 metabolites, a number that has since increased to over 1000 using a Time of Flight GC-MS (25, 27). Furthermore, the technique has benefited from recent technological advances in two-dimensional GC (28).
LC-MS provides metabolite separation by LC before detection by MS. Sample derivatization is generally not required because no prerequisite for volatility is necessary. Moreover, LC-MS can analyze a wider array of metabolites relative to GC-MS because it is not limited to volatile compounds or molecules that can be rendered volatile. In particular, thermolabile or large molecules such as di- and triphosphates, CoA adducts, peptides, and lipids are all amenable to analysis by LC-MS (29). Following separation by LC, the compounds are ionized, typically using electrospray ionization (26). The sample is passed through a thin metallic capillary tube that contains positively or negatively charged ions, dependent on the solvent used. Electrostatic charging of the sample then creates a cloud of charged droplets, and desolvation of these droplets results in ions that are sampled by the MS. Because electrospray ionization causes minimal fragmentation of the molecular ion, direct metabolite identification by comparison of mass spectra is often not possible. Structural information can be gained, however, by using tandem MS technologies, whereby sequential and selective fragmentations can be carried out. Moreover, compounds that contain a particular functional moiety can be analyzed by conducting neutral loss experiments.
Pietiläinen and coworkers (29) have used LC-MS-based lipidomics to examine whether acquired obesity was associated with different serum lipid profiles in young adult monozygotic twins who were either concordant or discordant (10-25 kg weight difference) for obesity. For the discordant twins, blood serum had increased concentrations of proinflammatory and proatherogenic lysophosphatidylcholines and decreases in the antioxidant ether phospholipids, which suggests that in these obese individuals the lipid profile was already such that it would promote atherogenesis, inflammation, and insulin resistance. LC-MS has also been used to follow changes in the aqueous fraction of tissue extracts (31), blood plasma, and urine (30) from rodent models of type II diabetes, but here the largest challenge is identifying the metabolites detected, and often such data has been largely used to classify samples as part of a metabolic fingerprinting study rather than to carry out biomarker identification.
Relative to GC-MS, the application of LC-MS within the metabolomics field is still at a preliminary stage. Reproducibility is a major concern, and true quantification can be hindered by ion suppression effects whereby one co-eluting metabolite affects the ionization of another (26). The lack of electrospray ionization mass spectral libraries also makes identification by LC-MS a particularly challenging problem. Nevertheless, the technique is developing fast and has benefited from several technological advancements such as the acquisition of accurate mass data by the use of ion cyclotron resonance Fourier transform MS (26).
It is possible to bypass the chromatography stage and use direct infusion of lipid extracts into the mass spectrometer as part of an approach referred to as shotgun lipidomics. Han and colleagues (32) using this approach demonstrated a profound decrease in cardiolipin in the myocardium following streptozotocin-induced diabetes proceeding accumulation of triglyceride in the heart. Similar modulations of cardiolipin metabolism were observed in the ob/ob mouse.
Note that an integral part of the metabolomic approach is the application of pattern recognition techniques to deduce what variation in a data set is associated with a given disease, genetic modification, or other manipulation of the system. Because of space limitations, it is not possible to discuss this area as part of this article, but we refer the reader to several excellent reviews (33, 34).
Applications of Metabolomics
Metabolomics has proved extremely versatile with a diversity of applications including the study of gene function, toxicology, plant metabolism, environmental analysis, clinical diagnostics, investigation of disease, and discrimination of organism genotypes. It is not possible to create a definitive list given the huge number and range of approaches that have now been published, but we have set out some key publications below to demonstrate the potential uses of metabolomics.
Study of gene function
One of the first major successes of metabolomics has been the definition of silent phenotypes in yeast where conventional growth rate analyses failed to distinguish different yeast mutants (12). By using 1H NMR spectroscopy, it was apparent that “silent” yeast mutants have compensatory changes in intracellular metabolites to allow the cells to grow at a normal rate. Mutants deleted for genes of related biological activity resulted in similar metabolic perturbations and, hence, metabolic profiles. Specifically in this study, two yeast strains, deleted for either one or other of the two redundant genes for 6-phosphofructo-2-kinase, clustered together according to their metabolic profiles, as did strains deleted for genes related to mitochondrial metabolism (12). The authors coined the term FANCY for functional analysis of coresponses in yeast (12) to describe this comparative metabolomics approach. Currently, FANCY is being used to predict gene function. Metabolic profiles of strains deleted for known genes are being compared with those deleted for unknown genes in the hope of assigning biological roles.
Plant metabolism
Some of the most significant developments in metabolic profiling, particularly those involving GC-MS, have been made in the plant sciences discipline (35) (Fig. 3). Plants are thought to be exceptionally metabolite-rich relative to mammals and yeast. This richness stems not only from the size of their genomes, which ranges from 20,000 to 50,000 genes, but also from multiple substrate specificities for many enzymes, subcellular compartmentation, and nonenzymatic reactions. Currently, 50,000 different compounds have been elucidated in plants (36), and the final number is set to increase to approximately 200,000 (28). Concomitantly, the tools and analytical techniques used in metabolomics have been advanced rapidly by researchers within this field. One major goal of plant metabolomics is to expand on the information about how plant biochemistry is composed and controlled. Metabolite profiling has been conducted on a diverse array of plant species, including Arabidopsis thaliana (25), tomato (37), potato (38), and rice (39). Efforts are being focused on characterizing silent plant phenotypes and trying to elucidate biological roles for genes of unknown function. For example, a modified potato plant line suppressed in expression of sucrose synthase isoform II displayed no overt phenotype in terms of morphology, yield, or growth rate compared with parental lines. However, by using GC-time-of-flight-MS to analyze the metabolic profiles, approximately 1000 compounds were quantified, and plant varieties were successfully discriminated (38). Similarly, other researchers have integrated transcriptomic and metabolomic analyses to investigate global responses to nutritional stress in Arabidopsis thaliana and have demonstrated that the genes and metabolites involved in glucosinolate metabolism are coordinately regulated in response to either sulphur or nitrogen deficiency (40). Metabolomics has also been employed in characterizing the regulatory synthesis of novel plant products; for example, modified health-related carotenoid and flavonoid-based antioxidants have been isolated in transgenic tomatoes (41). Plant-host interactions are also a popular target for metabolomic studies (42, 43).
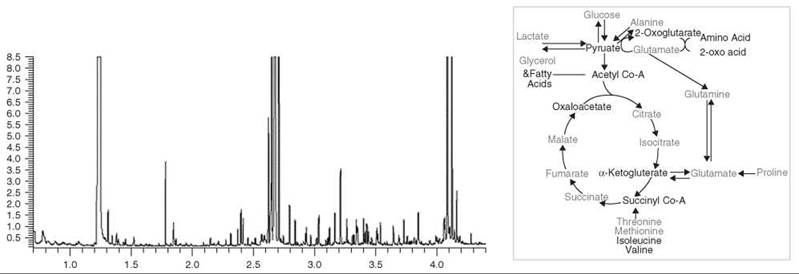
Figure 3. An example of the use of GC-MS for metabolic profiling. Left: A section of the total ion chromatogram from the analysis of TMS-derivatized aqueous tissue extracts from the liver of PPAR-α null mouse. Metabolites are identified from exact retention times and comparison of corresponding mass spectra with those in the NIST database. 97 metabolites were quantified. Right: Summary of metabolite differences in the tissues of the PPAR-α null mouse. Red- increased relative to control, blue- decreased relative to control. The increased/decreased width of certain arrows reflects relative increased/decreased concentrations across these pathways, respectively.
Environmental science
Metabolomics has made remarkable inroads into the environmental research community. Here, a major emphasis is to understand the impact that environmental stress, such as pollution and climate change, has on wildlife. Indeed, many government organizations monitor the prevalence of pollutants in certain species of wildlife as indicators of the exposure risk within the environment. Studies of Japanese medaka have been conducted to investigate the effects of trichloroethylene, a common environmental pollutant, and the pesticide dinoseb, on the development of fish embryos (44, 45). Similarly, cadmium toxicity has been examined in the bank vole and rat and has revealed changes in lipid metabolism that preceded classical nephrotoxicity (46, 47). Another study investigated the effects of environmental toxins on earthworms (48). In particular, the analysis of earthworm tissue extracts by 1H NMR spectroscopy identified maltose as a potential biomarker for ecotoxicity within a metal-contaminated site.
Clinical diagnostics
Metabolomics is well suited to clinical trials, and as more efficient and comprehensive analytical tools become available, a wealth of additional information can be gathered from costly clinical trials without increasing the level of discomfort to the patient or decreasing the practicality to the physician. The fact that all the technologies involved in metabolomics are relatively high-throughput means that large-scale clinical studies can be performed. Makinen and coworkers (49) examined the use of 1H NMR spectroscopy of blood serum to diagnose diabetic nephropathy in 182 sufferers of type 1 diabetes. This approach relied, in part, on modeling the different distributions of lipoprotein fractions and produced a test approximately as good as that used clinically. The group has since developed a Bayesian Markov chain Monte Carlo approach to modeling the constituents of the saturated lipid profile in blood serum (50) and is exploring the use of this approach to model complications associated with type 1 diabetes. Similarly, Brindle and colleagues described a 1H NMR spectroscopy-based method for potentially diagnosing coronary artery disease through blood serum as a potential replacement to angiography (51). However, data sets can be biased to the stratification of a particular population, and thus even in relatively large studies, one should view with caution the results produced. Kirschenlohr and colleagues (52) demonstrated that the diagnosis of CAD by 1H NMR spectroscopy of blood plasma was significantly complicated by related changes in the lipoprotein/saturated lipid region of the 1H NMR spectra associated with gender and statin treatment and that previous models had produced an artificially high classification of disease presence. Roussel and coworkers (53) similarly demonstrated that cardiovascular risk was poorly predicted by 1H NMR spectroscopy and multivariate analysis in a population of type 2 diabetics. Thus, as in epidemiology studies, it will be important to follow up metabolomic biomarker discovery studies in humans with more studies in other populations to detect potential confounding factors.
The discussed studies have largely focussed on NMR-based metabolomics, but many recent studies have also used mass spectrometry to either quantitate low-concentration metabolites in a targeted manner (54) or open profiling of the chromatogram (55). It is beyond the scope of this article to detail them all, but we would refer the reader to the reviews at the end of this article as well as a review by German and colleagues (56) that details how these technologies might be used for clinical diagnosis.
Toxicology
Metabolomics is increasingly being deployed in toxicology studies within the pharmaceutical industry, especially in the safety assessment of candidate drugs. Because of the high costs entailed in clinical trials, an effective early screen of toxicity is extremely desirable. Metabolomics is being directed at identifying novel biomarkers of toxicity and elucidating mechanisms of effect (5). Examples include the correlation between elevated creatine levels in serum and urine with hepatotoxicity and nutritional effects (9) and the characterization of urinary dicarboxylic aciduria being the result of impaired fatty acid metabolism (10). By measuring whole system metabolic responses in urine or plasma, as opposed to conventional assays that focus on selected organs, a metabolomic approach can improve our understanding of systemic toxic effects (5). Moreover, the technique easily can be incorporated into existing toxicology studies, and the results can be correlated with routinely measured end points from clinical chemistry and histopathology.
Investigation of animal models of disease and discrimination of organism genotypes
Metabolomics is ideally placed as a phenotyping tool in the exploration of naturally occurring and transgenic disease models. The refinement of knockout and knockin strategies combined with accumulating sequence data has accelerated the generation of accurate disease models. Moreover, large-scale mouse mutagenesis programs have been set up, which have produced thousands of mutants in need of analysis (57). Screening for congenital malformations and biochemical, hematological, and immunological defects is extremely labor-intensive and time-consuming. Furthermore, many models do not express the same phenotype as humans and often display a milder pathology such that it is difficult to assess the effects of genetic perturbation. This situation, in part, reflects the shorter life span of model organisms such as mice when compared with humans.
Metabolomics is becoming a prominent phenotyping aid to characterize tissues and organisms rapidly within a biological context. For example, using a metabolomics-guided screen of mutant mice, a mouse model with a novel enzyme deficiency that resembles human maple syrup urine disease has been isolated (58). Mice were identified as having elevated levels of branched chain amino acids in blood and increased concentrations of branched chain α-keto acids in urine. More investigation correlated these results with a deficiency in branched chain amino-transferase. Other researchers have used metabolomics to characterize the metabolic deficits associated with a particular disease process, for example, human metabolic syndrome. For example, the systemic effects of the PPARα mutation, a gene important in regulating the fed/fasting response, have recently been defined; using a combination of 1H NMR and GC-MS, metabolic changes have been followed in the heart, liver, skeletal muscle, and adipose tissue of the PPARα null mouse (31). Similar metabolic profiling approaches have also been conducted to investigate cardiac disease in the mdx mouse (10) and atherosclerosis in the ApoE3-Leiden mouse (59).
Metabolomic techniques have also been used widely to phenotype neurological disorders, with a diverse array of applications including the characterization of regional variation, brain tumors, and neurological disorders (60-64) (Fig. 4). Because the brain is heavily compartmentalized, a recent study used metabolic profiling to characterize distinct neuroanatomical regions in rats ex vivo by high-resolution magic angle spinning 1H NMR (62). Clear biochemical differences were defined between the brain stem, frontal cortex, cerebellum, and hippocampus. This study provides an invaluable baseline reference for more HRMAS 1H NMR spectroscopic studies to monitor disease and specific pharmacological insults within the brain. Furthermore, using HRMAS 1H NMR spectroscopy, it was possible to characterize an accumulation of polyunsaturated fatty acids in BT4C gliomas in rats during gene-therapy-induced apoptosis (61). Such lipids are easily detectable in vivo by magnetic resonance spectroscopy and could be used to monitor the efficacy of gene therapy in patients with glioma. As a complement to this study (65), the low molecular weight intermediate composition of the same rat gliomas was quantified subsequently, and it was demonstrated that myo-inositol, glycine, and taurine concentrations correlated with tumor cell density, whereas the overall concentration of choline-containing compounds was unaffected by cell loss (66). Another study has combined MRS with automated pattern recognition techniques to help radiologists categorize brain tumors according to histological type and grade (66). Using metabolic profiling, it was possible to discriminate between meningiomas, low-grade astrocytomas, and aggressive tumors such as glioblastomas and metastases. Spectral profiles prepared from intact tissue, tissue extracts, and biofluids have also proven to be highly discriminatory for several neurological diseases, including spinocerebellar ataxias and Huntington’s disease (58, 67).
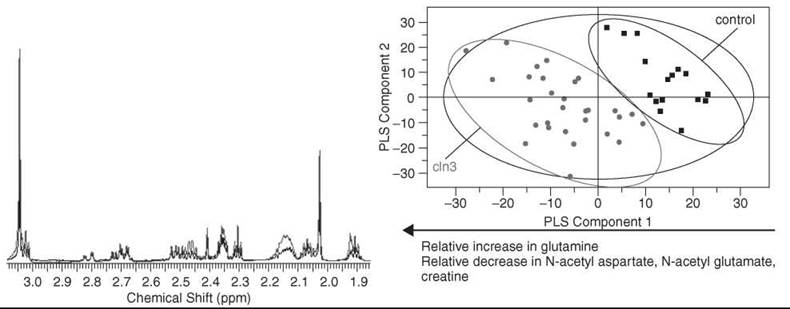
Figure 4. High-resolution 1H NMR spectroscopy has been used to monitor metabolic perturbations in a mouse model of Batten disease referred to as CLN3 54. Left: High-resolution solution state 1H NMR spectra of cortex tissue acquired at 700 MHz. Shown are typical spectra from cln3 (red) and control tissue (blue) along with assigned metabolites. Right: Score plot of a PLS-DA model that compares tissue from cln3 mice with respect to control mice. The model was formed by examining spectra from aqueous extracts of cortex tissue. Pareto scaling was used to scale the data. Major metabolites responsible for separation identified from examination of the corresponding loading plot to the score plot shown are listed below the plot.
Although the coining of the term metabolomics has been relatively new, an explosion of publications has occurred in this field plus a realization that many researchers already were doing similar studies, albeit without an “-omic” tag before the word. It has not been possible to review all the applications of metabolomics fully, and the applications will increase. In addition to the development of new applications, the development of the analytical approaches will also take center stage as researchers push back the limits of detection of NMR spectroscopy, mass spectrometry, and other analytical approaches. In addition to these “wet lab” developments, both the pattern recognition approaches used to process metabolomics and the metabolomic databases used to identify metabolites need to be developed or expanded. In this respect, an excellent place to start on the arduous journey to biomarker discovery through metabolomics is the current metabolomic databases found on the web that make standard spectra freely available (68-70).
1. Evans GA. Designer science and the “omic” revolution. Nat. Biotechnol. 2000; 18:127.
2. Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998; 16:373-378
3. Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999; 29:1181-1189.
4. Griffin JL. Metabolic profiles to define the genome: can we hear the phenotypes? Philos. Trans. R. Soc. 2004; 359:857-871.
5. Robertson DG. Metabonomics in toxicology: a review. Toxicol. Sci. 2005; 85:809-822.
6. Nicholson JK, Wilson ID. Opinion: understanding ‘global’ systems biology: metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003; 2:668-676.
7. Fiehn O. Metabolomics--the link between genotypes and phenotypes. Plant Mol. Biol. 2002; 48:155-171.
8. Griffin JL. The Cinderella story of metabolic profiling: does metabolomics get to go to the functional genomics ball? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006; 361:147-161.
9. Clayton TA, Lindon JC, Everett JR, Charuel C, Hanton G, Le Net JL, Provost JP, Nicholson JK. Hepatotoxin-induced hypercreatinaemia and hypercreatinuria: their relationship to one another, to liver damage and to weakened nutritional status. Arch. Toxicol. 2004; 78:86-96.
10. Mortishire-Smith RJ, Skiles GL, Lawrence JW, Spence S, Nicholls AW, Johnson BA, Nicholson JK. Use of metabonomics to identify impaired fatty acid metabolism as the mechanism of a drug-induced toxicity. Chem. Res. Toxicol. 2004; 17:165-173.
11. Jones GL, Sang E, Goddard C, Mortishire-Smith RJ, Sweatman BC, Haselden JN, Davies K, Grace AA, Clarke K, Griffin JL. A functional analysis of mouse models of cardiac disease through metabolic profiling. J. Biol. Chem. 2005; 280:7530-7539.
12. Tsang TM, Woodman B, McLoughlin GA, Griffin JL, Tabrizi SJ, Bates GP, Holmes E. Metabolic characterization of the R6/2 transgenic mouse model of Huntington’s disease by high-resolution MAS 1H NMR spectroscopy. J. Proteome Res. 2006; 5:483-492.
13. Raamsdonk LM, Teusink B, Broadhurst D, Zhang N, Hayes A, Walsh MC, Berden JA, Brindle KM, Kell DB, Rowland JJ, Westerhoff HV, van Dam K, Oliver SG. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 2001; 19:45-50.
14. Brindle KM. Pandora’s box or Aladdin’s cave? Biochemistry 2003; 25:15-17.
15. Griffin JL. Understanding mouse models of disease through metabolomics. Curr. Opin. Chem. Biol. 2006; 10:309-315.
16. Keun HC, Beckonert O, Griffin JL, Richter C, Moskau D, Lindon JC, Nicholson JK. Cryogenic probe 13C NMR spectroscopy of urine for metabonomic studies. Anal. Chem. 2002; 74:4588-4593.
17. Godbolt AK, Waldman AD, MacManus DG, Schott JM, Frost C, Cipolotti L, Fox NC, Rossor MN. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology 2006; 66:718-722.
18. Marjanska M, Curran GL, Wengenack TM, Henry PG, Bliss RL, Poduslo JF, Jack CR Jr, Ugurbil K, Garwood M. Monitoring disease progression in transgenic mouse models of Alzheimer’s disease with proton magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:11906-11910.
19. Shutter L, Tong KA, Holshouser BA. Proton MRS in acute traumatic brain injury: role for glutamate/glutamine and choline for outcome prediction. J. Neurotrauma 2004; 21:1693-1705.
20. Narayana PA. Magnetic resonance spectroscopy in the monitoring of multiple sclerosis. J. Neuroimaging 2005; 15:46S-57S.
21. Tate AR, Underwood J, Acosta DM, Majos C, Moreno-Torres A, Howe FA, van der Graaf M, Lefournier V, Murphy MM, Loosemore A, Ladroue C, Wesseling P, Luc Bosson J, Cabanas ME, Simonetti AW, Gajewicz D, Griffiths JR, Arus C. Development of a decision support system for diagnosis and grading of brain tumours using in vivo magnetic resonance single voxel spectra. NMR Biomed. 2006; 19:411-434.
22. Bollard ME, Murray AJ, Clarke K, Nicholson JK, Griffin JL. A study of metabolic compartmentation in the rat heart and cardiac mitochondria using high-resolution magic angle spinning 1H NMR spectroscopy. FEBS Lett. 2003; 553:73-78.
23. Griffin JL, Blenkiron C, Valonen PK, Caldas C, Kauppinen RA. High-resolution magic angle spinning 1H NMR spectroscopy and reverse transcription-PCR analysis of apoptosis in a rat glioma. Anal. Chem. 2006; 78:1546-1552.
24. Griffin JL, Troke J, Walker LA, Shore RF, Lindon JC, Nicholson JK. The biochemical profile of rat testicular tissue as measured by magic angle spinning 1H NMR spectroscopy. FEBS Lett. 2000; 486:225-229.
25. Fiehn O, Kopka J, Dormann P, Altmann T, Trethewey RN, Willmitzer L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000; 18:1157-1161.
26. Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst 2005; 130:606-625.
27. Nikiforova VJ, Kopka J, Tolstikov V, Fiehn O, Hopkins L, Hawkesford MJ, Hesse H, Hoefgen R. Systems rebalancing of metabolism in response to sulfur deprivation, as revealed by metabolome analysis of Arabidopsis plants. Plant Physiol. 2005; 138:304-318.
28. Fiehn, O. (2002) Metabolomics--the link between genotypes and phenotypes. Plant Mol Biol, 48, 155-171.
29. Pietilainen KH, Sysi-Aho M, Rissanen A, Seppanen-Laakso T, Yki-Jarvinen H, Kaprio J, Oresic M. Acquired obesity is associated with changes in the serum lipidomic profile independent of genetic effects - a monozygotic twin study. PLoS ONE. 2007; 2:e218.
30. Williams RE, Lenz EM, Evans JA, Wilson ID, Granger JH, Plumb RS, Stumpf CL. A combined (1)H NMR and HPLC-MS-based metabonomic study of urine from obese (fa/fa) Zucker and normal Wistar-derived rats. J. Pharm. Biomed. Anal. 2005; 38:465-471.
31. Atherton HJ, Bailey NJ, Zhang W, Taylor J, Major H, Shockcor J, Clarke K, Griffin JL. A combined 1H-NMR spectroscopy- and mass spectrometry-based metabolomic study of the PPAR-alpha null mutant mouse defines profound systemic changes in metabolism linked to the metabolic syndrome. Physiol. Genom. 2006; 27:178-186.
32. Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007; 46:6417-6428.
33. Holmes E, Antti H. Chemometric contributions to the evolution of metabonomics: mathematical solutions to characterizing and interpreting complex biological NMR spectra. Analyst 2002; 127:1549-1557.
34. Kell DB. Metabolomics, machine learning and modelling: towards an understanding of the language of cells. Biochem. Soc. Trans. 2005; 33(Pt 3): 520-524.
35. Hall RD, Brower ID, Fitzgerald MA. Plant metabolomics and its potential application for human nutrition. Physiol. Plant. 2008; 132:162-175.
36. De Luca V, St Pierre B. The cell and developmental biology of alkaloid biosynthesis. Trends Plant Sci. 2000; 5:168-173.
37. Schauer N, Zamir D, Fernie AR. Metabolic profiling of leaves and fruit of wild species tomato: a survey of the Solanum lycopersicum complex. J. Exp. Bot. 2005; 56:297-307.
38. Weckwerth W, Loureiro ME, Wenzel K, Fiehn O. Differential metabolic networks unravel the effects of silent plant phenotypes. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7809-7814.
39. Sato S, Soga T, Nishioka T, Tomita M. Simultaneous determination of the main metabolites in rice leaves using capillary electrophoresis mass spectrometry and capillary electrophoresis diode array detection. Plant J. 2004; 40:151-163.
40. Hirai MY, Yano M, Goodenowe DB, Kanaya S, Kimura T, Awazuhara M, Arita M, Fujiwara T, Saito K. Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:10205-10210.
41. Dixon RA. A two-for-one in tomato nutritional enhancement. Nat. Biotechnol. 2005; 23:825-826.
42. Choi YH, Kim HK, Linhorst HJ, Hollander JG, Lefeber AW, Erkelens C, Nuzillard JM, Verpoorte R NMR metabolomics to revisit the tobacco mosaic virus infection in Nicotiana tabacum leaves. J. Nat. Prod. 2006; 69:742-748.
43. Allwood JW, Ellis DI, Goodacre R Metabolomic technologies and their application to the study of plants and plant-host interactions. Physiol. Plant 2008; 132:117-135.
44. Viant MR, Bundy JG, Pincetich CA, de Ropp JS, Tjeerdema RS. NMR-derived developmental metabolic trajectories: an approach for visualizing the toxic actions of trichloroethylene during embryogenesis. Metabolomics 2005; 1:149-158.
45. Viant MR, Pincetich CA, Hinton DE, Tjeerdema RS. Toxic actions of dinoseb in medaka (Oryzias latipes) embryos as determined by in vivo 31P NMR, HPLC-UV and 1H NMR metabolomics. Aquat. Toxicol. 2006; 76:329-342.
46. Griffin JL, Walker LA, Shore RF, Nicholson JK. Metabolic profiling of chronic cadmium exposure in the rat. Chem. Res. Toxicol. 2001; 14:1428-1434.
47. Griffin JL, Walker LA, Troke J, Osborn D, Shore RF, Nicholson JK. The initial pathogenesis of cadmium induced renal toxicity. FEBS Lett. 2000; 478:147-150.
48. Bundy JG, Spurgeon DJ, Svendsen C, Hankard PK, Weeks JM, Osborn D, Lindon JC, Nicholson JK. Environmental metabonomics: applying combination biomarker analysis in earthworms at a metal contaminated site. Ecotoxicology 2004; 13:797-806.
49. Makinen VP, Soininen P, Forsblom C, Parkkonen M, Ingman P, Kaski K, Groop PH, and Ala-Korpela M; FinnDiane Study Group. Diagnosing diabetic nephropathy by 1H NMR metabonomics of serum. MAGMA 2006; 19:281-296.
50. Vehtari A, Makinen VP, Soininen P, Ingman P, Makela SM, Savolainen MJ, Hannuksela ML, Kaski K, and Ala-Korpela M. A novel Bayesian approach to quantify clinical variables and to determine their spectroscopic counterparts in 1H NMR metabonomic data. BMC Bioinformat. 2007; 8(suppl 2):S8.
51. Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HW, Clarke S, Schofield PM, McKilligin E, Mosedale DE, and Grainger DJ. Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using 1H-NMR-based metabonomics. Nat. Med. 2002; 8:1439-1444.
52. Kirschenlohr HL, Griffin JL, Clarke SC, Rhydwen R, Grace AA, Schofield PM, Brindle KM, and Metcalfe JC. Proton NMR analysis of plasma is a weak predictor of coronary artery disease. Nat. Med. 2006; 12:705-710.
53. Roussel R, Mentre F, Bouchemal N, Hadjadj S, Lievre M, Chatellier G, Menard J, Panhard X, Le Henanff A, Marre M, and Le Moyec L. DIABHYCAR Study Group. NMR-based prediction of cardiovascular risk in diabetes. Nat. Med. 2007; 13:399-400.
54. Sabatine MS, Liu E, Morrow DA, Heller E, McCarroll R, Wiegand R, Berriz GF, Roth FP, Gerszten RE. Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation 2005; 112:3868-3875.
55. Plumb RS, Johnson KA, Rainville P, Shockcor JP, Williams R, Granger JH, Wilson ID (2006) The detection of phenotypic differences in the metabolic plasma profile of three strains of Zucker rats at 20 weeks of age using ultra-performance liquid chromatography/orthogonal acceleration time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2006; 20:2800-2806.
56. German JB, Hammock BD, Watkins SM. Metabolomics: building on a century of biochemistry to guide human health. Metabolomics 2005; 1:3-9.
57. Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat. Genet. 2000; 25:444-447.
58. Wu JY, Kao HJ, Li SC, Stevens R, Hillman S, Millington D, Chen YT. ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J. Clin. Invest. 2004; 113:434-440.
59. Clish CB, Davidov E, Oresic M, Plasterer TN, Lavine G, Londo T, Meys M, Snell P, Stochaj W, Adourian A, et al. Integrative biological analysis of the APOE*3-leiden transgenic mouse. Omics, 2004; 8:3-13.
60. Griffin JL, Cemal CK, Pook MA. Defining a metabolic phenotype in the brain of a transgenic mouse model of spinocerebellar ataxia 3. Physiol. Genomics 2004; 16:334-340.
61. Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004; 4:551-561.
62. Tsang TM, Griffin JL, Haselden J, Fish C, Holmes E. Metabolic characterization of distinct neuroanatomical regions in rats by magic angle spinning 1H nuclear magnetic resonance spectroscopy. Magn. Reson. Med. 2005; 53:1018-1024.
63. Pears MR, Cooper JD, Mitchison HM, Mortishire-Smith RJ, Pearce DA, Griffin JL. High resolution 1H NMR-based meta- bolomics indicates a neurotransmitter cycling deficit in cerebral tissue from a mouse model of Batten disease. J. Biol. Chem. 2005; 280:42508-42514.
64. Griffin JL, Lehtimaki KK, Valonen PK, Grohn OH, Kettunen MI, Yla-Herttuala S, Pitkanen A, Nicholson JK, Kauppinen RA. Assignment of 1H nuclear magnetic resonance visible polyunsaturated fatty acids in BT4C gliomas undergoing ganciclovir-thymidine kinase gene therapy-induced programmed cell death. Cancer Res, 2003; 63:3195-3201.
65. Lehtimaki KK, Valonen PK, Griffin JL, Vaisanen TH, Grohn OH, Kettunen MI, Vepsalainen J, Yla-Herttuala S, Nicholson J, Kauppinen RA. Metabolite changes in BT4C rat gliomas undergoing ganciclovir-thymidine kinase gene therapy-induced programmed cell death as studied by 1H NMR spectroscopy in vivo, ex vivo, and in vitro. J. Biol. Chem. 2003; 278:45915-45923.
66. Tate AR, Majos C, Moreno A, Howe FA, Griffiths JR, Arus C. Automated classification of short echo time in vivo 1H brain tumor spectra: a multicenter study. Magn. Reson. Med. 2003; 49:29-36.
67. Tsang TM, Woodman B, McLoughlin GA, Griffin JL, Tabrizi SJ, Bates GP, Holmes E. Metabolic characterization of the R6/2 transgenic mouse model of Huntington’s disease by high-resolution MAS 1H NMR spectroscopy. J. Proteome Res. 2006; 5:483-492.
68. Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, et al. HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007; 35:D521-526.
69. Moco S, Bino RJ, Vorst O, Verhoeven HA, de Groot J, van Beek TA, Vervoort J, de Vos CH. A liquid chromatography-mass spectrometry-based metabolome database for tomato. Plant Physiol. 2006; 141:1205-1218.
70. Cui Q, Lewis IA, Hegeman AD, Anderson ME, Li J, Schulte CF, Westler WM, Eghbalnia HR, Sussman MR, Markley JL. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 2008; 26:162-164.
Further Reading
Beckonert O, Keun HC, Ebbels TM, Bundy J, Holmes E, Lindon JC, Nicholson JK. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat Protoc. 2007; 2:2692-2703.
Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst, 2005; 130:606-625.
Fiehn O. Metabolomics--the link between genotypes and phenotypes. Plant Mol. Biol. 2002; 48:155-171.
Griffin JL. The Cinderella story of metabolic profiling: does metabolomics get to go to the functional genomics ball? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2006; 361:147-161.
Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004; 4:551-561.
Kell DB. Metabolomics and systems biology: making sense of the soup. Curr. Opin Microbiol. 2004; 7:296-307.
Kell DB. Metabolomics, machine learning and modelling: towards an understanding of the language of cells. Biochem. Soc. Trans. 2005; 33:520-524.
Nicholson JK, Wilson ID. Opinion: understanding ‘global’ systems biology: metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003; 2:668-676.
Robertson DG. Metabonomics in toxicology: a review. Toxicol. Sci. 2005; 85:809-822.
See Also
Proteomics
Transcript Profiling, Tools for
Metabolic Profiling
Genotyping
Functional Genomics