CHEMICAL BIOLOGY
Depression, Chemical Mechanisms of
Glen B. Baker and Nicholas D. Mitchell, Neurochemical Research Unit, Department of Psychiatry, University of Alberta, Edmonton, Alberta, Canada
doi: 10.1002/9780470048672.wecb674
Although the etiology of depression remains to be elucidated, our knowledge of the neurobiology and biochemistry of this mental disorder has been updated increasingly. Early research focused on the biogenic amines norepinephrine (NE;noradrenaline) and 5-hydroxytryptamine (5-HT;serotonin). The biogenic amine (monoamine) hypothesis of depression states that depression is the result of a functional deficiency of NE and/or 5-HT at central synapses. However, it soon became obvious that other factors were also important in the neurobiology of depression. Dopamine is thought to be involved in various aspects, which include reward and locomotion. The amino acids gamma-aminobutyric acid (GABA) and glutamate, their receptors, and various neuroactive steroids that act as allosteric modulators at GABA-A and/or NMDA glutamate receptors have been proposed to have an important role. Stress and the hypothalamic-pituitary-adrenal (HPA) axis play a central role, and it has been proposed that increased secretion of corticotropin releasing factor (CRF) may be critical in producing the symptoms of depression. Various other neurochemicals have also been implicated in depression, and in several cases they have links to the HPA axis. Abnormal levels of Substance P have been reported in depression, which led to development of potential antidepressants that interact with neurokinin receptors. Abnormalities in the immune system, which presumably involve cytokines, have been reported in depression. Cell loss seems to occur in some brain areas (e.g., hippocampus) in depression, and such loss can be produced by stress and prevented by antidepressants (which also increase expression of various neurotrophic and transcription factors). Agonists at melatonin receptors have been proposed in recent years as effective antidepressants. The possible involvement of the various neurochemicals mentioned above in depression will be reviewed, and some other neurochemicals that are being examined will also be mentioned.
Introduction
Depression (referring here to major depressive disorder in the Diagnostic and Statistical Manual of the American Psychiatric Association, volume IV; DSM-IV) is a common, chronic, functionally limiting clinical syndrome that likely reflects a heterogenous group of underlying disorders (1, 2). In the DSM-IV (DSM-IV-TR is the most recent version), characteristic symptoms of a major depressive episode listed include five or more of the following symptoms, which have been present nearly every day in most cases during the same 2-week period and represent a change from previous functioning: 1) depressed mood; 2) markedly diminished interest or pleasure in all, or almost all, activities; 3) significant weight loss or gain; 4) insomnia or hypersomnia; 6) fatigue or loss of energy; 7) feelings of worthlessness or excessive or inappropriate guilt; 8) diminished ability to think or concentrate, or indecisiveness; and 9) recurrent thoughts of death or suicidal ideation without a specific plan, or a suicide attempt or a specific plan for committing suicide. Note: of the five symptoms, at least one is either 1) or 2) above. The lifetime prevalence of depression in the United States has been estimated at 5-12% in men and 10-25% in women (2). Depression has been shown to impact individual quality of life significantly (3), carry a heavy economic burden (4), and affect brain structure and function (5). Despite research into many potential causative factors, a putative etiology for depression remains elusive (6, 7). Research in the twentieth century initially focused on a decreased functional availability of the monoamine neurotransmitters norepinephrine (NE) and 5-hydroxytryptamine (5-HT) as the possible agents of depressive illness, and it was supported in part by the pharmacological properties of available antidepressants. However, a growing body of literature suggests that multiple contributory chemical systems, many of which are intertwined, are involved in producing the depressive phenotype (8-10).
Biogenic Amines
Endogenous biogenic amines in the brain include catecholamines [NE (noradrenaline, NA), dopamine (DA), epinephrine (adrenaline)] 5-HT, histamine, and the so-called trace amines (P-phenylethylamine, tyramine, tryptamine, and octopamine). These amines have in common a arylalkylamine structure, and all have been implicated in the etiology of one or more psychiatric disorders and/or in therapeutic and/or adverse effects of drugs used to treat such disorders. In this review on depression, the focus in the case of biogenic amines will be on 5-HT, NE, and DA, although epinephrine and histamine and trace amines have also been implicated (see the section on “Other Antidepressant Approaches and Targets”).
Early studies on the origins of depression focused on the monoamine neurotransmitters NE (a catecholamine) and 5-HT (an indoleamine) (for reviews, see References 7, 11, and 12) (Fig. 1). The antihypertensive and antipsychotic medication reserpine depletes central stores of NE, 5-HT, and DA and is known to produce symptoms of depression in some patients (13). Iproniazid is an antitubercular drug that was noted to produce mood elevation in tuberculosis patients and was shown to be an inhibitor of monoamine oxidase (MAO), which is a major catabolic enzyme for monoamines, including NE, 5-HT, and DA (another catecholamine neurotransmitter). In addition, studies in animals with drugs that caused depletions of NE and/or 5-HT demonstrated changes in locomotor activity, sleep, and sexual activity; these symptoms are similar to physiological symptoms observed in depressed patients. These observations led to catecholamine (14) and indoleamine (15, 16) hypotheses, which are now generally combined into the biogenic amine (monoamine) hypothesis that states that depression is the result of a functional deficiency of NE and/or 5-HT at specific synapses in the central nervous system (12). In addition to the effects of the MAO inhibitors (several of which became approved antidepressants), the hypothesis was supported by the actions of the tricyclic antidepressants (TCAs), which inhibit the reuptake of NE and 5-HT back into nerve terminals. Such reuptake is a major inactivation mechanism for the catecholamines and 5-HT, and inhibiton of this process leaves more NE and 5-HT available in the synaptic cleft between neurons to interact with postsynaptic receptors (Fig. 2). Indeed, most antidepressants developed subsequently inhibit NE and/or 5-HT reuptake and/or act on presynaptic receptors that affect synthesis and release of biogenic amine neurotransmitters (see Fig. 2).
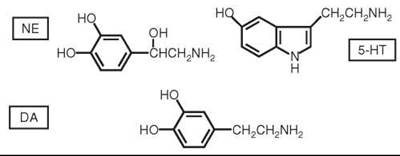
Figure 1. Structures of NE, 5-HT, and DA.

Figure 2. Sites of actions of antidepressants at the synapse (presynaptic nerve terminal, postsynaptic nerve cell, and synaptic cleft between the two are shown). MAO is present in mitochondria, and the shaded circles represent synaptic vesicles that contain neurotransmitter amines.
Serotonin (5-Hydroxytryptamine, 5-HT)
Regulation of mood, sleep, and aggression have all been shown to involve the serotoninergic system (17-19), and most antidepressant drugs currently being used inhibit 5-HT reuptake and/or act on 5-HT receptors (of which there are several subtypes). 5-HT is produced centrally from the amino acid tryptophan, and depressed mood can be induced experimentally by acute tryptophan depletion in healthy individuals. This effect is accentuated in those with a family history of depression (18-21). Similarly, depressive relapse can be initiated in individuals treated with MAO inhibitors or selective serotonin reuptake (SSRI) inhibitors by depleting tryptophan (22, 23).
A major problem with the biogenic amine hypothesis of depression is the discrepancy in the time course between relatively rapid biochemical and pharmacologic effects (e.g., inhibition of reuptake, inhibition of MAO) of the antidepressants and their clinical beneficial effects, which often require administration for 2-3 weeks or longer. This discrepancy led to the studies in both laboratory animals (receptor-binding studies and electrophysiological investigations) and humans (postmortem studies and in vivo neuroimaging studies) on possible dysregulation of receptors for the biogenic amines in depression and effects of antidepressants on that dysregulation (24-28). It has been suggested that antidepressants normalize the density and/or function of 5-HT1 and 5-HT2 receptors, but there are inconsistencies in the literature in this regard. Electroconvulsive shock in rats (an approximate animal model of electroconvulsive therapy in humans) seems to cause an opposite effect on 5-HT2 receptor regulation to that of several antidepressants (24, 25, for a review see Reference 26).
The 5-HT transporter protein (5-HTT) is critical for the reuptake of 5-HT into the presynaptic neuron. Decreased 5-HTT binding has been reported in depressed patients both in postmortem samples and in functional imaging studies (28, 29). Two common alleles of the gene that encode this protein have been identified, with the short (s) form of the allele being less active, which results in decreased transcription and reduced expression of 5-HTT. Studies have suggested that the s allele occurs more frequently in depressed patients as well as in suicide victims, and homozygous individuals are more likely to have family histories of depression. This finding seems paradoxical, as the s allele of the gene results in decreased serotonin reuptake, which thereby increases the duration of 5-HT in the synapse. It has been suggested that this observation may be explained by the lifelong duration of the genetic polymorphism compared with the acute effect of medication administration (28, 29).
Norepinephrine (Noradrenaline)
Investigations have also been conducted on the regulation of NE receptors in depression and after chronic administration of antidepressants (11, 30-32). In animal models, induced chronic stress has been used as a model for depression. Chronic stress has been reported to result in an increase in presynaptic inhibitory α2A adrenergic autoreceptors. Long-term exposure to drugs that block the α2receptor result in an increased levels of synaptic NE and has been shown to counter the effects of chronic stress in rats (11, 31). In humans and laboratory animals, contradictory evidence exists as to the role of noradrenergic receptors in depression (31, 32). Down regulation of β1-adrenoreceptor density occurs after chronic administration of some types of antidepressants, which coincides with clinical effect, but it does not occur with all antidepressants (for a review see References 25 and 32). It is not clear whether β-adrenoreceptor density is increased in victims of suicide; presynaptic α2A-adrenoreceptors have been reported to be increased in postmortem brains of depressed individuals who died by suicide compared with controls (30, 31).
NE is synthesized from the amino acid tyrosine, which can be depleted in the brain by administering a competitive inhibitor of its production, α-methyl-para-tyrosine. Tyrosine depletion has been shown to reverse antidepressant responses in individuals treated with antidepressants that inhibit NE reuptake, which suggests a direct role of this neurotransmitter in the course of depression (31).
Dopamine
Although the focus on monoamine neurotransmitters in depression has been on NE and 5-HT, it is generally considered that DA (Fig. 1) also plays a role, and readers are referred to several comprehensive papers that review basic science and clinical evidence for the involvement of DA (11, 33-35). Motivation, psychomotor speed, concentration, and anhedonia have all been linked to dopaminergic circuits. Most DA-producing neurons have nuclei located in the brain stem. Projections from dopaminergic neurons form three primary paths to the cortical and subcortical structures: The nigrostriatal pathway is involved in motor planning and execution; the mesocortical pathway is concerned with concentration and executive functions; and the mesolimbic pathway is important in motivation, pleasure, and reward. Mesolimbic DA dysfunction is observed in animal models of depression, with subsequent antidepressant use causing enhanced dopamine signaling (11, 33-35). Decreased levels of DA in the nucleus accumbens of rats are associated with a decreased response to rewards. Animals that experience learned helplessness have decreased levels of DA in the caudate nucleus and nucleus accumbens; exposure to antidepressant medications has been reported to increase the level of DA in the same regions (11, 33, 34).
In humans, genetic studies have shown that particular polymorphisms of the D3 and D4 DA receptors are associated with depressed phenotypes. In studies of CSF concentrations of homovanillic acid (HVA), which is a metabolite of DA, lower levels than controls have been reported in some depressed patients. Conversely, depressed individuals with psychotic symptoms have been reported to have increased CSF levels of HVA and DA compared with controls (33).
Classes of Antidepressants
The three neurotransmitter monoamines mentioned above do not operate independently, but rather they interact with one another and are influenced by various central and peripheral biological processes. The actions of most commercially available antidepressant medications continue to target the monoamine neurotransmitter systems. The structures of these antidepressants are shown in Fig. 3. The MAO inhibitors (tranylcypromine, phenelzine, and moclobemide) prevent metabolic breakdown of the amines; the TCAs (represented by imipramine and de- sipramine in Fig. 3, although several other structurally related TCAs are available) are inhibitors of reuptake of NE and 5-HT back into the presynaptic neuron, which results in increased levels of these amines in the synaptic cleft; the tetracyclic maprotiline is a NE reuptake inhibitor; bupropion and its metabolites inhibit reuptake of NE and DA; trazodone inhibits reuptake of 5-HT and acts on a adrenergic receptors; the selective serotonin reuptake inhibitors (SSRIs) are, as their name suggests, very selective for inhibiting serotonin (5-HT) reuptake into nerve terminals; venlafaxine, duloxetine, and milnacipran are inhibitors of NE and 5-HT reuptake; mirtazapine inhibits 5-HT reuptake and blocks presynaptic α2 adrenergic receptors; and reboxetine is a selective NE reuptake inhibitor. These medications have been shown to produce their pharmacological effects on NE and/or 5-HT very quickly (within hours or minutes), but clinical effects lag, which becomes apparent only after days to weeks. Up to 80% of patients respond to currently available therapies; however, only 50% of patients achieve complete remission (7). These phenomena suggest that biogenic amines alone are not sufficient targets for research into the etiology and treatment of depression. In recent years, extensive research has focused on other possible causative factors, and the literature in this area will now be reviewed.


Figure 3. Structures of antidepressants.
Beyond Biogenic Amines
γ-Aminobutyric Acid (GABA) and Glutamate
The amino acids GABA and glutamic acid (glutamate) (Figs. 4 and 5, respectively) are major inhibitory and excitatory neurotransmitters, respectively, in the central nervous system, and a requisite balance between the two operates in normal brain. Aberrations in the functions of one or both of these neurotransmitters have been implicated in the pathogenesis of several neurological and psychiatric disorders, which include depression (36-38).
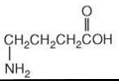
Figure 4. Structure of GABA.
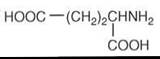
Figure 5. Structure of glutamic acid.
Modulators of alpha-amino-3-hydroxy-5-methyl-4-isoxazo-lepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) ionotropic (associated directly with ion channels) glutamate receptors as well as metabotropic (linked to G-proteins and second messenger systems) glutamate receptors have been investigated for antidepressant properties (39-42). NMDA receptor antagonists have shown effects similar to other antidepressants in animal models of depression. In humans, plasma levels of glutamate have been reported to be correlated with the severity of depression (43), and NMDA receptor abnormalities have been observed in postmortem brain tissue of suicide victims and individuals with depression. Clinical trials of intravenously administered ketamine, which is an NMDA receptor antagonist, have produced rapid diminishment of depressive symptoms after a single administration (41). Concern has been raised over the psychotogenic side effects of NMDA receptor antagonists; however, memantine, which is a lower-affinity NMDA receptor antagonist without these side effects, has been investigated and did not show clinical improvement (42). Less evidence exists for the role of the AMPA receptor in depression. Tricyclic antidepressants have affinity for this receptor, and in a rat model of depression, AMPA receptor density increases with chronic antidepressant treatment (44).
Decreased GABAergic effects have also been associated with depression. In vivo evidence of GABAergic dysfunction in patients with depression includes decreased levels of GABA in the CSF, plasma, and occipital cortex (45, 46). Premenstrual dysphoric disorder (PMDD), which is a condition of depressive symptoms prior to menstruation, has been associated with a reduced variability in cortical GABA levels across the menstrual cycle (47). The MAO inhibitor antidepressant phenelzine has been shown to also cause marked increases in brain levels of GABA when administered to rats (48, 49). Using magnetic resonance spectroscopy, SSRI antidepressants have been demonstrated to increase brain levels of GABA in humans (50).
Both GABA and glutamate are important beyond their direct effects as neurotransmitters. They interact with several other neurotransmitter and neuromodulatory systems and have effects on the regulation of the hypothalamic-pituitary-adrenal (HPA) axis, which may contribute to hyperactivity in this circuit in depression. The HPA axis will be discussed later in this article.
Neuroactive steroids
In recent years, a great deal of interest has focused on the possible involvement of several so-called neuroactive steroids (Fig. 6) in the etiology and pharmacotherapy of a variety of neurologic and psychiatric disorders, which include depression (51-53). These compounds are rapid-acting steroids that act as positive or negative allosteric modulators at many receptors, which include GABA-A and NMDA receptors. These steroids include pregananolone, pregnenolone, allopregnanolone, isoallopregnanolone, 3α,5β-tetrahydroprogesterone (THP), 3β,5α-THP, 3α,5α-tetrahydrodeoxycorticosterone (THDOC), dehydroepiandrosterone (DHEA), DHEA sulphate, and pregnenolone sulfate. Allopregnanolone and THDOC are strong positive modulators at the GABA receptor and are potent anxiolytics (Note: Most antidepressants drugs also have anxiolytic properties). The ratios of these various neuroactive steroids are altered in several psychiatric disorders, and allopregnanolone seems to be of particular interest with regard to depression. Allopreganolone has been reported to produce antidepressant-like actions in ovariectomized rats when given as an intra-accumbens infusion (54), and SSRI antidepressants have been shown to increase brain levels of allopregnanolone in rats at doses lower than those required to inhibit 5-HT reuptake (55). Blunted responses to allopreganaolone in women with PMDD correlate with the degree of depression in these subjects (56).

Figure 6. Structure of some neuroactive steroids.
The Hypothalamic-Pituitary-Adrenal (HPA) Axis
A vast amount of literature indicates a relationship between stress and depression, and in this regard it is interesting that the HPA axis, which is involved in the coordination of neuroendocrine responses to stress, has been shown to be hyperactive in many depressed patients (57, 58). In their diathesis-stress hypothesis of mood disorders, Stout et al. (58) propose that the HPA axis is the principal site where genetic and environmental influences converge to cause mood disorders.
In healthy subjects, stress activates the hypothalamus, which results in the release of corticotropin releasing factor (CRF), which in turn results in release of adrenocorticotropic hormone (ACTH). The ACTH activates the secretion of the glucocorticoid cortisol from the adrenal cortex (Fig. 7). The HPA axis has an autoregulating mechanism mediated via negative feedback by cortisol acting at glucocorticoid receptors in the hypothalmus and the pituitary; but in many depressed patients, this regulation is impaired, which results in higher circulating levels of CRF and cortisol. This impairment probably occurs as a result of a decrease in the number or sensitivity of glucocorticoid receptors, which can be affected by gene expression, monoamines, and early childhood expression (58, 59). It has been reported that depressed patients exhibit decreased ACTH secretion in response to exogenously administered CRF and increased secretion of cortisol in response to a given ACTH level (58, 59). Antidepressants have been reported to result in return of HPA functioning to control levels (59, 60), and it has been proposed (9) that normalization of the HPA axis is a requirement for the successful attenuation of depressive symptoms.
Chronic administration of corticosterone to rats results in behaviors associated with depression (61). Indeed, inhibitors of cortisol synthesis (e.g., ketoconazole and metyrapone) and glucocorticoid receptor antagonists (e.g., mifepristone) have been tested clinically. Although preliminary studies with these drugs are promising, the former drugs have an unfavorable side effect profile, and the latter suffer from lack of specificity (61, 62). The development of CRF1 receptor antagonists has been a major thrust by several researchers and pharmaceutical companies in recent years. Increased levels of CRF in the hypothalamus and CSF and an increased number of CRF neurons in the paraventricular nucleus of the hypothalamus have been observed in major depressive disorder (10). Intracranial administration of CRF to rats or overexpression of CRF in mice leads to sleep disturbances, reduced appetite, and diminished sexual activity. These symptoms appear in many depressed patients (58, 63). Researchers continue to discuss whether the CRF antagonists currently under investigation are acting primarily through blockade of prefrontal and limbic CRF1 receptors rather than having an effect on the HPA axis. In addition, CRF2 receptors are observed in the brain, and although their function is less clear, drugs that act at these receptors are also of investigative interest (58).
As indicated in other parts of this review, the HPA axis interacts with most neurochemicals that are thought to be important in depression, and many researchers consider that this axis plays a central role in depression.

Figure 7. Schematic diagram of the HPA axis.
The peptide Substance P acts primarily on neurokinin 1 (NK1) receptors that are coupled to the Gq subunit of G proteins. Substance P has been of interest with regard to depression because of its increased expression and that of NK1 receptors in fear-and anxiety-related circuits. Substance P is released in animals in response to fear-invoking stimuli and there is a high degree of colocalization of substance P with 5-HT or its receptors in human brain (9). Kramer et al. (64) reported that chronic treatment of humans with a nonpeptide NK1 receptor antagonist resulted in clinical improvement in depressed subjects; this finding was replicated by some groups but not by others (9). It stimulated additional research on NK1 antagonists. Although clinical findings with substance antagonists have been disappointing overall, this class of drug continues to be of interest, and recent studies in laboratory animals and humans suggest that some useful antidepressant agents may develop (66-69). Recent studies indicate that NK1 antagonists act through serotoninergic and noradrenergic neurons (70), and it has been suggested that these drugs may be useful agents in combination with traditional antidepressants (71).
Cytokines
In recent years, researchers have shown interest in the role of the immune system in depression (72, 73), and the cytokine theory of depression proposes that alterations in the immune response result in behavioral, cognitive, and neuroendocrine changes in depression. Cytokines can be proinflammatory or anti-inflammatory, and it has been postulated that depression may be caused by, or at least related to, excessive amounts of proinflammatory cytokines such as interleukin-1 and tumor necrosis factor-alpha (74). Indeed, it has been observed that antiviral treatment with the cytokine interferon results in “sickness behavior” characterized by symptoms such as weight loss, anorexia, depressed mood, sleep disturbances, social withdrawal, and fatigue, which are also observed in depression (9). It is also of interest that cytokines are potent stimulators of the HPA axis via activation of CRF release, and proinflammatory cytokines can also decrease 5-HT levels by increasing 5-HT secretion and diverting metabolism of tryptophan metabolism away from formation of 5-HT by tryptophan hydroxylase as well as increasing metabolism by the indolamine-2,3-dioxygenase (IDO) pathway (75). Although the evidence for abnormal cytokine levels and/or inflammatory responses in depression is inconsistent to date, this area is very interesting for future exploration and should lead to more studies on glial cells that synthesize and release cytokines.
Intracellular signaling cascades and neurotrophic factors
As the acute increase in synaptic monoamines produced by antidepressant drugs does not correlate with timing of clinical effects, the intracellular signaling pathways that their receptors they interact with have been investigated as targets for novel antidepressants. Several such pathways are observed, and an example is given in Fig. 8 (76).

Figure 8. Simplified diagram of a signaling cascade that involves NE, BDNF, and CREB after NE acts on the postsynaptic β-noradrenergic receptor. NE couples to a G protein (Gαs), which stimulates the production of cAMP from adenosine triphosphate (ATP). This reaction is catalyzed by adenylate cyclase (AC). cAMP activates protein kinase A (PKA). Inside the cell, PKA phosphorylates (P) the CREB protein, which binds upstream from specific regions of genes and regulates their expression. BDNF is one target of cAMP signaling pathways in the brain. CRE, cyclic AMP regulatory element; ER, endoplasmic reticulum. [reprinted from Reference 76 with permission of the author and the publisher, Canadian Medical Association].
Several serotoninergic and DA receptors and the p noradrenergic receptor activate the Gs pathway, whereas others activate the Gq pathway. These signaling cascades result in activation of protein kinases A and C, with a net effect of increasing phosphorylation of cyclic adenosine monophosphate (cAMP)-regulated element binding protein (CREB). CREB acts as a regulator in the expression of many genes, some of which have been implicated in neuroplasticity and neurogenesis. For example, transcription of brain-derived neurotrophic factor (BDNF), tyrosine kinase B (trkB) (which is a BDNF receptor), and the glucocorticoid receptor are activated by CREB. Inhibition of transcription of CRF and subunits of the NMDA receptor occur in the presence of CREB. Alterations in CREB levels have been observed in depressed individuals, and increases in CREB function have been associated with reduction or production of depressive symptoms in animals, which depends on the area of the brain involved (77, 78).
The most researched area of interest for antidepressant activity related to signaling pathways is the hippocampus (79-85). The volume of the hippocampus is reduced in patients with multiple episodes of major depression, as observed in imaging studies and in postmortem samples (81-84). Animal models of inescapable stress are associated with decreased hippocampal neurogenesis (84). Neurogenesis is also evident in the hippocampus of humans (85). The neurogenesis hypothesis of depression states that depression is a consequence of impaired neurogenesis in the hippocampus and that antidepressants exert their effect by stimulating neurogensis (86). Controversy exists as to whether neurogenesis is necessary for the efficacy of antidepressant medications in animal models (87), with some evidence suggesting that when neurogenesis is inhibited, antidepressant effects are lost (84, 88).
In the hippocampus, CREB seems to mediate antidepressant effects, and a variety of antidepressants, which includes electroconvulsive therapy, increases CREB expression (77). Experimentally increasing the expression of CREB in the hippocampi of depressed rats seems to have an antidepressant effect. Evidence suggests that the role of CREB in mediating antidepressant effects of medication is related to the expression of CREB-regulated neural growth factors, such as BDNF (79, 89). In humans, BDNF levels are reduced in postmortem samples of depressed individuals when compared with nondepressed controls (90). Antidepressant therapy increases serum BDNF levels in depressed humans (91). In animals, chronic stress models are associated with reduced expression of BDNF (79) and decreased cell proliferation in the hippocampus (83), which can be prevented or reversed by treatment with antidepressant drugs. However, researchers debate whether genetic disruption of the signaling pathways that involve BDNF and trkB causes depressive behavior (See Reference 92 for a review). It is also of interest that chronic administration of antidepressants has been reported to upregulate expression and activity of the neuroprotective enzyme superoxide desmutase and, depending on the dose and antidepressant, to increase immunostaining of BDNF and/or the antiapoptotic protein Bcl-2 (93, 94).
The picture with CREB is far from straightforward. Increased expression of CREB in the nucleus accumbens of rats produces depression-like effects, which include anhedonia (77, 89) and increased helplessness, in learned-helplessness models (81). Increases in BDNF expression in the mesolimbic DA system of rats are associated with induction of depressive effects in certain animal models. However, studies that use systemic administration of BDNF and activators of the cAMP-CREB cascade seem to have a net therapeutic effect in behavioral models of depression (90, 86, 89, 95).
Melatonin (N-acetyl-5-methoxytryptamine)
Depression and seasonal affective disorder, which is a form of depressive illness characterized by symptoms with onset and course related to season of the year, have been noted to be cyclic and possibly related to alterations in circadian rhythms (96). Alterations in the cyclical functioning of the HPA axis and the hypothalamic-pituitary-thyroid axis occur in some patients with major depression (96). Disruption in the sleep/wake cycle and insomnia are common in depression. Melatonin (Fig. 9), which is a hormone derived centrally in the pineal gland from serotonin, is instrumental in many biological functions with circadian variability. Several studies have shown that depressed individuals have a deficiency of melatonin secretion, but a body of evidence shows increased nocturnal melatonin production in the brains of depressed subjects with increased urinary excretion of its metabolite, 6-hydroxymelatonin sulfate (96, 97). This finding may reflect differences in subtypes of depression. As melatonin is secreted in a rhythmic fashion, the timing of the melatonin peak has been studied in depressed individuals. Phase shifts toward both earlier and later onset of peak secretion have been noted. Both treatment with antidepressant medications and electroconvulsive therapy have been associated with increases in melatonin excretion in depressed individuals. The pineal gland receives input from noradrenergic neurons, and it has been suggested that alterations in melatonin secretion merely reflect dysfunction in monoamine systems (96, 97). Agomelatine, which is a melatonin receptor agonist and 5HT2c receptor antagonist, has been shown to have antidepressant properties in animals and humans (98-101). Agomelatine targets the MT1/MT2 melatonin receptors and mimics melatonin in its effect on circadian rhythms. A combination of activity at melatonin receptors and monoaminergic receptors may represent a novel method of antidepressant drug action (97-100).

Figure 9. Structure of melatonin.
Other Antidepressant Approaches and Targets
Electroconvulsive therapy (ECT) is considered to be the most efficacious treatment for depression, although its side-effect profile limits its use, and it is associated with a high relapse rate (10, 102). The exact mechanism of action of ECT is still unknown, although effects on monoaminergic systems, CRF, neurotrophic factors, and neuroendocrine systems have been suggested (102). Because DA probably plays a role in the pathophysiology of depression, triple reuptake inhibitors that inhibit reuptake of NE, 5-HT, and DA have now been developed and are undergoing preclinical and clinical testing (10). Numerous reports in the literature suggest that trace amines (so-named because their absolute levels in the brain are much lower that those of the classic neurotransmitter amines) are involved in the etiology and pharmacotherapy of several neurologic and psychiatric disorders, which include depression (103-106). Researchers have shown an increased interest in these trace amines, which include β-phenylethylamine, tyramine, tryptamine, and octopamine, in recent years with the discovery of a family of G-protein coupled receptors that bind to and are activated by these amines (107, 108). Epinephrine is present in much lower concentrations than NE and DA in the brain, but it is a major stress hormone in the periphery and could be a contributing factor in depression (109). Histamine is present in lower concentrations in brain than NE, DA, or 5-HT as well, but it has also been implicated in the etiology of depression (110).
Reports in the literature indicate that atypical (second generation) antipsychotics are useful antidepressant agents when combined with standard antidepressants (10); several researchers suggest that in some cases they and/or their metabolites may be useful antidepressants in their own right. It is interesting that some of these atypical antipsychotics have been reported to have several neuroprotective actions, which include effects on neuroprotective enzymes and factors that affect apoptosis (programmed cell death) (111). Preliminary reports suggest similar effects of antidepressants (93, 94). Some other areas related to the chemistry of depression and its treatment that contain conflicting reports in the literature but are of interest include the following: the relationship of serum cholesterol and the use of statins (cholesterol-lowering drugs) to depression, anxiety, aggression, and suicide (112, 113); omega-3 fatty acids and mood disorders (114-118); the use of herbal products such as St. John’s wort extracts and S-adenosylmethionine as antidepressants (119-123); and the possible involvement of CB1 cannabinoid receptors, galanin, Neuropeptide Y, histone deacetylases, and tissue plasminogen activator in depression (See Reference 9 for a review). Other reported or putative neurobiological antidepressant treatments that we have not mentioned previously in this review include deep brain stimulation, vagus nerve stimulation, and transcranial magnetic stimulation; the reader is referred to the review of Holtzheimer and Nemeroff (10) and the references contained therein for details.
Concluding Remarks
Although a great deal has been learned about brain function in the search for newer antidepressants and some very interesting potential drug targets have been identified, our knowledge of the causes of depression remains inadequate. We still lack antidepressant drugs that are sufficiently rapid acting and effective in a large enough percentage of depressed patients. Problems that continue to make studies on depression difficult include the following: the heterogenous nature of depression itself, gender issues, the involvement of multiple interacting neurotransmitters and neuromodulators in the etiology of depression, the inadequacy of current animal models, the involvement of various brain regions in the symptomatology of depression, disagreements about the neurogensis hypothesis of depression and about the relative importance of the hippocampus in depression, regional differences in the effects of transcription factors such as CREB, and the differing effects of some antidepressants on the HPA axis.
Acknowledgments
Research findings provided by the Canadian Institutes of Health Research (CIHR), the Canada Research Chairs and Canada Foundation for Innovation programs, and the Davey Endowment. The assistance of Ms. Tara Checknita in preparing this manuscript is gratefully acknowledged.
References
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. 4th ed. 2000. American Psychiatric Association, Washington, DC.
2. Sadock BJ, Sadock VA. Mood disorders. In: Kaplan & Sadock’s Synopsis of Psychiatry: Behavioral Sciences, Clinical Psychiatry. 9th ed. Sadock BJ, Sadock, VA, eds. 2003. Lippincott Williams & Wilkins, Philadelphia, PA, pp. 534-590.
3. Papakostas GI, Petersen T, Mahal Y, Mischoulon D, Nierenberg AA, Fava M. Quality of life assessments in major depressive disorder: A review of the literature. Gen. Hosp. Psych. 2004;26:13-17.
4. Luppa M, Heinrich S, Angermeyer MC, Konig HH, Riedel-Heller SG. Cost of illness studies of major depression: a systematic review. J. Affec. Disorders. 2007; 98:29-43.
5. Maletic V, Robinson M, Oakes T, Iyengar S, Ball SG, Russell J. Neurobiology of depression: an integrated view of key findings. Int. J. Clin. Pract. 2007; 61:2030-2040.
6. Marchand WR, Dilda DV, Jensen, CR. Neurobiology of mood disorders. Hosp. Physician 2005; 41:17-26.
7. Nestler EJ, Barrot M, Dileone RJ, Eisch A, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron 2002; 34:13-25.
8. Shelton RC. The molecular neurobiology of depression. Psychiatr. Clin. N. Am. 2007; 1:1-11.
9. Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monamines. Nature Rev. Neurosci. 2006; 7:137-151.
10. Holtzheimer PE III, Nemeroff CB. Advances in the treatment of depression. J. Am. Soc. Exp. Neurotherapeutics 2006; 3:42-56.
11. Nutt DJ. The role of dopamine and norepinephrine in depression and antidepressant treatment. J. Clin. Psychiatry 2006;67(suppl 7): 3-8.
12. Baker GB, Dewhurst WG. Biochemical theories of affective disorders. In: The Pharmacotherapy of Affective Disorders: Theory and Practice. Dewhurst WG and Baker GB, eds. 1985. Croom Helm Ltd., London, pp. 1-59.
13. Freis ED. Mental depression in hypertensive patients treated for long periods with large doses of reserpine. N. Engl. J. Med. 1954; 251:1006-1008.
14. Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am. J. Psychiatry 1965; 122:509-522.
15. Lapin IP, Oxenkrug GF. Intensification of central serotonergic processes as a possible mechanism of thymoleptic effect. Lancet 1969; 1:132-136.
16. Coppen AJ. The biochemistry of affective disorders. Br. J. Psychiatry 1967; 113:1237-1264.
17. Firk C, Markus CR. Serotonin by stress interaction: a susceptibility factor for the development of depression? J. Psychopharmacol. 2007; 21:538-544.
18. Neumeister A. Tryptophan depletion, serotonin, and depression: Where do we stand? Psychopharmacol. Bull. 2003; 37:99-115.
19. Young SN, Smith SE, Pihl RO, Ervin, FR. Tryptophan depletion causes a rapid lowering of mood in normal males. Psychopharmacology 1985;87:173-177.
20. Klaasen T, Riedal WJ, van Someren A, Deutz NE, Honig A, van Praag HM. Mood effects of 24-hour tryptophan depletion in healthy first-degree relatives of patients with affective disorders. Biol. Psychiatry 1999; 46:489-497.
21. Delgado PL, Price L, Miller HL, Salomon RM, Aghajanian GK, Heninger GR, Charney DS. Serotonin and the neurobiology of depression. Effects of trypophan depletion in drug-free depressed patients. Arch. Gen. Psychiatry 1994; 51:865-874.
22. Delgado PL, Miller HL, Salomon RM, Licinio J, Krystal JH, Moreno FA, Heninger GR, Charney DS. Tryptophan-depletion challenge in depressed patients treated with desipramine or fluoxetine: implications for the role of serotonin in the mechanism of antidepressant action. Biol. Psychiatry 1999; 46:212-220.
23. Booij L, Van der Does AJW, Riedel WJ. Monoamine depletion in psychiatric and healthy populations: review. Mol. Psychiatry 2003; 8:951-973.
24. Blier P, de Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci. 1994; 15:220-226.
25. Bourin M, Baker GB. Do G proteins have a role in antidepressant actions? Eur Neuropsychopharmacol. 1996; 6:49-53.
26. Deakin JFW, Guimaraes FS, Wang M, Hensman R. Experimental tests of the 5-HT receptor imbalance theory of affective disturbance In: 5-Hydroxytryptamine in Psychiatry - A Spectrum of Ideas. Sandler, M, Coppen A, Harnett, S. eds., 1991. Oxford University Press, Oxford, UK, pp. 143-156.
27. Vetulani J, Lebrecht U, Pilc A Enhancement of responsiveness of the central serotonergic system and serotonin-2 receptor density in rat frontal cortex by electroconvulsive treatment. Eur. J. Pharmacol. 1981; 76:81-85.
28. Stockmeier CA. Involvement of serotonin in depression: evidence from postmortem imaging studies of serotonin receptors and the serotonin transporter. J. Psychiatric Res. 2003; 37:357-373.
29. Neumeister A, Young T, Stastny J. Implications of genetic research on the role of serotonin in depression: emphasis on the serotonin type 1A receptor and the serotonin transporter. Psychopharmacology 2004; 174:512-524.
30. Vetulani J, Sulser F. Action of various antidepressant treatments reduces reactivity of noradrenergic cylic AMP-generating system in limbic forebrain. Nature 1975; 257:495-496.
31. Brunello N, Mendlewicz J, Kasper S, Leonard B, Montgomery S, Nelson JC, Paykel E, Veriani M, Racagni G. The role of noradrenaline and selective noradrenaline reuptake inhibition in depression. Eur. Neuropsychopharmacol. 2002; 12:461-475.
32. Leonard BE. Noradrenaline in basic models of depression. Eur. Neuropsychopharmacology. 1997; 1(suppl 1): S11-16.
33. Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry. 2007; 64:327-337.
34. Willner P, Muscat R, Papp M. Chronic mild stress-induced anhedonia: A realistic animal model of depression. Neurosci. Biobehav. Rev. 1992; 16:525-534.
35. Fibiger HC. Neurobiology of depression: focus on dopamine. Adv. Biochem. Psychopharmacol. 1995; 49:1-17.
36. Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G, Epperson CN, Goddard A, Mason GF. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol. Psychiatry. 2002; 7:S71-80.
37. Paul IA, Skolnick P. Glutamate and depression: clinical and preclinical studies. Ann. NY Acad. Sci. 2003; 1003:250-272.
38. Coyle JT, Duman RS. Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron 2003; 38:157-160.
39. Matrisciano, F, Panaccione I, Zusso M, Giusti P, Tatarelli R, Iacovelli L, Mathe AA, Gruber SH, Nicoletti F, Girardi P. Group-II metabotropic glutamate receptor ligands as adjunctive drugs in the treatment of depression: A new strategy to shorten the latency of antidepressant medication? Mol. Psychiatry 2007; 12:704-706.
40. Belozertseva IV, Kos T, Popik P, Danysz W, Bespalov AY. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and mouse tail suspension tests. Eur. Neuropsychopharmacol. 2007; 17:172-179.
41. Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006; 63:856-864.
42. Zarate CA, Sing JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, Manji HK, Charney DS. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am. J. Psychiatry 2006; 63:153-155.
43. Mitani H, Shirayama Y, Yamada T, Maeda K, Ashby CR, Kawa- hara R. Correlation between plasma levels of glutamate, alanine, and serine with severity of depression. Progr. Neuro-Psychopharmacol. Biol. Psychiatry 2006; 30:1155-1158.
44. Martinez-Turrillas R, Frechilla D, Del Rio J. Chronic antidepressant treatment increases the membrane expression of AMPA receptors in rat hippocampus. Neuropharmacology 2002; 43:1230-1237.
45. Petty F. GABA and mood disorders: a brief review and hypothesis. J. Affec. Disorders 1995; 34:275-281.
46. Sanacora G, Mason GF, Rothman DL, Behar K L, Hyder F, Petroff OA, et al. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 1999; 56:1043-1047.
47. Epperson CN, Haga K, Mason GF, Sellers E, Gueorguieva R, Zhang W, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: A proton magnetic resonance spectroscopy study. Arch. Gen. Psychiatry 2002; 59:851-858.
48. Popov N, Matthies H Some effects of monoamine oxidase inhibitors on the metabolism of gamma-aminobutyric acid in rat brain. J. Neurochem. 1969; 16:899-907.
49. Baker GB, Wong JT, Yeung JM, Coutts RT. Effects of the antidepressant phenelzine on brain levels of gamma-aminobutyric acid (GABA). J. Affec. Disorders 1991; 21:207-211.
50. Bhagwagar Z, Wylezinska M, Taylor, M, Jezzard P, Matthews PM, Cowen PJ. Increased brain GABA concentrations following acute administration of a selective serotonin reuptake inhibitor. Am. J. Psychiatry 2004; 161:368-370.
51. Dubrovsky BO. Steroids, neuroactive steroids and neurosteroids in psychopathology. Prog. Neuro-Psychopharmacol. Biol Psychiatry, 2005; 29:169-192.
52. MacKenzie EM, Odontiadis J, Le Melledo JM, Prior TI, Baker GB. The relevance of neuroactive steroids in schizophrenia, depression, and anxiety disorders. Cell. Mol.Neurobiol. 2007; 27:541-574.
53. Marx CE, Stevens RD, Shampine LJ, Uzunova V, Trost WT, Butterfield MI et al. Neuroactive steroids are altered in schizophrenia and bipolar disorder: relevance to pathophysiology and therapeutics. Neuropsychopharmacology 2006; 31:1249-1263.
54. Molina-Hernandez M, Tellez-Alcantara NP, Garcia JP, Lopez JI, Jaramillo MT. Antidepressant-like actions of intra-accumbens infusions of allopregnanolone in ovariectomized wistar rats. Pharmacol. Biochem. Behav. 2005; 80:401-409.
55. Pinna G, Costa E, Guidotti A. Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake. Psychopharmacology 2006; 186:362-372.
56. Girdler SS, Klatzkin R. Neurosteroids in the context of stress: Implications for depressive disorders. Pharmacol. Therapeut. 2007; 116:125-139.
57. Carroll BJ, Martin FIR, Davies B. Pituitary-adrenal function in depression. Lancet 1976; I:1373-1374.
58. Stout SC, Owens MJ, Nemeroff CB. Regulation of corticotropinreleasing factor neuronal systems and hypothalamic-pituitary-adrenal axis activity by stress and chronic antidepressant treatment. J. Pharmacol. Exp. Ther. 2002; 300:1085-1092.
59. Schule C. Neuroendocrinological mechanisms of actions of antidepressant drugs. J. Neuroendocrinol. 2007; 19:213-226.
60. Young AH. Antiglucocorticoid treatments for depression. Aust. N.Z. Psychiatry 2006; 40:402-405.
61. Johnson SA, Fournier NM, Kalynchuk LE. Effect of different doses of corticosterone on depression-like behavior and HPA axis response to a novel stressor. Brain Behav. Res. 2006;168:280-288.
62. Ravaris CL, Sateia MJ, Beroza KW, Noordky DL, Brinck-Johnsen T. Effect of ketoconazole on a hypophysectomized, hypercortisolemic, psychotically depressed woman. Arch. Gen. Psychiatry 1988; 45:966-967.
63. van Gaalen MM, Marcel M, Stenzel-Poore MP, Holsboer F, Steckler T. Effects of transgenic overproduction of CRH on anxiety-like behavior. Eur. J. Neurosci. 2002; 15:2007-2015.
64. Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 1998; 281:1640-1645.
65. Deuschle M, Sander P, Herpfer I, Fiebich B L, Heuser I. Lieb K. Substance P in serum and cerebrospinal fluid of depressed patients: No effect of antidepressant treatment. Psychiatry Res. 2005; 136:1-6.
66. Malkesman O, Braw Y, Weller A. Assessment of antidepressant and anxiolytic properties of NK1 antagonists and substance P in wistar kyoto rats. Physiol. Behav. 2007; 90:619-625.
67. Dableh LJ, Yashpal K, Rochford J, Henry JL. Antidepressant-like effects of neurokinin receptor antagonists in the forced swim test in the rat. Eur. J. Pharmacol., 2005; 507:99-105.
68. Chenu F, Guiard BP, Bourin M, Gardier AM. Antidepressant-like activity of selective serotonin reuptake inhibitors combined with a NK1 receptor antagonist in the mouse forced swimming test. Behav. Brain Res. 2006; 172:256-263.
69. Kramer M S, Winokur A, Kelsey J, Preskorn SH, Rothschild AJ, Snavely D, Gosh K, Ball WA, Reines SA, Munjack D, Apter JT, Cunningham L, Kling M, Bari M, Getson A, Lee Y. Demonstration of the efficacy and safety of a novel substance P (NK1) receptor antagonist in major depression. Neuropsychopharmacology 2004; 29:385-392.
70. Gobbi G, Blier P. Effect of neurokinin-1 receptor antagonists on serotoninergic, noradrenergic and hippocampal neurons: Comparison with antidepressant drugs. Peptides 2005; 26:1383-1393.
71. Ryckmans T, Balancon L, Genicot C, Berton O., Lamberty Y Lallemand B, Pasau P, Pirlot N, Quere L, Talaga P. First dual NK(1) antagonists-serotonin reuptake inhibitors: synthesis and SAR of a new class of potential antidepressants. Bioorg. Med. Chem. Lett. 2002; 12:261-264.
72. Dunn AJ, Swiergiel AH, de Beaurepaire R. Cytokines as mediators of depression: What can we learn from animal studies? Neurosci. Biobehav. Rev. 2005; 29:891-909.
73. Kim YK, Na KS, Shin KH, Jung HY, Choi SH, Kim JB. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007; 31:1044-1053.
74. Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog. Neuro-Psychophramacol. Biol. Psychiat. 2005; 29:201-217.
75. O’Brien SM, Scott LV. Dinan TG. Cytokines: Abnormalities in major depression and implications for pharmacological treatment. Hum. Psychopharmacol. 2004; 19:397-403.
76. Young TL. Postreceptor pathways for signal transduction in depression and bipolar disorder. J. Psychiatr. Neurosci. 2001; 26:S17- S22.
77. Carlezon WA, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005; 28:436-445.
78. Nair A, Vaidya VA. Cyclic AMP response element binding protein and brain-derived neurotrophic factor: molecules that modulate our mood? J. Biosci. 2007; 31:423-434.
79. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006; 59:1116-1127.
80. Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav. Pharmacol. 2007; 18:391-418.
81. Pittenger C, Duman RS. Neuroplasticity and depression. Neuropsychopharmacol. Rev. 2008; 33:88-109.
82. Duman, R. S. Depression: a case of neuronal life and death? Biol. Psychiatry. 2004; 56:140-145.
83. Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, Uylings HBM, Freidman L, Rajkowska G. Cellular changes in postmortem hippocampus in major depression. Biol. Psychiat. 2004; 56:640-650.
84. Malberg JE, Duman RS. Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 2003; 28:1562-1571.
85. van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature 2002; 415:1030-1034.
86. Mackin P, Watson S, Ferrier N. The neurobiology of depression: focus on cortisol, brain-derived neurotrophic factor, and their interactions. Depression: Mind Body 2005; 2:11-18.
87. Sapolsky RM. Is impaired neurogenesis relevant to the affective symptoms of depression? Biol. Psychiatry 2004; 56:137-139.
88. Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003; 301:805-809.
89. Eisch AJ, Bolanos CA, de Wit J. Simonak RD. Pudiak CM, Barrot M, Nestler EJ. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol. Psychiatry. 2003; 54:994-1005.
90. Castren E, Voikar V, Rantamaki T. Role of neurotrophic factors in depression. Curr. Opinion Pharmacol. 2007; 7:18-21.
91. Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, Nazakato M, Watanabe H, Shinoda N, Okada S, Iyo M. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol. Psychiatry 2003; 54:70-75.
92. Martinowich K, Manji H, Bai L. New insights into BDNF function in depression and anxiety. Nature Neurosci. 2007; 10:1089-1093.
93. Kolla N, Wei Z, Richardson JS, Li XM. Amitriptyline and fluoxetine protect PC12 cells from cell death induced by hydrogen peroxide. J. Psychiatry Neurosci. 2005; 30:196-201.
94. Xu H, Steven Richardson J, Li XM. Dose-related effects of chronic antidepressants on neuroprotective proteins BDNF, bcl-2 and Cu/Zn-SOD in rat hippocampus. Neuropsychopharmacology 2003; 28:53-62.
95. Shiriyama Y, Chen ACH, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 2002; 22:3251-3261.
96. Srinivasan V, Smits M, Spence W, Lowe AD, Kayumov L, Pandi-Perumal SR, Parry B, Cardinali DP. Melatonin in mood disorders. World J. Biol. Sci. 2006; 7:138-151.
97. Pacchierotti C, Iapichino P, Bossini L, Pieraccini F, Castrogiovanni C. Melatonin in psychiatric disorders: a review of melatonin in psychiatry. Frontiers Neuroendocrinol. 2001; 22:18-32.
98. Olie JP, Kasper S. Efficacy of agomelatine, a MT1/MT2 receptor agonist with 5-HT2c antagonistic properties, in major depressive disorder. Int. J. Neuropsychopharmacol. 2007; 10:661-673.
99. Zupancic M, Guilleminault C. Agomelatine: a preliminary review of a new antidepressant. CNS Drugs 2006; 20:981-992.
100. Stahl SM. Novel mechanism of antidepressant action: norepinephrine and dopamine disinhibition (NDDI) plus melatonergic agonism. Int. J Neuropsychopharmacol 2007; 10:575-578.
101. Kennedy SH, Emsley R. Placebo-controlled trial of agomelatine in the treatment of major depressive disorder. Eur. Neuropsychopharmacol. 2006; 16:93-100.
102. Wahlund B, von Rosen D. ECT of major depressed patients in relation to biological and clinical variables: a brief overview. Neuropsychopharmacology 2003; 28(suppl 1): S21-6.
103. Sabelli HC, Mosnaim AD. Phenylethylamine hypothesis of affective behavior. Am. J. Psychiatry 1974; 131:695-699.
104. Sandler M, Ruthven CR, Goodwin BL, Coppen A. Decreased cerebrospinal fluid concentration of free phenylacetic acid in depressive illness. Clin. Chim. Acta 1979; 93:169-171.
105. Davis BA, Boulton AA. The trace amines and their acidic metabolites in depression-an overview. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1994; 18:17-45.
106. Berry MD. Mammalian central nervous system trace amines. pharmacologic amphetamines, physiologic neuromodulators. J. Neurochem. 2004; 90:257-271.
107. Borowsky B, Adham N, Jones KA, Raddatz R, Artymyshyn R, Ogozalek K L, Durkin MM, Lakhlani PP, Bonini JA, Pathirana S, Boyl A, Pu A., Kouranova E, Lichtblau H, Ochoa FY, Branchek TA, Gerad C. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8966-8971.
108. Lewin AH. Receptors of mammalian trace amines. AAPS J. 2006; 8:E138-145.
109. Stone EA, Lin Y, Rosengarten H, Kramer HK, Quartermain D. Emerging evidence for a central epinephrine-innervated a1- adrenoceptor system that regulates behavioural activation and is impaired in depression. Neuropsychopharmacology 2003; 28:1387-1399.
110. Kano M, Fukudo S, Tashiro A, Utsumi A, Tamura D, Itoh M, Iwata R, Tashiro M, Mochizuki K, Hongo M, Yanai K. Decreased histamine H1 receptor binding in the brain of depressed patients. Eur. J. Neurosci. 2004; 20:803-810.
111. Li XM, Xu H. Evidence for neuroprotective effects of antipsychotic drugs: Implications for the pathophysiology and treatment of schizophrenia. Int Rev Neurobiol. 2007; 77:107-142.
112. Tanskanen A, Tuomilehto J, Viinamaki H. Cholesterol, depression and suicide. Br. J. Psychiatry, 2000; 176:398-399;399-400.
113. Young-Xu, Y, Chan, KA, Liao JK, Ravid S, Blatt CM. Long-term statin use and psychological well-being. J. Am. Coll. Cardiol. 2003; 42:690-697.
114. Peet M, Stokes C. Omega-3 fatty acids in the treatment of psychiatric disorders. Drugs 2005; 65:1051-1059.
115. Freeman MP, Hibbeln JR, Wisner KL, Davis JM. Mischoulon D, Peet M, et al. Omega-3 fatty acids: Evidence basis for treatment and future research in psychiatry. J. Clin. Psychiatry 2006; 67:1954-1967.
116. Nieminen LR, Makino KK, Mehta N, Virkkunen M, Kim HY, Hibbeln JR. Relationship between omega-3 fatty acids and plasma neuroactive steroids in alcoholism, depression and controls. Prostaglandins, Leukotrienes, Essential Fatty Acids 2006; 75:309-314.
117. Lin PY, Su KP. A meta-analytic review of double-blind, placebo-controlled trials of antidepressant efficacy of omega-3 fatty acids. J. Clin. Psychiatry 2007; 68:1056-1061.
118. Young SN. Fish oils for depression? J. Psychiatry Neurosci. 2008; 33;(E1).
119. Hypericum Depression Trial Study Group. Effect of Hypericum perforatum (St John’s wort) in major depressive disorder: a randomized controlled trial. J. Am. Med. Assoc. 2002; 287:1807- 1814.
120. Linde K, Mulrow CD, Berner M, Egger M. St John’s wort for depression. Cochrane Database of Systematic Reviews (Online), 2005; (2) CD000448.
121. Kasper S, Anghelescu IG, Szegedi A, Dienel A, Kieser M. Superior efficacy of st. John’s wort extract WS 5570 compared to placebo in patients with major depression: a randomized, double-blind, placebo-controlled, multi-center trial. BMC Med. 2006; 4:14.
122. Mischoulon D, Fava, M. Role of S-adenosyl-L-methionine in the treatment of depression: a review of the evidence. Am. J. Clin. Nutrition 2002; 76:1158S-1161S.
123. Shippy RA, Mendez D, Jones K, Cergnul I, Karpiak SE. S-Adeno-sylmethionine (SAM-e) for the treatment of depression in people living with HIV/AIDS. BMC Psychiatry 2004; 4:38.
Further Reading
Anisman H, Merali Z, Stead JDH. Experiential and genetic contributions to depressive and anxiety-like disorders: clinical and experimental studies. Neurosci. Biobehav. Rev. 2008; In press.
Baghai TC, Volz HP, Moller HJ. Drug treatment of depression in the 2000s: An overview of achievements in the last 10 years and future possibilities. World J. Biol. Psychiatry 2006; 7:198-222.
Biol. Psychiatry 2008; 63:639-722. Theme issue devoted to “The Neurobiology and Therapeutics of Antidepressant-Resistant Depression” (12 papers).
Conti B, Maier R, Barr AM, Morale MC, Lu X, Sanna PP, et al. Region-specific transcriptional changes following the three antidepressant treatments electro-convulsive therapy, sleep deprivation and fluoxetine. Mol. Psychiatry 2007; 12:167-189.
De Souza E, Grigoriadis DE. Corticotrophin-releasing factor: physiology, pharmacology, and role in central nervous system disorders. In: Neuropsychopharmacology: The Fifth Generation of Progress. Davis KL, Charney D, Coyle JT, Nemeroff C, eds. 2002. Lippincott Williams & Wilkins, Philadelphia, PA, pp. 91-107.
Hasler G, Drevets WC, Manji HK, Harney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology 2004; 29:1765-1781.
Hauger RL, Risbrough V, Brauns O, and Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: New molecular targets. CNS Neurol. Disorders Drug Targets 2006; 5:453-479.
Maletic V, Robinson M, Oakes T. Iyengar S, Ball SG, Russell J. Neurobiology of depression: An integrated view of key findings. Int. J. Clin. Pract. 2007; 61:2030-2040.
Newport DJ, Stowe ZN, Nemeroff CB. Parental depression: animal models of an adverse life event. Am. J. Psychiatry 2002; 159:1265-1283.
Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 2008; 33:88-109.
Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc. Nat. Acad. Sci. U.S.A. 2006; 103:17501-17506.
Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav. Pharmacol. 2007; 18:391-418.
Simon NM, McNamara K, Chow CW, Maser RS. Papakostas GI, Pollack MH, et al. A detailed examination of cytokine abnormalities in major depressive disorder. Eur. Neuropsychopharmacol. 2008; 18:230-233.
Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res. Rev. 2005; 4:141-194.
Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nature Rev. Neurosci. 2007; 8:355-367.
Thomson F, Craighead M. Innovative approaches for the treatment of depression: Targeting the HPA axis. Neurochem. Res. 2007. 2008; 33(4):691-707.
Young LT, Bakish D, Beaulieu S. The neurobiology of treatment response to antidepressants and mood stabilizing medications. J. Psychiat. Neurosci. 2002; 27:260-265.