CHEMICAL BIOLOGY
Computational Approaches in Drug Discovery and Development
Honglin Li, Mingyue Zheng, Xiaomin Luo, Weiliang Zhu and Hualiang Jiang, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
doi: 10.1002/9780470048672.wecb098
Recently, computational approaches have been considerably appreciated in drug discovery and development. Their applications span almost all stages in the discovery and development pipeline, from target identification to lead discovery, from lead optimization to preclinical or clinical trials. In conjunction with medicinal chemistry, molecular and cell biology, and biophysical methods as well, computational approaches will continuously play important roles in drug discovery. Several new technologies and strategies of computational drug discovery associated with target identification, new chemical entity discovery, and lead optimization will be the focus of this review.
Drug research and development (R & D) is a comprehensive, expensive, and time-consuming enterprise, and it is full of risk throughout the process (1). Numerous new technologies have been developed and applied in drug R & D to shorten the research cycle and to reduce the expenses. Among them, computational approaches have revolutionized the pipeline of discovery and development (2). In the last 40 years, computational technologies for drug R & D have evolved very quickly, especially in recent decades with the unprecedented development of biology, biomedicine, and computer capabilities. In the postgenomic era, because of the dramatic increase of small-molecule and biomacromolecule information, computational tools have been applied in almost every stage of drug R & D, which has greatly changed the strategy and pipeline for drug discovery (2). Computational approaches span almost all stages in discovery and development pipeline, from target identification to lead discovery, from lead optimization to preclinical or clinical trials (see Fig. 1). In this review article, we highlight some recent advances of computational technologies for drug discovery and development; emphases are put on computational tools for target identification, lead discovery, and ADME/T (absorption, distribution, metabolism, excretion, and toxicity) prediction.
Target Identification
Target identification and validation is the first key stage in the drug-discovery pipeline (see Fig. 1). By 2000, only about 500 drug targets had been reported (3,4). The completion of human genome project and numerous pathogen genomes unveils that there are 30,000 to 40,000 genes and at least the same number of proteins; many of these proteins are potential targets for drug discovery. However, identification and validation of druggable targets from thousands of candidate macromolecules is still a challenging task (5).
Numerous technologies for identifying targets have been developed recently. Experimental approaches such as genomic and proteomic techniques are the major tools for target identification (6, 7). However, these methods have been proved inefficient in target discovery because they are laborious and time consuming. In addition, it is extremely difficult to get relatively clear information related to drug targets from enormous information produced by genomics, expression profiling, and proteomics (5). As complementarities to the experimental methods, a series of computational (in silico) tools have also been developed for target identification in recent two decades (6, 8-12). In general, they can be categorized into sequence-based approach and structure-based approach. Sequence-based approach contributes to the processes of the target identification by providing functional information about target candidates and positioning information to biological networks. Such methods include sequence alignment for gene selection, prioritization of protein families, gene and protein annotation, and expression data analysis for microarray or gene chip. Here, we only introduce a special structure-based method for target identification—reverse docking.
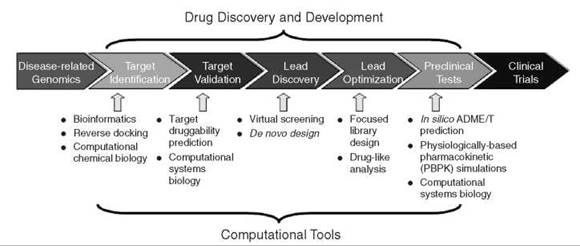
Figure 1. Computational approaches cover the drug-discovery pipeline, spanning from target identification to lead discovery to preclinical test.
Molecular docking has been a promising approach in lead discovery (2, 13) (also see discussion below). Reverse docking, which is opposite to the process of docking a set of ligands into a given target, is to dock a compound with certain biological activities into the binding sites of all the three dimensional (3-D) structures in a given protein database. The identified protein “hits” through this method then served as potential target candidates for further validation study. This computational approach in target identification has demonstrated to be efficient and cost effective (14-16). In general, identifying protein target by using reverse docking includes four steps: inverse docking of a small molecule to a series of selected proteins; hit proteins postprocessing through bioinformatics analysis to select candidates; experimental validation by using biochemical and/or cellular assays; and finally, if it is possible and necessary, determination of the X-ray crystal (or nuclear magnetic resonance) structures of the small molecule-protein complexes to verify the target at the atomic level. To this end, it requires a sufficient number of known protein structures that cover a diverse range of drug targets (17). The protein structures are usually selected from the Protein Data Bank (PDB) or constructed with protein structure prediction method.
Based on DOCK4.0, we developed a reverse docking method, TarFisDock (Target Fishing Docking) (16), and we constructed a corresponding drug target database with 3-D structures, potential drug target database (PDTD) (17). Based on TarFisDock and PDTD, we also developed a reverse docking web server (http://www.dddc.ac.cn/tarfisdock/). TarFisDock has proved to be a tool of great potential value for identifying target by more than 400 users from over 30 countries and regions. One typical example of TarFisDock application is that we found peptide deformylase (PDF) is a target for anti-H. pylori drugs.
Colonization of the human stomach by the bacterium H. pylori is a major causative factor for gastrointestinal illnesses and gastric cancer. However, discovering anti-H. pylori agents is a difficult task because of a lack of mature protein targets. Therefore, identifying new molecular targets for developing new drugs against H. pylori is obviously necessary. In a random screening of a diverse small-molecule and herbal extract library by using agar dilution method to identify active components against H. pylori, compound 1 (N-trans-caffeoyltyramine) isolated from Ceratostigma willmottianum, a folk medicine used to remedy rheumatism, traumatic injury and parotitis (18), showed inhibitory activity against H. pylori with MIC value of 180 μg/mL. Chemical modification on compound 1 afforded a number of analogs, among which, compound 2 ((E)-4-(3-oxo-3-(phenethylamino)-prop-1-enyl)-1,2-phenylene diacetate) is the most active one with improved MIC value of 100 μg/mL against H. pylori (see Fig. 2). Potential binding proteins of compound 1 were screened from PDTD using TarFisDock. Fifteen protein targets with interaction energies to compound 1 under —35.0kJ/mol were identified (19). Homology search indicated that only two proteins of the fifteen candidates, diaminopimelate decarboxylase (DC) and PDF, have homologous proteins in the genome of H. pylori. Enzymatic assay demonstrated compounds 1 and 2 are the potent inhibitors against the H. pylori PDF (HpPDF) with IC50 values of 10.8 and 1.25 μM, respectively. X-ray crystal structures of Hp PDF and the complexes of Hp PDF with compounds 1 and 2 were determined, which reveal that these two inhibitors exactly locate at the Hp PDF binding pocket (see Fig. 2). All these indicate that HpPDF is a potential target for screening new anti-H. pylori agents. In a similar approach, Paul et al. (15) successfully recovered the corresponding targets of four unrelated ligands with reverse docking method.
The advantage of reverse docking is obvious, in addition to identifying target candidates for active compounds, it is also possible to identify potential targets responsible for toxicity and/or side effects of a drug under the hypothesis that the target database contains all the possible targets (20). However, reverse docking still has certain limitations. The major one is that the protein entries in the proteins structure databases like PDB are not enough for covering all the protein information of disease related genomes. The second one is that this approach has not considered the flexibility of proteins during docking simulation. These two aspects will produce false negatives. Another limitation is that the scoring function for reverse docking is not accurate enough, which will produce false positives (16). One tendency to overcome these shortages is to develop new docking program including protein flexibility and accurate scoring function. Another tendency is to integrate sequence-based and structure-based approaches together.

Figure 2. Chemical structures of compounds 1 and 2 and their X-ray crystal structures in complex with HpPDF. Data inside the parentheses are inhibition activities (IC50 values) of the compounds.
Lead Discovery
Computational approaches for lead discovery are more mature than those for target identification. Some methods such as virtual screening and library design have become promising tools for lead discovery and optimization (2, 13). Here, we introduce two computational techniques, virtual screening, and focused combinatorial library design.
Virtual screening
The drugs developed in the past 100 years are found to interact with approximately 500 targets; in the same period, about 20,000,000 organic compounds including natural products have been synthesized or isolated. Moreover, the genomic and functional genomic projects have produced additional 1500 drug- gable targets for drug intervention to control human diseases (21). Therefore, it is believable that a large number of new drugs, at least many leads or hits, hide in the existing chemical mine. However, how to dig out this source is a hard task. Collecting all the existing compounds and screening them randomly against all the potential targets one by one are extremely unpractical, because it is intolerably expensive and time consuming. However, virtual screening shows a dawning to satisfy this requirement. Indeed, virtual screening has been involved into the pipeline of drug discovery as a practical tool (22).
In essence, virtual screening is designed for searching large-scale hypothetical databases of chemical structures or virtual libraries by using computational analysis for selecting a limited number of candidate molecules likely to be active against a chosen biological receptor (23). Therefore, virtual screening is a logical extension of 3-D pharmacophore-based database searching (PBDS) (24) or molecular docking (25), which is capable of automatically evaluating very large databases of compounds. Two strategies have been used in virtual screening (see Fig. 3): 1) using PBDS to identify potential hits from the databases, mostly in the cases that 3-D structures of the targets are unknown and 2) using molecular docking approach to rank the databases if the 3-D structures of the targets are available. Normally, these two approaches are used concurrently or sequentially, because the former can filter out the compounds quickly and the latter can evaluate the ligand-receptor binding more accurately.
For the pharmacophore-based screening, a 3-D-pharmacophore feature is constructed by structure-activity relationship analysis on a series of active compounds (26) or is deduced from the X-ray crystal structure of a ligand-receptor complex (27). Taking this 3-D-pharmacophore feature as a query structure, 3-D database search can be performed to select the molecules from the available chemical databases, which contain the pharmacophore elements and may conform to the pharmacophore geometric constraints. Then the selected compounds are obtained either from commercial sources or from organic synthesis for the real pharmacologic assays (see Fig. 3).
Docking-based virtual screening (DBVS) requires the structural information of both receptors and compounds. The general procedure of DBVS includes five steps: receptor modeling (virtual screening mode construction), compound database generation, computer screening, hit molecules postprocessing, and experimental bioassay. The core step of virtual screening is docking and scoring. Docking is a process to place each molecule from a 3-D small-molecule database into the binding site of a receptor protein, optimize the relative orientation and conformation for a ligand interacting with a protein, and select molecules from the database that may bind to the protein tightly. Billions of possible conformations can be created for a flexible ligand even with a few freedom degrees in the rotation space alone, current optimization algorithms cannot sample exhaustively for all conformations and orientations, needless to say to account for the flexibility of protein with thousands of degrees of freedom. Therefore, instead of searching exhaustively in the search space, an optimization algorithm should sample the solutions effectively and rapidly close to the global optimum (28). Basically, the optimization algorithms employed widely by molecular docking program can be divided into three categories: numerical optimization methods, random or stochastic methods, and hybrid optimization methods. Essentially, there are three types of scoring functions (29), which include force-field based scoring functions, empirical scoring functions, and knowledge-based scoring functions. However, no single scoring function can perform satisfactorily for every system because many physical phenomena determining molecular recognition were not fully accounted for, such as entropic or solvent effects (13). Since Kuntz et al. (30) published the first docking algorithm DOCK in 1982, more than 20 docking programs have been developed in recent two decades. Of the existing docking program, DOCK, FlexX, AutoDock, and GOLD are most frequently used, other programs such as Glide, ICM, and Surflex have been applied successfully in virtual screening. However, many limitations and challenges still exist for molecular docking, such as accurate prediction of the binding conformation and affinity, protein flexibility, entropy, and solvent effects. Without doubt, the molecular docking process is still a complex and challenging project of computational chemistry and biology (25, 31).
Virtual screening has discovered numerous active compounds and leads, more than 50 compounds have entered into clinical trials, and some have been approved as drugs (2). Virtual screening enriched the hit rate (defined as the quotient in percentage of the number of active compounds at a particular concentration divided by the number of all compounds experimentally tested) by about 100-fold to 1000-fold over random screening (22, 32). Additionally, virtual screening provides an alternative way in fleetly finding new leads of some targets, whereas the techniques for high-throughput screening remain in development (e.g. potassium ion (K+) channels). By using docking-based virtual screening in conjunction with electrophysiological assay, we discovered 10 new blockers of the eukaryotic Shaker K+ channels, 4 natural product blockers (33), and 6 synthetic compounds (34).

Figure 3. The flowchart for virtual screening.
Focused combinatorial library design
What big pharmas and medicinal chemists are seeking is new chemical entities (NCEs), which can be strictly protected by compound patents. Nevertheless, all hits discovered by screening the existing compounds databases can only find the new medical usages for the existing compounds or old drugs. At least two kinds of computational approaches are available for NCE discovery: de novo drug design (35, 36) and combinatorial library design (37). The de novo drug design does not start from a database of complete molecules but aims at building a complete molecule from molecular bricks (“building blocks”) to fill the binding sites of target molecule chemically. The complete chemical entries could be constructed through linking the “building blocks” together, or by growing from an “embryo” molecule with the guidance of evaluation of binding affinity. The “building blocks” could be either atoms or fragments (functional groups or small molecules). However, using atoms as “building blocks” is thought to be inefficient, which is seldom used currently. In the fragment-linking approach, the binding site is mapped to identify the possible anchor points for functional groups. Then, these groups are linked together to form a complete molecule. In the sequential-growing approach, the molecule grows in the binding site controlled by an appropriate search algorithm, which evaluates each growing possibility with a scoring function. Different from docking-based virtual screening, fragment based de novo design can perform sampling in whole compound space, which can obtain novel structures that are not limited in available databases. Nevertheless, the quality of a growing step strongly depends on previous steps. Any step chemically growing wrongly would lead to an unacceptable result. For the fragment linking approach, it remains a problem how to choose linkers to connect fragments together to form complete structures. The most remarkable drawback of this approach might be the synthetic accessibility of the designed structures.
To overcome the disadvantages of de novo design approaches, we developed a target-focused library design method to adopt the advantages of focused library and targeted library, as well as integrating technologies of docking-based virtual screening and drug-like (ADMET) analysis. A software package called LD1.0, was also developed (37) based on this technology. Starting with the structures of active hits and therapeutic target, the overall skeleton of potential ligands is split schematically into several fragments according to the interaction mechanism and the physicochemical properties of the binding site. Individual fragment library is constructed for each kind of fragment taking into account of the binding features of the fragments to the binding site. Finally, target-focused libraries regarding the studied target are constructed with the judgments from structural diversity, druglikeness (ADME/T) profiles, and binding affinities (37). During the target-focused library design, library-based genetic algorithm was applied to optimize our focused library, and our newly developed drug-likeness filter was used to predict the drug-like profile of the library. The molecular docking approach was employed to predict the binding affinities of the library molecules with the target.
One application example of target-focused library design is the discovery of new cyclophilin A (CypA) inhibitors (38, 39). CypA inhibitors are of therapeutic significance in organ transplantation. However, inhibitors of CypA are mainly derived from natural sources (such as cyclosporin A, FK506, rapamycin and sanglifehrin A) and peptide analogs, which are all structurally complex molecules, and little has been reported regarding the small-molecule CypA inhibitors. By using docking-based virtual screening, organic synthesis, and bioassay, we discovered dozens of binders of CypA with binding affinities under submicromolar or micromolar level, and 14 of them showed high CypA peptidyl-prolyl isomerase (PPIase) inhibition activities with IC50s of 2.5-6.2 μM (38). To discover new chemical entities of CypA inhibitors with more potent activities, a focused library was designed based on the structures of the 14 inhibitors and their binding modes to CypA. Based on binding mechanism, each inhibitor was divided into three fragments, and three fragment sublibraries were constructed with sizes of 5, 3, and 17 fragments, respectively. Therefore, 255 molecules could be formed by conventional combinatorial library approach. With LD1.0, a CypA-focused library was optimized with a size of 2 x 2 x 4. Thus, the 16 compounds in the library were synthesized for bioassay. All these 16 molecules were determined to be CypA binders with binding affinities (KD values) that ranged from 0.076 to 41.0 μM, and five of them (3a-3f) are potent CypA inhibitors with PPIase inhibitory activities (IC50values) of 0.25-6.43 μM (see Fig. 4). The hit rates for binders and inhibitors are as high as 100% and 31.25%, respectively. Remarkably, both the binding affinity and inhibitory activity of the most potent compound increase ~10 times than that of the most active compound discovered previously. The high hit rate and the high potency of the new CypA inhibitors demonstrated the efficiency of the strategy for focused library design and screening. In addition, the novel chemical entities reported in this study could be leads for discovering new therapies against the CypA pathway.
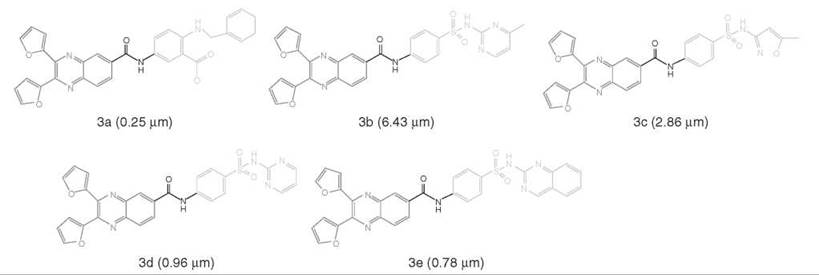
Figure 4. Chemical structures of the five CypA inhibitors discovered using focused library design. Left and right parts are the LD1.0 optimized fragments respectively binding to the small and large pockets of CypA, middle fragment is the LD1.0 optimized link binding to the saddle cleft between the two binding pockets of CypA (39). Data inside the parentheses are inhibition activities (IC50 values) of the compounds.
ADME/T Optimization
It has widely been appreciated that ADME/T should be involved in the early stage of drug discovery (lead discovery) in parallel with efficacy screening (40, 41). Many high-throughput technologies for ADME/T evaluation have been developed, and the accumulated data make it possible to construct models for predicting the ADME/T properties of compounds before structural modifications. These computational models may reduce the redesign-synthesize-test cycles. Hereinafter, we will show how computational approaches predict and model the most relevant pharmacokinetic, metabolic and toxicity endpoints, which thereby accelerate the pace of new drug development.
Prediction of intestinal absorption
Absorption of drugs from the gastrointestinal tract is a complex process that can be influenced by not only physicochemical and physiological factors but also by formulation factors. Some published models and commercially available computer programs have been developed for predicting the intestinal absorption. Among them, physiologically based approaches are of particular interest because the human physiological environment and the processes involved in drug absorption are simulated.
Mixing tank models describe the intestine as one or more well-mixed tanks, in which dissolved and solid forms of drug have a uniform concentration and are transferred between the mixing tanks by first-order transit kinetics. This model has been implemented in commercial program Intellipharm PK (http://www.intellipharm.com) and OraSpotter (http://www.zyxbio.com). Tube models assume the intestine to be a cylindrical tube with or without changing radius, which includes the mass balance models and the models used in PK-Map and PK-Sim (http://www.bayertechnology.com). In the mass balance model, the difference between the mass flows in and out of the tube represents the mass absorbed per time unit, and it is proportional to the permeability and the concentration of the drug in the intestines. In models of PK-Map and PK-Sim, a transit function was introduced and the intestinal permeability coefficient can be calculated solely by the lipophilicity and molecular weight of the investigated compound.
Yu et al. (42) developed an absorption and transit (CAT) model, which formed a basis of many models and commercially available computer programs, such as GITA (GI-Transit Absorption), iDEA (http://www.akosgmbh.de) and GastroPlus (http://www.simulations-plus.com). iDEA uses an absorption model proposed by Grass et al. (43), and GastroPlus uses the ACAT (advanced compartmental absorption and transit) model developed by Agoram et al. (44). Parrott et al. (45) compared these two commercial programs both in their ability to predict fraction absorbed for a set of 28 drugs and in terms of the functionality offered. The results suggested that iDEA may perform better with measured input data, whereas GastroPlus presents a more sophisticated user interface, which shows strengths in its ability to integrate additional data.
Prediction of blood-brain barrier (BBB) permeability
The blood-brain barrier (BBB) is a physiological barrier in the circulatory system that restrains substances from entering into the central nervous system (CNS). The orally administrated drugs that target receptors and enzymes in the CNS must cross the BBB to produce desired therapeutic effects. At the same time, the peripherally acting agents should not cross it to avoid the side effects (46). To date, various methods that have been applied for model generation include the traditional statistical approaches and state-of-art machine-learning techniques.
Generally, current BBB models have shown acceptable capabilities depending on the approaches used and particularly the type and the size of the dataset under investigation. Some insights into the molecular properties that determine the brain permeation have also been gained. However, most current in silico modeling of BBB permeability start with the assumption that most drugs are transported across the BBB by passive diffusion. Absence of data regarding active transport or P-glycoprotein (P-gp) efflux limits the development of accurate and reliable BBB models that can more closely mimic the in vivo situation. Recently, Adeno et al. (47) reported their studies based on a large heterogeneous dataset (~1700 compounds) including 91 P-gp substrates. Likewise, Garg and Verma (48) took the active transport of the molecules into consideration in the form of their probabilities of becoming a substrate to P-gp, and their results indicated that the P-gp indeed plays a role in BBB permeability for some molecules.
Prediction of CYP-mediated metabolism
The human cytochromes P450 (CYP450) constitute a large family of heme enzymes. In drug development, the CYP-mediated metabolism is of particular interest because it may profoundly affect the initial bioavailability, desired activity, and safety profile of compounds. A great variety of in silico modeling approaches have been applied to CYP enzymes. Connections and integrations between the “ligand-based,” “protein-based,” and “ligand-protein interaction-based” groups of methods have been extensively described in the review of Graaf et al. (49). Apart from this methodological categorization, current in silico models mainly address the following three aspects of metabolism: CYP inhibition, isoform specificity, and site of CYP-mediated metabolism.
Inhibition of CYP enzymes is unwanted because of the risk of severe side effects caused by drug-drug interactions. A novel Line-Walking Recursive Partitioning method was presented that uses only nine chemical properties of the shape, polarizability, and charge of the molecule to classify a compound as a 3A4, 2C9, or 2D6 inhibitor (50). Recently, Jensen et al. (51) constructed Gaussian kernel weighted k-NN models to predict CYP2D6 and CYP3A4 inhibition, resulting in classification models with over 80% of overall accuracy.
CYP isoform specificity predicts the subtype responsible for the main route of metabolism. This type of study started from the work by Manga et al. (52), in which the isoform specificity for CYP3A4, CYP2D6, and CYP2C9 substrates was investigated. Terfloth et al. (53) reported a similar study on the same dataset using ligand-based methods, and the accuracy of prediction was improved substantially.
The site of metabolism (SOM) refers to the place in a molecule where the metabolic reaction occurs, which is usually a starting point in metabolic pathway investigation. Recently, Sheridan et al. (54) reported a study addressing the SOM mediated by CYP3A4, CYP2C9, and CYP2D6. In this study, pure QSAR-model was constructed with only substructure and physical property descriptors. The cross-validated accuracies range from 72% to 77% for the investigated datasets, which are significantly higher than that of earlier models and seem to be comparable with the results from MetaSite (http://www.moldiscovery.com), which is a more mechanism- based method of predicting SOM.
Prediction of toxicity
At present, a variety of toxicological tests needs to be conducted by the drug regulatory authorities for safety assessment. Computational techniques are fast and cheap alternatives to bioassays as they require neither experimental materials nor physically available compounds. Some examples of available computer programs that predict toxicity are Deductive Estimation on Risk from Existing Knowledge (http://www.lhasalimited.org), Multiple Computer Automated Structure Evaluation (http://www.multicase.com), and Toxicity Prediction by Komputer Assisted Technology (current DS TOPKAT, http://accelrys.com/products/discovery-studio/toxicology). Recently, developments in artificial intelligence research and the improvement of computational resources have led to efficient data-mining methods that can automatically extract structure toxicity relations (STRs) from toxicity databases with structurally diverse compounds. Helma et al. (55) described some general procedures used for generating (Q)STR models from experimental data. Here, we briefly present a study made to model the mutagenic probability (56).
Mutagenicity is the ability of a compound to cause mutations in DNA, which is one of the toxicological liabilities closely evaluated in drug discovery and their toxic mechanisms have been relatively well understood. In the development of (Q)STR models, it is essential to select the structural or chemical properties most relevant to the endpoint of interest. For mutagenicity, we designed a molecular electrophilicity vector (MEV) to depict the electrophilicity of chemicals, which is a major cause of chemical/DNA interaction. A model for the classification of a chemical compound into either mutagen or nonmutagen was then obtained by a support vector machine together with a F-score based feature selection. For model construction, the SVM+MEV method shows its superior efficiency in the data fitting, with a concordance rate of 91.86%. For validation, a prediction accuracy of 84.80% for the external test set was yielded, which is close to the experimental reproducibility of the Salmonella assay. The F-score based feature weighting analyses highlight the important role of atomic partial charges in describing the noncovalent intermolecular interactions with DNA, which is usually a hard part in the mutagenicity prediction.
The Future
The technological progress of computational chemistry and biology brought a paradigm change to both pharmas and research institutions. Computational tools have been considerably appreciated in drug discovery and development, which play increasingly important roles in target identification, lead discovery, and ADME/T prediction. In the future, in addition to improving individual existing computational techniques, such as perfection of the accuracy and effectiveness of virtual screening, one major tendency is to integrate computational chemistry and biology together with chemoinformatics and bioinformatics. This research is leading to a new topic known as pharmacoinformatics, which will impact the pharmaceutical development process and increase the success rate of development candidates (57).
Another tendency is to use more accurate computational approaches, such as molecular dynamics simulation, to design specific inhibitors or activators against the pathway of target protein folding (58). By using this strategy, Broglia et al. (59) obtained inhibitors that may block HIV-1 protease monomer to fold into its native structure, which causes the inhibitors not to create resistance. Similarly, by targeting the C -terminal P-sheet region of an AP intermediate structure extracted from molecular dynamics simulations of AP conformational transition (60), we obtained a new inhibitor that abolishes AP fibrillation using virtual screening in conjunction with thioflavin T fluorescence assay and atomic force microscopy determination (61).
After the completion of the human genome and numerous pathogen genomes, efforts are underway to understand the role of gene products in biological pathways and human diseases and to exploit their functions for the sake of discovering new drug targets (62). Small and cell-permeable chemical ligands are used increasingly in genomics approaches to understand the global functions of genome and proteome. This approach is referred to as chemical biology (or chemogenomics) (63). Another emerging field is computational systems biology to model or simulate intracellular and intercellular events using data gathered from genomic, proteomic, or metabolomic experiments (64). This field highly impacts drug discovery and development.
References
1. DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J. Health. Econ. 2003; 22:151-185.
2. Jorgensen WL. The many roles of computation in drug discovery. Science 2004; 303:1813-1818.
3. Drews J. Drug discovery: a historical perspective. Science 2000; 287:1960-1964.
4. Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat. Rev. Drug Discov. 2008; 7:21-39.
5. Huang CM, Elmets CA, Tang DC, Li F, Yusuf N. Proteomics reveals that proteins expressed during the early stage of Bacillus anthracis infection are potential targets for the development of vaccines and drugs. Genomics Proteomics Bioinformat. 2004; 2:143-151.
6. Marton MJ, DeRisi JL, Bennett HA, Iyer VR, Meyer MR, Roberts CJ, Stoughton R, Burchard J, Slade D, Dai H, Bassett DE Jr., Hartwell LH, Brown PO, Friend SH. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 1998; 4:1293-1301.
7. Lindsay MA. Target discovery. Nat. Rev. Drug Discov. 2003; 2:831-838.
8. Orth AP, Batalov S, Perrone M, Chanda SK. The promise of genomics to identify novel therapeutic targets. Expert Opin. Ther. Targets 2004; 8:587-596.
9. Garcia-Lara J, Masalha M, Foster SJ. Staphylococcus aureus: the search for novel targets. Drug Discov. Today 2005; 10:643-651.
10. Fliri AF, Loging WT, Thadeio PF, Volkmann RA. Biospectra analysis: model proteome characterizations for linking molecular structure and biological response. J. Med. Chem. 2005; 48:6918-6925.
11. Lagunin A, Stepanchikova A, Filimonov D, Poroikov V. PASS: prediction of activity spectra for biologically active substances. Bioinformatics 2000; 16:747-748.
12. Stepanchikova AV, Lagunin AA, Filimonov DA, Poroikov VV. Prediction of biological activity spectra for substances: evaluation on the diverse sets of drug-like structures. Curr. Med. Chem. 2003; 10:225-233.
13. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004; 3:935-949.
14. Chen YZ, Zhi DG. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins 2001; 43:217-226.
15. Paul N, Kellenberger E, Bret G, Muller P, Rognan D. Recovering the true targets of specific ligands by virtual screening of the protein data bank. Proteins 2004; 54:671-680.
16. Li H, Gao Z, Kang L, Zhang H, Yang K, Yu K, Luo X, Zhu W, Chen K, Shen J, Wang X, Jiang H. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006; 34:W219-224.
17. Gao Z, Li H, Zhang H, Liu X, Kang L, Luo X, Zhu W, Chen K, Wang X, Jiang H. PDTD: a web-accessible protein database for drug target identification. BMC Bioinf. 2008; 9:104.
18. Yue JM, Xu J, Zhao Y, Sun HD, Lin ZW. Chemical components from Ceratostigma willmottianum. J. Nat. Prod. 1997; 60:1031-1033.
19. Cai J, Han C, Hu T, Zhang J, Wu D, Wang F, Liu Y, Ding J, Chen K, Yue J, Shen X, Jiang H. Peptide deformylase is a potential target for anti-Helicobacter pylori drugs: reverse docking, enzymatic assay, and X-ray crystallography validation. Protein Sci. 2006; 15:2071-2081.
20. Chen YZ, Ung CY. Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand-protein inverse docking approach. J. Mol. Graph Model 2001; 20:199-218.
21. Hopkins AL, Groom CR. The druggable genome. Nat. Rev. Drug Discov. 2002; 1:727-730.
22. Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002; 45:2213-2221.
23. Mclnnes C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007; 11:494-502.
24. Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007; 152:9-20.
25. Leach AR, Shoichet BK, Peishoff CE. Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J. Med. Chem. 2006; 49:5851-5855.
26. Kurogi Y, Miyata K, Okamura T, Hashimoto K, Tsutsumi K, Nasu M, Moriyasu M. Discovery of novel mesangial cell proliferation inhibitors using a three-dimensional database searching method. J. Med. Chem. 2001; 44:2304-2307.
27. Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model 2005;45:160-169.
28. Li H, Li C, Gui C, Luo X, Chen K, Shen J, Wang X, Jiang H. GAsDock: a new approach for rapid flexible docking based on an improved multi-population genetic algorithm. Bioorg. Med. Chem. Lett. 2004 14:4671-4676.
29. Boehm HJ, Stahl M. The Use of Scoring Functions in Drug Discovery Applicationg, volume 18. 2002. Wiley, New York.
30. Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982;161:269-288.
31. Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br. J. Pharmacol. 2007; 153:S7-26.
32. Shoichet BK. Virtual screening of chemical libraries. Nature 2004; 432:862-865.
33. Liu H, Li Y, Song M, Tan X, Cheng F, Zheng S, Shen J, Luo X, Ji R, Yue J, Hu G, Jiang H, Chen K. Structure-based discovery of potassium channel blockers from natural products: virtual screening and electrophysiological assay testing. Chem. Biol. 2003; 10:1103-1113.
34. Liu H, Gao ZB, Yao Z, Zheng SX, Li Y, Zhu WL, Tan XJ, Luo XM, Shen JH, Chen KX, Hu GY, Jiang HL. Discovering potassium channel blockers from synthetic compound database by using structure-based virtual screening in conjunction with electrophysiological assay. J. Med. Chem. 2007; 50:83-93.
35. Honma T. Recent advances in de novo design strategy for practical lead identification. Med. Res. Rev. 2003; 23:606-632.
36. Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug. Discov. 2005; 4:649-663.
37. Chen G, Zheng S, Luo X, Shen J, Zhu W, Liu H, Gui C, Zhang J, Zheng M, Puah CM, Chen K, Jiang H. Focused combinatorial library design based on structural diversity, druglikeness and binding affinity score. J. Comb. Chem. 2005; 7:398-406.
38. Li J, Chen J, Zhang L, Wang F, Gui C, Zhang L, Qin Y, Xu Q, Liu H, Nan F, Shen J, Bai D, Chen K, Shen X, Jiang H. One novel quinoxaline derivative as a potent human cyclophilin A inhibitor shows highly inhibitory activity against mouse spleen cell proliferation. Bioorg. Med. Chem. 2006; 14:5527-5534.
39. Li J, Zhang J, Chen J, Luo X, Zhu W, Shen J, Liu H, Shen X, Jiang H. Strategy for discovering chemical inhibitors of human cyclophilin a: focused library design, virtual screening, chemical synthesis and bioassay. J. Comb. Chem. 2006; 8:326-337.
40. van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat. Rev. Drug Discov. 2003; 2:192-204.
41. Selick HE, Beresford AP, Tarbit MH. The emerging importance of predictive ADME simulation in drug discovery. Drug Discov. Today 2002; 7:109-116.
42. Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv. Drug Deliv. Rev. 1996; 19:359-376.
43. Grass GM. Simulation models to predict oral drug absorption from in vitro data. Adv. Drug Deliv. Rev. 1997; 23:199-219.
44. Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001; 50:S41-S67.
45. Parrott N, Lave T. Prediction of intestinal absorption: comparative assessment of GASTROPLUS (TM) and IDEA (TM). Eur. J. Pharm. Sci. 2002; 17:51-61.
46. Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. Modelling of the blood-brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 2007; 6:650-661.
47. Adenot M, Lahana R. Blood-brain barrier permeation models: Discriminating between potential CNS and non-CNS drugs including P-glycoprotein substrates. J. Chem. Inf. Comput. Sci. 2004; 44:239-248.
48. Garg P, Verma J. In silico prediction of blood brain barrier permeability: an artificial neural network model. J. Chem. Inf. Model. 2006; 46:289-297.
49. de Graaf C, Vermeulen NPE, Feenstra KA. Cytochrome P450 in silico: an integrative modeling approach. J.Med. Chem. 2005; 48:2725-2755.
50. Hudelson MG, Jones JP. Line-walking method for predicting the inhibition of P450 drug metabolism. J. Med. Chem. 2006; 49:4367-4373.
51. Jensen BF, Vind C, Padkjaer SB, Brockhoff PB, Refsgaard HHF. In silico prediction of cytochrome P450 2D6 and 3A4 inhibition using Gaussian kernel weighted k-nearest neighbor and extended connectivity fingerprints, including structural fragment analysis of inhibitors versus noninhibitors. J. Med. Chem. 2007; 50:501-511.
52. Manga N, Duffy JC, Rowe PH, Cronin MTD. Structure-based methods for the prediction of the dominant P450 enzyme in human drug biotransformation: consideration of CYP3A4, CYP2C9, CYP2D6. Sar Qsar Environ. Res. 2005; 16:43-61.
53. Terfloth L, Bienfait B, Gasteiger J. Ligand-based models for the isoform specificity of cytochrome P450 3A4, 2D6, and 2C9 substrates. J. Chem. Inf. Model. 2007; 47:1688-1701.
54. Sheridan RP, Korzekwa KR, Torres RA, Walker MJ. Empirical regioselectivity models for human cytochromes p450 3A4, 2D6, and 2C9. J. Med. Chem. 2007; 50:3173-3184.
55. Helma C, Kazius J. Artificial intelligence and data mining for toxicity prediction. Curr. Comput.-Aid. Drug Des. 2006; 2:1-19.
56. Zheng M, Liu Z, Xue C, Zhu W, Chen K, Luo X, Jiang H. Mutagenic probability estimation of chemical compounds by a novel molecular electrophilicity vector and support vector machine. Bioinformatics 2006; 22:2099-2106.
57. Schuffenhauer A, Jacoby E. Annotating and mining the ligand-target chemogenomics knowledge space. Drug Discov. Today: BioSilico 2004; 2:190-200.
58. Broglia R, Levy Y, Tiana G. HIV-1 protease folding and the design of drugs which do not create resistance. Curr. Opin. Struct. Biol. 2008; 18:60-66.
59. Broglia RA, Tiana G, Sutto L, Provasi D, Simona F. Design of HIV-1-PR inhibitors that do not create resistance: blocking the folding of single monomers. Protein Sci. 2005; 14:2668-2681.
60. Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H. Conformational transition of amyloid beta-peptide. Proc. Natl. Acad. Sci. USA 2005; 102:5403-5407.
61. Liu D, Xu Y, Feng Y, Liu H, Shen X, Chen K, Ma J, Jiang H. Inhibitor discovery targeting the intermediate structure of beta-amyloid peptide on the conformational transition pathway: implications in the aggregation mechanism of beta-amyloid peptide. Biochemistry 2006; 45:10963-10972.
62. Kopec KK, Bozyczko-Coyne D, Williams M. Target identification and validation in drug discovery: the role of proteomics. Biochem. Pharmacol. 2005; 69:1133-1139.
63. Stockwell BR. Exploring biology with small organic molecules. Nature 2004; 432:846-854.
64. Materi W, Wishart DS. Computational systems biology in drug discovery and development: methods and applications. Drug Discov. Today 2007; 12:295-303.
Further Reading
Afzelius L, Arnby CH, Broo A, Carlsson L, Isaksson C, Jurva U, Kjellander B, Kolmodin K, Nilsson K, Raubacher F, Weidolf L. State-of-the-art tools for computational site of metabolism predictions: comparative analysis, mechanistical insights, and future applications. Drug Metab. Rev. 2007; 39:61-86.
Dutta A, Singh SK, Ghosh P, Mukherjee R, Mitter S, Bandyopadhyay D. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In. Silico. Biol. 2006; 6:43-47.
Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr. Opin. Chem. Biol. 2006; 10:194-202.
Helma C. In silico predictive toxicology: the state-of-the-art and strategies to predict human health effects. Curr. Opin. Drug Discov. Devel. 2005; 8:27-31.
Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov. Today 2006; 11:580-594.
Kumar N, Hendriks BS, Janes KA, de Graaf D, Lauffenburger DA. Applying computational modeling to drug discovery and development. Drug Discov. Today 2006; 11:806-811.
Leon D, Markel S. In silico technologies. In: Drug Target Identification and Validation (Drug Discoveries Series). 2006, Boca Raton, FL, CRC Press.
Lindsay MA. Target discovery. Nat. Rev. Drug. Discov. 2003; 2:831-838.
PDTD: http://www.dddc.ac.cn/pdtd
Sperandio O, Miteva MA, Delfaud F, Villoutreix BO. Receptor-based computational screening of compound databases: the main dockingscoring engines. Curr. Protein Pept. Sci. 2006; 7:369-393.
TarFisDock: http://www.dddc.ac.cn/tarfisdock
Villoutreix BO, Renault N, Lagorce D, Sperandio O, Montes M, Miteva MA. Free resources to assist structure-based virtual ligand screening experiments. Curr. Protein Pept. Sci. 2007; 8:381-411.
Wolber G, Seidel T, Bendix F, Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 2008; 13:23-29.
Zahler S, Tietze S, Totzke F, Kubbutat M, Meijer L, Vollmar AM, Apostolakis J. Inverse in silico screening for identification of kinase inhibitor targets. Chem. Biol. 2007; 14:1207-1214.
See Also
ADME Properties of Drugs
Computational Chemistry in Biology
Lead Optimization in Drug Discovery