CHEMICAL BIOLOGY
Cytochrome P450 Monooxygenases, Chemistry of
Ilia G Denisov, University of Illinois, Urbana, Illinois
doi: 10.1002/9780470048672.wecb110
Cytochromes P450 monooxygenases represent a large superfamily of heme enzymes, which require a dioxygen molecule and two electrons supplied by a NAD(P)H-dependent protein redox partner to form catalytically active high-valent ferryl-oxo intermediate. These heme enzymes are found in most organisms from all biological kingdoms and catalyze various oxidative chemical transformations important for the biosynthesis of steroid hormones, vitamins, signaling molecules, and many other compounds, as well as for the metabolism of xenobiotics.
Cytochromes P450 represent a superfamily of heme enzymes with a common fold and mechanism that requires an atmospheric dioxygen molecule, two electrons from NAD(P)H, and one or two protons available from solvent water to form an active catalytic ferryl-oxo intermediate (1). All cytochromes P450 have one heme b (protoporphyrin IX) buried deeply in the protein globule, with molecular masses in the range 45-60 kDa. In the resting state, heme iron is oxidized (Fe3+) and is coordinated with the thiolate sulfur atom of cysteine as the fifth axial ligand with the sixth position vacant or occupied by water. The iron serves as a sole redox center in the catalytic cycle, which accepts two electrons sequentially and channels them to coordinated dioxygen to form a (hydro)peroxo-ferric intermediate. Second protonation of the distal oxygen atom and transient formation of the iron coordinated oxo-water, Fe-OOH2, results in the heterolytic scission of O-O bond and formation of the ferryl oxo porphyrin-cation radical, which is termed Compound I, by analogy to the same active intermediate first documented in peroxidases and catalases (2).
Reactions Catalyzed By Cytochromes P450
Although they are termed “monooxygenases,” cytochromes P450 catalyze multiple chemical transformations that include hydroxylation, demethylation, epoxidation, C-C bond cleavage, deamination, and many others (3, 4). Most reactions performed by cytochromes P450 can be catalyzed effectively by Compound I, which is formally the same high-valent intermediate in all isozymes (5, 6). Substrate binding at the distal site of the heme brings the target chemical group into the close proximity of the reactive iron-oxygen catalytic intermediate, often in the regiospecific and stereospecific mode, to provide the selectivity of the chemical transformation. Thus, the broad range of metabolized substrates and the unprecedented variability of the chemical reactions catalyzed by cytochromes P450 can be attributed largely to the differences in substrate binding properties of different isozymes.
With respect to their functional roles in living organisms, cytochromes P450 can be separated tentatively into the enzymes essential for biosynthetic pathways and the xenobiotic metabolizing enzymes (1). The former group includes cytochromes P450 involved into the biosynthesis of steroid hormones, antibiotics, and the transformation of vitamins and cofactors such as retinoids, carotenoids, eicosanoids, and fatty acid derivatives as well as those involved in endogenous intracellular and extracellular signaling. Most reactions are catalyzed with narrow substrate specificity, and they afford products with high regioselectivity and stereoselectivity. As a result, some cytochromes P450 from this group are critical for normal life cycle, and deficiencies can lead to serious diseases (7). In contrast, most cytochromes P450 that belong to the latter group can metabolize a variety of substrates of different sizes through multiple chemical mechanisms and even can form several different products from one substrate. Some enzymes may also bind simultaneously two or three substrate molecules, same or different, and various cases of the resulting activation or inhibition of the product turnover were documented both in vivo and in vitro (4, 7). Cytochromes P450 from both groups are represented in all biological kingdoms; although in many cases, the information about their biological functions and underlying chemical mechanisms is incomplete and fragmented at best.
Despite the unprecedented variability of chemical reactions catalyzed by cytochromes P450, all the important features of their mechanism can be attributed to the oxidative transformations driven by the main catalytic intermediate, which is a ferryl-oxo heme complex with the porphyrin pi-cation radical, or Compound I (3, 4, 8). Some important exceptions involve the hydroperoxoferric complex, Compound 0, and perhaps the one-electron reduced Cpd I, ferryl oxo heme complex Compound II (1, 3, 4, 9). Nonclassic reactions catalyzed by some cytochromes P450, which do not use oxygen, and thus deviate from the main P450 mechanism, include direct reductive processes driven by the heme iron (conversion of nitric oxide to nitrous oxide by CYP55, reductive dehalogenation, reduction of azo compounds and quinines, etc.). Another important role of cytochrome P450 activity is that they can react with peroxides with no consumption of external redox equivalents and perform peroxidase-type chemistry with the O-O bond scission, or afford rearrangements of fatty acid hydroperoxides.
Hydroxylation of hydrocarbons
Oxygen atom from Cpd I is inserted into the C-H bond of saturated hydrocarbons (Scheme 1a) by means of hydrogen atom abstraction followed by recombination of the transient hydroxyl with the carbon radical [the so-called “oxygen rebound” mechanism proposed by Groves in 1976 (8, 10)]. Another possibility can be the concerted oxygen insertion into the C-H bond. Both pathways are rationalized by the two-state mechanism developed by Shaik et al. (6, 9), which describes different reactivities of Cpd I in the high-spin and low-spin states. The possibility of involvement of Cpd 0, which is the ferric-hydroperoxo intermediate, into the carbon hydroxylation is also debated in the literature, but this species is much weaker oxidant and may participate in catalysis only in rare cases when the barrier for hydroxylation by Cpd 0 is lower than the barrier for the Cpd I formation. Carbon hydroxylation is one of the most common reactions for all cytochromes P450, with substrates that include steroid hormones, fatty acids, vitamins, and antibiotics (5, 11).

Scheme 1. Typical reactions catalyzed by cytochromes P450: (a) hydroxylation of saturated hydrocarbons; (b) heteroatom oxidation and dealkylation; (c) olefin epoxidation; (d) aromatic hydroxylation; (e) desaturation; (f) aldehyde deformylation.
Oxidation and dealkylation of heteroatoms
Oxidative transformation of the heteroatom attached to the carbon (Scheme 1b) may result in direct electron abstraction from the former and formation of oxide (-NO, -SO, -PO, etc.) or may proceed through the hydrogen abstraction from the latter with subsequent hydroxylation as in the previous section. The second pathway is often followed by the heteroatom elimination and formation of carbonyl group. Substrates that contain nitrogen (i.e., alkylamines and amides) are easier to oxidize and give more various products in P450 catalyzed reactions than compounds that contain oxygen. An example is the reaction of N-demethylation of caffeine catalyzed by the human CYP1A2.
Olefin oxidation (epoxidation)
Insertion of oxygen atom from Cpd I into the carbon-carbon double bond with formation of epoxide (Scheme 1c) reveals features characteristic for a concerted process, although formation of radical intermediates is possible in many cases. A unified description of this alternative is also provided by the two-state mechanism of catalysis by Cpd I (see the section on Hydroxylation of hydrocarbons). Essentially, the concerted oxygen insertion represents a low-spin reaction surface, whereas the distinct radical intermediate is formed on the high-spin reaction pathway. In the latter case, the carbon radical may attack the nearby heme nitrogen and modify the heme covalently. This reaction is also an important inactivation pathway of cytochromes P450 during oxidative transformations of terminal double and triple bonds.
Aromatic oxidation and hydroxylation
Aromatic compounds may be oxidized either through epoxidation or via addition of oxygen atom from Cpd I (Scheme 1d). Usually, both pathways afford more stable phenols or quinones as the end product through rearrangements and/or addition of another nucleophile. Direct abstraction of an electron from aromatic moiety is viable in the presence of strong electron-donating substituents. The oxidative metabolism of polycyclic aromatic compounds is represented by CYP1A1 in humans.
Dehydrogenation and desaturation
Desaturation, or formation of double bonds after hydrogen abstraction (Scheme 1e), is an important class of transformations of steroids, flavones, and drugs with heterocyclic compounds. These reactions do not involve insertion of oxygen into the substrate molecule and deviate in this sense from the canonical stoichiometry of P450 monooxygenase catalysis. Desaturation reactions are common in the metabolism of heterocyclic xenobiotics and in biosynthesis of fatty acid derivatives. Aromatase CYP19 also performs a complex desaturation reaction at the third and last step of the transformation of androgens to estrogens. The aromatization reaction of the steroid ring A is catalyzed by the hydroperoxo-ferric intermediate, whereas Cpd I is responsible for the first two hydroxylation steps of this process.
Other oxidative transformations
Cleavage of C-C bonds can be catalyzed by cytochromes P450 involved in biosynthetic pathways and by xenobiotic metabolizing enzymes. The former case is exemplified by the conversion of cholesterol to pregnenolone catalyzed by the human CYP11A. This and other transformations of steroids with C-C bond cleavage involve three sequential steps with one dioxygen and one NADPH molecule consumed at each step. The mechanism similar to the last step is responsible for deformylation of aldehydes catalyzed by CYP2B4 and other hepatic enzymes (Scheme 1f). Oxidative decarboxylation reactions and decomposition of alcohols often afford C-C bond scission through rearrangement of various transient intermediates.
Atypical chemistry—reductive, peroxidative, and so on
In addition to the variety of oxidative transformations, cytochromes P450 can catalyze reduction reactions. Self-sufficient CYP55 (nitric oxide reductase) reduces nitric oxide NO to form nitrous oxide N2O and water via hydride transfer directly from NADH molecule to the Fe3+-NO complex and subsequent reaction of this intermediate with the second NO molecule. Ferrous cytochromes P450 can reduce azo compounds, N-oxides, quinines, and can perform reductive dehalogenation with the release of halide and concomitant hydrogenation or the formation of olefins. Another important class of reactions catalyzed by cytochromes P450 includes rearrangements of fatty acid peroxides (CYP74 family) and prostaglandin peroxides (CYP5 and CYP8 families), which usually do not involve NADPH and oxygen consumption.
Reaction Cycle and Intermediates
Substrate binding and spin shift
In the resting state 1 (Fig. 1) with no substrate at the active site of cytochrome P450, the low potential heme iron (-400 through -250 mV) is mostly in the low-spin state (S = 1/2) with the water coordinated as the sixth ligand trans to the proximal thiolate. When the substrate binds in the vicinity of the heme, water ligation is destabilized, and the heme iron may turn to the pentacoordinated configuration 2 with the concomitant shift of the spin equilibrium to the high-spin (S = 5/2). As a result of the loss of the sixth ligand by the heme iron, the cytochromes P450 saturated with substrates are reduced much faster than in the substrate-free state because of the positive shifts of the redox potential by ~ 100 mV. Such control over the rate of the first electron transfer by the substrate binding represents an important feature of the overall steady-state kinetics of many cytochromes P450; although some isozymes exist in a predominantly high-spin state, even without substrates, and they presumably lack this switch.
Some cytochromes P450 can bind two or three molecules of substrate or inhibitor simultaneously (CYP107, CYP158A1, CYP158A2, CYP2B4, CYP2C8, CYP3A4, CYP245) and as a result may reveal non-Michaelis turnover kinetics. Functional homotropic and heterotropic cooperativity (i.e., the perturbation of the activity of the enzyme with respect to one substrate molecule by the binding of another molecule of the same or different type, respectively) typically is observed for such cytochromes P450. This allosteric behavior of xenobiotic metabolizing cytochromes P450 constitutes the important aspect of the drug-drug interaction problem in the pharmaceutical and medicinal chemistry and industry.

Figure 1. Catalytic cycle of cytochromes P450. Main path (1) through (7) is shown in bold arrows forming a circle. Uncoupling pathways are shown in dashed lines. Reproduced with permission from the American Chemical Society from Reference 2, p. 2257.
First electron transfer—reduction of ferric heme
Reduction of the heme iron of cytochromes P450 to the ferrous state 3 is necessary for the binding and subsequent activation of atmospheric dioxygen. Initially, two electrons are derived from NAD(P)H by flavin adenine dinucleotide (FAD)-containing proteins and then are used sequentially via one-electron transfers. All cytochromes P450 can be divided into two main classes with respect to the reduction mechanism and the structure of their immediate redox partner. The first class includes most soluble bacterial cytochromes P450 and mitochondrial P450 systems. In these systems, the electron transfer from the FAD-containing reductase to the heme enzyme cytochrome P450 is mediated by the soluble iron-sulfur protein ferredoxin. In bacteria, all three proteins are soluble, whereas in mitochondria of eukaryotic cells, cytochromes P450 and reductases are associated with the inner membrane. The systems that belong to the second class include only two proteins, and the cytochrome P450 is reduced directly by the cytochrome P450 reductase (CPR), which contains both FAD and flavin mononucleotide. Variations of these systems include fusion enzymes in which a single polypeptide chain folds into two or three domains that correspond to the heme protein, iron-sulfur protein, and/or flavoprotein components of cytochrome P450 catalytic system (12). Many CPR flavoproteins strongly favor the usage of either NADH or NADPH, whereas some flavoproteins can use both efficiently.
Electron transfer in Class I systems is studied in detail for CYP101 and its redox partner putidaredoxin (2Fe-2S), which contains protein with molecular mass 12.6 kDa. Binding of putidaredoxin at the heme proximal site of CYP101 and formation of the transient electron transfer complex is accompanied by subtle conformational changes of both proteins and concomitant changes in their redox properties that stimulate the reduction of the heme iron to the ferrous state. The complex of CYP101-Pd is not very tight (Kd is in micromolar range), because facile dissociation is necessary to maintain the fast overall turnover. However, this complex is highly specific and can be perturbed significantly by single point mutations at the protein-protein interface. The apparent first-order rate of the first electron transfer in this system measured in kinetic experiments is in the range of 50-100 s-1.
Oxygen binding and autoxidation—first uncoupling
Ferrous cytochromes P450 bind dioxygen as the sixth ligand to the heme iron, and the resulting diamagnetic oxy-ferrous complex 4 is similar to that in myoglobin and hemoglobin. Quantum chemical studies show that the wave function of this complex predominantly is a mixture of approximately similar fractions of Fe2+- O2 (closed shell, Pauling configuration) and Fe3+- O2- (open shell singlet, Weiss configuration), with the minor contribution of other configurations. Results of vibrational spectroscopy reveal that both O-O and Fe-O bonds are slightly weaker in oxyP450 (1128-1139 cm-1 and 539-541 cm-1) than in oxygenated myoglobin and hemoglobin (1148 and 572 cm-1, respectively) (13). This difference is caused by more electron-rich thiolate ligand and the excess donation of electron density to the antibonding orbitals of Fe-O-O moiety in cytochrome P450. Similar effects were observed in nitric oxide synthase, which has the same thiolate ligand for the heme iron.
A comparison of available X-ray structures of oxy-ferrous complexes in CYP101, CYP107, and CYP158 reveals very similar structural arrangements of the heme iron and axial ligand with major differences concerning the detailed stereochemistry of the amino acid side chains and water molecules in the immediate vicinity of the bound dioxygen (14, 15). The main difference is caused by the replacement of the mostly conserved alcohol functionality of T252 residue in CYP101 by methyl groups of A245 (CYP107) and A245 in CYP158. The absence of T252 hydroxyl within the hydrogen bond distance to the distal oxygen atom of the coordinated dioxygen is compensated in the latter two enzymes by the specific positioning of hydroxyl groups on the bound substrates, which in turn stabilize water molecules assumed to be the ultimate source of protons for the oxygen activation (see below).
Usually, oxygen binding is faster than other steps in the enzymatic cycle, and it is not rate limiting in the overall substrate turnover. However, the lifetime of oxy-ferrous complex in cytochromes P450 at physiologic temperatures is in the range of seconds, because it decomposes spontaneously back to the resting ferric state with the formation of superoxide ion in an autoxidation reaction. Usually, the rate of autoxidation is lower by one or two orders of magnitude for the enzymes saturated with substrate. Higher stability of substrate-bound oxy-ferrous cytochromes P450 is explained in part by their higher redox potentials (i.e., by thermodynamic stabilization of the oxygenated heme enzyme in the enzyme-substrate complex). In addition, the presence of substrates in the active site was shown to impose dynamic constraints on the superoxide escape and to increase the kinetic stability of oxy-ferrous P450. Inhibition of autoxidation is an essential part of the regulatory role played by substrates in the overall optimization of the P450 cycle via minimization of this uncoupling reaction.
Second electron transfer and protonation, compound 0
Direct observation of the reduction of oxy-ferrous cytochrome P450 and formation of peroxo-ferric (5a) or hydroperoxo-ferric (5b) complex was never accomplished at ambient conditions because this intermediate, termed Compound 0, undergoes additional transformations and disappears faster than it is formed. However, it was isolated and studied by electron paramagnetic resonance (EPR), optical absorption, and resonance Raman spectroscopy at low temperatures using radiolytic reduction of oxy-ferrous cytochromes P450. As expected, the hydroperoxo-anion forms a relatively strong bond with the heme iron, and the resulting complex reveals the typical features of low-spin heme-thiolate complexes with the narrow span of g-values in EPR spectra and red-shifted Soret band (436-440 nm) (2). Spectroscopic and theoretical studies reveal that the length and strength of the O-O bond in Compound 0 are similar to those observed in the low-spin oxygen activating nonheme (hydro)peroxo-ferric complexes. The presence of the distinct hydroperoxo-ferric heme intermediate in the frozen solutions and in crystals of cytochromes P450 and several other heme proteins indicates that the spontaneous homolysis or heterolysis of O-O bond is unfavorable. Thus, the efficient formation of the main active intermediate Compound I requires catalytic delivery of the second proton to the distal oxygen atom.
Second protonation and O-O- scission. peroxide dissociation-second uncoupling
The second protonation of Compound 0 at the distal oxygen atom catalyzes fast scission of O-O bond with the leaving water molecule. In cryoradiolytic experiments, Compound 0 is stable below the glass transition temperature, 180-190 K, which means that the second proton delivery requires sufficient mobility as well as possibly relaxation and diffusion of the solvent molecules. Experiments with the native CYP101 and D251N mutant proved that at higher temperatures, Compound 0 disappears with formation of Compound I (6) and concomitant product formation (16). For the T252A mutant CYP101, in which the native proton transfer mechanism is perturbed, the dissociation of peroxide with no product formation is the dominant path of Compound 0 decomposition. The latter reaction is considered as the main source of reactive oxygen species in the poorly coupled P450 systems, in which the redox equivalents from NADPH oxidation are not used efficiently for the product formation. In general, the coupling efficiency measured by the ratio of the product molecules formed per one consumed NADPH molecule can be very different for the same cytochrome P450 with different substrates. The efficient proton delivery requires specific positioning and stabilization of water molecules near dioxygen moiety, which can be perturbed significantly by variations in the structure of substrate.
Compound I—properties and activity
Compound I (6) is the main catalytically active intermediate. A description of the main chemical reactions is given above. Compound I has yet to be characterized on the natural P450 pathway, although the transient formation of the short-lived ferryl-oxo intermediate with porphyrin-cation radical has been documented using reaction with peroxides. The electronic configuration of this intermediate, as evaluated by quantum chemical methods, represents it as a triradicaloid with two possible spin states S = 1/2, 3/2, with closely spaced energies. In addition to the almost degenerate ground state with doublet and quartet relative stability regulated by the protein matrix, the sextet S = 5/2 can also be involved. The presence of high spin and low spin states of Cpd I provides variability in the reaction pathways and product distributions. This concept of two-state and multistate reactivity of Cpd I in cytochromes P450 developed by Shaik et al. (6, 9) provides basis for the unified explanation of the plethora of apparently contradictory experimental results in P450 chemistry.
Structures—Common Fold and Variations
All cytochromes P450, from soluble enzymes in archae and bacteria to the mammalian integral membrane proteins, have essentially the same fold, which is specific for this superfamily despite their functional diversity (17) (Fig. 2). Only several enzymes with this fold do not follow classic P450 mechanism based on activation of dioxygen (i.e., nitric oxide reductase CYP55 and CYP152, which uses hydrogen peroxide). Approximately 50% of the residues are located in helical fragments, which are designated by letters A to L with the addition of B’, F’, G’, and J’ in some isozymes, and 7-10% are found in small beta-sheets interspersed with flexible loops. As compared with the soluble bacterial enzymes, membrane-bound proteins usually have an extra N-terminal fragment that is thought to be incorporated into the membranes and longer loops with one or two small additional helical fragments.
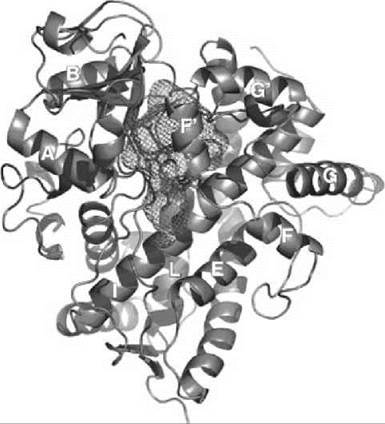
Figure 2. Structure of the human hepatic cytochrome P450 CYP3A4 (18) is shown from the distal site of the heme in cartoon representation. The spacious substrate-binding cavity is represented as a mesh. The heme is shown in sticks, which can be observed under the mesh grid. Helices are labeled by letters according to the common notation for all cytochromes P450 that share the same fold (17). Prepared using PyMol (DeLano Scientific LLC, San Carlos, CA)
The heme (protoporphyrin IX, heme b) is buried in the protein matrix between the long I-helix on the distal side and L helix on the proximal side. The loop at the N-terminus of L-helix contains absolutely conserved cysteinate with the deprotonated sulfur atom that serves as the proximal axial ligand of the heme iron. Three hydrogen bonds with this sulfur atom are formed by the amide groups of the highly conserved neighboring residues. These bonds are necessary to raise the redox potential of cytochrome P450 to the range accessible for the ultimate physiologic reductant NAD(P)H. Hydrogen bonds between propionic carboxyls and the side chains of protein amino acids prevent the loss of the heme and stabilize the native conformation.
Another important and highly conserved feature in all structures of cytochromes P450 with only a few exceptions is the presence of the acid-alcohol pair (i.e., sequential acid and alcohol side chains at the I-helix just next to the dioxygen coordinated to the heme iron). An acidic side chain is provided by Asp or Glu residue (n), whereas alcohol (n + 1) is from Thr or Ser. The hydroxyl of the latter is very often hydrogen bonded to the peptide carbonyl of the Gly (n - 3) forming a kink, which interrupts the regular a-helical structure and is thought to be essential for the efficient oxygen activation. Structures of the oxy-ferrous complexes resolved for CYP101, CYP107, and CYP158 reveal the appearance of specifically positioned water molecules, which are stabilized by the appropriate rearrangement of the side-chains of the acid-alcohol pair described above. These water molecules play an important role in the directed proton delivery provided by protein for the formation of Compound 0 and Compound I, although the details of the mechanism and specific pathway of this proton delivery are still debated.
Helices B, B’, F, F’, G’, and G and connecting loops are involved in substrate binding and in general are much more variable and flexible (upper part of the substrate binding cavity shown in Fig. 2). These parts have different length in different classes of P450, with longer disordered insertions typical for eukaryotic enzymes and shorter and more compact structures characteristic for the proteins from extremophilic organisms. In all classes, however, the protein has to experience a large-scale conformational change to open access to the substrate-binding cavity. Comparison of the X-ray structures of cytochromes P450 with and without substrates and the results of molecular dynamics studies suggest that these conformational changes involve concerted movement of the fragments, which form the substrate-binding cavity that includes helices F and G and fragments between them, as well as B and C helices. Such an “opening” movement was first observed in the X-ray structures of CYP101 with the “wired” substrates (19). In these structures, the diaminoalkane chain traced the tentative substrate access channel that connects adamantane positioned in the substrate-binding pocket and the fluorescent group on the surface of the protein. However, in many cases, the conformations of cytochromes P450 with the bound substrates or inhibitors are even more compact than in substrate-free state, in which the flexible fragments rearrange to provide additional interactions with the hydrophobic molecule bound in the active center. In general, the high plasticity of P450 fold provides basis for their ability to bind multiple organic molecules with different sizes and chemical structures, which is critically important for the efficient metabolism of xenobiotics.
References
1. Ortiz de Montellano PR, ed. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd edition. 2005. Kluwer Academic/ Plenum Publishers, New York.
2. Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005; 105:2253-2278.
3. Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem. Rev. 1996; 96:2841-2887.
4. Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001; 14:611-650.
5. Ortiz de Montellano PR, De Voss JJ. Substrate oxidation by cytochrome P450 enzymes. In: Cytochrome P450: Structure, Function, Genetics. 3rd edition. Ortiz de Montellano PR, ed. 2005. Kluwer Academic/Plenum Publishers, New York. pp. 183-245.
6. Shaik S, Hirao H, Kumar D. Reactivity patterns of cytochrome P450 enzymes: multifunctionality of the active species, and the two states-two oxidants conundrum. Nat. Prod. Rep. 2007; 24:533-552.
7. Guengerich FP. Human cytochrome P450 enzymes. In: Cytochrome P450: Structure, Function, Genetics. 3rd ed. Ortiz de Montellano PR, ed. 2005. Kluwer Academic/Plenum Publishers, New York. pp. 377-530.
8. Groves JT, Han Y. Models and mechanisms of Cytochrome P450 Action. In: Cytochrome P450: Structure, Function, Genetics. 3rd edition. Ortiz de Montellano PR, ed. 2005. Kluwer Academic/Plenum Publishers, New York. pp. 1-43.
9. Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Theoretical perspective on the structure and mechanism of cytochrome P 450 enzymes. Chem. Rev. 2005; 105:2279-2328.
10. Groves JT, McClusky GA. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J. Am. Chem. Soc. 1976; 98:859-861.
11. Munro AW, Girvan HM, McLean KJ. Variations on a (t)heme-novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 2007; 24:585-609.
12. Hannemann F, Bichet A, Ewen KM, Bernhardt R. Cytochrome P450 systems—biological variations of electron transport chains. Biochim. Biophys. Acta 2007; 1770:330-344.
13. Kincaid JR Resonance Raman spectra of heme proteins and model compounds. In: Porphyrin Handbook, Volume 7. Kadish KM, Smith KM, Guilard R, eds. 2000. Academic Press, New York. pp. 225-291.
14. Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000; 287:1615-1622.
15. Poulos TL. Structural biology of P450-oxy complexes. Drug Metab. Rev. 2007; 39:557-566.
16. Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM. Hydroxylation of camphor by reduced oxy-cytochrome P450cam: mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc. 2001; 123:1403-1415.
17. Poulos TL, Johnson EF. Structures of cytochrome P450 enzymes. In: Cytochrome P450: Structure, Function, Genetics. 3rd edition. Ortiz de Montellano PR, ed. 2005. Kluwer Academic/Plenum Publishers, New York. pp. 87-114.
18. Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by x-ray crystallography to 2.05-A resolution. J. Biol. Chem. 2004; 279:38091-38094.
19. Gray HB, Stout CD, Goodin DB. Conformational states of cytochrome P450cam revealed by trapping of synthetic molecular wires. J. Mol. Biol. 2004; 344:455-469.
Further Reading
Bernhardt R. Cytochrome P450: structure, function, and generation of reactive oxygen species. Rev. Physiol. Biochem. Pharmacol. 1996; 127:137-221.
Bernhardt R. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 2006; 124:128-145.
Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004; 104:3947-398O.
Ortiz de Montellano PR, ed. Cytochrome P450: Structure, Mechanism, and Biochemistry. 2nd edition. 1995. Plenum Press, New York.
Sigel A, Sigel H, Sigel RKO, eds. The Ubiquitous Roles of Cytochrome P450 Proteins, Volume 3: Metal Ions in Life Sciences. 2007. John Wiley & Sons, Ltd., Chichester, UK.
See Also
Drug Metabolizing Enzymes
Hemes in Biology
Inorganic Chemistry in Biology
Metallo-Enzymes and Metallo-Proteins, Chemistry of
Oxygen-Activating Enzymes, Chemistry of