CHEMICAL BIOLOGY
Small Molecule Inhibitors, Design and Selection of
Jianwei Che and Yi Liu, Genomics Institute of the Novartis Research Foundation, San Diego, California
Nathanael S. Gray, Harvard Medical School, Dana Farber Cancer Institute, Biological Chemistry and Molecular Pharmacology, Boston, Massachusetts
doi: 10.1002/9780470048672.wecb123
The discovery and optimization of small-molecule inhibitors for the treatment of human disease has been the major focus of the pharmaceutical industry for more than a century. In the last decade, they have also attracted increased attention from academia because they provide a powerful means to interrogate biologic systems in an acute fashion. Although pharmaceutical companies have identified many small-molecule inhibitors for drug targets, to find a desirable small-molecule inhibitor for a specific target is still a major challenge for drug discovery and for chemical biology. Here, we present an overview of the computational approaches that can be exploited to discover small-molecule inhibitors for protein targets of interest.
Small molecule inhibitors are of special value to chemical biology and to drug discovery. Thousands of small molecules have been developed as drugs to treat various human diseases. Increasingly, small-molecule inhibitors are used to perturb the function of proteins to elucidate their biologic functions (1, 2). Complementary to the conventional genetics, small molecules allow rapid, conditional, and tunable inhibition. Furthermore, small molecules can often be identified that alter specifically one particular function of a multifunctional protein. The recent completion of the human genome sequencing project has resulted in the identification of a plethora of new protein targets for which small-molecule modulators could be developed as potential therapeutics and as biologic “tools.” Identifying a selective small-molecule inhibitor for each protein in human genome is a formidable challenge for drug discovery and for chemical biology. Experimentally, high-throughput screening is the most widely used technology currently to identify hits for a target. On the other hand, computational methods, such as virtual screening and structure-based design, have become equally important to identify and to optimize small-molecule inhibitors. In Fig. 1, we illustrate how computational and experimental approaches can complement each other in the compound discovery and optimization process. The numbers of compounds associated typically with each method is indicated. At early stages of hit identification, high throughput methods such as virtual ligand screening (VLS) and ultra high-throughput screening (uHTS) are used frequently. At later stages, high accuracy algorithms combined with biologic assays can be used to guide the selection and the synthesis of a limited set of analogs. As shown in Fig. 1, computational and experimental approaches are not mutually exclusive processes. They are best exploited by using computation and experiment in an iterative fashion to optimize compounds progressively for a particular target of interest. Without these interactions, none of the methods can achieve its maximum potential. In this review, we focus on computational approaches depicted in Fig. 1. As will become clear later in the article, most computational methods are not associated exclusively with predefined processes or protocols. Significant redundancies exist in terms of when and what methods should be applied. The structure of the article follows the common flow of identifying small-molecule inhibitors. We will discuss the particular computational methodologies used at different stages of the process in subsequent sections.

Figure 1. Overview of computational methods used in the process of identifying small-molecule inhibitors.
Virtual Screening in Selecting Small Molecule Inhibitors
Virtual screening (VS) uses computational or theoretic methods to select molecules with desired biologic properties. It has broad applications in pharmaceutical industry. In this article, we limit ourselves to VS to identify biologically active small molecules. The estimated chemical space for small organic molecules is over 1060, and the registered organic and inorganic molecules in CAS are already over 30 million. Despite advances in experimental high throughput screening technologies, currently it is impossible to screen more than a few million compounds. With the rapid improvement of computational power and algorithms, VS methods have become an important complementary technique to experimental uHTS. It has many distinct advantages, such as investigating virtual molecules that can cover a broader area of chemical space. Compared with uHTS, VS is also a relatively inexpensive means to probe many molecular structures.
Generally, two types of virtual screening are used. One is receptor based, such as docking, and the other is ligand based, such as similarity search. In practice, both methods are often used synergistically. In Fig. 2, we illustrate how these two types of methods are used to select “hits” from a given small-molecule library.

Figure 2. Workflow of computational algorithms used for identifying ''hit'' molecules from a compound library. The top panel indicates the compound collection from which the ''hit'' molecules are to be identified. The middle panel illustrates two virtual screening strategies. One is based on a known receptor 3-D structure (i.e., docking and scoring) and the other is based on active ligand molecules alone (i.e., pharmacophore searching and ranking). The docking/scoring uses existing 3-D structures of receptors to construct binding models, and the pharmacophore searching/ranking uses known active small molecules to construct pharmacophore models.
Receptor-Based VLS
In receptor-based VLS, the atomic coordinates of receptor molecules are known experimentally or theoretically (e.g., homology models). The interactions between a small molecule and a protein are often characterized by a “lock-key” mechanism, in which high affinity small molecules often fit the protein-binding site well. The shape complementarities provide important enthalpy contributions from favorable van der Waals interactions. In addition, correctly positioned charges, hydrogen bonds, and hydrophobic contacts usually are required for higher affinity. Based on computational efficiency, receptor-based VLS can be divided into three tiers. The first tier is the popular high-throughput docking and scoring that can be computed for each ligand molecule within a few minutes on a typical desktop computer. In the rest of this article, we refer to high-throughput docking as docking and scoring for simplicity. The second tier includes the medium throughput methods that calculate the interactions more thoroughly and accurately. It often includes algorithms that account more accurately for desolvation energies, entropic penalties, and receptor flexibility. For example, detailed implicit solvent models usually are applied in these calculations. It normally takes tens of minutes for each calculation. The third tier represents the slowest method but ultimately offers more accurate results. The typical examples are atomistic molecular dynamics (MD) or Monte Carlo simulations with explicit water molecules. The details of these calculations are out of the scope of this review. Interested readers can consulate recent reviews on these specific topics (3).
Among the three classes of methods, docking is the most widely used method in pharmaceutical industry. Many high throughput docking software packages are available publicly, such as Dock, Autodock, Gold, Glide, FlexX, QXP, and ICM. In addition, many companies have built their own proprietary docking technologies. A docking based VLS calculation usually consists of three stages: preparing, computing binding modes, and scoring. Currently, most docking programs treat receptors as rigid molecules. Under this approximation, a receptor field can be calculated for the binding site. A receptor field is the interaction potential created by receptor atoms. It consists of van der Waals, electrostatic, hydrogen bonding, and solvation contributions. Without atom thermal motions, the receptor field is also static, so it can be precalculated in the absence of ligand molecules. This feature provides enormous savings of computational time over explicit atom models. Different programs employ very different algorithms to sample ligand conformations, to orient small molecules in the binding site, to retain preferred binding modes, and to increase the computational efficiency (4-7). The predicted binding configuration is selected based on molecular interaction energies or an empirical scoring function. Finally, docking ranks all ligand molecules based on binding poses that possess the highest score, and virtual “hit” molecules are selected from the top of the list. This method allowed Hopkins et al. (8) to identify submillimolar Kinesin inhibitors from an 110,000 compound library. Novel casein kinase II inhibitors (9) and BCR-ABL tyrosine kinase inhibitors (10) were also discovered by docking. Docking can also be applied to discover small-molecule inhibitors for protein-protein interactions. Bonacci et al. (11) used docking (FlexX) to screen the chemical diversity set of 1990 compounds. Among 85 virtual hits, 9 of them inhibited SIGK binding with Gβ1γ2 with IC50 that ranged from 100 nM to 60 μM. It was also used by Trosset et al. (12) to identify β-Catenin inhibitors for Wnt signal pathway from 177,000 compounds. Among 22 tested virtual hits, 3 hits were confirmed as binders to β-Catenin and Tcf4 competitors. Without experimental receptor structures, docking can be carried out using carefully constructed model structures. Becker et al. (13) and Salo et al. (14) reported the successful identification of small-molecule inhibitors of several G-protein coupled receptors (GPCR) systems based on docking.
One major challenge in docking-based VLS is the accuracy of scoring functions. Conceptually, binding free energy is a rigorous measure for correct binding conformation and compound affinity. However, the inherent errors in classic force fields and the extremely lengthy computation required preclude its immediate use for VLS. In practice, an empirical function (i.e., a scoring function) normally is used to estimate free energy of binding. A few examples of commonly used scoring functions are PMF (15), Chemscore (16), Drugscore (17), Goldscore (18), and Glidescore (19). The specific mathematic forms of Chem- score, Goldscore, and PMF are shown here to highlight their similarity and difference. Other scoring functions can be found in respective references.
![]()
where ∆G0 is a fitting constant, ∆Ghbond is hydrogen bond energy between ligand and receptor, ∆Gmetai is the energy between ligand atoms and metal ions in a receptor active site, ∆Glipo is the ligand receptor lipophilic interaction, and ∆Grot is for the rotatable bond penalty.
![]()
where ∆Ehbond is the hydrogen bond energy between ligand and receptor, and ∆Evdw is the van der Waals energy of ligand and receptor, and ∆Einternal is the van der Waals energy and torsional strain of the ligand molecule.
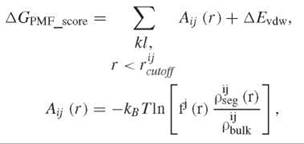
where kl is a ligand receptor atom pair of type ij, fj(r) is ligand volume correction factor, pijseg(r) is the number density of a ligand receptor atom pair of type ij at inter atomic distance r, pijbulk is the reference number density of ligand receptor pair of type ij, and ∆Evdw is the van der Waals interaction between ligand and receptor. Although common features exist within different scoring functions, they often emphasize different aspects of the protein-ligand interaction. These empirical scoring functions bear significant errors because of crude approximations. Currently, no scoring functions exist that are able to rank diverse compound libraries consistently and accurately. The performance of docking based VLS varies considerably from system to system and from program to program (20). Although some programs tend to perform better than others do, it is difficult to predict the performance a priori on a specific system. In addition to ranking molecules based upon a single scoring function, combination of several scores (i.e., consensus score) or statistical methods (e.g., Bayesian statistics) are also used to select virtual hits.
Often, rigid receptor models provide a reasonable approximation and offer tremendous practical value. The majority of docking calculations used in industry adopt this approximation. In reality, however, receptor atoms undergo constant thermal motions, and some proteins have significant plasticity. One example is inactive and active conformations of kinases in which the activation loop moves in and out of active sites (21) with some atoms that move well over 10 A. It is common to see a variety of conformational states for a given protein in PDB databank. It is also possible that such conformations observed crystallographically represent only a fraction of the transient dynamic states that may exist in solution. Although several methods (22-24) have been developed to account for protein flexibility, considerably more computational and experimental work will be required to create algorithms that can incorporate receptor flexibility accurately. In addition to receptor plasticity, another complicating factor is the difficulty to account accurately for the enthalpic and entropic contributions of discrete water molecules in the ligand bound and unbound states. Water molecules have been observed crystallographically to play important roles in mediating hydrogen bonds between protein atoms and between protein atoms and ligands. In most docking calculations, the water molecules are removed from binding sites, which assumes that to displace these water molecules by small molecules would give favorable entropic contribution. However, the entropic gain comes at the expense of enthalpy loss if the water molecules make strong hydrogen bonds to receptor atoms or mediate ligand-protein interactions. Several programs exist that attempt to include movable water molecules in docking calculations, however, often this results in a significant loss of computational efficiency.
Besides virtual screening of ligand libraries, docking is extremely useful for lead optimization. Successful docking can elucidate the important interactions between lead molecules and the target, explain important features of the structure activity relationships (SAR), and consequently help to design new molecules. Other than docking, useful receptor based VLS tools include MD simulation, MC simulation, and QM/MM calculation.
Ligand-Based Virtual Screening (2-D Similarity Search, 3-D Pharmacophore Search)
Another category of VLS methods is to identify ligands for targets that have unknown 3-dimensional (3-D) structures. Generally, these methods are called ligand-based VLS. The most simple and probably the most used searches involve a physical, chemical, or topological similarity search based on one or several active compounds. The similarity between two molecules can be defined in various ways. Two-dimensional (2-D) methods refer to those that only consider atom connection tables to describe molecular structures, because atom connection tables are best represented by 2-D pictures such as the ones in the top panel of Fig. 2. These methods consider topological relationships between atoms in a molecule regardless their actual 3-Dl coordinates. The simplicity of these methods allows computationally efficient comparisons. It can screen millions of molecules in seconds. Here, we outline a few representative examples.
1. Molecular 2-D fingerprints are a class of methods that use chemical substructures to characterize similarities between molecules. A fingerprint is a signature that encodes the chemical topological information of a small molecule. It is usually a long binary bit string in which each bit represents the presence 1 or absence 0 of a particular chemical substructure. For example, an ATP molecule has substructures such as a pyrimidine and imidazole heterocycle but not benzene; therefore, the bits that represent pyrimidine and imidazole would be set to “1” and the bit that represents benzene would be set “0” in the fingerprint bit string of ATP. Many variations in detailed definitions of 2-D fingerprints exist. In one straightforward way, molecular similarities are measured by the number of bits two compounds have in common.
2. Pharmacophores are defined as essential structural features in a molecule that are responsible for its binding activity, and they can also be used to generate 2-D fingerprints. Examples of commonly used pharmacophores are specific molecular fragments (i.e., an isopropyl group), hydrogen bond donors and acceptors, and negatively and positively charged atoms or groups. In two dimensions, the minimum number of bonds between two pharmacophores can be used as a distance metric. For example, the exocyclic amino group of ATP is located 3, 4, and 5 bonds away to the three acceptor nitrogens in the purine ring. This information is coded readily in a fingerprint bit string.
3. In addition to 2-D fingerprints, the topological relationships of atoms in a molecule can be explored in high feature dimensions and nonlinear fashion by other means, such as kernel methods (25), support vector machines (26), and self-organizing maps (27).
However, molecular recognition occurs in a 3-D world. It is well known that minor chemical modifications can alter the activity of a molecule drastically. On the other hand, very different chemical structures can have similar activity against the same target. The mere existence of particular pharmacophores and/or their topological relations are not sufficient for a molecule to be active against its target. To account for 3-D information, various VLS techniques based on activities of small molecules without receptor structures have been developed, such as 3-D quantitative structure activity relationship (QSAR) models. It is commonly accepted that a small molecule must exhibit a high degree of shape complementarity to bind to its protein target with high affinity. Finding the correct superposition of active ligand molecules is the key to a successful 3-D QSAR model. Of course, shape alone does not account for all contributions of small molecules. Other factors such as locations of hydrogen bond donors and acceptors, types of functional groups, electrostatic properties, and polarizabilities all play important roles for small-molecule activities. One attempt to capture this information is 3-D pharmacophore method. Like docking, many software programs exist to develop 3-D pharmacophore models, such as DISCO (28), Catalyst (28), GASP (28), PHASE (29), and many others (30, 31). Pharmacophore features are not mutually exclusive; for instance, a hydrogen bond acceptor (i.e., donating electron pair) can be negatively charged (e.g., side chain carboxylate from Asp or Glu). Most programs provide users with the flexibility to define their own pharmacophore features. The main merit of 3-D pharmacophore models is the ability to identify lead compounds that are diverse structurally.
Although software packages differ in how they construct pharmacophore models, they go through the following common steps: generate conformers, enumerate pharmacophore features, generate a hypothesis, and screen the ligand library. Usually, the active conformation of a small molecule is unknown without experimental evidence. A key component of a good pharmacophore method is to sample active conformations rapidly. Studies have shown that the active conformation of a small molecule may not necessarily be the same as the lowest energy one in the unbound state (32). Therefore, a set of possible conformers within a certain energy window (e.g., 15 kcal/mol above the lowest energy conformer) are collected and clustered. The feature-enumerating step is relatively straightforward. It identifies all the possible pharmacophore features in small molecules. Hypothesis generation is another challenging step in building pharmacophore models. In essence, this step involves finding a proper alignment of active molecules with a high degree of overlap in 3-D space in terms of their pharmacophore features. During this step, the spatial arrangements of pharmacophore features are identified so that they are common for all or a significant subset of active molecules. This step assumes that active molecules must satisfy a common pattern of pharmacophore features. However, it should not require a molecule to have all the features defined in a model to be active. The proper alignment of active molecules determines the quality of a 3-D pharmacophore model. Several algorithms have been developed to identify pharmacophore patterns (33-36) among the set of possible conformations for each active molecule. Pharmacophore hypothesis algorithms try to find and to rank the most probable pharmacophore models for known active molecules. It should also be noted that active molecules do not necessarily share a common pharmacophore pattern. Distinct binding modes have been observed in cocrystal structures (37). Moreover, different small molecules may bind to different binding sites of a receptor. Although a few algorithms exist to identify multiple pharmacophore patterns within a set of actives (29, 36), more robust methods are needed. The conformations of small molecules can be precomputed or sampled “on the fly.” Once a valid pharmacophore model is constructed, it can be used to screen millions of small molecules. This strategy was used successfully by Singh et al. to identify some potent and novel VLA-4 antagonists (38). Three-dimensional pharmacophore models are used commonly for GPCR systems because generally, these membrane-localized target structures are unknown. A three-point pharmacophore model was constructed for Urotensin II receptor (39) and was used to search Aventis compound collection. Of 500 virtual “hits,” 10 hits were active with the most potent compound exhibiting an IC50 of 400 nM. Recently, the first GPR30 specific agonist was discovered by 2-D and shape similarity in combination with a pharmacophore search (40).
Not only can active molecules be used in pharmacophore modeling, structures of inactive molecules can also provide valuable information. In common pharmacophore models, information provided by inactive compounds is captured in the concept of “excluded volume” (29, 41). An “excluded volume” is a region that is so close to the receptor that no ligand atoms are tolerated. “Excluded volume” can be obtained automatically or defined manually. The method relies on the assumption that the binding modes of the compound series are similar to each other. Although many examples from cocrystal structures and SAR have confirmed this assumption, many exceptions to this empirical observation exist. Chemical similarity does not always predict similarity in biologic activities. When active compounds consist of multiple scaffolds because of different modes of action, it is very challenging to develop pharmacophore models and to classify the actives into proper categories automatically. In addition, a compound that satisfies a good pharmacophore model is not guaranteed to have activity. For example, entropic contributions, solubility, cell permeability, and stability can also have important implications on a compound’s biologic activity.
Even though most pharmacophore models are built without receptor structures, binding site structures do help to construct proper pharmacophore models. The critical residues provide a complementary image of ligand pharmacophore field. This “negative image” can be converted to a ligand pharmacophore model. One advantage of receptor structure based pharmacophore models is the natural definition of “excluded volume.” The pharmacophore model derived from a receptor structure can be used not only to search small-molecule libraries directly, but also to guide docking calculations (42). Similarly, docking can help to derive receptor based pharmacophore models as well. For instance, docking active compounds can help to identify the important pharmacophore features. Submicromolar tRNA-guanine transglycosylase inhibitors were discovered by combinations of pharmacophore searching and docking (43). Pharmacophore models can also be used for de novo design (44, 45) and building 3-D QSAR models (29, 46).
Design of Small Molecule Inhibitors
VLS is a cost-effective and time-efficient approach to identify inhibitors for a given target from a small-molecule library. However, the hits from virtual screening usually are of low affinity and have properties that are not ideal for drug development or even for use as research tools (such as lacking cell permeability, potency, selectivity, and solubility). Furthermore, VLS is often performed using existing small-molecule libraries, which are biased heavily toward thoroughly explored regions of chemical space. An alternative and very important method to identify small-molecule inhibitors is through design. Two strategies are used in lead design. One strategy is based on known scaffolds or fragments from VLS, HTS, and fragment based screening to improve potency and desired pharmacological properties. The other strategy is de novo design, which generates small molecule leads from scratch. Figure 3 shows the relationships between these strategies and shows when they can be used appropriately.

Figure 3. Workflow of computational algorithms used to design lead molecules. The top panels indicate various sources of starting points on which ''lead'' design can be based. The middle panels illustrate the corresponding design strategies under the different starting conditions. The details of each method are described in the main text.
Scaffolds and Fragments-Based Lead Design
Computational methods offer ways to investigate chemical modifications automatically and rapidly that may be beneficial to the small-molecule binding. Based on a core fragment or scaffold of a hit, substituents are introduced to the core structure from a small fragment library to the core structure. The selection of substituents is biased toward favorable binding interactions. For each modification, a local energy optimization or full docking can be carried out. The new molecule is then evaluated based on a scoring function that measures interactions with the protein-binding site. The scoring function is similar to the one used in docking with necessary modifications to accommodate additional property requirements. The substituents can be added continuously until a given criteria, such as the number of iterations, desired score, or maximum size of a molecule, is satisfied. LUDI (47) by Accelrys, Rachel and LeapFrog by Tripos, and molecular evolution developed internally at GNF are examples of the method. Programs differ in the details of how molecular structures are modified, optimized, and ranked. This strategy has been used to improve activities of HIV protease inhibitors (47) and other molecules (48).
Besides obtaining scaffolds from VLS, experimental fragm ent-based screening by NMR and crystallography offers a good starting point to design novel small-molecule inhibitors. The key to this method is cocrystal structures of the target protein and low-molecular-weight compounds, typically limited to molecular weights of between 120-250 Da to allow for subsequent additions. Usually, these compounds have weak affinities (Kds = 10 μM-10 mmol/L). To explore modifications that have the potential to increase binding affinities, the aforementioned computational methods can be applied readily. For structures with a single small molecule in a complex, the small compound can be extended automatically to maximize its interaction with target protein. During the progressive modifications, the binding mode for the initial fragment can be altered. However, certain constraints are applied to avoid drastic changes to the binding modes unless substantial modifications are introduced to the original structure. When evidence exists that multiple ligands bind to different pockets of an extended binding site in one or multiple crystal structures, computational methods can be used to propose proper linkers or fuse-disjointed fragments to yield the synergistic effects. Gill et al. (49) have used this strategy to design P38a MAP kinase inhibitors based on hits with affinities in the millimolar range. For example, they identified a lead molecule with IC50 = 65 nM by extending the base fragment to acquire additional interactions, and also to achieve a 100-fold increasing in potency rapidly by cojoining two overlapping fragment hits for an indole-derived compound. The structures are depicted in Fig. 4. It should be emphasized that crystal structures are extremely important in fragment-based design iterations. Because fragment hits are usually small and possess low affinity for the targets, to predict the correct binding modes by docking is difficult. In addition, the modifications to the base fragment may alter its binding modes fundamentally.

Figure 4. Inhibitor design based on low-molecular-weight compounds. In the upper panel, the base fragment is shown in black and the added groups are shown in red. In the lower panel, a common core fragment for cojoining two fragments is shown in black and the different substituents are shown in red and light blue.
Hybrid Design Based on Known Inhibitors
Through many years of drug discovery efforts, pharmaceutical companies have synthesized millions of small-molecule inhibitors. A large amount of information has been accumulated on how these inhibitors bind to their protein targets and to the QSAR of these inhibitors. Hybrid design is an emerging technique that attempts to take full advantage of this information to design new inhibitors for existing or new protein targets (50). The essence of the hybrid design is to recombine known inhibitors for a particular target in a rational way to create new inhibitors for the same target or for a new target. In a manner analogous to the evolutionary recycling of protein domains, the hope for chemical evolution through hybrid design is to create new inhibitors by recombining pharmacophores derived from different classes of compounds. Hybrid design starts by using inhibitor-receptor binding information obtained experimentally from crystallography or computationally by docking to overlay multiple known ligands, and then to predict how to recombine inhibitor substructures to create new compounds. This strategy is illustrated in Fig. 5, in which a potent Aurora A kinase inhibitor can be thought of as a hybrid of two inhibitors developed originally for two other kinases. This strategy has also been applied successfully to design novel kinase inhibitors that bind to a specific inactive kinase conformation (51, 52). Gleevec is a Bcr-Abl inhibitor that has been demonstrated crystallographically to bind to the nucleotide binding cleft of Abl, which uses both the adenine binding region and an adjacent hydrophobic pocket created by the activation loop being in a unique inactive conformation (53). A hybrid strategy involved appending a 3-trifluoromethylbenzamide group to a known ATP binding site inhibitor (51). Of the hybrid compounds thus created, potent and selective inhibitors have been identified for Abl, c-Kit, p38, PDGFR, and Aurora kinases (52). Figure 5 shows how a benzamide group is attached to a quinazoline scaffold to create an inhibitor that binds to an inactive Aurora kinase conformation similar to that observed on Abl-Gleevec complex.
A computer program named BREED automates the process for hybrid design (54). The method imitates the common medicinal chemistry practice of joining fragments of two known ligands to generate a new inhibitor. The known active ligands are superimposed in their active conformation to identify all overlapping bonds, and the fragments on each side of each matching bond are swapped to generate a large set of novel inhibitors. This method has been demonstrated to have a high rate of success to identify novel inhibitors for HIV protease and protein kinases (54).

Figure 5. Illustration of hybrid design strategy for small-molecule inhibitors. The activation loops of the proteins are colored differently to indicate active (blue) and inactive (orange) states. The important fragments of inhibitors are colored blue and orange to highlight the hybrid design concept.
De Novo Design
Small molecule inhibitors can also be generated by de novo design without the prior knowledge of other active ligands. In essence, the procedures are similar to those mentioned previously. For example, LeapFrog can start without a given small molecule. It will select a core randomly and will grow a ligand from its fragment library. Although it is still rare for a computer program to generate practical and active molecules from scratch, reports of novel molecules from “raw” output of automated computer program exist. SkelGen (55) was able to generate small-molecules inhibitors for estrogen receptor binding sites without hints from known actives. Among the 17 highest scoring structures, 5 structures were active with 4 structures that posses novel structures. We have also implemented a de novo ligand design algorithm in our in-house software GModE using evolutionary algorithm. A molecule is represented by a unique genetic code that is subject to normal genetic operations such as mutation, addition, deletion, and translocation. The population of ligand molecules is evolved toward increasing fitness. Our retrospective studies have shown that the method can produce lead molecules that are extremely similar to experimental nanomolar inhibitors.
If the target binding site structure is unavailable, de novo design can be carried out based on known active molecules. This circumstance may occur because none of the known actives that pose desired properties or that pose novel inhibitors are sought for patentability. However, the known inhibitors provide a basis to construct 3-D pharmacophore and other models such as Comparative Molecular Field Analysis (CoMFA) that can provide a template to guide the computational algorithm to maximize the fitness of the molecule “population” toward a given model. For example, LeapFrog can use CoMFA models in place of target binding site structures. It is also straightforward to apply pharmacophore models to novel molecular design with and without starting fragments. Novel structures are chosen based on its fitness to the given model. Unlike de novo design with known binding site structures, it is important to minimize the possibilities for resulting molecules to have steric clashes with receptor atoms. Even though these programs have produced promising results, the major drawback for such automated computer algorithms is that the “raw” computer derived molecules often are undesirable synthetically or implausible chemically. Some programs (55) have implemented chemistry rules to guide the generation of new molecules, but it is still far from satisfactory. Moreover, because a drug molecule interacts with its intended target in a complex biologic environment, a good lead molecule is selected by not only its binding affinity to its intended target, but also other properties such as crucial physical chemical properties, ability to expand SAR, selectivity relative to undesired off targets such as cytochrome P450 enzymes, and ion channels such as HERG. All are important considerations to advance a molecule toward clinical development. Various computational methods have been developed to predict these properties; however, normally they are not integrated into automated de novo design program.
Hit and Lead Optimization
Computational algorithms and modeling have advanced significantly over the last two decades in terms of accuracy, efficiency, and ease of use. Many modeling procedures are automated; however, they are far from replacing the intuition and knowledge of an experienced computational chemist. Although automated programs are used widely in early stages of lead discovery with limited human intervention, lead optimization is a process in which a computational chemist makes most contributions out of his knowledge, insight, experience, and intuition. Before lead optimization, computational algorithms are designed to explore wide areas of chemical space and a wide range of activity data. During lead optimization, focus is placed on one or a few particular compound series. Small modifications are introduced to investigate local SAR and to improve other pharmacologic properties. Incremental improvements are often the objectives at this stage. Whereas the automated computational methods described previously are still important, they are not sufficiently precise to distinguish between modifications that result in 2kcal/mol changes in binding affinities. Manual intervention, such as visual inspection and manual docking from an experienced chemist, are often of great importance for this phase.
Structure Based
As we can observe from previous discussion, protein structures have played invaluable roles in small-molecule inhibitor selection and design. Currently, over 40,000 biomacromolecular structures are deposited in RCSB protein databank. Over 5000 new structures were deposited in 2006. The atomic level structural information provides an enormous wealth of mechanistic insights with regard to protein function. Approximately 6500 protein structures in the database are in complex with an inhibitor or substrate to provide chemists with important insights into the mechanism of ligand recognition. Because biologic systems are very diverse, each system requires specialized knowledge and treatment. Successful ligand design requires an intimate knowledge of all known inhibitors, which include how their activity and selectivity is changed as a function of chemical modification (SAR), rules for which interactions are required for high affinity binding, knowledge about the potential for conformational rearrangements, and insight into what is tractable synthetically. Success therefore requires the close collaboration of computational chemists, medicinal chemists, and structural biologists. Here, we will attempt only to describe a few common techniques that computational chemists use to optimize the activity of a small-molecule inhibitor.
Visual inspection is probably one of the oldest and most powerful methods in computer modeling. Large amounts of information can be acquired by looking at the 3-D structures of an inhibitor and a protein complex, such as important hydrogen bonds, hydrophobic contacts, and strong electrostatic interactions. It allows one to design a derivative to make stronger or additional hydrogen bonds, to explore unoccupied pockets, and even to alter the scaffold while maintaining important interactions. Of course, regular molecular modeling methods are used here to ensure the proposed molecules possess sensible conformations. In contrast to the automatic molecular generating programs mentioned previously, the modifications here are proposed directly by chemists; therefore, the molecules are tractable synthetically and often follow a particular experimentally derived SAR series. The iterative use of model building, synthesis, and biologic evaluation is the most powerful and efficient means to obtain small molecules with a desired biologic activity. When multiple cocrystal structures with different ligands exist, one can gain even more insights into conserved interactions. This insight can be very useful to identify key interactions and to help design new compounds to maximize possible key interactions. An algorithm developed by Deng et al. (56) uses a binary string to represent ligand protein interactions. By clustering the binary strings of different small-molecule binders, the authors were able to illustrate the similarity and diversity of small-molecule binding modes. This information can be used to select correct binding modes as well as to propose new molecules.
“Anchored docking” is another common technique used in lead optimization. Unlike automated docking, “anchor” points such as particular hydrogen bonding are specified by the user to limit the configuration space sampled by docking program. This limit is to ensure that the knowledge of a chemist about the system is enforced, because usually automated docking is sufficiently accurate to find correct binding modes consistently. The “anchor” points can be defined for a receptor, a small molecule, or both. The correct definition of “anchor” points largely relies on the users’ knowledge of the system.
Qsar Based
Even with amount of protein structures available, many interesting protein targets without 3-D structural information exist. GPCRs are typical examples. In this case, only SAR derived from the biologic assay can be used to guide optimization. Under these circumstances, QSAR has been an important tool. It is probably one of the earliest tools for computer aided drug design. Properly choosing descriptors for QSAR models is much like an art and requires a deep understanding of the system of interest. Most common descriptors are based on physical, chemical, geometric, and topological properties. Three-dimensional QSAR models may require active ligands to be aligned properly just like the pharmacophore model building. In fact, 3-D QSAR models can be generated directly by pharmacophore models, and many pharmacophore-generating programs provide this capability. One method developed early and widely used for 3-D QSAR is CoMFA.
Design of Selective Small Molecule Inhibitors
For a small-molecule inhibitor to be useful as a therapeutic drug or as a tool for chemical biology study, it must have specificity for its intended target. A small-molecule drug often causes side effects or toxicity if it also modulates other proteins in the body (i.e., off-targets). Nonselective inhibitors will confound the analysis of resulting phenotypes. Therefore, the selectivity is a very important feature when considering design and selection of small-molecule inhibitors. Because ~30,000 proteins are encoded in the human genome, which can possess multiple ligand binding sites and can be present in vastly different concentrations, finding specific inhibitors can be an extremely difficult task. Analyzing the sequence and structural differences between a targeted protein and its closely related homologs is an effective way to design selectivity rationally.
Targeting the Differences in the Binding Pocket
The binding affinity of a ligand is determined by its complementarity to the size, shape, and physicochemical properties of the protein-binding site. To design selective small-molecule inhibitors rationally, the 3-D structure of the target-binding site is required. In principle, the structure of a protein-binding pocket is determined by its amino acid sequence, and it is straightforward to identify unique amino acids by sequence alignment. Once the unique residues are identified, they can be analyzed in the context of small-molecule binding to determine the residues that are important for protein ligand interaction. The inhibitors can then be designed to target them specifically for selectivity. Taunton et al. have used this approach to design selective inhibitors for p90 ribosomal protein S6 kinases (RSKs) (57). They identified two key selective determinants for RSKs relative to other kinases: One determinant is a small “gatekeeper” The amino acid, and the other is a Cys in the glycine-rich loop. Targeting these two residues, they were able to make very potent and selective inhibitors for RSKs.
Although successful for RSKs, it is often very difficult to identify unique residues suitable for selectivity for all proteins in a family. In many cases, a mono-selective inhibitor among the protein family members is not feasible, so selectivity against a subset of the family members is desired. In this case, selectivity can be achieved by targeting the sequence differences in the binding sites between the target protein and the unwanted proteins. This strategy has been applied successfully to design selective inhibitors for targets within a closely related subfamily, such as p38 kinase α, β, γ, and δ isoforms (58); protein kinase CDK2, and CDK4 (59); and COX-1 and COX-2 (60).
For protein targets that are closely related, the binding sites may look the same and no sequence difference is around the binding pocket. Therefore, a small molecule that binds to this primary site has very little selectivity. In these cases, a commonly used strategy is to search for a secondary site near the primary binding pocket. If it is less conserved, selective inhibitors can be designed by targeting the secondary site. For example, it is very difficult to develop a selective protein tyrosine phosphatase-1 B (PTP-1B) inhibitor, but a crystal structure of PTP-1B in complex with bis-(para-phosphophenyl) methane reveals a secondary aryl phosphate-binding pocket adjacent to the active site that can be targeted to design selective PTP-1B inhibitors (61).
Many protein structures are highly dynamic, and induced-fit effects are well known in small-molecule and protein binding (21). Although two proteins can have very similar binding pockets in static form, they can have different plasticity. Although protein flexibility is one major obstacle to predict the binding mode and the affinity of a ligand accurately, it provides a structural basis to design selective inhibitors. Usually, the conformations of the active enzymes within a family are more alike, but the inactive conformations are more different. For example, the ATP binding pockets of protein kinases in active states are highly similar, but considerable conformational variability exists among kinases in the inactive state that can be exploited for selective inhibitor design. The unique “DFG-out” inactive conformation has been applied already to the design of selective kinase inhibitors, which includes the already approved drug Gleevec and Nexavar. Lapatinib, one of the most selective kinase inhibitors, achieves its high selectivity by binding to an inactive conformation of EGFR with a methionine in the a-C helix moved away from its normal position to accommodate the benzyloxy group of lapatinib (62). Exploiting differentially accessible conformational states among closely related targets has also been the structural basis for observed high selectivity of PI3δ kinase inhibits (63).

Figure 6. Flowchart of methods employed for designing selectivity of small-molecule inhibitors.
Engineering Selectivity Using
Chemical
Genetics
Identifying a selective small-molecule inhibitor can be time consuming. For a target in a large protein family, it is almost impossible to achieve monoselectivity. Inhibiting multiple targets by a small molecule leads to a significant complication in the analysis of target validation and studies of protein functions. To overcome the difficulty of identifying monoselective small-molecule inhibitors, a chemical genetic approach is applied to design as orthogonal receptor-ligand pairs (64, 65). In this approach, the targeted protein is mutated in the active site to create a structural distinction between the target and all other members of proteins in the same family. Then, a small molecule is designed to target the distinct structure so that it only binds the mutated protein. The first step is to study the binding mode of a small molecule to its target and identify an important interaction that is highly conserved for the ligand binding to all members in the protein family. Then, the ligand is modified in the conserved interaction region to disrupt the interaction so that it cannot bind effectively to any wild type protein, which includes the target. Two modifications are used commonly. One is to add a bulky substitute (a “bump”) at the interacting position to create steric clashes, and the other is to disrupt the critical hydrogen bonding between the ligand and the protein. The next step is to mutate the residues around the bump to smaller ones (which creates a “hole”) to accommodate the “bump” or to mutate residues to restore the critical hydrogen bonds. Although both approaches have introduced orthogonal protein-ligand pairs successfully, the “bump-hole” strategy is more suitable for design and is applicable to many different protein families. Shokat’s lab has successfully applied the “bump-hole” strategy to generate monospecific inhibitors for engineered kinases (64). The monospecific inhibitors have been used widely in kinase signaling pathway elucidation and target validation (66). Figure 6 outlines the strategies and workflows that we discussed for selective inhibitor design, in which the method details of each step in the flow chart have to be tailored towards specific systems.
Summary
In this review, we described briefly some common computational techniques to select and to design small-molecule inhibitors. It is not a comprehensive list of all methods available. Instead, we focused on basic methods that are used commonly in drug discovery research. Because of the size limitation, it is also impossible to discuss all the details in each method. We hope that this short review can provide some fundamental concepts used in virtual screening and rational design, and can provide the interested reader with an entry point to explore the field of computational inhibitor design and selection. Although we described the computational methods in a linear flow, the methods used in one stage of small-molecule inhibitor discovery process can be applied at different stages whenever appropriate. To choose the proper methods for different purposes at different stages is crucial for computational modeling to be effective. In addition, computational modeling should always interact tightly with experimental investigation in order to make significant contributions. VS and rational design is also an ever-changing field. New algorithms and strategies are emerging continuously. We believe that computational methods will play increasingly important roles in discovering new small-molecule inhibitors.
References
1. Shogren-Knaak MA, Alaimo PJ, Shokat KM. Ann. Rev. Cell Dev. Biol. 2001; 17:405-433.
2. Schreiber SL. Nat. Chem. Biol. 2005; 1:64-66.
3. Adcock SA, McCammon JA. Chem. Rev. 2006; 106:1589-1615.
4. Kuntz ID, Blaney JM, Oatley S, Langridge R, Ferrin TJ. Mol. Biol. 1982; 161:269-288.
5. Morriss GP, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson A. J. Comp. Chem. 1998;19:1639-1662.
6. Abagyan R, Totrov M, Kuznetsov DJ. Comp. Chem. 1994; 15:488-506.
7. Che J. Chem. Theory Comput. 2005; 1:634-642.
8. Hopkins SC, Vale RD, Kuntz ID. Biochemistry 2000; 39:2805-2814.
9. Vangrevelinghe E, Zimmermann K, Schoepfer J, Portmann R, Fabbro D. Furet PJ. Med. Chem. 2003; 46:2656-2662.
10. Peng H, Huang N, Qi J, Xie P, Xu C, Wang J, Yang C. Bioorg. Med. Chem. Lett. 2003; 13:3693-3699.
11. Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Science 2006; 312:443-446.
12. Trosset J, Dalvit C, Knapp S, Fasolini M, Veronesi M, Mantegani S, Gianellini LM, Catana C, Sundstro m M, Stouten PFW, Moll J. Proteins: Struct. Funct. Bioinfo. 2006; 64:60-67.
13. Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, Bar-Haim S, Warshaviak D, Fichman M, Noiman S. Proc. Nat. Sci.Acad. U.S.A. 2004; 101:11304-11309.
14. Salo OMH, Raitio KH, Savinainen JR, Nevalainen T, Lahtela-Kakkonen M, Laitinen JT, Jarvinen T, Poso AJ. Med. Chem. 2005; 48:7166-7171.
15. Muegge IJ. Med. Chem. 2006; 49:5895-5902.
16. Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RPJ. Comput. Aided Mol. Des. 1997;11.
17. Gohlke H, Hendlich M, Klebe GJ. Mol. Biol. 2000; 295:337-356.
18. Verdonk ML, Cole JC, Hartshorn M, Murry C, Taylor R. Proteins 2003; 52:609-623.
19. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DTJ. Med. Chem. 2006; 49:6177-6196.
20. Kontoyianni M, McClellan LM, Sokol GSJ. Med. Chem. 2004; 47:558-565.
21. Huse M, Kuriyan J. Cell 2002; 109:275-282.
22. Lorber D, Udo M, Shoichet BK. Protein Sci. 2002;11.
23. Kairys V, Gilson MKJ. Comp. Chem. 2002; 23:1656.
24. Sherman W, Day T, Jacobson MP, Friesner RA, Farid RJ. Med. Chem. 2006; 49:534-553.
25. Mahe P, Ueda N, Akutsu T, Perret J, Vert JJ. Chem. Inf. Model. 2005; 45:939-951.
26. Jorissen RN, Gilson MKJ. Chem. Inf. Model. 2005; 45:549-561.
27. Schneider G, Nettekoven MJ. Comb. Chem. 2003; 5:233-237.
28. Patel Y, Gillet VJ, Bravi G, Leach AR. J. Comp. Aided Molec. Design 2002; 16:653-681.
29. Dixon SL, Smondyrev AM, Rao SN. Chem. Biol. Drug Des. 2006; 67:370-372.
30. Handschun S, Wagener M, Gasteiger JJ. Chem. Inf. Comput. Sci. 1997; 38:220-232.
31. Holliday J, Willett PJ. Mol. Graph. Model. 1997; 15:203-253.
32. Nicklaus M, Wang S, Driscoll J, Milne G. Bioorg. Med. Chem. 1995; 3:337-469.
33. Jones G, Willett P, Glen RC. In: Pharmacophore Perception, Development, and Use in Drug Design. 2000. International University Line, La Jolla, CA. pp. 85-106.
34. Campos-Martinex J, Waldeck JR, Coalson RDJ. Chem. Phys. 1992; 96:3613.
35. Martin YC, Bures MG, Danaher EA, DeLazzer J, Lico I, Pavlik PA. J. Comp. Aided Molec. Design 1993; 7:83-102.
36. Feng J, Sanil A, Young SJ. Chem. Inf. Comput. Sci. 2006; 46:1352-1359.
37. Muchmore SW, Smith RA, Stewart AO, Cowart MD, Gomtsyan A, Matulenko MA, Yu H, Severin JM, Bhagwat SS, Lee CH, Kowaluk EA, Jarvis MFLJC. J. Med. Chem. 2006; 49:6726-6731.
38. Singh J, van Vlijmen H, Liao Y, Lee W, Cornebise M, Harris M, Shu I, Gill A, Cuervo JH, Abraham WM, Adams SP. J. Med. Chem. 2002; 45:2988-2993.
39. Flohr S, Kurz M, Kostenis E, Brkovich A, Fournier A, Klabunde TJ. Med. Chem. 2002; 45:1799-1805.
40. Bologa C, Revankar C, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Nat. Chem. Biol. 2006; 2:207-212.
41. Kurogi Y, Guner O. Curr. Med. Chem. 2001; 8:1035-1055.
42. Goto J, Kataoka R, Hirayama N. J. Med. Chem. 2004; 47:6804-6811.
43. Brenk R, Naerun L, Gradler U, Gerber H, Garcia G, Reuter K, Stubbs MT, Klebe G. J. Med. Chem. 2003; 46:1133-1143.
44. Tschinke V, Cohen N. J. Med. Chem. 1993; 36:3863-3870.
45. Bohm HJ. J. Comp. Aided Molec. Design 1994; 8:243-356.
46. McGregor MJ, Muskal SM. J. Med. Chem. 1999; 39:569-574.
47. Bohm H-J. J. Comp. Aided Mol. Design 1992; 6.
48. Pisabarro MT, Ortiz AZ, Palomer A, Cabre F, Garcia L, Wade RC, Gago F, Mauleon D, Carganico G. J. Med. Chem. 1994; 37:337-341.
49. Gill AL, Frederickson M, Cleasby A, Woodhead SJ, Carr MG, Woodhead AJ, Walker MT, Congreve MS, Devine LA, Tisi D, et al. J. Med. Chem. 2005; 48:414-426.
50. Lazar C, Kluczyk A, Kiyota T, Konishi Y. J. Med. Chem. 2004; 47:6973-6982.
51. Okram B, Nagle A, Adrian FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Xia G, Spraggon G, Warmuth M, Liu Y, Gray NS. Chem. Biol. 2006; 13:779-786.
52. Liu Y, Gray NS. Nat. Chem. Biol. 2006; 2:358-364.
53. Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Science 2000; 289:1938-1942.
54. Pierce AC, Rao G, Bemis GW. J Med. Chem. 2004; 47:2768-2775.
55. Firth-Clark S, Willems HMG, Williams A, Harris W. J. Chem. Inf. Comput. Sci. 2006; 46:642-647.
56. Deng Z, Chuaqui C, Singh J. J. Med. Chem. 2004; 47:337-344.
57. Cohen MS, Zhang C, Shokat KM, Taunton J. Science 2005; 308:1318-1321.
58. Wilson KP, McCaffrey PG, Hsiao K, Pazhanisamy S, Galullo V, Bemis GW, Fitzgibbon MJ, Caron PR, Murcko MA, Su MS. Chem. Biol. 1997; 4:423-431.
59. Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S, Morishima H. J Med. Chem. 2001; 44: 4628-4640.
60. Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. J. Biol. Chem. 1996; 271:15810-15814.
61. Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang ZY. Proc. Nat. Acad. Sci. U.S.A. 1997; 94:13420-13425.
62. Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, et al. Cancer Res. 2004; 64:6652-6659.
63. Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. Cell 2006; 125:733-747.
64. Bishop A, Buzko O, Heyeck-Dumas S, Jung I, Kraybill B, Liu Y, Shah K, Ulrich S, Witucki L, et al. Annu. Rev. Biophys. Biomol. Struct. 2000; 29:577-606.
65. Clemons PA, Gladstone BG, Seth A, Chao ED, Foley MA, Schreiber SL. Chem. Biol. 2002; 9:49-61.
66. Shokat K, Velleca M. Drug Discov. Today 2002; 7:872-879.
Further Reading
Brooijmans N, Kuntz I. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003; 32:335-373.
Tollenaera J, De Winter H, Langenaeker W, Bultinck P, eds. Computational Medicinal Chemistry and Drug Discovery. 2004. Marcel Dekker, New York.
See Also
Computation and Modeling of Molecular Recognition
Intermolecular Interactions in Molecular Recognition
Mechanisms of Enzyme Inhibition
Protein-Ligand Interactions
Structural and Functional Aspects of Molecular Recognition