CHEMICAL BIOLOGY
Chemistry of DNA Damage Repair
Erin J. Heenan and Aziz Sancar, university of North Carolina School of Medicine, Chapel Hill, North Carolina
doi: 10.1002/9780470048672.wecb131
DNA repair is the elimination of covalent DNA modifications and the correction of base mismatches. There are six basic repair categories: direct repair, base excision repair, nucleotide excision repair, recombination, cross-link repair, and mismatch repair.
Direct Repair
In direct repair the chemical modification that constitutes the lesions is reversed without removing and replacing nucleotides. There are four known direct repair enzymes: photolyase, spore photoproduct lyase, methylguanine DNA methyltransferase, and AlkB family oxidative demethylases (1).
Photolyase. Photolyase harnesses blue light (400-500 nm) energy to repair ultraviolet (UV) (200-300 nm)-induced DNA damage. Photolyase is a 55-65-kDa flavoprotein that contains two noncovalently bound chromophores (2, 3). These chromophores are a flavin in the form of FADH- and a pterin in the form of methenyltetrahydrofolate (5,10-MTHF) in most photolyases and in rare cases 8-hydroxydeazariboflavin. There are two types of photolyases: cyclobutane photolyase, which repairs cyclobutane pyrimidine dimers (Pyr<>Pyr), and 6-4 photolyase, which repairs pyrimidine 6-4 pyrimidone photoproducts. The structures and mechanisms of action of both types of enzymes are similar (2). They bind damaged DNA in a light-independent reaction and carry out catalysis by light-initiated cyclic electron transfer. In cyclobutane photolyase, MTHF, which has a high extinction coefficient that absorbs a blue light photon, transfers the excitation energy to FADH- in the active site. The excited state FADH-* transfers an electron to Pyr<>Pyr to generate a radical pair, FADH° and Pyr<>Pyr-°. The cyclobutane radical undergoes bond rearrangement to yield two canonical pyrimidines concomitant with back electron transfer to regenerate the catalytically competent FADH-. Then the enzyme dissociates from the repaired DNA (Fig. 1). The (6-4) photolyase acts by a similar mechanism except that the (6-4) photoproduct is first converted to a four-membered oxetane ring thermally (1, 3) (Fig. 1). Although humans and other placental mammals do not possess photolyase, they do possess two proteins with a high degree of structural similarity to photolyase but no repair activity (1). These proteins, named cryptochrome 1 and 2, are essential for the regulation of the circadian clock (4, 5).
Spore Photoproduct Lyase. The DNA in spores is in A-form because of dehydration. As a consequence, when spores are exposed to UV, the stereochemistry of the bases is not conducive to the formation of cyclobutane pyrimidine dimers or (6-4) photoproducts. Instead, UV induces the formation of 5-thyminyl-5,6-dihydrothymine or spore photoproduct (SP). This lesion is repaired by a 40-kDa enzyme called SP lyase. The enzyme is an iron-sulfur [4Fe-4S] protein that employs S-adenosyl-methionine (AdoMet) as a catalytic cofactor and carries out repair by a radical mechanism (6) (Fig. 2). In this mechanism, the reduced [Fe-S] center cleaves AdoMet to generate a 5'-deoxyadenosyl radical intermediate and methionine. The radical then abstracts an H-atom from C-6 of the SP. The resulting substrate radical undergoes bond cleavage to generate a product radical. The latter abstracts an H-atom from the 5'deoxyadenosine to form canonical thymines and a 5'deoxyadenosyl radical. Finally, the catalytic cycle is closed by electron transfer back to the [Fe-S] cluster concomitant with the formation of AdoMet (6).
Methylguanine DNA Methyltransferase. Methylguanine DNA methyltransferase (MGMT), which repairs the O6methylguanine and O4methylthymine, is a 20-30-kDa protein that has no cofactors and is ubiquitous in nature (1). It recognizes DNA damage either by three-dimensional diffusion or, as recently claimed, by two-dimensional diffusion along the duplex with 3' to 5' scanning bias, and it forms a low stability complex with the backbone of the DNA at the active site (7). It then flips out the O6methylguanine base into its active site (7). The methyl group is then transferred to a nucleophilic cysteine residue in the active site in a SN2 manner (8). The repair reaction generates methyl-cysteine and inactivates the enzyme (Fig. 3) (9-11). The protein cannot be regenerated and is degraded after acting in the reaction. In the reaction the enzyme acts as an alkyl transfer reagent and not as a true enzyme (12). The MGMT encoded by the ada gene is largely responsible for the so-called “Adaptive Response” in Escherichia coli (13).
AlkB Family Oxidative Demethylases. These enzymes repair 3-methylcytosine and 1-methyladenine that are introduced by SN2-type methylating agents. They use molecular oxygen, 2-oxoglutarate, and Fe(II) to oxidize the methyl group and release it in the form of formaldehyde. The enzyme is also capable of repairing RNA. It has been shown that the human homologues of AlkB, ABH2 and ABH3, prefer dsDNA and ss-DNA, respectively, and that in the case of RNA, ABH2 prefers double-stranded substrates, whereas ABH3 prefers singlestranded substrates. AlkB is widespread in nature, and at least eight genes encoding the enzyme have been identified in humans (Fig. 4) (14).
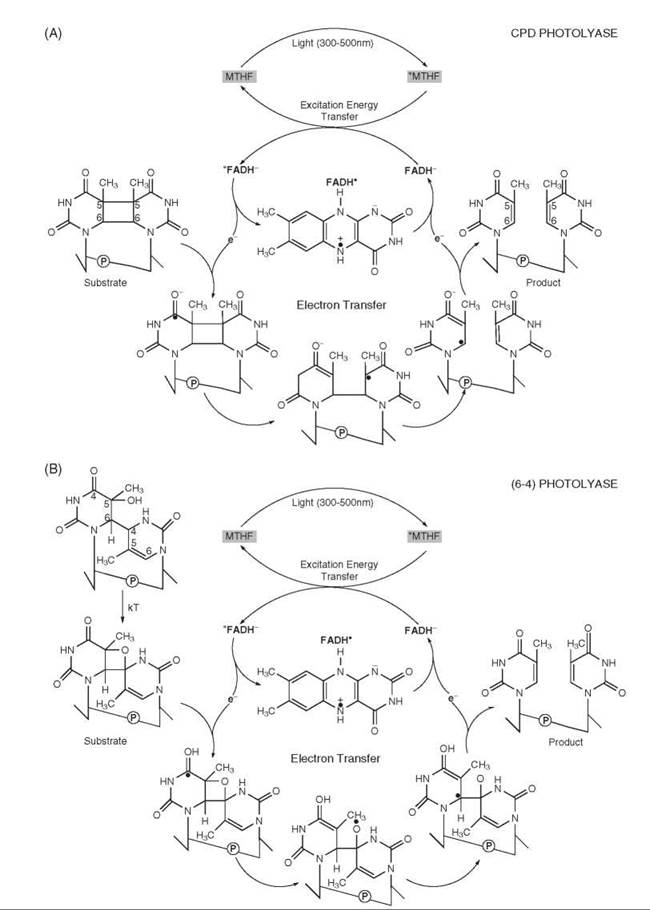
Figure 1. Reaction mechanism of DNA photolyases: (A) mechanism of cyclobutane photolyase and (B) mechanism of (6-4) photolyase. Both photolyases harness blue light energy to remove UV-induced damage and contain two noncovalently bound chromophores. They bind UV-damaged DNA in a reaction that is light independent and carry out catalysis in a light-initiated cyclic electron transfer. In (6-4) photolyase, the (6-4) photoproduct is converted to a four-membered oxetane ring thermally (kT) before the photochemical reaction.
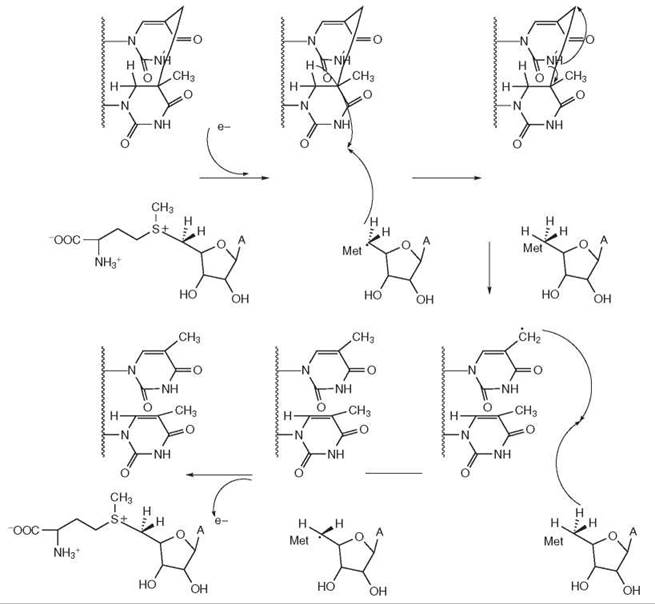
Figure 2. Reaction mechanism of spore photoproduct lyase. Spore photoproduct lyase removes the UV damage spore photoproduct. The enzyme is an iron sulfur protein that uses AdoMet as a cofactor and repairs the damage through a radical mechanism.
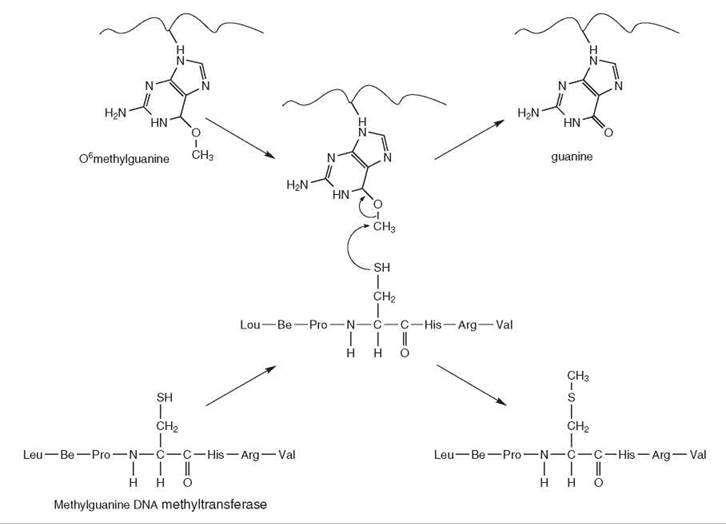
Figure 3. Repair of O6methylguanine by methylguanine DNA methyltransferase. Methylguanine DNA methyl transferase has no cofactors and recognizes DNA cooperatively. It flips the O6methylguanine into its active site and transfers the methyl group to a nucleophilic cysteine in an SN2 reaction.
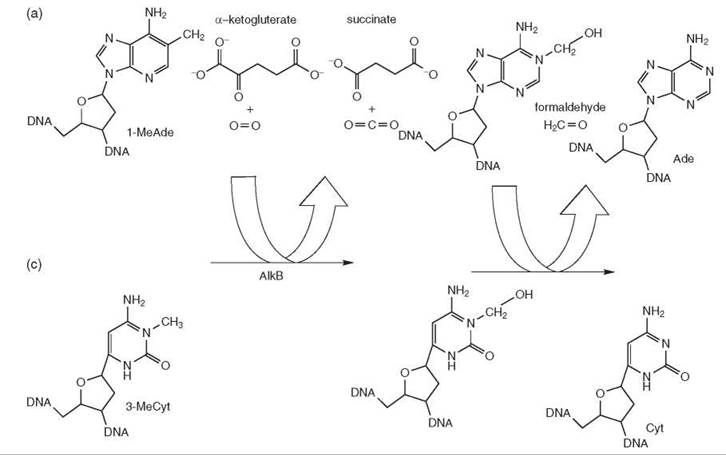
Figure 4. Direct repair through oxidative demethylation by the AlkB protein. AlkB uses molecular oxygen, 2-oxoglutarate, and Fe(II) to oxidize the methyl group and then releases it as formaldehyde.
Base Excision Repair
In base excision repair, the damaged nucleotide is removed in two steps. First, a glycosylase cleaves the glycosidic bond of the damaged base and releases it; then a combination of AP lyase/AP endonucleases cleaves the phosphodiester bonds on either side of the resulting abasic site (AP = apurinic/apyrimidinic site) to release the deoxyribose. The resulting gap is filled in by a polymerase and sealed by DNA ligase. Base excision repair as a general rule works on non-bulky DNA lesions that do not cause gross structural alterations in the duplex (15).
Some glycosylases are simple glycosylases, which only catalyze the hydrolytic removal of a base to form an abasic site, whereas other glycosylases cleave off the base using a lyase mechanism and catalyze an AP lyase reaction. In this AP lyase reaction, a Schiff’s base is formed from the 1' aldehyde of the AP deoxyribose. This labilizes the 2' hydrogen, resulting in a β-elimination reaction, and leaving a 5'-phosphomonoester and a 3' unsaturated sugar phosphate residue. Lyase reactions are usually associated with glycosylases that remove oxidized bases, but not with glycosylases that remove alkylated or normal bases (15).
Depending on the initial events in the removal of the damage, in humans repair may proceed by two possible mechanisms: short patch repair or long patch repair. In short patch repair, the repair patch is a single base; in long patch repair, it is 2-10 nucleotides long. In the short patch monofunctional glycosylase pathway, APE1 makes a 5' incision at an AP site and the 5'-baseless sugar phosphate is trimmed by the dRP lyase activity of Polβ to generate a ligatable 5' end, which is ligated by the Ligase III /XRCC1 complex. Long patch repair occurs when hydrolytic glycosylases or spontaneous base loss generates the AP site. In long patch repair, APE1 cleaves the 5' phosphodiester bond, and then repair synthesis and nick translation are carried out by DNA Polymerase δ or ε aided by RFC/PCNA clamp loader/polymerase clamp. DNA synthesis displaces several nucleotides. The flap that is generated is cleaved off by the Fen1 endonuclease and the repaired DNA is ligated by Ligase I (1, 16, 17) (Fig. 5).
The damage recognition mechanism of DNA glycosylases is similar to the mechanism of recognition of DNA photolyase. The initial recognition by DNA glycosylases is by diffusion. The distortions and H-bond changes that occur as a result of DNA damage are recognized with a low affinity/specificity. This recognition is followed by base-flipping, which forms a high affinity/specificity complex (16, 18). The glycosylic bond is then attacked by the active site residues. To achieve specificity, the DNA glycosylases employ multiple proofreading mechanisms, including initial recognition of the helix deformity, base shape and charge, base flipping, and the chemical step. Despite these safeguards, many glycosylases, especially alkylbase DNA glycosylases, release all four normal bases at low, but measurable and biologically relevant, rates (19).
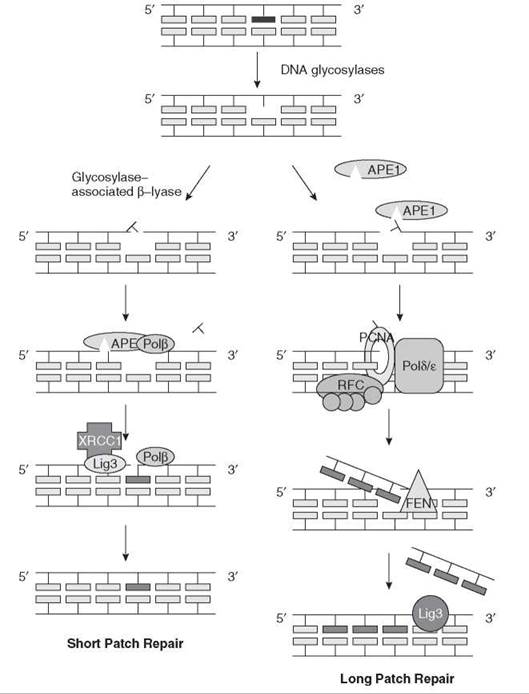
Figure 5. Base excision repair in mammalian cells. In base excision repair, the damaged nucleotide is removed in a two-step process. A glycosylase cleaves the glycosidic bond of the damaged base, releasing it, and then AP lyase and AP endonuclease cleave the phosphodiester bonds on either side of the abasic site, which releases the deoxyribose.
Nucleotide Excision Repair
Nucleotide excision repair is the primary repair system for the removal of DNA damage caused by chemicals that produce bulky adducts or by the UV component of sunlight, which produces cyclobutane pyrimidine dimers (Pyr<>Pyr) and (6-4) photoproducts in DNA (1). These lesions, as well as other bulky lesions induced by chemical carcinogens, are removed by the universally conserved repair mechanism called nucleotide excision repair. Nucleotide excision repair can be used to repair almost all single base lesions in DNA. As a result, it recognizes an extremely diverse substrate range, and rather than recognizing the specific damage itself, the excision nuclease tends to recognize the backbone conformations created by the damage (1).
The basic steps of nucleotide excision repair are as follows: 1) damage recognition, 2) dual incisions, 3) release of the excised oligomer, 4) repair synthesis to fill in the gap, and 5) ligation. In E. coli, nucleotide excision repair is carried out by three proteins, UvrA, UvrB, and UvrC, which excise a 12-13 nucleotide-long oligomer containing the damaged base. A (UvrA)2(UvrB)1 complex traces along the DNA backbone until UvrA recognizes a damaged base. The complex then binds to the damaged base. This binding activates the helicase function of UvrB, which unwinds the DNA around the damage (about 5 bp on either side of the damage). The resulting single stranded DNA leads to a tighter binding of UvrB to the single-stranded DNA and a dissociation of the UvrA from the tight UvrB-DNA complex. Therefore, UvrA is referred to as the “Molecular Matchmaker” (1). The resulting UvrB-damaged DNA complex then recruits UvrC, which initiates the dual incisions of the DNA. The 3' incision is performed first at the fourth or fifth phosphodiester bond to the lesion by the GIY-YIC endonuclease domain of UvrC. In a concerted but asynchronous reaction, the second active site in the C-terminal half of UvrC makes the 5' incision. Helicase II (UvrD) releases the 12-13 nucleotide-long oligomer carrying the lesion along with UvrC. The dissociation of UvrC leaves a free hydroxyl group at the 5' incision. DNA Pol I binds to this hydroxyl group and fills in the gap while releasing UvrB. DNA ligase then completes the repair reaction (Fig. 6) (20).
In humans, excision repair is carried out by six factors: RPA, XPA, XPC, TFIIH, XPG, and XPF-ERCC1, which are comprised of 15 proteins. These act in a coordinated manner to make dual incisions bracketing the lesion and remove the damage in 24-32 nucleotide-long oligomers (13). Damage is recognized by RPA, XPA, and XPC acting in a cooperative manner, followed by recruitment of TFIIH by XPA and XPC. The resulting complex is called pre-incision complex 1 (PIC1). Within this complex, the DNA is unwound by about 25 bp around the damage site by the XPB and XPD helicase subunits of TFIIH in a reaction that requires ATP hydrolysis. Then, XPC is displaced concurrent with binding of the XPG nuclease to form PIC2; finally XPF-ERCC1 is recruited to form PIC3. In PIC3, XPG makes the 3' incision at the 6th ± 3 phosphodiester bond and XPF makes the 5' incision at the 20th ± 5 phosphodiester bond and the excised 24-32 nucleotide-oligomer is released and the complex dissociates. The excision gap is filled in by DNA polymerases and then ligated (21, 22) (Fig. 6).
Both the prokaryotic and the eukaryotic excision nucleases employ thermodynamic discrimination and kinetic proofreading to achieve high specificity. However, despite these elaborate discriminatory mechanisms, excision nucleases do not have absolute specificity and “gratuitous repair” of undamaged DNA occurs at a low but potentially deleterious level (23-25).
Finally, in addition to nucleotide excision repair and base excision repair, an alternative repair pathway exists to remove Pyr<>Pyr and (6-4) photoproducts in some organisms, including Schizosaccharomyces pombe. In this alternative pathway, an enzyme called UV dimer endonuclease cleaves DNA immediately 5' to the damaged lesion. The damage is then removed by a 5' to 3' exonuclease (26, 27). Additionally, AP endonucleases, like Nfo in E. coli and APE1 in humans, have the ability to incise immediately 5' to nonbulky lesions caused by oxidative damage and initiate excision of the lesion by a 5' to 3' exonuclease (28).
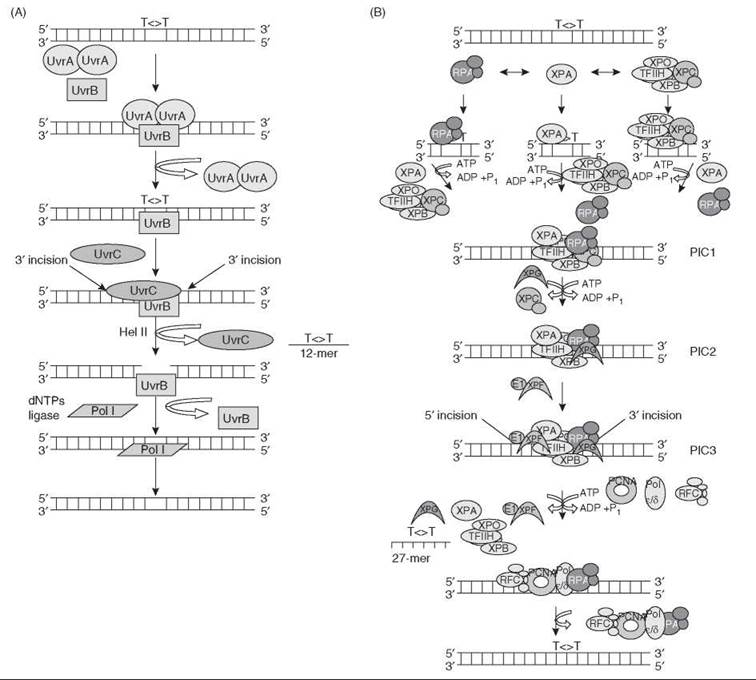
Figure 6. Nucleotide excision repair in: (A) E. coli and (B) humans. There are five basic steps of nucleotide excision repair: (1) damage recognition, (2) dual incisions, (3) release of the excised oligomer, (4) repair synthesis to fill in the gap, and (5) ligation.
Double-Strand Break Repair and Recombinational Repair
Double-strand breaks occur naturally in V(D)J recombination to generate antibodies and during the immunoglobulin class switching reaction, but they also occur improperly during replication as a result of the replication fork stalling and collapsing. Double-strand breaks may also be caused by ionizing radiation, chemicals, and reactive oxygen species. Homologous recombination (HR) and non-homologous end joining (NHEJ) are the two mechanisms used to repair double-strand breaks (1).
Homologous Recombination (HR)
In homologous recombination, an intact homologous duplex is used to retrieve information and fix the duplex with strand breaks. The three basic steps in homologous recombination are strand invasion, branch migration, and Holliday junction formation (Fig. 7). Strand invasion and branch migration are carried out by RecA in prokaryotes (29) and Rad51 in eukaryotes (30). The Rad51 paralogs (XRCC2, XRCC3, Rad51B, Rad51 C, and Rad51D) along with BRCA2 are considered the mediators during homologous recombination in mammalian cells. Before the initiation of strand invasion by Rad51, the Mre11/Rad50/NSB1 (MRN) complex processes the termini of double-strand break to facilitate strand invasion. After strand invasion and branch migration, the two duplexes are linked through the Holliday junction, which is the key intermediate of homologous recombination. In the Holliday junction, the two strands that are being recombined are joined together covalently by single-strand crossovers. A heterodimeric resolvase, Mus81/Mms4, then cleaves the Holliday junction to separate the two duplexes (31) (Fig. 7). A key feature of HR is that any information that is lost by double-strand break is retrieved using the information from a sister chromatid or homologous chromosome.
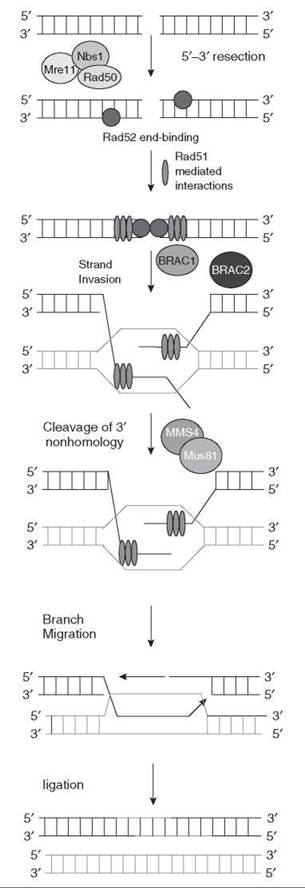
Figure 7. Repair of the double-strand break by homologous recombination in mammalian cells. In homologous recombination, an intact homologous chromosome is used to retrieve information and repair double-strand breaks in the duplex. The three basic steps of homologous recombination are strand invasion, branch migration, and Holliday junction formation.
Nonhomologous End Joining
In NHEJ in eukaryotes, the Ku heterodimer binds to both ends of the double-strand break (32) and recruits DNA-PKcs and the ligase IV-XRCC4 heterodimer. These then ligate both ends of the double-strand break regardless of whether they actually come from the same chromosome (33). In NHEJ, Artemis is the end-processing nuclease (34) (Fig. 8).
HR is important for the rescue of collapsed replication forks. NHEJ is the physiological recombination mechanism in V(D)J recombination and seems to be the major pathway of the repair of double-stranded breaks induced by ionizing radiation and radiomimetic agents in mammals.
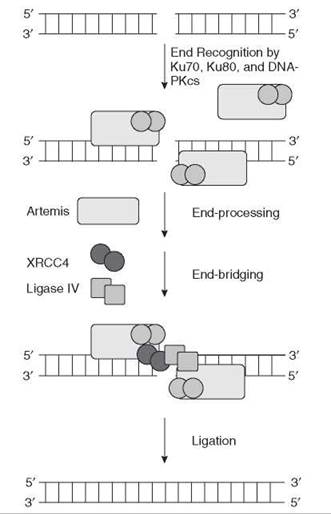
Figure 8. Repair of double-strand breaks by nonhomologous end joining in mammalian cells. In nonhomologous end joining, the ends of the double-strand break are brought together by DNA-PKcs (light blue rectangle) in conjunction with the Ku70-Ku80 (dark blue circles) heterodimer. After end processing by the Artemis nuclease and some DNA polymerases, the ends are ligated by the XRCC4-Ligase IV complex.
Cross-Link Repair
Chemotheraputic drugs such as cisplatin, psoralen, and mitomycin C and other DNA-damaging agents cause interstrand DNA cross-links. These lesions pose unique problems for the cellular machinery because both base excision repair and nucleotide excision repair rely on the redundant information in the undamaged strand to correctly fill in the gap resulting from the damage removal. However, in cross-linked DNA, both strands are damaged and the genetic information is therefore lost. The cell overcomes this difficulty by using multiple repair systems to repair the damage. Nucleotide excision repair and homologous recombination work either coordinately or in tandem to remove these lesions in E. coli and Sacchharomyces cerevisiae (35, 36), and the mechanism of removal of cross-links in mammalian cells is not well understood.
E. coli overcomes the dilemma of cross-link repair by combining nucleotide excision repair with homologous recombination. First, dual incisions are made in one strand by the Uvr(A)BC excision nuclease. Second, through homologous pairing of the incised duplex with the intact sister chromosome, a segment of the intact duplex is paired with the gap, generating a three-strand noncovalent intermediate. The transferred strand is ligated to form a three-stranded covalent intermediate. Third, the “excised oligomer” remaining linked to the duplex through the cross-link is recognized as a bulky adduct and is excised by dual incisions. The gaps generated by repair and recombination in the damaged and sister duplex, respectively, are filled in by DNA polymerases and the newly synthesized DNA is ligated (35) (Fig. 9). Genetic evidence indicates that the yeast S. cerevisiae also employs combinations of nucleotide excision repair and recombination to eliminate cross-links (37, 38).
The role of nucleotide excision repair in cross-link repair in humans is not currently clear. Replication fork collapse due to cross-links can induce double-strand breaks during replication both in vivo and in vitro (39). Mutations in XPF and ERCC1 render mammalian cells extremely vulnerable to DNA cross-linking agents (40). This has led to the conclusion that XPF●ERCCl, which is a structure-specific endonuclease, plays a significant role in cross-link repair that is distinct from its role in nucleotide excision repair. At the double-strand break induced by replication, XPF●ERCCl degrades one of the cross-linked strands 3' to 5' though the cross-link (38). Recombination proteins, which include Rad51, Rad52, XRCC2, XRCC3, and RPA then act to generate a Holliday junction intermediate, which would be resolved as described previously (40).
It has been found that patients with Fanconi’s anemia (FA) are extremely susceptible to cross-links. Evidence has been found that suggests that the FA complex (which is composed of FANC-A, C, E, and F, 4 of the 12 Fanconi anemia proteins) along with the M/R/N complex and BRAC1/BRAC2 use homologous recombination to participate in both cross-link repair and double-strand break repair (41).
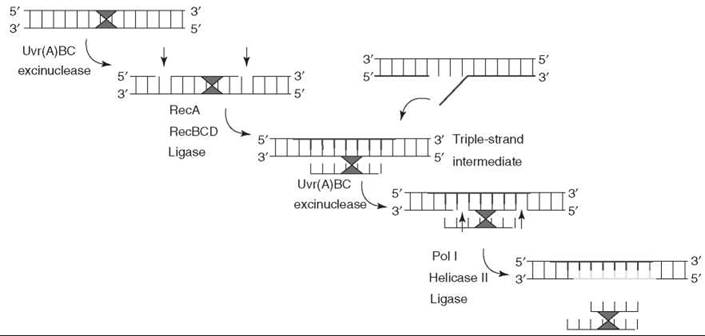
Figure 9. Cross-link repair in E. coli. In cross-link repair, nucleotide excision repair and homologous recombination work coordinately or in tandem to remove cross-links.
Mismatch Repair
Mismatch repair removes mismatched bases from newly synthesized DNA. The general strategy of mismatch repair follows. The repair reaction is usually coupled to DNA replication and corrects errors introduced by DNA polymerases in the newly synthesized strand. The mismatch is recognized by a mismatch-specific protein, and the mismatched nucleotide in the newly synthesized strand is removed by an exonuclease that initiates hydrolyses of the newly synthesized strand at a nick that may be located up to 1000 nucleotides away from the mismatch and that may be 3' or 5' to the mismatch. Exonucleolytic degredation of DNA past the mismatch is coupled to DNA synthesis to replace the excised DNA and substitute the correct nucleotide in place of the mismatched one. The mismatch repair system is evolutionarily conserved among organisms ranging from E. coli to humans. The mismatch is recognized by MutS in E. coli and by MutSa and MutS|8 in humans (heterodimers of MutS paralogs MSH2 and MSH6 and MSH2 and MSH3, respectively). The strand-discriminating nick is introduced by MutH in E. coli. This enzyme recognizes the transiently hemimethylated GATC sequence in the newly synthesized strand. This sequence is methylated by deoxyadenine methylase (Dam) at the A residue, and shortly after synthesis, the newly synthesized DNA is transiently unmethylated. In a hemimethylated GATC site, MutH makes a nick at the 5' side of G in the unmethylated GATC. In humans there is no methyl-directed mismatch repair and the enzyme responsible for introducing nicks is not known (42, 43). It is thought that the terminus of the elongating leading strand and the termini of the Okazaki fragments of the lagging strand may constitute sites of entry of the mismatch removing exonuclease. In both E. coli and in humans, a protein called MutL provides communication between the nick and the mismatch and helps direct the exonucleolytic degradation from the nick to the mismatch. MutL is a homodimer in E. coli , and there are two heterodimers of three MutL homologs in humans (MutLα = MLH1 plus PMS2 and MutSβ = MLH1 plus PMS1). The DNA located between the nick and the mismatch is removed by 3' to 5' or 5' to 3' exonucleases depending on whether the nick is 3' or 5' to the mismatch. The gap is then filled by DNA Pol III in E. coli (Fig. 10) by DNA Pol δ in humans and is ligated. Mismatch repair increases the spontaneous mutation rate in E. coli and yeast. Humans defective in mismatch repair exhibit a cancer-prone syndrome called hereditary nonpolyposis colon cancer (HNPCC), and sporadic mutations of mismatch repair genes are also associated with up to 50% of all human cancers (42-44).
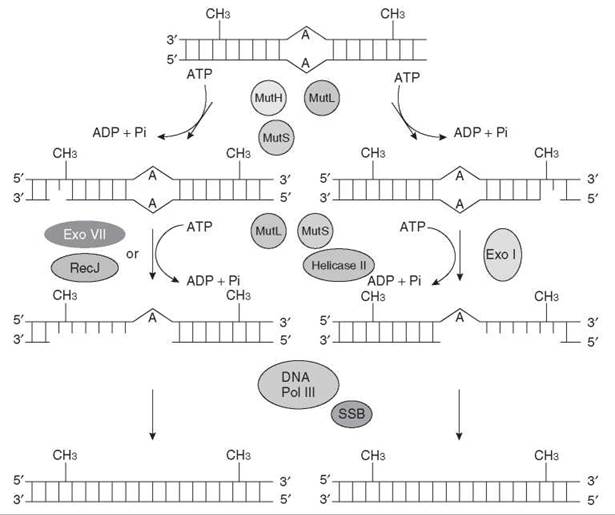
Figure 10. Methyl-directed mismatch repair in E. coli. The mismatch repair reaction is usually coupled to DNA replication and serves to correct errors in the newly synthesized strand that are introduced by DNA polymerase.
References
1. Sancar A, Lindsey-Boltz LA, Unsal-KaAcmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004; 73:39-85
2. Sancar A. Structure and function of DNA photolyase ad cryptochrome blue-light photoreceptors. Chem. Rev. Rom. 2003; 103(6):2203-2238.
3. Kao Y, Saxena C, Wang L, Sancar A, Zhong D. Direct observation of thymine dimer repair in DNA by photolyase. Proc. Natl. Acad. Sci. U.S.A. 2005; 102(45):16126-16132
4. Sancar A. Regulation of the mammalian circadian clock by cryptochrome. J. Biol. Chem. 2004; 279(33):34079-34082.
5. Gauger MA, Sancar A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005; 65(15):6828-6834.
6. Buis JM, Cheek J, Kalliri E, Broderick JB. Characterization of an active spore photoproduct lyase, a DNA repair enzyme in the radical S-Adenosylmethionine Superfamily. J. Biol Chem. 2006; 281;25994-26003.
7. Daniels DS. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nature Struct. Molec. Biol. 2004; 11:714-720.
8. Lindahl T, Sedgwick B, Sekiguchi M, Nakabeppu Y. Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem. 1988; 57:133-157.
9. Begley TJ, Samson LD. AlkB mystery solved: oxidative demethylation of N1-methyladenine and N3-methylcytosine adducts by a direct reversal mechanism. Trends Biochem. Sci. 2003; 28(1):2-5.
10. Pegg A, Byers T. Repair of DNA containing 06-alkylguanine. FASEB J. 1992; 6(6):2302-2310.
11. Kawate H, Sakumi K, Tsuzuki T, Nakatsuru Y, Ishikawa T. Separation of killing and tumorigenic effects of an alkylating agent in mice defective in two of the DNA repair genes. Proc. Natl. Acad. Sci. U.S.A. 1998; 95(9):5116-5120.
12. Mishina Y, Duguid EM, He C. Direct reversal of DNA alkylation damage. Chem. Rev. 2006; 106:215-232.
13. Landing P, Volkert MR. Regulatory responses of the adaptive response to alkylation damage: a simple regulon with complex regulatory features. J. Bacteriol. 2000; 182(23):6543-6549.
14. Falnes PO, Bj0ras M, Aas PA, Sundheim O, Seeberg E. Substrate specificities of bacterial and human AlkB proteins. Nucleic Acids Res. 2004; 32(11):3456-3461.
15. McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu. Rev. Biochem. 1999; 68:255-285.
16. Roberts RJ, Cheng X. Base flipping. Annu. Rev. Biochem. 1998; 67:181-198.
17. Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996; 271(16):9573-9578.
18. Mol CD, Parikh SS, Putnam CD, Lo TP, Tainer JA. DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu. Rev. Biophys. Biomol. Struc. 1999; 28:101-128.
19. Berdal KG, Johansen RF, Seeberg E. Release of normal bases from intact DNA by a native DNA repair enzyme. EMBO J. 1998; 17(2):363-367.
20. Petit C, Sancar A. Nucleotide excision repair: from E. Coli to man. Biochimie 1999; 81(1-2):15-25.
21. Reardon JT, Sancar A. Nucleotide excision repair. Progress Nucl. Acid Res. Mol. Biol. 2005; 79:183-235.
22. Reardon JT, Sancar A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003; 17(20):2539-2551.
23. Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Nat. Acad. Sci. U.S.A. 1974; 71(10):4135-4139.
24. Branum ME, Reardon JT, Sancar A. DNA repair excision nuclease attacks undamaged DNA. A potential source of spontaneous mutations. J. Biol. Chem. 2001; 276(27):25421-426.
25. Sancar A. DNA excision repair. Annu. Review. Biochem. 1996; 65:43-81.
26. Alleva JL, Zuo S, Hurwitz J, Doetsch PW. In vitro reconstitution of the Schizosaccharomyces pombe alternative excision repair pathway. Biochemistry 2000; 39(10):2659-2666.
27. McCready SJ, Osman F, Yasui A. Repair of UV damage in the fission yeast Schizosaccharomyces pombe. Mutat. Res. 2000; 451(3):197-210.
28. Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damge. Nature. 2002; 415(6868):183-187
29. Kowalczkowski SC. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 2000; 25(4):156-165.
30. Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast Rad51 protein. Science 1994; 265(5176):1241-1243.
31. Chen YB, Melchionaa R. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol Cell 2001; 8(5):1117-1127.
32. Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 1993; 72(1):131-142.
33. Ramsden DA, Gellert M. Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J. 1998; 17(2):609-614.
34. Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, Koiwai O, Hsieh CL, Schwarz K, Lieber MR. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 2004; 6:701-713.
35. Van Houten B, Gamper H, Holbrook SR, Hearst JE, Sancar A. Action mechanism of ABC excision nuclease on a DNA substrate containing a psoralen crosslink at a defined position. Proc. Natl. Acad. Sci. U.S.A. 1986; 83(21):8077-8081.
36. Jachymczyk WJ, von Borstel RC, Mowat MR, Hastings PJ. Repair of interstrand cross-links in DNA of Saccharomyces cerevisiae requires two systems for DNA repair: the RAD3 system and the RAD51 system. Mol. Gen. Genet. 1981; 112(2):196-205.
37. Thompson LH, Salazar EP, Brookman KW, Hoy CA. Hypersensitivity to cell killing and mutation induction by chemical carcinogens in an excision-repair deficient mutant of CHO cells. Mutat. Res. 1983; 45(6):1737-1743.
38. Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat. Res. 2001; 486:217-247.
39. Mu D, Bessho T, Nechev LV, Chen DJ, Harris TM, Hearst JE, Sancar A. DNA interstrand cross-links induce futile repair synthesis in mammalian cell extracts. Mol. Cell. Biol. 2000; 20(7):2446-2454.
40. Greenberg RB, Alberti M, Hearst JE, Chua MA, Saffran WA. Recombinational and mutagenic repair of psoralen interstrand crosslinks in Saccharomyces cerevisiae. J. Biol Chem. 2001; 276(34):31551-31560.
41. D’Andrea AD, Grompe M. The fanconi anaemia/BRCA pathway. Natl. Rev. Cancer 2003; 3(1):23-34.
42. Lahue R, Modrich P. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996; 65:101-133.
43. Modrich P. Strand-specific mismatch repair in mammalian cells. J. Biol. Chem. 1997; 272(40):24727-24730.
44. Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem. Rev. 1996; 106(2):302-323.