CHEMICAL BIOLOGY
Enzymatic Synthesis of Carbohydrate-Containing Biomolecules
Hai Yu and Xi Chen, Department of Chemistry, University of California-Davis, California
doi: 10.1002/9780470153672.wecb153
As important biomolecules in living organisms, carbohydrates have received increasing attention in recent years. Their important roles in biologic events are being continuously unraveled. The development of synthetic methodologies, including both chemical and enzymatic methods, contributes greatly to the advance of the field of glycoscience. The involvement of regio- and stereo-selective enzymes in the synthesis of complex carbohydrate-containing molecules has become an indispensable approach. Many enzymes involved in the biosynthesis and biodegradation of carbohydrates have been characterized and have been applied for the production of carbohydrate-containing biomolecules, including oligosaccharides, polysaccharides, glycoproteins, glycolipids, and glycosylated natural products. A range of strategies for enzymatic synthesis have also been developed, such as protein engineering of glycosidases and glycosyltransferases by site-directed mutagenesis or directed evolution, one-pot multiple-enzyme synthesis, sugar nucleotide regeneration, solid-phase enzymatic synthesis, synthesis using immobilized enzymes, and cell-based synthesis. Enzymatic synthesis will continue to play critical roles in obtaining complex carbohydrate-containing biomolecules. Future efforts should focus on identifying synthetically useful enzymes such as those with flexible or novel substrate specificity and those that can form new bonds. This identification can be achieved by functional genomics and mutagenesis studies. Development of novel enzymatic synthetic methods is also critical to access diverse naturally occurring and non-natural derivatives of carbohydrates and glycoconjugates.
Biomolecules are chemical compounds that naturally occur in living organisms. They primarily contain carbon and hydrogen atoms, some with nitrogen, oxygen, phosphorus, and sulfur. A wide range of biomolecules exists. Major types are carbohydrates, nucleic acids, lipids, and peptides/proteins. Due to their important roles in bioprocesses and their potential use as drugs or drug targets, synthesis of biomolecules has been a subject of enormous interest. Most active biomolecules are chiral compounds. Although new synthetic reagents, catalysts, and strategies have been developed, chemical synthesis of complex biomolecules continues to pose substantial challenges. Therefore, environmentally benign, highly efficient, and highly selective enzymatic synthesis has been and will continue to play indispensable roles in obtaining many biomolecules. This is especially true for the synthesis of complex carbohydrates or glycoconjugates. The enzymatic synthesis of carbohydrate and carbohydrate-containing biomolecules will, therefore, be the subject of discussion here.
Enzymatic Synthesis of Oligosaccharides
Terminal carbohydrate moieties of naturally occurring glycoconjugates are usually the oligosaccharide synthetic targets. They are generally the key determinates recognized by carbohydrate-binding proteins and are directly involved in many important biologic and pathologic processes, including cell adhesion, differentiation, development, and regulation (1). Enzymatic synthesis of these molecules has been extensively studied. Both glycosidases and glycosyltransferases have been widely applied.
Glycosidase-catalyzed synthesis of oligosaccharides
Glycosidases, including endoglycosidases and exoglycosidases, are enzymes that catalyze the cleavage of glycosidic bonds. Under certain conditions, glycosidases can be used for the formation of glycosidic links in which a carbohydrate hydroxyl acts as a more efficient nucleophile than as a water molecule. Oligosaccharide formation catalyzed by wild-type glycosidases can be accomplished by either a thermodynamically (shifting the equilibrium by using a large excess of acceptors, adding organic solvent, or increasing reaction temperature) or a kinetically (using activated glycosyl donors and exogenous nucleophiles) controlled process (2, 3). Many wild-type glycosidases have been studied. Some are readily available and with low cost. They are relatively stable and have been used in the enzymatic synthesis of oligosaccharides. More recently, with a better understanding of the catalytic mechanism of glycosidases, mutants of glycosidases with high efficiency in catalyzing the formation of glycosidic bonds have been constructed. These mutants, which are mainly of retaining-type glycosidases, are called glycosynthases. They have been increasingly used in synthesis.
Endoglycosidase-catalyzed synthesis of oligosaccharides
Endoglycosidases have mainly catalyzed the hydrolysis of internal glycosidic bonds. Many endoglycosidases, however, have transglycosylation activity and have been used for the synthesis of oligosaccharides of varied lengths. For example, β1,3-D-glucanases (laminaranases) purified from the culture medium of Oerskovia sp. and homogenate of the marine mollusk Spisula sachalinensis have been used in transglycosylation reaction to synthesize several 4-methylumbelliferyl β1,3-linked D-gluco-oligosacchairdes containing two to six glucose residues (4). In this case, polymers laminaran and curdlan were used as donors and 4-methylumbelliferyl β-glucoside was an acceptor. More recently, E. freundii endo-β-galactosidase has been used as a transglycosidase in the synthesis of a series of GlcNAc-terminated poly-N-acetyllactosamine β-glycosides: GlcNAcβ1,3(Galβ1,4GlcNAc1,3)1-4Galβ1,4GlcNAcβ-pNP from GlcNAcβ1,3Galβ1,4GlcNAcβ-pNP (serves as both donor and acceptor) (5). More important application of endoglycosidases in the synthesis of N-linked and O-linked glycoproteins will be discussed in later sections.
Exoglycosidase-catalyzed synthesis of oligosaccharides
The native function of exoglycosidases is to cleave a terminal monosaccharide unit, but exoglycosidases can be used to add a monosaccharide in the oligosaccharide synthesis. Many types of exoglycosidases from bacterial, fungal, animal, and plant sources have been reported and have been used in the formation of glycosidic bonds, including sialidases, galactosidases, glucosidases, fucosidases, mannosidases, xylosidases, N-acetyl hexosaminidases, and more.
Sialidases
Sialic acids are mainly found as terminal carbohydrate units of glycoproteins and glycolipids in vertebrates or as components of capsular polysaccharides or lipooligosaccharides in pathogenic bacteria. As the outermost residues, sialic acid residues are directly involved in many biologically important molecular recognition and interaction events (6-8).
Sialidases from Vibrio cholerae, Clostridium perfringens, Salmonella typhimurium, and Newcastle disease virus have been used in the synthesis of α2,3- and α2,6-linked sialosides (sialic acid-terminated oligosaccharides) in a kinetic-controlled manner in which a p-nitrophenyllated α-N-acetylneuraminic acid was used as a donor (9). In this study, yields of 10-30% were obtained, and the galactoside acceptors were synthesized using a bovine testes β-galactosidase. Sialidase from Vibrio cholerae has also been immobilized on a polymeric carrier for the synthesis of an array of α2,3- and α2,6-linked sialosides with different acceptors (10). More efficient syntheses have been obtained by using trans-sialidases. For example, Trypanosoma cruzi trans-sialidase was used in the synthesis of 3'-sialyl-lacto-N-biose I (Neu5Acα2,3Galβ1,3GlcNAc) in a yield of 35%. The disaccharide donor lacto-N-biose I (Galβ1,3GlcNAc) was obtained using p-nitrophenyl β-D-galactopyranoside (GalβpNP) as the donor for Xanthomonas manihotis β-D-galactosidase (11). In a different study, α2,3- sialylactose was used as a donor. The yield of the formation of Neu5Acα2,3Galβ1,3GlcNAc was increased from 45% to 75% by decreasing the concentration of by-product lactose using an Escherichia coli β-galactosidase (12).
Galactosidases
N-Acetyllactosamine is one of the core structures in the carbohydrate moieties of glycoproteins. β1,4-Galactosidases have been widely studied and used to synthesize β1,4-linked oligosaccharides, including LacNAc, Galβ1,4Gal. Kragl et al. used 25% of 1,3-di-methyl-imidazolmethyl sulfate (a water-miscible ionic liquid) in water to decrease the hydrolysis of the formed product in a Bacillus circulans β-galactosidase-catalyzed synthesis of N-acetyllactosamine from lactose and GlcNAc. They could double the yield to about 60%, and they reused enzyme several times without the loss of activity by ultrafiltrating the reaction mixture (13). Another Bacillus circulans β-galactosidase, however, was specific for the cleavage of β1,3-links. It was used to synthesize Galβ1,3GlcNAc, Galβ1,3GalNAc, and their pNP derivatives in 10-46% yields using Galp NP as a donor (14).
β-Galactosidases from Penicillium multicolor, Aspergillus oryzae, Bifidobacterium bifidum, and Streptococcus 6646 K have been used to catalyze the transfer of Gal from GalpNP to GalNAc or GlcNAc for the formation of β-linked galactosides (15).
In another case, Thiem et al. used a β-galactosidase from bovine testes in the transfer of a galactose from lactose to glucose or galactose-containing structures with yields ranging from 7% to 44% (16). The enzyme showed a preference for the formation of β1,3-linkage.
Galα1,3Gal is the terminal disaccharide of α-Gal epitopes that are found in almost all mammals except human, apes, and Old World monkeys. Humans naturally produce anti-Gal antibodies that specifically recognize α-Gal epitopes (17). Although in low yields, α-galactosidases from A. oryzae, A. parasiticus, A. flavipes, A. terreus, Talaromyces, and coffee beans have been used to construct Galα1,3Gal (18-20). A highly specific α-galactosidase from Penicillium multicolor was able to catalyze the synthesis of α-Gal disaccharides and trisaccharides from GalαpNP donor in 25-46% yields. The yield of the reaction can be increased by decreasing the concentrations of the substrates (21). Interestingly, a coffee bean and a Thermomyces lanuginosus α-galactosidases could catalyze the galactosylation of a cyclic glucotetrasaccharide to form an α1,6-linkage (22).
Li et al. used a CLONEZYME thermophilic glycosidase library (Diversa Corporation, San Diego, CA) in the synthesis of Galβ1,4Gal, Galβ1,6Gal, Galα1,6Gal, and Galα1,3Gal. Some enzymes can use lactose as a donor and transfer two Gal residues in tandem to the hydroxyl group of the acceptor (23).
Glucosidases
Glucosidases have been commonly used as catalysts for the formation of alkyl glucosides, which are surface-active compounds that have applications in the pharmaceutical and the food industry. Alcohols are usually used as both solvent and acceptors. For example, glucoamylase and β-glucosidase have been used to catalyze the synthesis of α- and β-glucosides, respectively. Maximum yields were obtained when the reaction solution consisted 10% (vol/vol) of water in primary alcohol or 15% (vol/vol) of water in diol (24). Kosary et al. used an immobilized β-glucosidase in preparative scale synthesis of alkyl and aryl β-D-glucopyranosides in moderate yields ranging from 12% to 19%. They found that the alcohol solvent component of the reaction mixture can be replaced by 1,2-diacetoxyethane (25). An α-glucosidases from Xanthomonas campestris was used to synthesize α-arbutin (hydroquinone-O-α-glucopyranoside), an important cosmetic ingredient, in 93% yield using maltose as donor and hydroquinone as acceptor (26).
Glucosidases have also been widely used as catalysts for the formation of various oligosaccharides. A thermostable β-glucosidase from Thermus thermophilus was used to transfer the glucose or fucose residue from GlcβpNP or FucβpNP to a glucose acceptor for the synthesis of Glcβ1,3Glc and Fucβ1,3Glc in an 88% and a 58% yield, respectively (27). β-Glucosidases from Sclerotinia sclerotiorum and Aspergillus niger were both used to synthesize gluco-oligosaccharides, such as gentiobiose, cellotriose, and cellotetraose, from cellobiose (28). α-Glucosidases from a different source can catalyze the formation of gluco-oligosaccharide with different links. For example, an α-glucosidase from Aspergillus nigher favors the formation of an α1,6-linkage; a Penicillium purpuro- genum enzyme yields mostly α1,4-linked maltotriose from maltose; an α-glucosidase purified from fungus Paecilomyces lilacinus prefers to catalyze the synthesis of α1,2- and α1,3-linked gluco-oligosaccharides from maltose (29). A thermostable α-glucosidase from Bacillussp. strain SAM1606 shows very broad substrate specificity. Site-directed mutagenesis of this enzyme generates mutants with altered specificity for oligosaccharide formation (30).
Mannosidases
Mannose-terminated oligosaccharides are commonly found in the carbohydrate moieties of N-linked glycoproteins of the eukaryotic system. For example, Manα1,2Man and Manα1,3Man are terminal structures of high mannose type and hybrid type N-glycans. Athanasopoulos et al. (31) reported that incubating mannose with a novel α-D-mannosidases from A. phoenicis afforded Manα1,6Man and Manα1,6Mana1,6Man in 21% and 5% yield, respectively.
β-Mannoside links, which are considered one of the most challenging synthetic glycosidic bonds, can be achieved by the catalysis of β-D-mannosidases from Aspergillus orizae and Helix pomatia (32).
Jack bean α-mannosidase has also been able to catalyze the transglycosylation of rhamnose residue. It has been used in the synthesis of a disaccharide Rhaα1,2RhaαSEt in 32.1% yield from RhaαpNP and ethyl 1-thio-α-D-rhamnopyranoside. The disaccharide product is a derivative of the common oligosaccharide unit of antigenic lipopolysaccharides from Pseudomonas (33).
Fucosidases
α-L-Fucose is commonly found as the terminal unit in the carbohydrate moiety of many important glycoconjugates involved in a variety of biologic events. For example, α1,2-fucosylated LacNAc is the determinant of blood-group type O antigen. Disaccharides Fucα1,3GlcNAc and Fucα1,4GlcNAc are part of Lex and Lea antigens, respectively.
By using α-L-fucosidases from Penicillium multicolor (34), Aspergillus niger (35, 36), Corynebacterium sp. (35), and other sources (37-39), α1,3-linked L-fucosides have been obtained. By using an α-L-fucosidase from Ampullaria niger (35), α1,6-linked disaccharide Fucα1,6GalβOMe has been produced in 14% yield from FucαpNP and GalβOMe. With FucαpNP as the donor, α1,4-linked disaccharide Fucα1,4(6OBn)GlcNH2 βSEt has been obtained in 50% yield using an α-L-fucosidase from bovine kidney (40) and in 33% yield using an enzyme from bovine testes (41). Compared with α-L-fucosidases, only a few β-D-fucosidases were reported to form β-D-fucosides (42-44). For example, β-glucosidase from Thai rosewood and almond both catalyse transfucosylation of alcohols to synthesize alkyl fucosides with high yields using pNP-Fuc as the donor (43).
N-Acetyl Hexosaminidases
N-Acetyl hexosaminidases can be used to transfer aminosugars such as GlcNAc and GalNAc to various acceptors (45, 46). Two disaccharides, GlcNAcβ1,4GlcNAc and GlcNAcβ1,6GlcNAc, have been synthesized from GlcNAcβpNP and GlcNAc using a β-N-acetylhexosaminidase from Aspergillus oryzae (47). β-N-Acetylhexosaminidase from Penicillium oxalicum was also used to construct a β1,4-linked to 6-benzyloxyhexy-β-N-acetylglucosaminide in 32% yield using N-acetyl chitotriose as a donor, which is a useful intermediate in drug carrier production (48). The derivatives of N-acetyl hexosamine can also be transferred. For example, Uzawa et al. have shown that a β-N-acetylhexosaminidase from A. oryzae can transfer sulfated N-acetylglucosamine residue from its donor pNP-β-D-6-SO3-GlcNAc to GlcNAc and to various galacosides to afford sulfated disaccharides with β1,3-, β1,4-, or β1,6-links (49).
Xylosidases
β-Xylosidases are being used commonly in paper industry for complete degradation of xylans. The enzymes used in the synthesis of xylose-containing compounds have also been reported. For example, Eneyskaya et al. (50) have used p-nitrophenyl β-D-xylopyranosides (XylβpNP) as a donor and a β-xylosidase from Aspergillus sp. in the synthesis of several β1,4-D-xylooli-gosaccharides as β-xylanase substrates.
Also α-linked xylosides have been synthesized. For example, α1,3-linked xylosyl disaccharides were obtained from XylapNP (donor) and glucose or GlcβpNP (acceptor) in 10-18% yield using an α-xylosidase from archaeon Sulfolobus solfataricus (51). An α1,6-linked trisaccharide unit of xylogucan resulted in 15% yield when α-xylosyl fluoride was used as a glycosyl donor and pNP-β-cellobioside was used as an acceptor.
Glycosynthases—glycosidase mutants
Glycosylation or transglycosylation reactions catalyzed by wild- type glycosidases inevitably suffer from low yields and unpredictable regioselectivity because of the hydrolytic nature of the glycosidases and the lack of regioselectivity of many glycosidases. The creation of glycosynthases, a class of glycosidase mutants, has greatly advanced the field of glycosidase-catalyzed synthesis of oligosaccharide.
Glycosynthases are a new class of glycosidase mutants developed based on the mechanistic understanding of glycosidases. Glycosidases hydrolyze glycosidic links with net retention or inversion of stereochemistry, and thus they are classified into retaining glycosidases and inverting glycosidases. Inverting glycosidases proceed via a general acid/base-catalyzed direct displacement, whereas retaining glycosidases use a doubledisplacement mechanism in which a covalent glycosyl-enzyme intermediate is involved (Fig. 1a and b). A glycosynthase is formed by replacing a single active site nucleophile (usually a glutamate or an aspirate residue) of a glycosidase with a neutral alanine or serine residue. An activated glycosyl donor, usually a glycosyl fluoride of the opposite anomeric configuration to that of the natural substrate, is used for the glycosynthase-catalyzed formation of the glycosidic bond (Fig. 1c). These mutated enzymes have increased activity for the synthesis of oligosaccharides but no hydrolytic activity. Since the first glycosynthase was reported by Withers et al. on a single catalytic carboxylate nulceophile mutant of a retaining glycosidase Agrobacterium sp. 3-glucosidase (Abg) E358A mutant (52), many glycosynthases have been generated by Withers et al. and others. The first glycosynthase of an endoglycosidase was a E134A mutant of the retaining 1,3-1,4-β-glucanase from Bacillus licheniformis reported by Malet and Planas (53). Currently, about 20 glycosynthases from ten different glycosidase families have been produced. Except for a recently reported mutant of an exo-oligoxylanase (Rex) from Bacillus halodurans (54), which is the first glycosynthase derived from an inverting enzyme, all others are mutants of retaining glycosidases. Despite the development of glycosynthases, many naturally occurring carbohydrate structures have not been obtained by the glycosynthase-catalyzed reaction, because the strategy relies on the availability of gene sequence and on the understanding of the catalytic mechanism of the glycosidases, and this information is not available for all glycosidases known to date.

Figure 1. Catalytic mechanisms for wild-type and engineered glycosidases. (a) Retaining glycosidase, to hydrolyze glycosidic links using the double-displacement mechanism; (b) inverting glycosidase, to hydrolyze glycosidic links via a general acid/base-catalyzed direct displacement; (c) glycosynthase, mutation of the catalytic nucleophile allows transfer an activated sugar donor (such as a glycosyl fluoride) to suitable acceptors; (d) thioglycoligase, mutation of the catalytic acid/base allows transfer of an activated p-glycoside to suitable acceptors; (e) thioglycosynthase, mutation of both the catalytic nucleophile and the acid/base residues allows transfer of glycosyl fluoride donor to acceptors.
Glycosyltransferase-catalyzed synthesis of oligosaccharides
Leloir-type glycosyltransferases are enzymes that catalyze the transfer of a monosaccharide from activated sugar nucleotide donor to an acceptor. Because of their high efficiency and regio- and stereo-specificities, they offer significant advantages in the formation of glycosidic bonds, and thus, they have been widely used in the synthesis of carbohydrate-containing structures. Glycosyltransferases have been classified based on the carbohydrate residue that they transfer.
Sialyltransferases
Chemical synthesis of sialosides is considered one of the most difficult glycosylation reactions because of a hindered tertiary anomeric carbon and the lack of a participating auxiliary functionality in the carbon next to the anomeric carbon in sialic acids (55, 56). Sialyltransferase-catalyzed glycosylation is believed to be the most efficient approach for the production of sialic acid-containing structures.
Sialyltransferases catalyze the transfer of a sialic acid residue from CMP-sialic acid to a galactose, GalNAc, or another sialic acid residue. Sialyltransferases from bacterial and mammalian sources have been extensively studied and used in the enzymatic synthesis of sialosides, sialoglycoconjugates, and enzymatic modification of cell surface.
Rat liver α2,3SiaT and α2,6SiaT have been the most widely used mammalian sialyltransferases for the synthesis of sialo-sides. Both enzymes have broad donor and acceptor substrate specificity. They can tolerate a variety of modifications at the Neu5Ac moiety of CMP-Neu5Ac (57-59) and Gal on acceptors (60-62). Four SiaTs from rat liver or porcine submaxillary glands were studied for their abilities to transfer synthetic 9-substituted sialic acid analogs onto N- or O-linked glycoprotein glycans. They all accepted CMP-9-azido-Neu5Ac as the donor substrate. In contrast, 9-amino-Neu5Ac was only accepted by α2,6SiaT form rat liver (63).
Wong et al. have studied the acceptor specificity of a recombinant α2,3SiaT from Neisseria gonorrhoeae by using several synthetic oligosaccharides, glycolipids, and glycopeptides. Lactose, its allyl, thiophenyl glycosides, sulfated oligosaccharides, and the PSGL-1 glycopeptide carrying a sulfotyrosine residue were all excellent acceptors for the enzyme. However, most glycolipids were poor substrates for this bacterial enzyme (55).
Using genes cloned from Neisseria meningitides, a fusion protein has been constructed that has both CMP-Neu5Ac synthetase and α2,3SiaT activities. This fusion protein could sialylate various oligosaccharide acceptors with Neu5Ac, as well as Neu5Gc and N-propionyl-neuraminic acid, in high yields. A 100 gram-scale of α-2,3-sialyllactose was produced using this protein with the regeneration of sugar nucleotide CMP-Neu5Ac (64). Very recently, the fusion protein and a recombinant human α2,6-sialyltransferase (hST6Gal-I) have been used in gram-scale synthesis of sialosides (65).
A bifunctional sialyltransferase Cst-II from C. jejuni OH4384 carrying both α2,3- and α2,8-sialyltransferase activities was used in the enzymatic synthesis of ganglioside mimic GD3 (66). Another multifunctional bacterial enzyme that has been reported recently is Pasteurella multocida sialyltransferase (PmST1 or tPm0188Ph) (67, 68). It has four different activities, including an α2,3SiaT, α2,6SiaT, α2,3-sialidase, and trans-sialidase activities. PmST1 is a highly active sialyltransferase with broad donor and acceptor substrate specificity; therefore it is a powerful tool in synthesizing diverse sialosides that contain structurally modified sialic acids.
As more than 50 different sialic acids derivatives have been found in nature, sialyltransferases with flexible donor and acceptor specificity are preferred by chemists. An α2,6SiaT cloned from Photobacterium damsela (Pd2,6ST) can efficiently transfer Neu5Ac, KDN, Neu5Gc, and their derivatives with extensive modifications at C-5 and C-9 in the sialic acid residues from their CMP-activated forms to the acceptors (8). Pd2,6ST also has very relaxed acceptor specificity. For example, this bacterial enzyme has been applied for the enzymatic sialylation of the TN glycopeptide with GalNAc α-linked to the serine residue in the sequence with 71% yield. More interesting, Pd2,6ST has shown activity in transferring sialic acid to N- and O-linked glycoproteins (69).
Galactosyltransferases
Galactosyltransferases are a family of enzymes that catalyze the addition of a galactose residue from activated sugar nucleotide donor uridine diphosphate galactose (UDP-Gal) to different acceptor substrates in different links.
β1,4-Galactosyltransferases-1(β1,4GalT-1) is one of the most exhaustively studied glycosyltransferases and has been the most commonly used glycosyltransferase in the synthesis of oligosaccharides and glycoconjugates (70). The 31,4GalT exhibits polymorphic donor specificity and can transfer modified galactose from their activated UDP form to acceptors to afford corresponding LacNAc analogs (71, 72). The β1,4GalT is equally relaxed in respect to its acceptor specificity, and the modified GalNAc acceptor can be tolerated by the β1,4GalT. As this enzyme has a highly relaxed substrate specificity, it has been exploited in the synthesis of many non-natural LacNAc-based structures. β1,4GalT from bacterial sources such as H. pylori (73) and N. meningitides (74) have also been cloned, but examples of their application in the synthesis of galactosides have been limited (75).
Carbohydrate structures bearing a Galα1,3Galβ terminus are α-Galactosyl epitopes. The interaction of these epitopes on the surface of animal cells with anti-α-galactosyl antibodies in human serum is believed to be the main cause in antibody-mediated hyperacute rejection in xenotransplantation. The unique enzyme responsible for the formation of α-Gal epitopes is α1,3galactosyltransferase (α1,3GalT). Fang et al. (76) reported that a truncated bovine α1,3GalT (80-368) can be produced as a soluble recombinant enzyme on a large scale with highly specific activity. A variety of α1,3-galactosylated epitopes were synthesized using such a recombinant enzyme.
The 2" and 6"-deoxy analogs of globotriose Galα1, 4Galβ1,4GlcβOR, which is the known receptor for Shiga and Shiga-like toxins, were prepared using UDP-2-deoxy-Gal and UDP-6-deoxy-Gal along with α1,4GalT from N. meningitidis in 11% and 95% yield, respectively (77). The inexpensive galactosyl fluoride has also been used as a donor for an α1,4GalT to produce an α1,4-linked galactoside in the presence of a catalytic amount of UDP (78).
Various β1,3GalTs have been cloned and expressed from bacterial (66, 79) and mammalian sources (80-82). Core 1, Galβ1,3GalNAcα1-serine/threonine, is the major constituent of O-glycan core structures in many cells. The core 1 structure is also called T antigen in pathologic studies. Core 1 β1.3-galactosyltransferases (core 1 β1,3GalT), which are responsible for the synthesis of core 1 disaccharide, have been purified recently (83, 84).
Compared with galactopyranosyltransferases, a few studies on galactofuranosyltransferase (Galf T), which is responsible for the biosynthesis of a galactan core in the major structural component of mycobacterial cell wall, have been reported (85). Galf T transfers Galf residues from UDP-galactofuranose (UDP-Galf), instead of UDP-Gal, to the growing galactan chain. Very recently, Lowary et al. (86) reported the high-level expression and purifaction of a novel bifunctional recombinant galactofuranosyltransferase from Mycobacterium tuberculosis H37Rv, which can produce both β1,5- and β1,6-galactofuranose links in an alternating fashion.
N-acetylgalactosaminyltransferase
The human blood group A glycosyltransferase (α1,3-N-acetylgalactosaminyltransferase) is responsible for the biosynthesis of blood group A antigens. It catalyzes the transfer of a GalNAc from UDP-GalNAc to the C-3 hydroxyl of Gal residue in (Fucα1,2)GalβOR to form blood group A antigen GalNAcα1,3(Fucα1,2)GalβOR. β1,3GalNAcT from N. meningitidis was reported to transfer a GalNAc to 1-thio-β-D-lactose to produce a trisaccharide, which was further used to synthesize lacto-N-neotetraose (87). Wang et al. (88) had constructed β1,3-N-acetylgalactosaminyltransferase/UDP-N-acetylglucosamine C4 epimerase fusion protein and had synthesized a variety of globotetraose and isoglobotetraose derivatives using this fusion protein and UDP-GlcNAc. This construction alters the donor substrate requirement of the reaction from the high-cost UDP-GalNAc to UDP-GlcNAc. Recently, gram-scale synthesis of GD3, GT3, GM2, GD2, GT2, and GM1 ganglioside oligosaccharides was reported by Blixt et al. using β1,4-N-acetylgalactosaminyltransferase (β1,4GalNAcT) from Campylobacter jejuni along with other glycosyltransferases (89).
N-Acetylglucosaminyltransferases
N-Acetylglucosaminyltransferases (GlcNAcTs) play important roles in the synthesis of O-glycan core structures and in the branching and subsequent elaboration of N-linked glycans on glycoproteins. GlcNAcTs I-VI (differing in their specificities and glycosidic bonds formed with trimannose core-containing N-glycans) have been studied on their substrate specificity and their application. For example, Core 2 GlcNAcT (β1,6GlcNAcT) has been used in the synthesis of O-linked core 2-type sLex epitope (90). Norberg et al. (91) had investigated the donor and acceptor substrate specificity of β1,3GlcNAcT from N. meningitidis. This enzyme can use both UDP-GlcNAc and UDP-GalNAc as donors. It is also capable of tolerating deoxy derivatives at any position other than C-3 of the Gal residue in Galβ1,4GlcβOPh acceptor.
Fucosyltransferases
Several mammalian fucosyltransferases (FucTs) have been characterized, and their application to the synthesis of fucosecontaining oligosaccharides has been well studied. Recently Drouillard et al. (92) developed an efficient bioengineering method for large-scale production of fucosyl α1,2-linked oligosaccharides from lactose. 2'-Fucosyllactose and lacto-N-neo-fucopentaose-1 (LNnF-1, an H-2 antigen oligosaccharide) were produced by expressing Helicobacter pylori α1,2-fucosyltrans- ferase in metabolically engineered E. coli cells. Trisaccharide 2'-fucosyllactose could be synthesized industrially on a multiton scale for nutraceutic applications. The pentasaccharide LNnF-1, which contains the H-2 antigen structure, could be used as a precursor for the synthesis of other antigens of the ABH histo-blood-group system.
Mannosyltransferases
The β1,4-mannosidic linkage is common to most asparagine-linked oligosaccharides (N-glycans) in glycoproteins as part of their pentasaccharide core structure. The formation of the glycosidic link in ManP1,4GlcNAc is particularly challenging for chemical synthesis. Glycosidases have been used, but they require a large excess of the expensive chitobiose acceptor and produce low yields. A highly active recombinant β1,4-mannosyltransferase was reported (93), and this enzyme was used to produce the core trisaccharide of N-glycans with a yield of 80% using UDP-mannose as donor and the synthetic chitobiosyl phospholipid as acceptor. A recombinant α1,2-mannosyltransferase was recently reported to use GDP-5-thio-Man as a sugar nucleotide donor to produce the corresponding 5S-mannosides (94). 5S-Glycosides are expected to have potentially stronger affinity for receptors compared with their natural counterparts, and they may also find other applications in hydrolase-resistant vaccines.
Enzymatic formation of S-glycosidic bonds
Natural occurring carbohydrates contain monosaccharides linked through O-glycosidic bonds. Thioglycosides, in which the glycosidic oxygen atom has been replaced by the sulfur atom, can be tolerated by most biologic systems and are less susceptible to acid/base or enzyme-mediated hydrolysis. They are valuable in the studies of glycosidases and are gaining interest as targets for pharmaceutical industry. Many efforts have been focused on the synthesis of thioglycosides by conventional chemical synthetic methods. Only recently, mutants of glycosi- dases and glycosyltransferase have been constructed and applied to form S-linked oligosaccharides using thiolated acceptors.
Glycosidase mutants (thioglycoligase and thioglycosynthase) in the synthesis of S-linked glycosides
A glycosidase mutant, in which the acid/base carboxyl residue was replaced by a catalytically inactive residue, was recently developed by Withers et al. as a novel strategy for the synthesis of S-linked oligosaccharides (95). This new class of glycosidase mutants is named “thioglycoligases” (Fig. 1d). Two different alanine acid/base mutants of retaining β-glycosidases, a β-glucosidase from Agrobacterium sp. Abg E171A and a P-mannosidase from Cellulomonas fimi Man2A, were used for the formation of thioglycosidic bonds. The readily available dinitrophenyl glycoside donor DNP-Glc or DNP-Man was incubated with the mutant enzyme and the acceptor to provide corresponding thiodisaccharides in good yields. With this new methodology, Stick and Stubbs (96) have synthesized various types of S-linked disaccharides using glycosidase mutant Abg 171A. Incubation of the glucose donor DNP-Glc and various thiol acceptors with Abg 171A provided corresponding β1,4-, β1,3- and β1,6-S-linked disaccharides, but it did not work for the formation of β1,2 thio-linkage. More recently, the generation of α-thioglycosides has been successfully achieved for the first time using thioglycoligases derived from glycoside hydrolase Family 31 S. solfataricus α-glucosidase and E. coli α-xylosidase (97).
The thioglycoligase technology was further extended with the advent of thioglycosynthases (Fig. 1e), double-mutants of retaining glycosidases which lack both catalytic nucleophile and catalytic acid/base residues and can efficiently catalyze the thioglycosidic bond formation using glycosyl fluoride donors and thioglycoside acceptors (98). Reaction of the synthesized α-glycosyl fluoride with the acceptors in the presence of the double mutant Abg E171A, E358G at neutral pH afforded two β-1,4-S-linked disaccharides with 51% and 45% yields, respectively.
Glycosyltransferase mutants in the synthesis of S-linked glycosides
Rich et al. (99) have reported a new enzymatic method for the synthesis of thiooligosaccharides using glycosyltransferase. Incubation of a mixture of thiol acceptor and UDP-Gal with unit quantities of a recombinant bovine α1,3-galactosyltransferase in the presence of DTT afforded the thio-linked tetrasaccharide in 92% yield instead of the expected trisaccharide product because of the second glycosyl transfer that occurred after the initial transfer of a galactosyl residue to the thiolated acceptor. The desired trisaccharide, which is an analog of the Clostridium difficile toxin A binding ligand, was obtained in near-quantitative yield after treatment with an α-galactosidase from green coffee beans. Furthermore, their initial results confirmed that a β1,3-N-acetylglucosaminyltransferase from N. meningitides can catalyze the transfer of UDP-GlcNAc to a thiol acceptor to afford a thio-linked trisaccharide. This is the first example of using glycosyltransferases in synthesizing thiooligosaccharides. Such thiooligosaccharides could serve as important immunogens and components of conjugate vaccines.
Enzymatic Synthesis of Polysaccharides
Polysaccharides, along with nucleic acids and proteins, belong to three major important classes of naturally occurring biopolymers. Polysaccharides are important biomacromolecules with unique physical properties that lead to advanced biomaterials and biomedical applications (100). For example, glycosamino- glycans (GAGs) are linear polysaccharides containing repeating disaccharide units of a hexosamine and a uronic acid. Heparin/heparan, hyaluronan, and chondroitin are three prevalent glycosaminoglycans. Vertebrates use glycosaminoglycans in structural, recognition, adhesion, and signaling roles. Chemical synthesis of naturally occurring polysaccharides is considered to be impractical. Most polysaccharides, especially those from bacteria origins, are obtained by purification from natural sources or from cell culture, enzymatic approaches have been increasingly applied to obtain some structures.
Heparin/Heparan Sulfate
Heparin and heparan sulfate are linear polysaccharides with a repeating disaccharide unit of 1,4-linked uronic acid (D-glucuronic or L-iduronic acid) and D-glucosamine residues. Both uronic acid and glucosamine can contain sulfo groups at different positions including 2-O-sulfo substitution of the uronic acid residue and 2-N-, 3-O-, and 6-O-sulfo substitution in the glucosamine residue. Heparin and heparan sulfate are structurally related glycosaminoglycans that participate in numerous important biologic processes, such as blood coagulation, viral and bacterial entry and infection, angiogenesis, and cancer development.
The enzymatic synthesis of heparin and heparan sulfate is currently under extensive investigation using glycosyltransferases of E. coli (101, 102) and heparosan synthase of P. multocida (103, 104). Rosenberg et al. reported an approach to rapidly assemble anticoagulant III-binding classic and nonclassic anticoagulant heparan polysaccharides using the enzymes involved in the heparan sulfate biosynthesis (105). Recently Chen et al. (106) have described a method for the enzymatic sulfation of multimilligram amounts of heparan sulfate with specific functions using immobilized sulfotransferases combined with a 3'-phosphoadenosine 5'-phosphosulfate regeneration system. Because the recombinant sulfotransferases are expressed in bacteria and the method uses a low cost sulfo donor, it can be readily used to synthesize large quantities of anticoagulant heparin drug or other biologically active heparan sulfates.
Hyaluronan
Hyaluronan (hyaluronic acid, HA) is a highly anionic unbranched linear polymer containing a -GlcAβ1,4GlcNAcβ1,4-repeating unit, which plays important roles in modulating cell adhesion, signaling, and motility. Several enzymes responsible for HA synthesis, namely Hyaluronic Acid Synthases, have been cloned from bacteria and mammals (107, 108).
HA synthase from Streptococcus equisimilis was employed for milligram-scale synthesis of HA (109). In this reaction system, UDP-sugars (UDP-GlcA and UDP-GlcNAc) were effectively regenerated by the catalyses of several enzymes, synthetic HA was produced in 90% yield. In addition, mutated HA synthase from Type A Pasteurella multocida (PmHAS 1-703 aa) was recently used for the stepwise synthesis of HA, which has a monodispersed molecular mass of up to 20 sugar units (110).
On the other hand, using hyaluronidase (HAase) to synthesize HA has also been reported (111, 112). HA oligomers of up to 22 sugar units were synthesized by enzymatic reconstruction of HA chains using the transglycosylation reaction of Bovine Testicular HAase (111).
Chondroitin
Chondroitin (Ch) and chondroitin sulfate (ChS) are naturally occurring heteropolysaccharides belonging to the family of GAGs. Ch is a nonsulfated derivative of ChS, which consists of a GlcAβ1,3GalNAc disaccharide repeating unit connected through β1,4-glycosidic bonds. ChS exists predominantly as polysaccharide side chains of proteoglycans in extracellular matrixes where it plays important roles in the bioactivities of living systems. Ch is widely used as a therapeutic material for the prevention or alleviation of symptoms of diseases.
The enzymatic polymerization to provide synthetic Ch and its derivatives catalyzed by hyaluronidase has been reported by Kobayashi et al. (113). Synthetic oxazoline monomers were recognized and catalyzed by ovine testicular HAase (OTH) to produce Ch and the polymerization behaviors greatly depend on the reaction conditions. The Mn value of synthetic Ch reached 4600, which corresponds to that of naturally occurring Ch.
Polysialic acids
Polysialic acids (PSAs) are linear homopolymers of N-acetylneuraminic acid and N-glycolylneuraminic acid joined by α2,8-, α2,9-, or α2,8/2,9-ketosidic linkages. Polysialic acids play many important biologic roles. For example, polysialylation of mammalian neural cell adhesion molecules affects cell-cell adhesive interactions during embryogenesis (114). Polysialic acid is also a component of many bacterial capsular polysaccharides that are important virulence factors. Sialyl-transferases catalyze the addition of sialic acid to form diverse carbohydrate molecules. The substrate specificity of α2,8 and α2,8/2,9-polySiaT toward glycolipids and sialylated oligosaccharides has been characterized (115-117). For example, the neuS gene product from E. coli K92 that exhibits α2,8/2,9-poly-sialyltransferase activity both in vitro and in vivo has been described for the first time by Wong et al. (116).
Cellulose
Cellulose is a linear polysaccharide of β1,4-linked dehydrated β-glucose repeating units. Cellulose is the most abundant polysaccharide on earth. It is a major component in higher plant cell walls and has been used as raw material in paper, fibers, and lumber industries.
Enzymatic synthesis of cellulose has been achieved by cellulase. For example, incubation of β-cellobiosyl fluoride with a cellulase from Trichoderma viride can produce cellulose in 54% yield with DP around 22 after 12 h. In addition, change of the reaction conditions (substrate concentration or organic solvent concentration) enabled the selective synthesis of the water-soluble cellooligosaccharides (118).
Amylose
Amylose is a linear polymer of glucose mainly linked with an α1,4-glycosidic bond. Maltooligosaccharides have been effectively prepared by polycondensation of α-D-maltosyl fluoride using an α-amylase from Aspergillus oryzae as the catalyst in a mixed solvent of methanol-phosphate buffer (119). Amylose was also prepared via in vitro polymerization of D-glucosyl phosphate catalyzed by a potato phosphorylase (120). A large excess amount of Glc-1-β is required in this equilibrium-controlled reaction.
Xylan
Xylan, a xylose polymer having a β1,4-glycosidic linkage in the main chain, is the important component of hemicellulose in plant cell walls. Kobayashi et al. (121) reported that synthetic xylan that consists exclusively of xylose units can be prepared by the crude cellulase (containing xylanase) polymerization of β-xylobiosyl fluoride as a monomer in a mixed solvent of acetonitrile/ acetate buffer. The reaction proceeded to produce β1,4-linked synthetic xylan with a degree of polymerization of 23.
Chitin
Chitin is a β1,4-linked polymer of N-acetyl-D-glucosaminopyranose. It is used as structural material in nature, such as the main component in the cell walls of fungi, the exoskeletons of insects and other arthropods, as well as in some animals. It attracts much interest in several scientific and application areas as a multifunctional substrate.
Chitin was prepared by the catalysis of chitinase. Incubation of a chitobiose oxazoline monomer with chitinase form bacillus sp. in phosphate buffer afforded chitin in quantitative yield (122). Using regiospecific 3-O- and/ or 3'-O-methylated derivatives of a chitobiose oxazoline as new substrate monomers, 3-O-methylated chitin oligomers were produced by enzymatic oligomerization using chitinase (123).
Enzymatic Synthesis of Glycoproteins
Many biologically important proteins are carbohydrate-containing glycoproteins. Two major types of glycoproteins are O-linked glycoproteins (a sugar moiety is linked to the β-hydroxyl group of either a serine or a threonine residue in the polypeptide) and N-linked glycoproteins (an oligosaccharide is linked to the amide-side chain of asparagine residue of the polypeptide). For O-linked glycosylation, three different ways can occur: 1) A single GlcNAc residue may be reversibly added to proteins in the cytoplasm or nucleus, 2) initiation of synthesis of proteoglycans may occur by the addition of xylose in the Golgi, and 3) mucin-type O-glycosylation is initiated in the Golgi, which involves the addition of GalNAc to Ser/Thr. N-linked glycosylation is initiated in the ER. As glycoproteins usually present a mixed population of glycan structures at different glycoslylation sites, the isolation of homogeneous glycoproteins from natural sources in significant quantity is impractical. The chemical method for the de novo synthesis of large glycoproteins is currently unavailable. Although challenging, enzymatic synthesis of structurally defined glycoproteins is feasible.
O-linked glycoproteins
O-GlcNAc-modified glycoproteins
Many nuclear and cytosolic proteins are β-O-GlcNAc modified, which is the covalent attachment of β-GlcNAc to Ser/ Thr residues in the proteins. Nearly 80 proteins bearing the O-GlcNAc group have been identified to date (124). Understanding the functional roles of O-GlcNAc requires the development of new strategies for the detection and study of O-GlcNAc-modified proteins. Khidekel et al. (125) reported the direct, high-throughput analysis of O-GlcNAc-glycosylated proteins using a chemoenzymatic approach. An engineered galactosyltransferase was exploited to selectively label O-GlcNAc proteins with a ketone-biotin tagged Gal, which permited the enrichment of low-abundance O-GlcNAc species and the localization of the modification site. Another chemical strategy was developed by Bertozzi et al. for identifying O-GlcNAc-modified proteins from living cells. O-GlcNAcase is capable of transfer azidoacetylglucosamine (GlcNAz) from UDP-GlcNAz to known protein substrates, such as recombinant nuclear pore protein p62. These O-GlcNAz-modified proteins can be covalently derivatized with various biochemical probes at the site of protein glycoslyation using the Staudinger ligation reaction (126). Such a strategy for in vitro modification of target proteins can provide a rapid means for the identification of sites of O-GlcNAc modification on purified recombinant protein. However, so far no reports exist on enzymatic synthesis of O-GlcNAc-linked glycoproteins by directly transferring GlcNAc to proteins.
Zhang et al. (127) developed a novel strategy for the generation of homogeneous glycoprotein by selective incorporation of glycosylated amino acids (GlcNAc-β-(1-O)-serine) into proteins. An orthogonal M. Jannaschii tRNA synthetase (TyrRS) mutant was identified that selectively incorporated GlcNAc-β-(1-O)-serine into myoglobin. With the evolved TyrRS synthetase, milligram quantities of homogeneous glycoprotein was obtained upon coexpression of the synthetase, suppressor tRNA, and TAG-mutated myoglobin genes in E. coli in medium that contained the glycosylated amino acid. This approach has advantages of high selectivity, efficiency, and high yield without the generation of other glycoforms or unmodified forms of myoglobin.
O-GalNAc-modified glycoproteins
Mucin-type O-linked glycoproteins that are involved in varieties of biologic interactions in higher eukaryotes are α-O-GalNAc modified. Strategies for the detection and study of O-GalNAc-modified proteins have been developed (128-131). Bertozzi et al. have developed a metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation, a unique azide chemical tag introduced permits the identification of O-GalNac modified proteins from complex cell lysates (128).
O-GalNAc-linked glycopeptides have been synthesized using glycosidases. Ajisaka et al. (132) recently used the transglycosylation activity of an endo-α-N-acetylgalactosaminidase from Streptomyces sp. for the synthesis of O-linked glycopeptides. Using Galβ1,3GalNAcβpNP as the glycosyl donor, this enzyme can transfer the disaccharide to a serine in a hexapeptide and produced a Galβ1,3GalNAc-linked hexapeptide in 11% yield. The synthesis of α-GalNAc derivatives has been achieved by an exoglycosidase α-N-acetylgalactosaminidases from beef liver and Aspergillus. GalNAc-α-(1-O)-serine was prepared in 5% yield using GalNAcapNP as the donor substrate and free serine as the acceptor (133).
Another strategy is a glycosyltransferase-involved chemoenzymatic method; mono- or disaccharide glycopeptide is synthesized chemically using SPPS (solid-phase peptide synthesis) and appropriate enzymes are then used to elaborate the core mono- or disaccharide to provide targeted glycopeptides. Glycosyl- transferases are also often used in the elaboration of the existing glycans on glycopeptides and glycoproteins. For example, a sulfated N-terminal fragment of PSGL-1 (P-selection glycoprotein ligand 1, which is an important cell surface glycoprotein counter-receptor) has been prepared using this method (134). A disaccharide-sulfated octapeptide was synthesized followed by the catalysis of a series of glycosyltransferases to provide the final product as a sulfated glycopeptide carrying a pentasaccharide side chain. Two glycoforms of PSGL-1 have also been obtained from a monosaccharide glycopeptide by using six gly- cosyltransferases and one sulfotransferase (135).
Modification of protein drugs by covalent attachment of polyethylene glycol (PEG) can prolong the half-life and enhance the pharmacodynamics of therapeutic proteins. DeFrees et al. (136) recently developed a novel approach for site-directed PE-Gylation using glycosyltransferases to attach PEG to O-glycans. The process involves enzymatic GalNAc glycosylation at specific serine and threonine residues in non-glycosylated proteins expressed in Escherichia coli, followed by the enzymatic transfer of sialic acid conjugated with PEG to the introduced GalNAc residues. The strategy was applied to three therapeutic polypeptides, G-CSF, IFN-α2b, and GM-CSF. Selective addition of sialic acid-PEG to O-linked GalNAc on a protein provides a novel, highly site-selective mechanism for PEGylation, which enables the manufacturing of long-acting protein drugs with greater structural homogeneity as compared with PEGylated proteins prepared by conventional chemical methods (137).
N-linked glycoproteins
N-linked glycoproteins are found in a wide range of organisms ranging from archaea to mammals and other eukaryotes (138). The major method for enzymatic synthesis of N-linked glycopeptides/glycoproteins is to elaborate the existing sugar moieties on the proteins.
Glycosidases are used to trim the existing glycan structures, other sugars can then be put back on by glycosyltransferases (this process is called glycoprotein remodeling). For example, the synthesis of homogeneous unnatural glycoform of ribonuclease B (Rnase-B, which presents a mixture of high-mannose glycoforms) was achieved by endo H degradation to GlcNAcβ-Asn followed by elaboration with a galactosyltransferase, a fucosyltransferases, and a sialyltransferase to form sLex glycoform (139).
Some endo-β-N-acetylglucosaminidase (ENGase), such as Endo-A from Arthrobacter protophormiae (140) and Endo-M from Mucor hiemalis (141), have transglycosylation activities. They can transfer a large intact oligosaccharide to a GlcNAc-peptide acceptor in a single step. This methodology has become very attractive for the production of complex glycopeptides. For example, Endo A-catalyzed transglycosylation has been applied to transfer the Man9GlcNAc from naturally derived Man9GlcNAc2-Asn to the partially deglycosylated Rnase-B (142), a native N-link GlcNAc-Asn containing pentapeptide (143), and to a nonnative C-linked analog (144). However, a low transglycosylation yield (generally 5-20%) and the product hydrolysis limit its application.
A recent development has advanced enzymatic synthesis of N-linked glycoproteins. Fujita et al. (145) reported that the disaccharide oxazoline of Manβ1,4GlcNAc could serve as a substrate for ENGase-catalyzed transglycosylation. Wang et al. then explored sugar oxazolines as donor substrates for Endo A and Endo M in the N-glycopeptide synthesis using large peptides and partially deglycosylated Rnase B as model systems (146-148). They found that Endo-A and Endo-M both can effectively catalyze the reaction between the synthetic oligosaccharide oxazoline and the GlcNAc-heptapeptide to form the glycopeptide in 82% and 78% yields, respectively. The newly formed glycosidic link was indicated to be a β1,4-type by NMR analysis, which confirms that the ENGase-catalyzed transglycoslation using sugar oxazoline as the glycosyl donor proceeded in a stereo- and regiospecific manner to form the desired glycopeptide (147). The ENGase-catalyzed transglycosylation method has also been successfully employed in constructing glycoproteins. Incubation of the tetra- or hexasaccharide oxazoline with homogeneous GlcNAc-Rnase B in the presence of Endo-A afforded the glycoproteins in 82% and 96% yield, respectively (146).
Hamilton et al. (149) reported a new approach to produce complex human N-glycosylation glycoproteins using a yeast-based protein expression system. After eliminating some endogenous yeast glycosylation pathways, five active eukaryotic proteins were properly expressed and localized in the yeast Pichia pastoris. Targeted localization of the enzymes enabled the generation of human glycoproteins with homogeneous N-Glycan structures. This is a big step toward producing therapeutic glycoproteins. Glycoengineered yeast Pichia pastoris was also employed to produce human antibodies with specific human N-glycan structures (150) and human glycoproteins with complex terminally sialylated N-glycans (151).
Enzymatic Synthesis of Glycolipids
Glycosphingolipids (GSL) are amphipathic molecules that are synthesized by sequential actions of glycosyltransferases. Regarding the structural variation of GSLs, the expression of enzymes acting on lactosylceramide (LacCer) is the ratedetermining step. Several genes that code for enzymes responsible for the synthesis of the carbohydrate moiety of glycosphingolipids have been recently identified.
Sialic acid-containing glycosphingolipids, called gangliosides, have various important biologic functions. In vertebrates, almost all gangliosides are synthesized from a common precursor, ganglioside GM3 (Neu5Acα2,3LacCer). GM3 is a major glycosphingolipid in the plasma membrane and is widely distributed in vertebrates. CMP-N–acetylneuraminic acid:lactosylceramide α2,3-sialyltransferase (GM3-synthase) specifically catalyzes the transfer of sialic acid to the nonreducing terminal sugar of lactosylceramide to form GM3. Studies on substrate specificity indicated that the purified GM3 synthase from rat liver exhibited a broader substrate specificity: the preferred acceptors have the general structure of saccharide β1,1ceramide, a disaccharide is preferred to a monosaccharide (152). The enzyme from rat brain could use galactosylceramide, asialoganglioside GM3 (GA2) as well as lactosylceramide as acceptors (153). In contrast, among all the glycosphingolipids tested only lactosylcermide serves as an acceptor for the sialylation catalyzed by the clone human GM3 synthase (154).
Carbohydrate chains on SGGLS (the HNK-1 epitope, named sulfoglucuronylglycolipid SGGL-1 and -2) are extended by stepwise reaction catalyzed by glycosyltransferases. Lc3Cer synthase is the key enzyme for the expression of SGGLS in developing rat brain; it catalyzes the transfer of GlcNAc to the Gal residue of LacCer with a β1,3-linkage to give Lc3Cer (GlcNAcβ1,3Galβ1,4Glcβ1,1Cer) (155). A new member of the UDP-N-acetylglucosamine: β-galactose β1,3-GlcNAcT (β3Gn-T) family was cloned from rat and human cDNA libraries and named β3Gn-T5 based on its position in phylogenetic tree. β3Gn-T5 exhibited strong activity in transferring GlcNAc to glycolipid substrates, such as lactosylceramide (LacCer) and neolactotetraosylceramide (nLc4Cer; paragloboside), to produce Lc3Cer and neolactopentaosylceramide (nLc5Cer), respectively (156).
Globotriaosylceramide (Gb3Cer) is synthesized from LacCer by α1,4-galactosyltransferases. This glycolipid has been characterized on red blood cells as the Pk antigen of the P blood group system. The cloning of globotriaosylceramide (Gb3)/CD77 synthase (α1,4-galactosyltransferase) was achieved by Furukawa et al. The enzyme showed α1,4-galactosyltransferase activity only on lactosylceramide and galactosylceramide (157). The cloning of an α1,3-galactosyltransferase (iGb3 synthase) from a rat placental cDNA expression library was reported by Keusch et al. (158). The iGb3 synthase acts on lactosylceramide to form iGb3 (Galα1,3Galβ1,4Glcβ1,1Cer), initiating the synthesis of the isoglobo-series of GSL. This enzyme also has the ability to act on two other substrates found in different GSL series (the globoseries and the galactosylceramides).
Glycosphingolipids are a class of therapeutically valuable compounds that are extremely difficult to obtain through either chemical or traditional chemoenzymatic methods. Recently, glycosynthases have been generated from Thodococcus sp. endoglycoceramidase II, a β-endoglycosidase that cleaves the glycosidic link between the glycan and lipid moieties of glycosphingolipids, by substituting the nucleophilic residue E351 with Ser, Ala, and Gly. The E351 S mutant has been used for the synthesis of several lyso-glycosphingolipids, including GM1 and GM3 gangliosides, in yields up to 95% by condensing sphingolipids with a series of oligosaccharyl fluorides (159).
Glycosylation of Natural Products
Many biologically active natural products are glycosides. It is becoming more and more obvious that glycosylation affects bioactivity or selectivity of natural products, such as anticancer drugs and antibiotics (160, 161). Altering glycans has become a focus within natural product chemistry and pharmaceutical sciences. Recent developments in molecular glycobiology make it possible to develop new and more effective glycodrugs.
The glycopeptides vancomycin and teicoplanin are clinically important antibiotics. As the carbohydrate portions of these molecules affect biologic activity, developing efficient strategies to make carbohydrate derivatives is of interest in searching for vancomycin and teicoplanin analogs. Glycosyltransferases from Amycolatopsis orientalis were used by the Walsh group to produce variant sugar forms on both vancomycin and teicoplanin classes of glycopeptide antibiotics using nucleotide diphosphosugar (NDP-sugars) as the glycosyl donors (162). Although GalTs are generally perceived as unidirectional catalysts, very recently, Thorson et al. (163) reported that four glycosyltransferases from two distinct natural product biosynthetic pathways (calicheamicin and vancomycin) readily catalyzed reversible reactions, allowing sugars and aglycons to be exchanged with ease. Using these new reactions, more than 70 differentially glycosylated calicheamicin and vancomycin variants were produced.
The glycosyltransferase DesVII can catalyze the attachment of TDP-D-desosamine onto 12- and 14-membered macrolactone rings to make methymycin/neomethymycin and nar- bomycin/pikromycin, respectively, in Streptomyces venezuelae. The purified DesVII is active only in the presence of another protein, DesVIII, at high pH. YC-17 (10-deoxymethymycin) was synthesized in preparative scale using TDP-D-desosamine as the donor and 10-deoxymethynolide as aglycon acceptor (164).
Steroidal glycosides constitute a structurally and biologically diverse class of molecules such as cardenolides or saponins. They have received considerable recent attention because of their physiologic and pharmacologic activities. Synthesis of steroidal glycosides via enzymatic systems is still rare as most enzymes are not available. β-Galactosidase from Aspergillus oryzae was used in the synthesis of various cardiac glycosides which are unstable under basic or acidic condition during chemical synthesis (165). A periplogenin β-D-glucoside was prepared in 37% yield using the biotransformation of digitoxigenin by cultured Strophanthus hybrid cells (166). Thiem et al. have reported the enzymatic synthesis of the β-glucuronides of estradiol and ethynylestradiol on a preparative scale by incubating bovine liver UDP-glucuronyl transferase with corresponding phenolic aglycone substrates (167).
Peptidoglycan is a polymer of carbohydrate chains connected by peptide crosslinks. It is the major component in bacterial cell wall. The enzymes that synthesize peptidoglycan layers have received special attention because many known antibiotics function by blocking peptidoglycan synthesis (168). Among these enzymes, bacterial transglycosylases (TGases) are located on the external surface of the bacterial membrance where they polymerize lipid II, a disaccharide anchored to the membrane by a 55 carbon undecaprenyl chain (169, 170). Terrak et al. reported that the penicillin-binding protein (PBP) 1b of E. coli catalyzes the conversion of C55H89 lipid-transported disaccharide pentapeptide units into polymeric peptidoglycan (171). This bifunctional enzyme catalyzes both glycosylation for the formation of the carbohydrate backone of murein and transpeptidation for the formation of the interstrand peptide links (172).
Strategies in Enzymatic Synthesis of Carbohydrates and Glycoconjugates
Protein engineering of glycosidases and glycosyltransferases
Other than protein crystal structure-based construction of glycosidase mutants, such as glycosynthases, thioglycosynthases, and thioglycoligases discussed above, for the synthesis of carbohydrate-containing structures, protein crystal structurebased rational design of glycosylatransferase with altered substrate specificity has also been performed. In addition, directed evolution has emerged as a powerful tool in generating mutants with designed function.
Crystal structure-based rational design of glycosyltransferase mutants
Many X-ray crystal structures have been reported for glyco- syltransferases, which makes the structure-based redesign of glycosyltransferases feasible.
For example, crystallographic analysis of the Y289L mutation of P1,4GalT predicted that such a mutation should provide space for a C-2 N-acetyl group (173). Using the GalT tolerating the C-2 donor sugar substituents, Khidekel et al. (174) reported that Y289L mutant P1,4GalT could efficiently transfer a Gal analog with a ketone functionality at C-2 from donor to the O -GlcNAc glycosylated protein CREB. The ketone functionality on the Gal was used as a tag to identify O-GlcNAc posttranslational modified proteins.
Directed evolution
Wild-type enzymes are powerful tools in the synthesis of carbohydrates. Enzymatic synthesis using wild-type enzymes, however, is limited by the enzyme instability and the restriction on the substrates that can be recognized by the wild-type enzymes. Protein crystal structure-based rational design and mutants generated by site-directed mutagenesis provide some solution for the problems. Some properties of the mutants, however, cannot be obtained by rational design because of the limited information available about the structure-function relationship of proteins. Directed evolution has emerged as a promising approach to obtain enzymes with broader or altered substrate specificity. The key for a successful directed evolution process involves an efficient screening system to identify randomly generated mutants with desired properties. This has been a challenge for applying directed evolution approaches for glycosyltransferases. A recent report by Mayer et al. describes the development of a novel agar plate-based coupled-enzyme screen to select an improved glycosynthase form a library of mutants (175). Withers et al. developed a new fluorescence-based high-throughput screening methodology for the directed evolution of sialyltransferases. Using this methodology, a library of >106 SaiT mutants was screened and a new sialyltransferase variant was discovered, which had more than 400-fold higher catalytic activities than the parent enzyme (176).
Feng et al. (177) reported the converting a β-glycosidase of Thermus thermophilus to a β-transglycosidase by directed evolution. Mutants possessing high transferase activity are identified by using a simple screening procedure. Using these mutants, self-condensation of nitrophenyl glycosides can reach nearly quantitative yield, whereas transglycosylation of maltose and cellobiose can reach 60% and 75%, respectively.
One-pot multiple-enzyme synthesis
Glycosyltransferases have been used in combination with other enzymes such as sulfotransferases, proteases, lipases, and acetyl- transferases to synthesize complex oligosaccharides. Most of these enzymatic reactions can be successfully achieved under similar conditions, which makes it possible to carry out a multiple-enzyme reaction in one-pot system to produce oligosaccharide and their derivatives. One-pot multiple-enzyme reaction can simplify the product purification process without the necessary of isolation of intermediates, thus avoiding the compound loss during the multiple purification steps. More important, it avoids the use of high cost sugar nucleotides and their analogs by using less-expensive starting materials.
Yu et al. (8, 67) recently reported a highly efficient and convenient one-pot three-enzyme chemoenzymatic approach for the synthesis of libraries of α2,6-linked and α2,3-linked sialosides containing naturally occurring as well as non-natural sialic acids. In this method, sialic acid modifications can be chemically introduced at the very beginning, onto the six carbon sugar precursors (ManNAc or mannose) of sialic acids. These ManNAc or mannose analogs can then be directly converted to naturally occurring and non-natural sialosides in one-pot using three enzymes, including a sialic acid aldolase, a CMP-sialic acid synthetase, and a sialyltransferase, without the isolation of intermediates (Fig. 2). Such process takes advantage of the relaxed substrate specificity of all the enzymes involved in the synthesis.
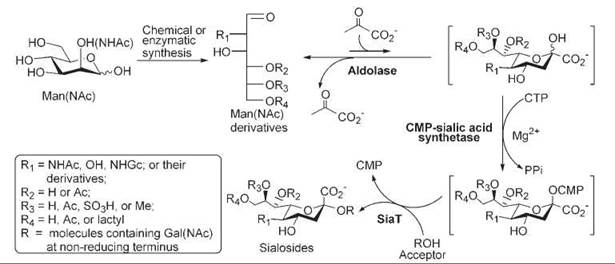
Figure 2. One-pot three-enzyme chemoenzymatic synthesis of sialosides containing sialic acid modifications. In this strategy, mannose or ManNAc derivatives are chemically or enzymatically synthesized. These compounds are then used by a recombinant E. coli K-12 sialic acid aldolase to obtain sialic acids and their derivatives followed by an N. meningitidis CMP-sialic acid synthetase for the formation of CMP-sialic acids. From which, sialic acids can be transferred to lactose, LacNAc, galactose, GalNAc, or their derivatives by a multifunctional P. multocida sialyltransferase (PmST1) or a P. damsela α2,6-sialyltransferase (Pd2,6ST) to form α2,3- or α2,6-linked sialosides in one pot without the isolation of intermediates.
Sugar nucleotide regeneration
The application of glycosyltransferases in the glycosidic link formation is limited because of the high cost of sugar nucleotides. This limitation has led to the development of sugar nucleotide recycling systems in enzymatic glycosylations. These regeneration systems require the use of only catalytic quantities of the sugar nucleotides, which can be regenerated continuously from inexpensive precursors, making the large-scale enzymatic synthesis of complex carbohydrates economically viable.
Sugar nucleotide regeneration systems have been developed by mimicking their biosynthetic pathways. For example, Wang et al. reported the incorporation of a UDP-GalNAc-4-epimerase in a UDP-GalNAc regeneration system, which in combination with a GalNAcT, was used in the synthesis of globotetraose and its derivatives (Fig. 3) (88).
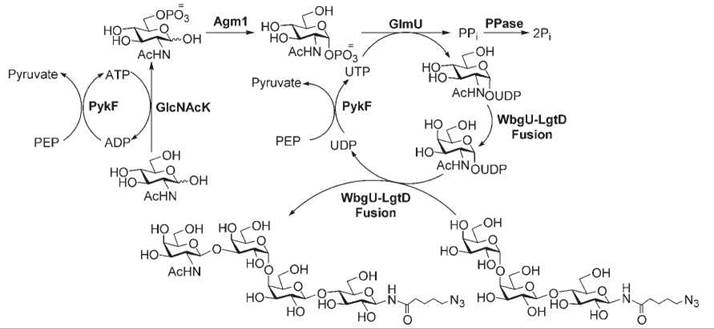
Figure 3. Enzymatic synthesis of globotetraose with in situ UDP-GlcNAc regeneration. The system contains UDP-GlcNAc pyrophosphorylase (GlmU), pyruvate kinase (PykF) and inorganic pyrophosphatase (PPase) from E. coli K12; GlcNAc phosphate mutase (Agm1) from S. cerevisiae; and GlcNAc kinase (GlcNAcK) from C. albicans.
Other examples of multi-enzyme systems with sugar nucleotide regeneration for large-scale economic synthesis of many oligosaccharides have also reported (76, 178). Trisaccharide Galα1,3Galβ1,4GlcNAcβO(CH2)8CO2Me was enzymatically synthesized by combining four enzymes (sucrose synthase, UDP-Glc 4'-epimerase, β1,4-GalT, and α1,3-GalT) in one pot using acceptor GlcNAcβ1-O(CH2)8CO2Me with in situ UDP-Gal regeneration (Fig. 4). This is an efficient and convenient method for the synthesis of Galα1,3Galβ1,4GlcNA epitope, which plays an important role in various biologic and immunologic processes.
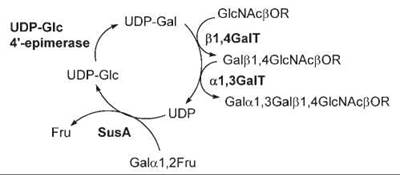
Figure 4. Enzymatic synthesis of Galα1,3Galβ1,4GlcNAcpOR with in situ UDP-Gal regeneration. Four enzymes are used in the synthesis: sucrose synthase (SusA), UDP-Glc 4'-epimerase, β1,4GalT, and α1,3GalT.
Solid-phase enzymatic synthesis
Solid-phase synthesis of oligosaccharides is a practical and convenient method as it simplifies the product purification and makes combinatorial process feasible (179). Combining the highly efficient enzyme-catalyzed glycosylation with solid-phase techniques offers a particularly convenient approach for the synthesis of oligosaccharides and glycoconjugates. The solid-phase enzymatic synthesis has been applied for sLex tetrasaccharide (180) and a biomedically important tetrasaccharide Neu5Acα2,3Galβ1,4GlcNAcβ1,3Gal (an inhibitor of the attachement of H. pylori to mucous cells) (181). Other polymer supported syntheses of oligosaccharides have also been reported. Nishimura et al. reported a new approach for highly efficient chemoenzymatic synthesis of glycopeptide using a combination of solid-phase and water-soluble supports (Fig. 5) (182). This approach was efficient and versatile for the synthesis of a glycopeptide library.

Figure 5. Chemoenzymatic synthesis of glycopeptide using a combination of solid-phase and water-soluble polymer supports.
Immobilized enzymes in the synthesis—superbeads
The use of immobilized enzymes in bioprocesses offers greater productivity because the same enzyme molecules can be used multiple times over a long period of time. Wang et al. reported a novel approach that transfers in vitro multiple enzyme sugar nucleotide regeneration systems onto solid beads (the superbeads) that can be used and reused as common synthetic reagents for production of glycoconjugates. First the multiple enzymes involved in the biosynthetic pathway for the regeneration of UDP-Gal were expressed as N-terminal His-tagged enzymes; these enzymes were then co-immobilized on Ni-NTA beads by taking the advantage of the affinity of the histdine tag and nickel-nitrilotriacetic acid (NTA) resin. The sugar nucleotide regeneration superbeads can then be conveniently combined with glycosyltransferases for the synthesis of specific oligosaccharides. Using the UDP-Gal regeneration beads (containing galactokinase, galactose-1-phosphate uridylyltrans- ferase, glucose-1-phosphate uridylyltransferase, and pyruvate kinase, which are required for the regeneration of UDP-Gal from UDP) with a truncated bovine α1,3-galactosyltransferase, the trisaccharide Galα1,3LacOBn was synthesized in 72% yield in gram-scale (183). Those beads were reused three times during a three-week period and still retained 90% enzyme activity. Also the nickel beads can be recharged for more use after removing the deactivated enzymes. The superbeads can also be used in combination with multiple immobilized glycosyltransferases to generate target oligosaccharides. For example, using 2 equiv of galactose as the starting sugar and GlcNAcβ1,3Galβ1,3GlcN3 as the glycosyl acceptor, a combination of α1,3GalT and β1,4GalT immobilized on the UDP-Gal superbeads was used to produce a pentasaccharide with an overall yield of 76% (183). Combined with galactosyltransferase, the superbeads become a very useful reagent to synthesize a variety of oligosaccharides and their derivatives.
Other examples of enzyme immobilization have also been reported. For example, the recombinant human α1,3/1,4-fucosyltransferase were immobilized on Ni2+-agarose through a 6His tag and exhibited a remarkable stability. It was exploited in the synthesis of Lea and Lex trisaccharides (184).
Cell-based synthesis
Metabolically engineered bacteria for the large-scale synthesis of a variety of oligosaccharides have been developed (185-187). Whole bacterial cells (single strain or coupled multiple strains), which overexpress the glycosyltransferases involved in the synthesis of the oligosaccharide and the genes involved in the sugar nucleotide synthesis, are used as catalysts.
A very efficient and economical whole cell-based bacterial coupling method for the synthesis of many sugar nucleotide donors and oligosaccharides has been developed (188). In the system for synthesizing globotriose, the C. ammoniagenes strain was engineered to synthesize UTP from inexpensive orotic acid. Coupling this strain with E. coli strains engineered to overexpress UDP-Gal biosynthetic genes and GalT resulted in the accumulation of globotriose in the reaction solution in high concentration (Fig. 6). This methodology has also been applied to the synthesis of sugar nucleotides and other oligosaccharides (75, 189, 190).
Drouillard et al. (92) developed a “living factory” to produce oligosaccharides in a single bacterium that overexpressing and maintaining the level of sugar nucleotides using the cellular machinery of E. coli. By overexpressing different glycosyl-transferases, various oligosaccharides have been synthesized, including fucosylated oligosaccharides, sialylated oligosaccharides, chitooligosaccharides, and human milk oligosaccharides.
Another approach by Chen et al. (186) used a single product-producing E. coli strain (superbug) containing all the genes required for the sugar nucleotide regeneration and oligosaccharide production assembled in a single plasmid. This superbug technology has been used in the synthesis of αGal epitope, P1 trisaccharide antigen, globotriose, and their derivatives. The superbug technology is cost effective because only catalytic amounts of the high energy phosphates are required. The use of a single bacterial strain instead of multiple strains avoids transport of reaction intermediates among strains and prevents the complication of maintaining the growth of different strains.
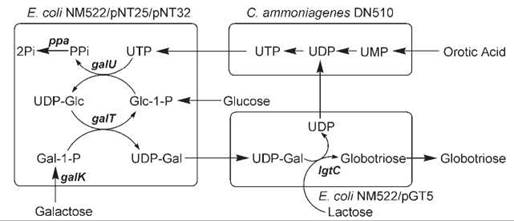
Figure 6. The production system of UDP-Gal and globotriose. NM522/pNT25/pNT32 cells express galactose-1-phosphate uridyltransferase (galT), galactokinase (galK), glucose-1-phosphate uridyltransferase (galU), and pyrophosphatase (ppa). NM522/pGT5 cells express α1,4-galactosyltransferase gene (IgtC). C. ammoniagenes cells produce uridine 5'-triphosphate (UTP) from orotic acid.
Conclusion And Perspective
Significant progress has been made over the past two decades for the application of enzymes in the synthesis of complex carbohydrate-containing biomolecules. Challenges are still exist. Taking advantage of the increasingly available genomic data, advancement in enzymatic synthesis of biomolecules relies on the identification and characterization of enzymes with better stability and novel or more flexible substrate specificity, a better understanding of the enzyme mechanism, genetic manipulation for tailor-made enzymes, and new methodology development.
References
1. Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 1993; 3:97-130.
2. Wong CH, Whitesides GM. Glycosidases. In Wong CH, Whitesides GM, eds. Enzymes in Synthetic Organic Chemistry. 2000 Science, Pergamon, Oxford, pp. 283-290.
3. Chen X, Kowal P, Wang PG. Large-scale biosynthesis of oligosaccharides. Curr. Opin. Drug. Discov. Devel. 2000; 3:756- 763.
4. Borriss R, Krah M, Brumer H, Kerzhner MA, Ivanen DR, Eneyskaya EV, Elyakova LA, Shishlyannikov SM, Shabalin KA, Neustroev KN. Enzymatic synthesis of 4-methylumbelliferyl (1→3)-β-D-glucooligosaccharides-new substrates for β-1,3-1,4- D-glucanase. Carbohydr. Res. 2003; 338:1455-1467.
5. Murata T, Honda H, Hattori T, Usui T. Enzymatic synthesis of poly-A-acetyllactosamines as potential substrates for endo-β-galactosidase-catalyzed hydrolytic and transglycosylation reactions. Biochim. Biophys. Acta 2005; 1722:60-68.
6. Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem. Rev. 2002; 102:439-469.
7. Schauer R. Achievements and challenges of sialic acid research. Glycoconj. J. 2000; 17:485-499.
8. Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Highly efficient chemoenzymatic synthesis of naturally occurring and non-natural α-2,6-linked sialosides: a P. damsela α-2,6-sialyltransferase with extremely flexible donor-substrate specificity. Angew. Chem. Int. Ed. Engl. 2006; 45:3938-3944.
9. Schmidt D, Sauerbrei B, Thiem J. Chemoenzymatic synthesis of sialyl oligosaccharides with sialidases employing transglycosylation methodology. J. Org. Chem. 2000; 65:8518-8526.
10. Thiem J, Sauerbrei B. Chemoenzymatic syntheses of sialy-loligosaccharides with immobilized sialidase. Angew. Chem. Int. Ed. Engl. 1991; 30:1503-1505.
11. Vetere A, Miletich M, Bosco M, Paoletti S. Regiospecific glycosidase-assisted synthesis of lacto-A-biose I (Galβ1-3GlcNAc) and 3’-sialyl-lacto-A-biose I (NeuAcα2-3Galβ1-3GlcNAc). Eur. J. Biochem. 2000; 267:942-949.
12. Lee S, Kim B. trans-Sialidase catalyzed sialylation of β-galacto-syldisaccharide with an introduction of β-galactosidase. Enzyme Microb. Technol. 2001; 28:161-167.
13. Kaftzik N, Wasserscheid P, Kragl U. Use of ionic liquids to increase the yield and enzyme stability in the β-galactosidase catalysed synthesis of A-acetyllactosamine. Org. Proc. Res. Dev. 2002; 6:553-557.
14. Fujimoto H, Miyasato M, Ito Y, Sasaki T, Ajisaka K. Purification and properties of recombinant beta-galactosidase from Bacillus circulans. Glycoconj. J. 1998; 15:155-160.
15. Yoon JH, Ajisaka K. The synthesis of galactopyranosyl derivatives with beta-galactosidases of different origins. Carbohydr. Res. 1996; 292:153-163.
16. Schroder S, Schmidt U, Thiem J, Kowalczyk J, Kunz M, Vogel M. Synthesis of oligosaccharides as potential novel food components and upscaled enzymatic reaction employing the β-galactosidase from bovine testes. Tetrahedron 2004; 60:2601-2608.
17. Chen X, Andreana PR, Wang, PG. Carbohydrates in transplantation. Curr. Opin. Chem. Biol. 1999; 3:650-658.
18. Vic G, Scigelova M, Hastings JJ, Howarth OW, Crout DHG. Glycosidase-catalysed synthesis of oligosaccharides: trisaccharides with the α-D-gal-(1→3)-D-gal terminus responsible for the hyperacute rejection response in cross-species transplant rejection from pigs to man. Chem. Commun. 1996; 12:1473-1474.
19. Matsuo I, Fujimoto H, Isomura M, Ajisaka K. Chemoenzymatic synthesis of Gal alpha 1-3Gal, Gal alpha 1-3Gal beta 1-4GlcNAc, and their PEG-conjugates. Bioorg. Med. Chem. Lett. 1997; 7:255-258.
20. Weignerova L, Sedmera P, Hunkova Z, Halada P, Kren V, Casali M, Riva S. Enzymatic synthesis of iso-globotriose from partially protected lactose. Tetrahedron Lett. 1999; 40:9297-9299.
21. Singh S, Scigelova M, Crout DHG. Glycosidase-catalysed synthesis of α-galactosyl epitopes important in xenotransplantation and toxin binding using the α-galactosidase from Penicillium multicolor. Chem. Commun. 1999: 2065-2066.
22. Biely P, Puchart V, Cote GL. Enzymic α-galactosylation of a cyclic glucotetrasaccharide derived from alternan. Carbohydr. Res. 2001; 332:299-303.
23. Li J, Robertson DE, Short JM, Wang PG. Chemical and enzymatic synthesis of glycoconjugates. 5: one-pot regioselective synthesis of bioactive galactobiosides using a CLONEZYME thermophilic glycosidase library. Bioorg. Med. Chem. Lett. 1999; 9:35-38.
24. Laroute V, Willemot RM. Glucoside synthesis by glucoamylase or glucosidase in organic solvents. Biotechnol. Lett. 1992; 14:169-174.
25. Balogh T, Boross L, Kosary J. Novel reaction systems for the synthesis of O-glucosides by enzymatic reverse hydrolysis. Tetrahedron 2004; 60:679-682.
26. Kurosu J, Sato T, Yoshida K, Tsugane T, Shimura S, Kirimura K, Kino K, Usami S. Enzymatic synthesis of alpha-arbutin by alpha-anomer-selective glucosylation of hydroquinone using lyophilized cells of Xanthomonas campestris WU-9701. J. Biosci. Bioeng. 2002; 93:328-330.
27. Fourage L, Colas B. Synthesis of beta-D-glucosyl- and beta-D-fucosyl-glucoses using beta-glycosidase from Thermus thermophilus. Appl. Microbiol. Biotechnol. 2001; 56:406-410.
28. Smaali MI, Michaud N, Marzouki N, Legoy MD, Maugard T. Comparison of two β-glucosidases for the enzymatic synthesis of β-(1-6)-β-(1-3)-gluco-oligosaccharides. Biotechnol. Lett. 2004; 26:675-679.
29. Kobayashi I, Tokuda M, Hashimoto H, Konda T, Nakano H, Kitahata S. Purification and characterization of a new type of a-glucosidase from Paecilomyces lilacinus that has transgluco- sylation activity to produce α-1,3- and α-1,2-linked oligosaccharides. Biosci. Biotechnol. Biochem. 2003; 67:29-35.
30. Okada M, Nakayama T, Noguchi A, Yano M, Hemmi H, Nishino T, Ueda T. Site-specific mutagenesis at positions 272 and 273 of the Bacillus sp SAM1606 alpha-glucosidase to screen mutants with altered specificity for oligosaccharide production by transglucosylation. J. Mol. Catal. B: Enzym. 2002; 16:265-274.
31. Athanasopoulos VI, Niranjan K, Rastall RA. Regioselective synthesis of mannobiose and mannotriose by reverse hydrolysis using a novel 1,6-α-D-mannosidase from Aspergillus phoenicis. J. Mol. Catal. B: Enzym. 2004; 27:215-219.
32. Vic G, Hastings JJ, Crout DHG. Glycosidase-catalysed synthesis of glycosides by an improved procedure for reverse hydrolysis: Application to the chemoenzymatic synthesis of galactopyranosyl-(1→4)-O-alpha-galactopyranoside derivatives. Tetrahedron-Asymmetry 1996; 7:1973-1984.
33. Nishio T, Hoshino S, Kondo A, Ogawa M, Matsuishi Y, Kitagawa M, Kawachi R, Oku T. alpha-Mannosidase-catalyzed synthesis of a (1→2)-alpha-D-rhamnodisaccharide derivative. Carbohydr. Res. 2004; 339:1389-1393.
34. Ajisaka K, Fujimoto H, Miyasato M: An alpha-L-fucosidase from Penicillium multicolor as a candidate enzyme for the synthesis of a (1→3)-linked fucosyl oligosaccharides by transglycosylation. Carbohydr. Res. 1998; 309:125-129.
35. Ajisaka K, Shirakabe M. Regioselective synthesis of α-L-fucosyl-containing disaccharides by use of α-L-fucosidases of various origins. Carbohydr. Res. 1992; 224:291-299.
36. Vetere A, Galateo C, Paoletti S. All-aqueous, regiospecific transg-lycosylation synthesis of 3-O-alpha-L-fucopyranosyl-2-acetamido-2-deoxy-D-glucopyranose, a building block for the synthesis of branched oligosaccharides. Biochem. Biophys. Res. Commun. 1997; 234:358-361.
37. Svensson SCT, Thiem J. Purification of α-fucosidase by C-glycosylic affinity chromatography, and the enzymic synthesis of α-fucosyl disaccharides Carbohydr. Res. 1990; 200:391-402.
38. Zeng XX, Murata T, Usui T. Glycosidase-catalyzed synthesis of fucosyl di- and trisaccharide derivatives using alpha-L-fucosidase from Alcaligenes sp. J. Carbohydr. Chem. 2003; 22:309-316.
39. Cobucci-Ponzano B, Trincone A, Giordano A, Rossi M, Moracci M. Identification of the catalytic nucleophile of the family 29 alpha-L-fucosidase from Sulfolobus solfataricus via chemical rescue of an inactive mutant. Biochemistry 2003; 42:9525-9531.
40. Nilsson KGI, Eliasson A, Larssonforek U. Production of glucosamine containing disaccharides of the lewis-A and lewis-X types employing glycosidases. Biotechnol. Lett. 1995; 17:717-722.
41. Nilsson KGI, Pan HF, Larsson-Lorek U. Syntheses of modified carbohydrates with glycosidases: Stereo- and regiospecific syntheses of lactosamine derivatives and related compounds. J. Carbohydr. Chem. 1997; 16:459-477.
42. Kobayashi T, Adachi S, Matsuno R. Synthesis of alkyl fucosides through beta-glucosidase-catalyzed condensation of fucose and 1-alcohols. Biotechnol. Lett. 1999; 21:105-109.
43. Lirdprapamongkol K, Svasti J. Alkyl glucoside synthesis using Thai rosewood beta-glucosidase. Biotechnol. Lett. 2000; 22:1889-1894.
44. Srisomsap C, Subhasitanont P, Techasakul S, Surarit R, Svasti J. Synthesis of homo- and hetero-oligosaccharides by Thai rosewood beta-glucosidase. Biotechnol. Lett. 1999; 21:947-951.
45. Scigelova M, Crout DHG. Microbial β-N-acetylhexosaminidases and their biotechnological applications. Enzyme Microb. Technol. 1999; 25:3-14.
46. Scigelova M, Singh S, Crout DHG. Glycosidases-a great synthetic tool. J. Mol. Catal. B: Enzym. 1999; 6:483-494
47. Singh S, Packwood J, Samuel CJ, Critchley P, Crout DHG. Glycosidase-catalysed oligosaccharide synthesis: preparation of N-acetylchitooligosaccharides using the β-N-acetylhexosamini- dase of Aspergillus oryzae. Carbohydr. Res. 1995; 279:293-305.
48. Kadowaki S, Saskiawan I, Watanabe J, Yamamoto K, Bunno M, Ichihara Y, Kumagai H. Transglycosylation activity of β-N-acetylhexosaminidase from Penicillium oxalicum and its application to synthesis of a drug carrier. J. Ferm. Bioeng. 1997; 83:341-345.
49. Uzawa H, Zeng X, Minoura N. Synthesis of 6’-sulfodisaccharides by β-N-acetylhexosaminidase-catalyzed transglycosylation. Chem. Commun. 2003; 34:100-101.
50. Eneyskaya EV, Brumer H, Backinowsky LV, Ivanen DR, Kulminskaya AA, Shabalin KA, Neustroev KN. Enzymatic synthesis of β-xylanase substrates: transglycosylation reactions of the β-xylosidase from Aspergillus sp. Carbohydr. Res. 2003; 338:313-325.
51. Moracci M, Ponzano BC, Trincone A, Fusco S, De Rosa M, van der Oost J, Sensen CW, Charlebois RL, Rossi M. Identification and molecular characterization of the first alpha-xylosidase from an Archaeon. J. Biol. Chem. 2000; 275:22082-22089.
52. Mackenzie LF, Wang QP, Warren RAJ, Withers SG. Glycosynthases: Mutant glycosidases for oligosaccharide synthesis. J. Am. Chem. Soc. 1998; 120:5583-5584.
53. Malet C, Planas A. From beta-glucanase to beta-glucansynthase: glycosyl transfer to alpha-glycosyl fluorides catalyzed by a mutant endoglucanase lacking its catalytic nucleophile. FEBS Lett. 1998; 440:208-212.
54. Honda Y, Kitaoka M. The first glycosynthase derived from an inverting glycoside hydrolase. J. Biol. Chem. 2006; 281:1426-1431.
55. Izumi M, Shen GJ, Wacowich-Sgarbi S, Nakatani T, Plettenburg O, Wong CH. Microbial glycosyltransferases for carbohydrate synthesis: α-2,3-sialyltransferase from Neisseria gonorrheae. J. Am. Chem. Soc. 2001; 123:10909-10918.
56. Boons GJ, Demchenko AV. Recent advances in O-sialylation. Chem. Rev. 2000; 100:4539-4566.
57. Chappell MD, Halcomb RL. Enzyme-catalyzed synthesis of oligosaccharides that contain functionalized sialic acids. J. Am. Chem. Soc. 1997; 119:3393-3394.
58. Gross HJ, Bunsch A, Paulson JC, Brossmer R. Activation and transfer of novel synthetic 9-substituted sialic acids. Eur. J. Biochem. 1987; 168:595-602.
59. Dufner G, Schworer R, Muller B, Schmidt RR. Base- and sugar-modified cytidine monophosphate N-acetylneuraminic acid (CMP-Neu5Ac) analogues - synthesis and studies with (2-6)- sialyltransferase from rat liver. Eur. J. Org. Chem. 2000; 14671482.
60. Baisch G, Ohrlein R. Glycosyl-transferase catalyzed assemblage of sialyl-Lewis(x)-saccharopeptides. Bioorg. Med. Chem. 1998; 6:1673-1682.
61. Wlasichuk KB, Kashem MA, Nikrad PV, Bird P, Jiang C, Venot AP. Determination of the specificities of rat liver Gal(beta 1→4)GlcNAc alpha 2,6-sialyltransferase and Gal(beta 1→3/4) GlcNAc alpha 2,3-sialyltransferase using synthetic modified acceptors. J. Biol. Chem. 1993; 268:13971-13977.
62. Schwardt O, Gao G, Visekruna, T, Rabbani S, Gassmann E, Ernst B. Substrate specificity and preparative use of recombinant rat ST3Gal III. J. Carbohydr. Chem. 2004; 23:1-26.
63. Gross HJ, Rose U, Krause JM, Paulson JC, Schmid K, Feeney RE, Brossmer R. Transfer of synthetic sialic acid analogues to N - and O -linked glycoprotein glycans using four different mammalian sialyltransferases. Biochemistry 1989; 28:7386-7392.
64. Gilbert M, Bayer R, Cunningham AM, DeFrees S, Gao Y, Watson DC, Young NM, Wakarchuk WW. The synthesis of sialylated oligosaccharides using a CMP-Neu5Ac synthetase/sialyltransfer- ase fusion. Nat. Biotechnol. 1998; 16:769-772.
65. Vasiliu D, Razi N, Zhang Y, Jacobsen N, Allin K, Liu X, Hoffmann J, Bohorov O, Blixt O. Large-scale chemoenzymatic synthesis of blood group and tumor-associated poly-N-acetyllactos- amine antigens. Carbohydr. Res. 2006; 341:1447-1457.
66. Gilbert M, Brisson JR, Karwaski MF, Michniewicz J, Cunningham AM, Wu Y, Young NM, Wakarchuk WW. Biosynthesis of ganglioside mimics in Campylobacter jejuni OH4384. Identification of the glycosyltransferase genes, enzymatic synthesis of model compounds, and characterization of nanomole amounts by 600-mhz (1)h and (13)c NMR analysis. J. Biol. Chem. 2000; 275:3896-3906.
67. Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. A multifunctional Pasteurella multocida sialyltransferase: a powerful tool for the synthesis of sialoside libraries. J. Am. Chem. Soc. 2005; 127:17618-17619.
68. Ni L, Sun M, Yu H, Chokhawala H, Chen X, Fisher AJ. Cytidine 5’-monophosphate (CMP)-induced structural changes in a multifunctional sialyltransferase from Pasteurella multocida. Biochemistry 2006; 45:2139-2148.
69. Yamamoto T, Nagae H, Kajihara Y, Terada I. Mass production of bacterial alpha 2,6-sialyltransferase and enzymatic syntheses of sialyloligosaccharides. Biosci. Biotechnol. Biochem. 1998; 62:210-214.
70. Ohrlein R. Glycosyltransferase-catalyzed synthesis of non-natural oligosaccharides. Top. Curr. Chem. 1999; 200:227-254.
71. Srivastava G, Hindsgaul O, Palcic MM. Chemical synthesis and kinetic characterization of UDP-2-deoxy-D-lyxo-hexose(UDP-2-deoxy-D-galactose), a donor-substrate for β-(1→4)-D-galacto- syltransferase. Carbohydr. Res. 1993; 245:137-144.
72. Kajihara Y, Endo T, Ogasawara H, Kodama H, Hashimoto H. Enzymatic transfer of 6-modified D-galactosyl residues - synthesis of biantennary penta-saccharides and hepta-saccharides having 2,6-deoxy-D-galactose residues at the nonreducing end and evaluation of 6-deoxy-D-galactosyl transfer to glycoprotein using bovine β-(1→4)-galactosyltransferase and UDP-6-deoxy-D- galactose. Carbohydr. Res. 1995; 269:273-294.
73. Logan SM, Conlan JW, Monteiro MA, Wakarchuk WW, Altman E. Functional genomics of Helicobacter pylori: identification of a β-1,4 galactosyltransferase and generation of mutants with altered lipopolysaccharide. Mol. Microbiol. 2000; 35:1156-1167.
74. Wakarchuk WW, Cunningham A, Watson DC, Young NM. Role of paired basic residues in the expression of active recombinant galactosyltransferases from the bacterial pathogen Neisseria meningitidis. Protein. Eng. 1998; 11:295-302.
75. Endo T, Koizumi S, Tabata K, Kakita S, Ozaki A. Large-scale production of N-acetyllactosamine through bacterial coupling. Carbohydr. Res. 1999; 316:179-183.
76. Fang J, Li J, Chen X, Zhang Y, Wang J, Guo Z, Zhang W, Yu L, Brew K, Wang PG. Highly efficient chemoenzymatic synthesis of a-Galactosyl epitopes with a recombinant α(1→3)-galactosyl- transferase. J. Am. Chem. Soc. 1998; 120:6635-6638.
77. Sujino K, Uchiyama T, Hindsgaul O, Seto NOL, Wakarchuk WW, Palcic MM. Enzymatic synthesis of oligosaccharide analogues: UDP-Gal analogues as donors for three retaining a- galactosyltransferases. J. Am. Chem. Soc. 2000; 122:1261-1269.
78. Lougheed B, Ly HD, Wakarchuk WW, Withers SG. Glycosyl fluorides can function as substrates for nucleotide phosphosugar-dependent glycosyltransferases. J. Biol. Chem. 1999; 274:37717- 37722.
79. Linton D, Gilbert M, Hitchen PG, Dell A, Morris HR, Wakarchuk WW, Gregson NA, Wren BW. Phase variation of a β-1,3 galactosyltransferase involved in generation of the ganglioside GM1-like lipo-oligosaccharide of Campylobacter jejuni. Mol. Microbiol. 2000; 37:501-514.
80. Hennet T, Dinter A, Kuhnert P, Mattu TS, Rudd PM, Berger EG. Genomic cloning and expression of three murine UDP-galactose: β-N-acetylglucosamine β1,3-galactosyltransferase genes. J. Biol. Chem. 1998; 273:58-65.
81. Okajima T, Nakamura Y, Uchikawa M, Haslam DB, Numata SI, Furukawa K, Urano T, Furukawa K.Expression cloning of human globoside synthase cDNAs. Identification of beta 3Gal-T3 as UDP-N-acetylgalactosamine: globotriaosylceramide beta 1,3-N-acetylgalactosaminyltransferase. J. Biol. Chem. 2000; 275:40498-40503.
82. Ju T, Brewer K, D’Souza A, Cummings RD, Canfield WM. Cloning and expression of human core 1 β1,3-galactosyltrans- ferase. J. Biol. Chem. 2002; 277:178-186.
83. Ju T, Cummings RD, Canfield WM. Purification, characterization, and subunit structure of rat core 1 β1,3-galactosyltransferase. J. Biol. Chem. 2002; 277:169-177.
84. Kudo T, Iwai T, Kubota T, Iwasaki H, Takayma Y, Hiruma T, Inaba N, Zhang Y, Gotoh M, Togayachi A, Narimatsu H. Molecular cloning and characterization of a novel UDP-Gal:GalNAc(alpha) peptide p1,3-galactosyltransferase (C1Gal-T2), an enzyme synthesizing a core 1 structure of O-glycan. J. Biol. Chem. 2002; 277:47724-47731.
85. Kremer L, Dover LG, Morehouse C, Hitchin P, Everett M, Morris HR, Dell A, Brennan PJ, McNeil MR, Flaherty C, Duncan K, Besra GS. Galactan biosynthesis in mycobacterium tuberculosis. Identification of a bifunctional UDP-galactofuransoyltransferase. J. Biol. Chem. 2001; 276:26430-26440.
86. Rose NL, Completo GC, Lin SJ, McNeil M, Palcic MM, Lowary TL. Expression, purification, and characterization of a galactofuranosyltransferase involved in Mycobacterium tuberculosis arabinogalactan biosynthesis. J. Am. Chem. Soc. 2006; 128:6721-6729.
87. Renaudie L, Daniellou R, Auge C, Le Narvor C. Enzymatic supported synthesis of lacto-N-neotetraose using dendrimeric polyethylene glycol. Carbohydr. Res. 2004; 339:693-698.
88. Shao J, Zhang J, Kowal P, Lu Y, Wang PG. Efficient synthesis of globoside and isogloboside tetrasaccharides by using β(1→3) N-acetylgalactosaminyltransferase/UDP-N-acetylglucosamine C4 epimerase fusion protein. Chem. Commun. 2003:1422-1423.
89. Blixt O, Vasiliu D, Allin K, Jacobsen N, Warnock D, Razi N, Paulson JC, Bernatchez S, Gilbert M, Wakarchuk W. Chemoen- zymatic synthesis of 2-azidoethyl-ganglio-oligosaccharides GD3, GT3, GM2, GD2, GT2, GM1, and GD1a. Carbohydr. Res. 2005; 340:1963-1972.
90. Gutierrez Gallego R, Dudziak G, Kragl U, Wandrey C, Kamerling JP, Vliegenthart JF. Enzymatic synthesis of the core-2 sialyl Lewis X O-glycan on the tumor-associated MUC1a’ peptide. Biochimie 2003; 85:275-286.
91. Westerlind U, Hagback P, Tidback B, Wiik L, Blixt O, Razi N, Norberg T. Synthesis of deoxy and acylamino derivatives of lactose and use of these for probing the active site of Neisseria meningitidis N -acetylglucosaminyltransferase. Carbohydr. Res. 2005; 340:221-233.
92. Drouillard S, Driguez H, Samain E. Large-scale synthesis of H-antigen oligosaccharides by expressing Helicobacter pylori α1,2-fucosyltransferase in metabolically engineered Escherichia coli cells. Angew. Chem. Int. Ed. Engl. 2006; 45:1778-1780.
93. Watt GM, Revers L, Webberley MC, Wilson IB, Flitsch SL. The chemoenzymatic synthesis of the core trisaccharide of N-linked oligosaccharides using a recombinant beta-mannosyltransferase. Carbohydr. Res. 1997; 305:533-541.
94. Tsuruta O, Yuasa H, Hashimoto H, Sujino K, Otter A, Li H, Palcic MM. Synthesis of GDP-5-thiosugars and their use as glycosyl donor substrates for glycosyltransferases. J. Org. Chem. 2003; 68:6400-6406.
95. Jahn M, Marles J, Warren RA, Withers SG. Thioglycoligases: mutant glycosidases for thioglycoside synthesis. Angew. Chem. Int. Ed. Engl. 2003; 42:352-354.
96. Stick RV, Stubbs KA. From glycoside hydrolases to thioglycoligases of thioglycosides. Tetrahedron: Asymmetry 2005; 16:321-335.
97. Kim YW, Lovering AL, Chen H, Kantner T, McIntosh LP, Strynadka NC, Withers SG. Expanding the thioglycoligase strategy to the synthesis of alpha-linked thioglycosides allows structural investigation of the parent enzyme/substrate complex. J. Am. Chem. Soc. 2006; 128:2202-2203.
98. Jahn M, Chen H, Mullegger J, Marles J, Warren RA, Withers SG. Thioglycosynthases: double mutant glycosidases that serve as scaffolds for thioglycoside synthesis. Chem. Commun. 2004; 274-275.
99. Rich JR, Szpacenko A, Palcic MM, Bundle DR. Glycosyltransferase-catalyzed synthesis of thiooligosaccharides. Angew. Chem. Int. Ed. Engl. 2004; 43:613-615.
100. Mano JF, Koniarova D, Reis RL. Thermal properties of thermoplastic starch/synthetic polymer blends with potential biomedical applicability. J. Mater. Sci. Mater. Med. 2003; 14:127-135.
101. Griffiths G, Cook NJ, Gottfridson E, Lind T, Lidholt K, Roberts IS. Characterization of the glycosyltransferase enzyme from the Escherichia coli K5 capsule gene cluster and identification and characterization of the glucuronyl active site. J. Biol. Chem. 1998; 273:11752-11757.
102. Hodson N, Griffiths G, Cook N, Pourhossein M, Gottfridson E, Lind T, Lidholt K, Roberts IS. Identification that KfiA, a protein essential for the biosynthesis of the Escherichia coli K5 capsular polysaccharide, is an alpha-UDP-GlcNAc glycosyltransferase. The formation of a membrane-associated K5 biosynthetic complex requires KfiA, KfiB, and KfiC. J. Biol. Chem. 2000; 275:27311-27315.
103. DeAngelis PL, White CL. Identification and molecular cloning of a heparosan synthase from Pasteurella multocida type D. J. Biol. Chem. 2002; 277:7209-7213.
104. DeAngelis PL. Microbial glycosaminoglycan glycosyltransferases. Glycobiology 2002; 12:9R-16R.
105. Kuberan B, Beeler DL, Lech M, Wu ZL, Rosenberg RD. Chemoenzymatic synthesis of classical and non-classical anticoagulant heparan sulfate polysaccharides. J. Biol. Chem. 2003; 278:52613-52621.
106. Chen J, Avci FY, Munoz EM, McDowell LM, Chen M, Pedersen LC, Zhang L, Linhardt RJ, Liu J. Enzymatic redesigning of biologically active heparan sulfate. J. Biol. Chem. 2005; 280:42817-42825.
107. DeAngelis PL, Jing W, Drake RR, Achyuthan AM. Identification and molecular cloning of a unique hyaluronan synthase from Pasteurella multocida. J. Biol. Chem. 1998; 273:8454-8458.
108. Hoshi H, Nakagawa H, Nishiguchi S, Iwata K, Niikura K, Monde K, Nishimura S: An engineered hyaluronan synthase: characterization for recombinant human hyaluronan synthase 2 Escherichia coli. J. Biol. Chem. 2004; 279:2341-2349.
109. De Luca C, Manfred L, Martini I, Crescenzi F, Shen GJ, O’Regan M, Wong CH. Enzymic synthesis of hyaluronic acid with regeneration of sugar nucleotides. J. Am. Chem. Soc. 1995; 117:5869-5870.
110. DeAngelis PL, Oatman LC, Gay DF. Rapid chemoenzymatic synthesis of monodisperse hyaluronan oligosaccharides with immobilized enzyme reactors. J. Biol. Chem. 2003; 278:35199- 35203.
111. Saitoh H, Takagaki K, Majima M, Nakamura T, Matsuki A, Kasai M, Narita H, Endo M. Enzymic reconstruction of glycosaminoglycan oligosaccharide chains using the transglycosylation reaction of bovine testicular hyaluronidase. J. Biol. Chem. 1995; 270:3741-3747.
112. Kobayashi S, Morii H, Itoh R, Kimura S, Ohmae M. Enzymatic polymerization to artificial hyaluronan: a novel method to synthesize a glycosaminoglycan using a transition state analogue monomer. J. Am. Chem. Soc. 2001; 123:11825-11826.
113. Kobayashi S, Fujikawa S, Ohmae M. Enzymatic synthesis of chondroitin and its derivatives catalyzed by hyaluronidase. J. Am. Chem. Soc. 2003; 125:14357-14369.
114. Cunningham BA, Hemperly JJ, Murray BA, Prediger EA, Brackenbury R, Edelman GM. Neural cell adhesion molecule: structure, immunoglobulin-like domains, cell surface modulation, and alternative RNA splicing. Science 1987; 236:799-806.
115. Cho JW, Troy FA. Polysialic acid engineering: synthesis of polysialylated neoglycosphingolipids by using the polysialyltransferase from neuroinvasive Escherichia coli K1. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:11427-11431.
116. Shen GJ, Datta AK, Izumi M, Koeller KM, Wong CH. Expression of alpha2,8/2,9-polysialyltransferase from Escherichia coli K92. Characterization of the enzyme and its reaction products. J. Biol. Chem. 1999; 274:35139-35146.
117. McGowen MM, Vionnet J, Vann WF. Elongation of alternating alpha 2,8/2,9 polysialic acid by the Escherichia coli K92 polysialyltransferase. Glycobiology 2001; 11:613-620.
118. Kobayashi S, Kashiwa K, Kawasaki T, Shoda S. Novel method for polysaccharide synthesis using an enzyme: the first in vitro synthesis of cellulose via a nonbiosynthetic path using cellulase as catalyst. J. Am. Chem. Soc. 1991; 113:3079-3084.
119. Kobayashi S, Shimada J, Kashiwa K, Shoda S. Enzymic polymerization of α-D-maltosyl fluoride using α-amylase as catalyst: a new approach for synthesis of maltooligosaccharides. Macromolecules 1992; 25:3237-3241.
120. Ziegast G, Pfannemuller B. Phosphorolytic syntheses with di-, oligo- and multifunctional primers. Carbohydr. Res. 1987; 160:185-204.
121. Kobayashi S. Wen X, Shoda S. Specific preparation of artificial xylan: a new approach to polysaccharide synthesis by using cellulase as catalyst. Macromolecules 1996; 29:2698-2700.
122. Kobayashi S. Ethylenimine polymers. Prog. Polym. Sci. 1990; 15:751-734.
123. Sakamoto J, Kobayashi S. Enzymatic synthesis of 3-O-Methylated chitin oligomers from new derivatives of a chitobiose oxazoline. Chem. Lett. 2004; 33:698-699.
124. Whelan SA, Hart GW. Proteomic approaches to analyze the dynamic relationships between nucleocytoplasmic protein glycosylation and phosphorylation. Circ. Res. 2003; 93:1047-1058.
125. Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13132-13137.
126. Vocadlo DJ, Hang HC, Kim EJ, Hanover JA, Bertozzi CR. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9116-9121.
127. Zhang Z, Gildersleeve J, Yang YY, Xu R, Loo JA, Uryu S, Wong CH, Schultz PG. A new strategy for the synthesis of glycoproteins. Science 2004; 303:371-373.
128. Hang HC, Yu C, Kato DL, Bertozzi CR. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:14846-14851.
129. Hang HC, Bertozzi CR. The chemistry and biology of mucin-type O-linked glycosylation. Bioorg. Med. Chem. 2005; 13:5021-5034.
130. Pratt MR, Hang HC, Ten Hagen KG, Rarick J, Gerken TA, Tabak LA, Bertozzi CR. Deconvoluting the functions of polypeptide N-alpha-acetylgalactosaminyltransferase family members by glycopeptide substrate profiling. Chem. Biol. 2004; 11:1009-1016.
131. Kato K, Takeuchi H, Kanoh A, Mandel U, Hassan H, Clausen H, Irimura T. N-acetylgalactosamine incorporation into a peptide containing consecutive threonine residues by UDP-N-acetyl-D-galactosaminide:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 2001; 11:821-829.
132. Ajisaka K, Miyasato M, Ishii-Karakasa I. Efficient synthesis of O-linked glycopeptide by a transglycosylation using endo alpha-N-acetylgalactosaminidase from Streptomyces sp. Biosci. Biotechnol. Biochem. 2001; 65:1240-1243.
133. Naundorf A, Ajisaka K. Purification of alpha-N-acetyl-galactosaminidase from Aspergillus niger and its use in the synthesis of GalNAc-alpha-(1→O)-serine. Enzyme Micro. Technol. 1999; 25:483-488.
134. Koeller KM, Smith MEB, Huang RF, Wong CH: Chemoenzymatic synthesis of a PSGL-1 N-terminal glycopeptide containing tyrosine sulfate and alpha-0-linked sialyl Lewis X. J. Am. Chem. Soc. 2000; 122:4241-4242.
135. Leppanen A, White SP, Helin J, McEver RP, Cummings RD. Binding of glycosulfopeptides to P-selectin requires stereospecific contributions of individual tyrosine sulfate and sugar residues. J. Biol. Chem. 2000; 275:39569-39578.
136. DeFrees S, Wang ZG, Xing R, Scott AE, Wang J, Zopf D, Gouty DL, Sjoberg ER, Panneerselvam K, Brinkman-Van der Linden EC, Bayer RJ, Tarp MA, Clausen H. GlycoPEGylation of recombinant therapeutic proteins produced in Escherichia coli. Glycobiology 2006; 16:833-843.
137. Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv. Drug. Deliv. Rev. 2002; 54:459-476.
138. Lechner J, Wieland F. Structure and biosynthesis of prokaryotic glycoproteins. Annu. Rev. Biochem. 1989; 58:173-194.
139. Witte K, Sears P, Martin R, Wong CH. Enzymatic glycoprotein synthesis: Preparation of ribonuclease glycoforms via enzymatic glycopeptide condensation and glycosylation. J. Am. Chem. Soc. 1997; 119:2114-2118.
140. Takegawa K, Yamaguchi S, Kondo A, Iwamoto H, Nakoshi M, Kato I, Iwahara S. Transglycosylation activity of endo-beta-N-acetylglucosaminidase from Arthrobacter protophormiae. Bio- chem. Int. 1991; 24:849-855.
141. Yamamoto K, Kadowaki S, Watanabe J, Kumagai H. Transglycosylation activity of Mucor hiemalis endo-beta-N-acetyl-glucosaminidase which transfers complex oligosaccharides to the N-acetylglucosamine moieties of peptides. Biochem. Biophys. Res. Commun. 1994; 203:244-252.
142. Takegawa K, Tabuchi M, Yamaguchi S, Kondo A, Kato I, Iwahara S. Synthesis of neoglycoproteins using oligosaccharide-transfer activity with endo-beta-N-acetylglucosaminidase. J. Biol. Chem. 1995; 270:3094-3099.
143. Deras IL, Takegawa K, Kondo A, Kato I, Lee YC. Synthesis of a high-mannose-type glycopeptide analog containing a glucose-asparagine link. Bioorg. Med. Chem. Lett. 1998; 8:1763-1766.
144. Wang LX, Tang M, Suzuki T, Kitajima K, Inoue Y, Inoue S, Fan JQ, Lee YC. Combined chemical and enzymatic synthesis of a C-glycopeptide and its inhibitory activity toward glycoamidases. J. Am. Chem. Soc. 1997; 119:11137-11146.
145. Fujita M, Shoda S, Haneda K, Inazu T, Takegawa K, Yamamoto K. A novel disaccharide substrate having 1,2-oxazoline moiety for detection of transglycosylating activity of endogly- cosidases. Biochim. Biophys. Acta. 2001; 1528:9-14.
146. Li B, Song H, Hauser S, Wang LX. A highly efficient chemoen- zymatic approach toward glycoprotein synthesis. Org. Lett. 2006; 8:3081-3084.
147. Li B, Zeng Y, Hauser S, Song H, Wang LX. Highly efficient endoglycosidase-catalyzed synthesis of glycopeptides using oligosaccharide oxazolines as donor substrates. J. Am. Chem. Soc. 2005; 127:9692-9693.
148. Li H, Li B, Song H, Breydo L, Baskakov IV, Wang LX. Chemoenzymatic Synthesis of HIV-1 V3 Glycopeptides Carrying Two N-Glycans and Effects of Glycosylation on the Peptide Domain. J. Org. Chem. 2005; 70: 9990-9996.
149. Hamilton SR, Bobrowicz P, Bobrowicz B, Davidson RC, Li H, Mitchell T, Nett JH, Rausch S, Stadheim TA, Wischnewski H, Wildt S, Gerngross TU. Production of complex human glycoproteins in yeast. Science 2003; 301:1244-1246.
150. Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, Bobrowicz P, Choi BK, Cook WJ, Cukan M, Houston-Cummings NR, Davidson R, Gong B, Hamilton SR, Hoopes JP, Jiang Y, Kim N, Mansfield R, Nett JH, Rios S, Strawbridge R, Wildt S, Gerngross TU. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat. Biotechnol. 2006; 24:210-215.
151. Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi BK, Hopkins D, Wischnewski H, Roser J, Mitchell T, Strawbridge RR, Hoopes J, Wildt S, Gerngross TU. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 2006; 313:1441-1443.
152. Melkerson-Watson LJ, Sweeley CC. Purification to apparent homogeneity by immunoaffinity chromatography and partial characterization of the GM3 ganglioside-forming enzyme, CMP-sialic acid:lactosylceramide alpha 2,3-sialyltransferase (SAT-1), from rat liver Golgi. J. Biol. Chem. 1991; 266:4448-4457.
153. Preuss U, Gu X, Gu T, Yu RK. Purification and characterization of CMP-N-acetylneuraminic acid: lactosylceramide (alpha 2-3) sialyltransferase (GM3-synthase) from rat brain. J. Biol. Chem. 1993; 268:26273-26278.
154. Ishii A, Ohta M, Watanabe Y, Matsuda K, Ishiyama K, Sakoe K, Nakamura M, Inokuchi J, Sanai Y, Saito M. Expression cloning and functional characterization of human cDNA for ganglioside GM3 synthase. J. Biol. Chem. 1998; 273:31652-31655.
155. Chou DK, Jungalwala FB. Characterization and developmental expression of lactotriosylceramide: galactosyltransferase for the synthesis of neolactotetraosylceramide in the nervous system. J. Neurochem. 1994; 62:307-314.
156. Togayachi A, Akashima T, Ookubo R, Kudo T, Nishihara S, Iwasaki H, Natsume A, Mio H, Inokuchi J, Irimura T, Sasaki K, Narimatsu H. Molecular cloning and characterization of UDP-GlcNAc:lactosylceramide beta 1,3-N-acetylglucosaminyl-transferase (beta 3Gn-T5), an essential enzyme for the expression of HNK-1 and Lewis X epitopes on glycolipids. J. Biol. Chem. 2001; 276:22032-22040.
157. Kojima Y, Fukumoto S, Furukawa K, Okajima T, Wiels J, Yokoyama K, Suzuki Y, Urano T, Ohta M, Furukawa K. Molecular cloning of globotriaosylceramide/CD77 synthase, a glycosyl- transferase that initiates the synthesis of globo series glycosphingolipids. J. Biol. Chem. 2000;275:15152-15156.
158. Keusch JJ, Manzella SM, Nyame KA, Cummings RD, Baenziger JU. Expression cloning of a new member of the ABO blood group glycosyltransferases, iGb3 synthase, that directs the synthesis of isoglobo-glycosphingolipids. J. Biol. Chem. 2000; 275:25308-25314.
159. Vaughan MD, Johnson K, DeFrees S, Tang X, Warren RA, Withers SG. Glycosynthase-mediated synthesis of glycosphingolipids. J. Am. Chem. Soc. 2006; 128:6300-6301.
160. Thorson JS, Hosted TJ, Jiang J, Biggins JB, Ahlert J. Natures carbohydrate chemists the enzymatic glycosylation of bioactive bacterial metabolites. Curr. Org. Chem. 2001; 5:139-167.
161. Dwek RA, Butters TD, Platt FM, Zitzmann N. Targeting glycosylation as a therapeutic approach. Nat. Rev. Drug. Discov. 2002; 1:65-75.
162. Losey HC, Peczuh MW, Chen Z, Eggert US, Dong SD, Pelczer I, Kahne D, Walsh CT. Tandem action of glycosyltransferases in the maturation of vancomycin and teicoplanin aglycones: novel glycopeptides. Biochemistry 2001; 40:4745-4755.
163. Zhang C, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li L, Thorson JS: Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science 2006; 313:1291- 1294.
164. Borisova SA, Zhao L, Melancon IC, Kao CL, Liu HW. Characterization of the glycosyltransferase activity of desVII: analysis of and implications for the biosynthesis of macrolide antibiotics. J. Am. Chem. Soc. 2004; 126:6534-6535.
165. Yasuhiro TH, Mitsuo N, Satoh T. Enzymic synthesis of chemically unstable cardiac glycosides by β-galactosidase from Aspergillus oryzae. Tetrahedron Lett. 1984; 25:2241-2244
166. Kawaguchi K, Koike S, Hirotani M, Fujihara M, Furuya T, Iwata R, Morimoto K. Biotransformation of digitoxigenin by cultured Strophanthus hybrid cells. Phytochemistry 1998; 47:1261-1265.
167. Werschkun B, Wendt A, Thiem T. Enzymic synthesis of β-glucuronides of estradiol, ethynylestradiol and other phenolic substrates on a preparative scale employing UDP-glucuronyl transferase. J. Chem. Soc., Perkin Trans 1. 1998; 3021-3023.
168. Wong KK, Pompliano DL. Peptidoglycan biosynthesis. Unexploited antibacterial targets within a familiar pathway. Adv. Exp. Med. Biol. 1998; 456:197-217.
169. Holtje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998; 62:181-203.
170. Schiffer G, Holtje JV/ Cloning and characterization of PBP 1C, a third member of the multimodular class A penicillin-binding proteins of Escherichia coli. J. Biol. Chem. 1999; 274:32031-32039.
171. Terrak M, Ghosh TK, van Heijenoort J, Van Beeumen J, Lampilas M, Aszodi J, Ayala JA, Ghuysen JM, Nguyen- Disteche M. The catalytic, glycosyl transferase and acyl transferase modules of the cell wall peptidoglycan-polymerizing penicillin-binding protein 1b of Escherichia coli. Mol. Microbiol. 1999; 34:350-364.
172. Schwartz B, Markwalder JA, Seitz SP, Wang Y, Stein RL. A kinetic characterization of the glycosyltransferase activity of Eschericia coli PBP1b and development of a continuous fluorescence assay. Biochemistry 2002; 41:12552-12561.
173. Ramakrishnan B, Qasba PK. Structure-based design of β1,4-galactosyltransferase I (β4Gal-T1) with equally efficient N-acetylgalactosaminyltransferase activity: point mutation broadens β4 gal-T1 donor specificity. J. Biol. Chem. 2002; 277:20833- 20839.
174. Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, Hsieh-Wilson LC. A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. J. Am. Chem. Soc. 2003; 125:16162-16163.
175. Mayer C, Jakeman DL, Mah M, Karjala G, Gal L, Warren RA, Withers SG. Directed evolution of new glycosynthases from Agrobacterium beta-glucosidase: a general screen to detect enzymes for oligosaccharide synthesis. Chem. Biol. 2001; 8:437-443.
176. Aharoni A, Thieme K, Chiu CP, Buchini S, Lairson LL, Chen H, Strynadka NC, Wakarchuk WW, Withers SG. High-throughput screening methodology for the directed evolution of glycosyl- transferases. Nat. Methods 2006; 3:609-614.
177. Feng HY, Drone J, Hoffmann L, Tran V, Tellier C, Rabiller C, Dion M. Converting a β-glycosidase into a β-transglycosidase by directed evolution. J. Biol. Chem. 2005; 280:37088-37097.
178. Hokke CH, Zervosen A, Elling L, Joziasse DH, VandenEijnden DH. One-pot enzymatic synthesis of the Gal alpha 1→3Cal beta 1→4GlcNAc sequence with in situ UDP-Gal regeneration. Glycoconjugate. 1996; 13:687-692.
179. Seeberger PH, Haase WC. Solid-phase oligosaccharide synthesis and combinatorial carbohydrate libraries. Chem. Rev. 2000; 100:4349-4394.
180. Blixt O, Norberg T. Solid-phase enzymatic synthesis of a sialyl lewis X tetrasaccharide on a sepharose matrix. J. Org. Chem. 1998; 63:2705-2710.
181. Halcomb RL, Huang HM, Wong CH. Solution- and solid-phase synthesis of inhibitors of H. pylori attachment and E-Selectin-mediated leukocyte adhesion. J. Am. Chem. Soc. 1994; 116: 11315-11322.
182. Fumoto M, Hinou H, Matsushita T, Kurogochi M, Ohta T, Ito T, Yamada K, Takimoto A, Kondo H, Inazu T, Nishimura S. Molecular transporter between polymer platforms: highly efficient chemoenzymatic glycopeptide synthesis by the combined use of solid-phase and water-soluble polymer supports. Angew. Chem. Int. Ed. Engl. 2005; 44:2534-2537.
183. Chen X, Fang J, Zhang J, Liu Z, Shao J, Kowal P, Andreana P, Wang PG. Sugar nucleotide regeneration beads (superbeads): a versatile tool for the practical synthesis of oligosaccharides. J. Am. Chem. Soc. 2001; 123:2081-2082.
184. Auge C, Malleron A, Lubineau A, Tahrat H, Marc A, Goergen JL, Cerutti M, Steelant WFA, Delannoy P. Outstanding stability of immobilized recombinant (1→3/4)-fucosyltransferases exploited in the synthesis of Lewis A and Lewis X trisaccharides Chem. Commun. 2000: 2017-2018.
185. Herrmann GF, Wang P, Shen GJ, Garciajunceda E, Khan SH, Matta KL, Wong CH. Large-Scale Production of Recombinant α-1,2-Mannosyltransferase from Escherichia-Coli for the Study of Acceptor Specificity and Use of the Recombinant Whole Cells in Synthesis. J. Org. Chem. 1994; 59:6356-6362.
186. Chen X, Zhang JB, Kowal P, Liu Z, Andreana PR, Lu YQ, Wang PG. Transferring a biosynthetic cycle into a productive Escherichia coli strain: large-scale synthesis of galactosides. J. Am. Chem. Soc. 2001; 123:8866-8867.
187. Samain E, Drouillard S, Heyraud A, Driguez H, Geremia RA: Gram-scale synthesis of recombinant chitooligosaccharides in Escherichia coli. Carbohydr. Res. 1997; 302:35-42.
188. Koizumi S, Endo T, Tabata K, Ozaki A. Large-scale production of UDP-galactose and globotriose by coupling metabolically engineered bacteria. Nat. Biotechnol. 1998; 16:847-850.
189. Tabata K, Koizumi S, Endo T, Ozaki A. Production of UDP-N-acetylglucosamine by coupling metabolically engineered bacteria. Biotechnol. Lett. 2000; 22:479-483.
190. Endo T, Koizumi S, Tabata K, Kakita S, Ozaki A. Large-scale production of the carbohydrate portion of the sialyl-Tn epitope, α-Neup5Ac-(2→6)-D-GalpNAc, through bacterial coupling. Carbohydr. Res. 2001; 330:439-443.
Further Reading
Davis BG. Synthesis of glycoproteins. Chem. Rev. 2002; 102:579-601.
Jahn M, Withers SG. New approaches to enzymatic oligosaccharide synthesis: glycosynthases and thioglycoligases. Biocatal. Biotransform. 2003; 21:159-166.
Liu L, Bennett CS, Wong, CH. Advances in glycoprotein synthesis. Chem. Commun. 2006; 1:21-33.
Perugino G, Cobucci-Ponzano B, Rossi M, Moracci M. Recent advances in the oligosaccharide synthesis promoted by catalytically engineered glycosidases. Adv. Synth. Catal. 2005: 347; 941-950.
Rowan AS, Hamilton CJ. Recent developments in preparative enzymatic syntheses of carbohydrates. Nat. Prod. Rep. 2006; 23:412-443.
Ruffing A, Chen RR. Metabolic engineering of microbes for oligosaccharide and polysaccharide synthesis. Microb. Cell Fact. 2006; 5:25.
Trincone A, Giordano A. Glycosyl hydrolases and glycosyltransferases in the synthesis of oligosaccharides. Curr. Org. Chem. 2006; 10:1163-1193.
See Also
Enzyme Catalysis, Chemical Strategies for
Glycan Biosynthesis in Mammals
Glycosyltransferases, Chemistry of
Glycolipids, Synthesis of
Glycopeptides and Glycoproteins, Synthesis of