CHEMICAL BIOLOGY
Epigenetic Modifications
Dori Huertas, Cancer Epigenetics and Biology Program, Catalan Institute of Oncology (ICO), Barcelona, Catalonia, Spain
Manel Esteller, Cancer Epigenetics Laboratory, Spanish National Cancer Centre (CNIO), Madrid, Spain
doi: 10.1002/9780470048672.wecb161
Epigenetic modifications play a fundamental role in chromatin structure and function. Histone modifications and DNA methylation regulate important biologic processes such as heterochromatin formation, genomic imprinting, X-chromosome inactivation, and transcriptional activation and silencing. Epigenetic mechanisms are responsible for a considerable part of the phenotype of complex organisms. The understanding of epigenetic modifications in chromatin may offer some clues to solve the mechanisms of cellular identity, tumorigenesis, stem cell plasticity, regeneration, and aging. These processes are of interest in the fields of human biology and human diseases. Here, we describe the principal components involved in epigenetic modifications, such as the enzymes that modify chromatin, the protein domains that recognize modified chromatin, some best-characterized downstream effectors, and the tools and techniques for studying epigenetics. However, the biological mechanism of the majority of components is still poorly understood, and the analysis of these components is fundamental to our understanding of epigenetics.
The identity of cells within an organism is determined by its heritable information, which is encoded by genetic and epigenetic information. Genetic information is the ordered sequence of nucleotides present in the genome of all cells of a given organism. Epigenetic information is based on epigenetic modifications, which are chromatin-covalent modifications responsible for maintaining stable states of gene expression through mitotic divisions without alterations in DNA sequence. Although genetic information is homogenous throughout all cells within an organism, epigenetic modifications vary according to developmental programs that define the transcriptome of each cell type and ultimately determine the identity of cells (1). The epigenetic information of cells is stored as covalent modifications of DNA and histones. The most common epigenetic modification of DNA is the methylation of the 5' position of cytosines in the context of CpG dinucleotide (2). In addition, epigenetic modifications of histones include a variety of complex post-translational modifications, of which lysine acetylation and methylation and serine phosphorylation are the best characterized.
In the last decade, remarkable progress has been made to characterize epigenetic modifications and to associate specific patterns of modification with chromatin functionality. In this review, we will discuss the nature of DNA and histone epigenetic modifications. We will describe the modifying enzymes responsible for creating or eliminating each modification and the proteins that read such epigenetic modifications and recruit functional protein complexes. These are the downstream effectors that regulate chromatin structure and DNA accessibility.
The functionality of some chromatin modification patterns is well established; however, the vast majority remains poorly understood. Given that histones are subject to more than 100 post-translational modifications, considerable efforts to characterize the mammalian epigenome are ongoing.
Biological Background
Studies of a wide range of systems have contributed to our understanding of epigenetic processes in various model organisms. The majority of epigenetic processes are associated with gene silencing, for example, heterochromatin formation (3), genomic imprinting (4), X-chromosome inactivation (5), and Polycomb repression (6). In addition, the activation of transcription by the Trithorax group of genes is also an epigenetic mechanism (7).
Constitutive heterochromatin
The DNA within the cell exists as a nucleoprotein complex termed chromatin. Chromatin allows the packaging of DNA within the nucleus while at the same time is flexible enough to facilitate the access to this DNA of factors essential for transcription, repair, replication, and segregation. Two types of chromatin exist: euchromatin and heterochromatin. Most active genes are found in euchromatin, whereas heterochromatin includes the portion of chromatin that is transcriptionally silenced. Heterochromatin may be classified into two more types: constitutive and facultative. Constitutive heterochromatin is permanently condensed throughout development and provides structural support to important structures, such as the centromeres and telomeres. These structures regulate essential functions necessary to preserve the chromosome, such as the segregation of chromosomes in mitosis (centromeres) and the protection of chromosome ends during DNA replication (telomeres). The centromeric sequences differ between species but can assemble the kinetochore and acquire centromeric functionality by epigenetic mechanisms (8).
Genomic imprinting and X-chromosome inactivation
Facultative heterochromatin is silenced transiently during development. The silencing involves regions that switch from heterochromatin to euchromatin and vice versa, according to developmental requirements. This switching is associated with epigenetic modifications such as those that occur in genomic imprinting, which is the mechanism of monoallelic silencing of either the maternally or paternally inherited genes. Imprinted genes are clustered, and their allelic expression depends on specific DNA methylation, which determines gene silencing during oogenesis and spermatogenesis (4).
The genes present in one female X chromosome are silenced to balance the gene dose of the X male in mammals. This phenomenon is called X-chromosome inactivation and is mediated by Xist. Xist is a non-coding RNA expressed from the X chromosome that will become silenced. This RNA transcript coats the entire inactive X chromosome, which leads to the gene silencing of the whole chromosome by recruitment of the epigenetic machinery (5).
Regulation of homeotic gene expression by the trithorax and polycomb groups of proteins
The expression of homeotic genes is regulated spatially and temporally throughout development to ensure the correct structures along the anterior-posterior axis of the body organism. The Trithorax group (TrG) of proteins maintains the active expression of homeotic genes in the tissues in which they should be expressed (7), whereas those of the Polycomb group (PcG) repress their expression (6). Both types of protein maintain the cellular memory by epigenetic mechanisms. Proteins of the PcG, in addition to controlling development, regulate cellular differentiation and tumorigenesis (9).
The biologic processes described above and cytosine methylation, fit the “epigenetic” definition well because they are heritable. However, whether the entire repertoire of histone modifications is heritable remains to be established. In fact, it is likely that only a subset will have epigenetic inheritance. Whether they are “epigenetic” in this sense or not is a matter of semantics, and the term may need to be redefined. Indeed Allis, Jenuwein, and Reinberg have proposed (10, 11) that epigenetics be defined as “the sum of the alterations to the chromatin template that collectively establish and propagate different patterns of gene expression (transcription) and silencing from the same genome.”
This definition includes transient chromatin modifications, which are associated with DNA repair or cell-cycle stages, and stable chromatin modifications that are maintained across multiple cell generations.
DNA methylation and the Polycomb/Trithorax system are the paradigms of epigenetic heritable systems. Cavalli and Paro (12) have shown that an activated state of the Fab-7 element, incorporated in transgenic flies, is inheritable mitotically through development and can be transmitted to the subsequent generations through female meiosis. This finding means that the Polycomb/Thrithorax system can memorize gene expression patterns that have been set up by other cellular mechanisms.
The evidence that DNA methylation is heritable is self-sustaining. Maintenance DNA methylases recognize hemi-methylated DNA, the product of replication of the fully methylated DNA, and add methyl groups to the unmethylated DNA strand.
The study of epigenetic modifications is of great interest. Perturbations of chromatin structure can cause inappropriate gene expression and genomic instability that results in cellular transformation and malignant outgrowth. Proteins that control chromatin organization therefore constitute key players in cancer pathogenesis.
Histone Modifications
The DNA within the cell exists in the form of chromatin. The basic repeating unit of chromatin is the nucleosome, a structure consisting of 147 bp of DNA wrapped around an octamer of the core histones H2A, H2B, H3, and H4. The histone proteins are composed of a globular domain and unstructured and protruding N- and C-terminal tails. A striking feature of the tail histones is that they are subjected to several posttranslational modifications, such as acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, deimination, and proline isomerization (Table 1). Certain combinations of histone modifications affect the organization of the chromatin to modulate its involvement in important nuclear functions such as DNA transcription, repair, and replication. The possibility that such combinations could modify chromatin behavior led to the hypothesis that they might constitute a molecular “histone code” (13) that is read by other proteins to mediate unique cellular responses.
Acetylation of the lysine residues of histones H3 and H4 was one of the first modifications of chromatin to be described (Fig. 1) (14). It is associated with the establishment of an open chromatin state that is active transcriptionally. This modification contrasts with hypoacetylation, which is associated with a compacted chromatin structure that is inaccessible to transcription machinery. The majority of all lysine residues of the N-terminal tails of histones H3, H4, H2A, and H2B have the ability to be acetylated (15-17). Some evidences indicate that acetylated histone H3 plays a key role in gene-expression regulation (18) and that acetylated histone H4 is involved mainly in chromatin structure and histone deposition during DNA replication (19). Following this hypothesis, it has been shown that acetylated H4K16 inhibits the formation of higher orders of chromatin structure and modulates functional interactions between a nonhistone protein and the chromatin fiber (20). SirT2, a member of the Sir2 family of NAD+-dependent protein deacetylases that is involved in a caloric restriction-dependent life span extension, has a strong preference for histone H4K16Ac in its deacetylation activity. Mouse embryonic fibroblasts (MEFs) deficient for SirT2 show higher levels of H4K16Ac in mitosis when compared with the normal levels exhibited by SirT1-deficient MEFs (21). The enzymatic conversion of H4K16Ac to its deacetylated form may be pivotal to the formation of condensed chromatin. Thus, SirT2 is a major contributor to the inheritance of epigenetic marks through mitosis (21).

Figure 1. Histone modifications. The best-characterized human histone modifications are shown, which include the acetylation of lysines (Ac), the methylation of lysines and arginines (Me), the phosphorylation of serine and threonines (Ph), and the ubiquitination of lysines (Ub). The vast majority of modifications are within the N-terminal domain of the histone tail, but ubiquitination occurs at the C-terminal domain.
Table 1. A catalog of posttranslational chemical modifications of the histone proteins
|
Histone |
Residue |
Modification |
Biologic function |
|
H2A |
S1 |
Phosphorylation |
Chromosome condensation |
|
|
K5, K9, K13 |
Acetylation |
Transcriptional activation |
|
|
K119 |
Ubiquitylation |
Polycomb silencing |
|
H2B |
K5, K12, K15, K20 |
Acetylation |
Transcriptional activation |
|
|
K123 |
Ubiquitylation |
Transcriptional activation |
|
H3 |
T3 |
Phosphorylation |
Metaphase alignment |
|
|
T11 |
Phosphorylation |
Mitosis |
|
|
S10, S28 |
Phosphorylation |
Mitosis, transcriptional activation |
|
|
K9, K14, K17, K23, K27 |
Acetylation |
Transcriptional activation |
|
|
R2, R8, R17, R26 |
Methylation |
Gene expression regulation |
|
|
K4 |
Methylation |
Transcriptional activation |
|
|
K9 |
Methylation |
Transcriptional repression |
|
|
K27 |
Methylation |
Polycomb repression |
|
|
K36 |
Methylation |
Transcriptional activation |
|
|
K79 |
Methylation |
Transcriptional activation |
|
H4 |
S1 |
Phosphorylation |
DNA repair |
|
|
K5, K8, K12, K16 |
Acetylation |
Histone deposition, transcriptional activation |
|
|
R3 |
Methylation |
Transcriptional activation |
|
|
K20 |
Methylation |
Transcriptional repression |
The methylation of histones can occur in various lysines of histone H3, such as K4, K9, K27, K36, and K79, and at lysine 20 of histone H4. Lysines can be mono-, di-, or trimethylated at all these sites. This modification could be associated with either the repression or activation of DNA transcription, depending on the site and methylation status (mono-, di-, or tri-) of the histone lysine residues. Chromatin immunoprecipitation (ChIP) experiments have shown that three methylation sites on histones are involved in transcriptional activation: methylation at lysine 4 (H3K4), at lysine 36 (H3K36), and at lysine 79 (H3K79) (22, 23). Three lysine methylation sites are connected with transcriptional repression: methylation of lysine 9 (H3K9) and lysine 27 (H3K27) of histone H3 and methylation of lysine 20 of histone H4 (H4K20). These modifications are epigenetic modifications of a repressed chromatin state (24).
Histone arginine methylation can be either mono- or dimethylated at residues R2, R8, R17, and R26 of histone H3 and at R3 of histone H4. It plays a role in transcriptional activation and repression. The biological mechanism of each histone arginine modification is mainly unknown; however some data implicates H4R3, H3R17, and H3R26 modifications with transcriptional activation. In contrast, H3 R8 and H3 R2 are associated with repression (25, 26). Furthermore, two different groups have recently provided the first mechanistic insight into the function of arginine methylation on chromatin (27, 28). They have shown that H3 R2 is enriched throughout all silenced locus in budding yeast and that in all cases the pattern of H3 R2me2 is mutually exclusive with the trimethylation of H3K4 (27), which marks active genes. The role of H3 R2me2 in controlling H3K4me3 also is conserved in humans (28).
The phosphorylation of histones can occur at serine and threonine residues. The function of histone phosphorylation is related largely to either chromosome condensation and/or DNA repair of double-stranded breaks (DSBs). Phosphorylation at serine 10 of histone 3 (H3S10) plays a key role in chromosome condensation during mitosis (29). Phosphorylation at threonine 3 of histone 3 (H3 T3) is required for normal metaphase chromosome alignment (30). Other lesser-known residues, such as phosphorylation at serine 1 of histone 4 (H4S1) and at serine 14 of histone H2B (H2BS14), also regulate chromosome condensation in budding yeast (31). Serine 139 of the histone variant H2AX is phosphorylated in response to DNA DSBs and seems to be an early step in the response to DNA damage (32). Phosphorylation at other sites, such as serine 1 of histone H4 (H4S1), serine 129 of histone H2A (H2AS129), and serine 14 of histone H2B (H2BS14), also contributes to sense DSBs during DNA repair (33). In addition, the phosphorylation of histone H3 is involved in transcriptional activation through H3 S10 or H3 S28 (34).
Histone-modifying enzymes
The modification of histones is a dynamic process with contributing enzymes that can either direct or remove the modification. In the last 10 years, enzymes that play a role in both directions have been identified (Table 2).
Table 2. Representative capture strategies for preparing microarrays
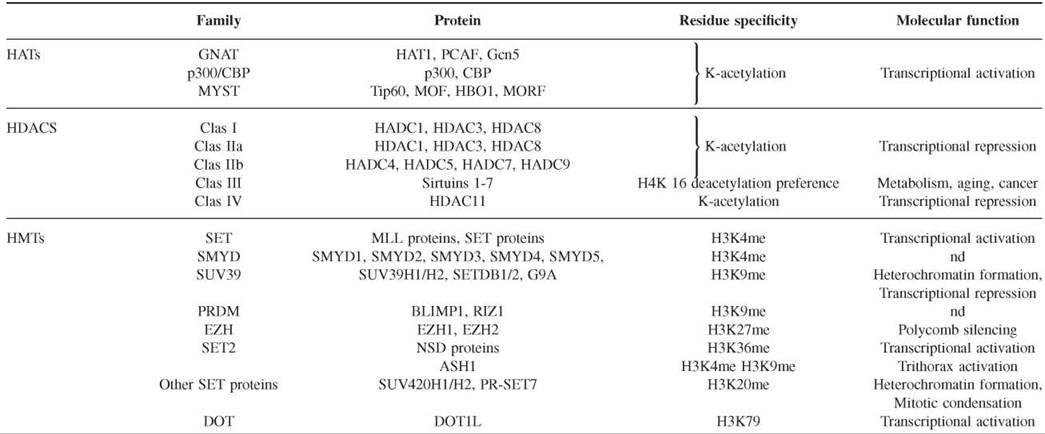
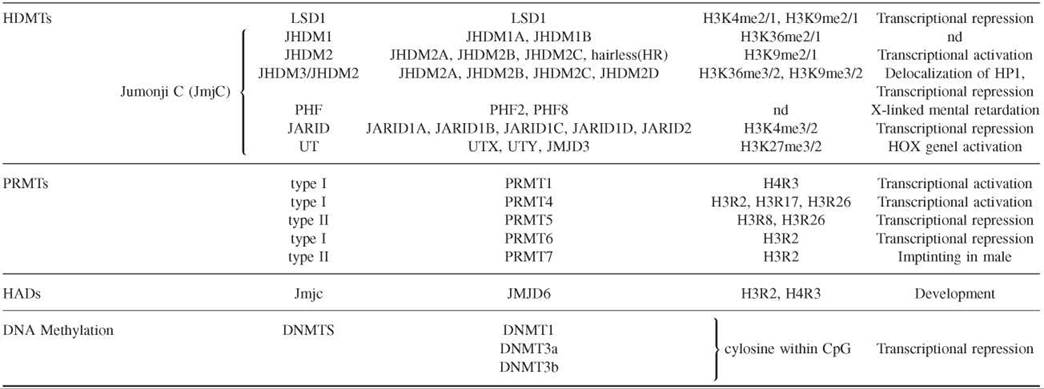
Histone deacetylases (HDACs), histone acetyltransferases (HMTs), histone demethylases (HDMTs), protein arginine methyltransterases (PRMTs), histone arginine demethylases (HADs), and DNA methyltransterases (DNMTs).
Histone acetyltransferases (HATs) form an extended family of enzymes that can be grouped into three families on the basis of sequence homology. The first of these families is the GNAT family (Gcn5-related N-acetyltransferase), which includes HAT1, the yeast Gcn5, and its human ortholog, PCAF. The second family is p300/CBP, which includes the two human paralogs p300 and CBP. The third family is the MYST family, which includes MOZ, Ybf2/Sas3, Sas2, and Tip60. Although HATs in general acetylate more that one lysine, some specificity has been detected. Structural comparison between the three families has shown that HATs contain a conserved core for Ac-CoA binding and that different domains fold around this core to facilitate substrate specificity (35). However, more analyses are necessary to elucidate the possible specificity of HATs for lysine residues.
Histone deacetylases (HDACs) are divided into four families on the basis of phylogenetic analysis. The class I family includes human HDAC1, HDAC3, and HDAC8, which are homologous to the yeast Rpd3. The class II family is homologous to the yeast Hdal and is subdivided into the IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and IIb (HDAC6 and HDAC10) subfamilies on the basis of their sequence homology. The class III family includes the NAD+-dependant enzymes of the Sir (or sirtuin) family. Its components are yeast Hst proteins 1-4 and human Sirtuins 1-7. The class IV family contains the human HDAC 11. In general, HDACs are not specific for a particular acetyl group, but the sirtuin family seems to prefer deacetylating the lysine 16 of histone H4 (36). The catalytic mechanism of the Sirtuin family differs from those of other families in its use of NAD+ as a cofactor.
One important feature of HAT and HDAC enzymes is that they are integrated frequently within multiprotein complexes in which the noncatalytic subunits tend to regulate the substrate specificity that contributes to the molecular functionality in a cellular context.
Histone methyltranferases (HMTs) of lysines contain a catalytic SET domain of about 130 amino acids, with the exception of those of the Dot family. SET owes its name to the shared domain of the first three HMTs to be identified in Drosophila: Suppressor of position-effect variegation Su(var)3-9, Enhancer of Zeste E(Z), and Trithorax (TRX). The HMTs can be classified into several families on the basis of the sequence homology within the SET domain and adjacent sequences (24, 37). The SET1 family includes the MLL family of proteins, which are homologous to the TRX proteins of Drosophila, and the Set1 protein of yeast. They specifically methylate lysine 4 of histone H3 linked to the activation of transcription. The SMYD family proteins also methylate H3K4 but include an MYND domain that binds to specific DNA sequences. The SUV39 family includes the SUV39H1 and SUV39H2 histone methyltransferases, SETDB1, and G9A, which specifically methylate lysine 9 of histone H3. The PRDM family contains a PR SET domain that is considered a subclass of the SET domain. The catalytic activity of the PRDM family is controversial. However, it includes crucial proteins such as RIZ1 and BLIMP1 that are associated with the methylation of lysine 9 of histone H3, which is important in regulating the gene transcription of many biological processes. The EZH family includes the two homologs of Enhancer of zeste, EZH1 and EZH2, which methylate lysine 27 of histone H3 that is associated with transcriptional repression linked to the Polycomb group proteins. The SET2 family contains the NSD proteins, which specifically methylate lysine 36 of histone H3, and ASH1, which methylates H3K4, H3K9, and H4K20. Other SET domain-containing proteins include SUV420H1 and SUV420H2, which specifically dimethylate and trimethylate lysine 4 of H4K20, and PR-SET7, which monomethylates H4K20. The Dot1L family does not contain a SET domain and is responsible for the methylation of lysine 79 of histone H3.
Histone demethylases have been identified recently (38). Lysine-specific demethylase 1 (LSD1) was the founding member of the first family of histone demethylases. The LSD catalytic domain reverses histone H3K4 and H3K9 methylation. The second and largest family is that of the demethylase enzymes that contain a Jumonji C (JmjC) domain as a catalytic domain. These demethylases can remove all three histone lysine methylation states (mono-, di-, and trimethylation). The family is divided into several subfamilies, including JHDM1, JHDM2, JHDM3/JMJD2, PH2/PH8, JARID1/JARID2, and UTX/UTY. Histone demethylases have been associated with important roles in chromatin structure and development. For example, JHDM3 and JMJD2 demethylate H3K9 and H3K36, which antagonizes HP1 recruitment to chromatin and regulates gene expression. Moreover, very recent studies have confirmed the important role of histone demethylases in histone modification dynamics. It has been shown that four proteins of the JARID family possess the demethylase activity specific to H3K4 and contribute to cell-fate determination (39). Two more studies have established that members of the UTX/UTY family possess demethylase activity specific to H3K27 and are involved in HOX gene regulation and development (40, 41).
Histone arginine methylation is achieved by protein arginine methyltransferases (PRMTs), which are evolutionarily conserved from yeast to humans. PRMT are classified as type I and II based on the nature of the methylation introduced. Type I catalyses symmetric dimethylation of arginine, and type II catalyses the asymmetric one. Eleven PRMTs have been identified in humans based on sequence homology; however their substrate specificity and their biologic functions still remain unknown. PRMTs can methylate many cellular proteins, and some of them also can methylate histones, such as PRMT1, PRMT4, PRMT5, and PRMT7 (26). PRMT1 methylates H4R3 contributing to transcriptional activation. PRMT4 or CARM1 can methylate H3 R17 and H3 R26, which are marks linked to gene activation. PRMT4 also can methylate H3 R2 in vitro with low efficiency. PRMT5 methylates H3R8 and H4R3, and it has been copurified with chromatin remodeling complexes such as hSWI/SNF and NURD. It has been implicated also in transcriptional repression of cell cycle regulator and tumor suppressor genes. PRMT7 mediates H4R3 methylation at the imprinting loci in embryo male germ cells. Very recently, it has been shown that PRMT6 catalyses asymmetric H3 R2 dimethylation in human cells thereby preventing the recruitment of MLL methyltransferase complexes and H3K4 methylation. Reciprocally, H3K4me3 prevents H3 R2 methylation by PRMT6 (28).
Of special interest is the finding of the first histone arginine demethylase, the Jumonji domain-containing 6 protein (JMJD6). It shares homology with the JmjC domains found in the histone lysine demethylases. JMJD6 can demethylate H3 R2 and H4R3 in biochemical and cell-based assays (42). The recent data that are emerging place histone arginine methylation as a key player of cell growth and cell proliferation processes.
Reading the histone modifications
Histone modifications generate two types of downstream effects on chromatin organization. The first type is produced mainly by the acetylation of lysine residues, which neutralize positive charges of lysine and allow the unfolding of the chromatin through an electrostatic mechanism. The DNA then is more accessible to the transcription machinery. The second effect arises from the generation of docking sites that are read by specific proteins with the purpose of tethering enzymatic activities to the chromatin.
The acetylation of lysines is recognized by the bromodomains, which are small domains included in an extensive family of proteins. The first bromodomain was identified in the Drosophila Brahma protein. Bromodomains were later found widely distributed among different enzymes that acetylate, methylate, or remodel chromatin (43). The bromodomain is present in the members of different families of histone acetyl-transferases, such as the GCN5/PCAF and p300/CBP families. They also are part of some histone methyltransferase enzymes such as Ash1, RIZ, and MLL. Bromodomains are important components of the remodeling enzymes that use ATP to remodel chromatin structure, such as BRM or members of the SWI/SNF complexes. Sequence-specific lysine acetylation recognition by a bromodomain is dependent on how the bromodomain recognizes residues flanking the acetylated lysine, but currently the exact specificity is poorly understood.
Different protein domains recognize the methylation of lysine: the chromodomain, the Tudor domain, the WD40 repeat, the MBT domain, and the PHD finger (24, 37). The methylated lysine 9 of histone H3, H3K9, is recognized by the chromodomain of HP1 within heterochromatin. HP1 can recruit other heterochromatic proteins as the histone methyltransferases Su(var)3-9 and deacetylases. Methylation of lysine 27 of histone H3 is recognized specifically by the chromodomain of Polycomb proteins that are part of the polycomb repressor complex within the homeotic genes. The JMJD2A lysine demethylase can bind the methyl H3K20 via its Tudor domain but also can bind methyl H3K4. The two chromodomains of Chd1 can bind the methyl H3K4, which helps the recruitment of the SAGA complex. Different domains can recognize methyl H3K4 within different biologic backgrounds. For instance, WDR5, a component of the histone methyltransferase MLL complex, binds methyl H3K4 via its WD40 repeat, and the PHD finger of BPTF, a component of NURF, also targets methyl H3K4 modification (24, 32).
Biologic function of histone modifications
As mentioned above, histone modifications generate two broad effects. Firstly, histone modification can alter the global charge of histones and disrupt contacts between nucleosomes to cause chromatin unfolding. Secondly, histone modification can create a docking site that can be read by a protein domain. This protein may act as an independent effector or alternatively tether other complexes with different enzymatic activities that collectively modify the chromatin environment in response to histone modification. In this section, we will discuss some best-characterized biologic processes that involve specific histone modifications.
Histone modification is of paramount importance to the establishment of constitutive heterochromatin at centromeres (Fig. 2). The histone methyltransferase SUV39H1/2 trimethylates lysine 9 of histone H3, which generates targets for the chromodomain-containing proteins HP1a and HP1P HP1 molecules recruit other histone methyltransferases, like SUV39H1/2 and SUV420H1/2, that di- and trimethylate histones of adjacent sequences spreading the heterochromatin (44) (Fig. 2a). The initial targeting of SUV39H1/2 to heterochromatin involves components of the RNAi silencing machinery, as has been shown in the centromeres (Fig. 2b) of Schizosac-charomyces pombe. Centromeric transcripts can generate siRNA that are loaded within the RITS (RNA-induced initiation of transcriptional gene silencing) complex to target heterochromatin, probably by base-pairing homology with centromeric transcripts. The RITS recruit Clr4, the S. pombe homolog of SUV39, that will methylate lysine 9 of histone H3, acting as recognition sites of the S. pombe homolog of HP1, Swi6, and favoring heterochromatin spreading (45).

Figure 2. Constitutive heterochromatin at centromeres. a) Mammalian centromere model. SUV39H1/2 produces H3K9me3 residues, which are recognized by the chromodomain protein HP1 a and HP1 p. Both proteins interact with other HMTs, such as SUV39H and SUV420H1/2, that methylate adjacent histones that lead to the spreading of heterochromatin. Mammalian heterochromatin is enriched in H3K9me3 and H4K20me3 residues. b) S. pombe centromere model. The assembly of centromeric heterochromatin in S. pombe is very well established. The components of the RNAi machinery play an essential role. Dicer produces siRNA from the centromeric dsRNA transcripts. siRNA are loaded within the RITS (RNA-induced transcriptional gene silencing) complex to target heterochromatin by recruiting Clr4, which methylates lysine 9 of histone H3. H3K9me2 will be recognized by Swi6 and Chp1. Chp 1 is part of the RITS complex that also includes Ago1 and with the RDRC complex (which contains Rdp1, an RNA-dependent RNA polymerase) and dicer process nascent transcripts into siRNA. Clr4 will be recruited in a positive feedback loop that propagates the heterochromatin. Methylation of lysine 9 is accomplished after recruiting histone deacetylases (HDAC) complexes to heterochromatin to deacetylate lysine 9 residues.
DNA methylation also is present within tandem-repetitive DNA sequences in the heterochromatin. They also are thought to inhibit recombination between homologous repeats, which could lead to genomic instability (46). In fact, mutations of the DNMT3b cause centromeric instability. In addition, HDACs lie at the heart of heterochromatin pathways and can be recruited by transcriptional repressor or by methyl-DNA binding proteins.
The epigenetic gene silencing by Polycomb protein complexes is one of the best examples of how epigenetics modulate gene transcription across generations. When the Polycomb repressive complex 2 (PRC2) is recruited to the Polycomb target genes, its histone methyltransferase component, EZH2, trimethylates lysine 27 residues of histone H3 (Fig. 3). The Polycomb repressive complex 1 (PRC1) can recognize trimethylated H3K27 through the chromodomain of the Polycomb protein, a component of the PRC1. This interaction might bring adjacent nucleosomes into the proximity of the PRC2 complex to facilitate widespread methylation over extended chromosomal regions. PRC1 mediates recruitment of DNA methyltransferases, chromatin compaction, and ubiquitylation of the lysine 119 of histone H2A, which is thought to contribute to transcriptional inhibition, but the precise mechanism of gene silencing is unknown (6). Very important contributions to this biologic process have been made recently. The proteins UTX and JMJD3 can demethylate H3K27me3 within the HOX genes, which leads to transcriptional activation. Moreover, UTX can associate with the H3K4me3 histone methyltranferase MLL2. This association is consistent with a model in which the coordinated removal of repressive marks, the polycomb group displacement, and the deposition of activating marks are important for the stringent regulation of the transcription during cellular differentiation (40, 41).

Figure 3. Epigenetic gene regulation of HOX genes by Polycomb complexes and H3K27 demethylases. The HMT EZH2 is a component of the PRC2 complex, and its binding to the HOX genes leads to H3K27me, which is recognized by the PRC1 complex through its Polycomb member. The polycomb binding produces chromatin compaction and transcriptional repression. The mechanism of transcriptional repression is unknown but might be achieved either by the ubiquitylation of the H2AK119 through the ubiquitin E3 ligase activity present in the PRC1 complex or by the recruitment of DNMTs by the PRC2 complex. Recently, two H3K27 demethylases, UTX and JMJD3, have been identified. Moreover, UTX associates with MLL2, an H3K4me3 HMT. These results suggest a model in which H3K27 demethylases associated with MLL2 can reverse the transcriptional silencing mediated by Polycomb. The complex would remove H3K27me3 residues and create H3K4me3 marks that lead to the activation of transcription. In this model, the hypothetical association of the PRC2 complex with H3K4me3 demethylases facilitates transcriptional silencing.
The best-characterized model that describes the activation of transcription at a molecular level has been developed in Saccharomyces cerevisiae (47). Activator factors recruit the Rad-Bre1 complex, which is loaded into RNA polymerase II (RPNII) and catalyzes the ubiquitylation of lysine 123 of histone H2B (Fig. 4). This ubiquitylation favors the recruitment of the histone methyltransferases Set1, Set2, and probably Dot1. Ubiquitylation is necessary for methylation of H3K4 and H3K79 by Set1 and Dot1, respectively. During elongation, the C-terminal domain of RNPII is phosphorylated at serine 2 and Set1 dissociates from RNPII, whereas Set2 methylates H3K36. H3K4 serves as a binding site for the recruitment of histone acetylation SAGA complex through a specific interaction between methyl H3K4 and the chromodomain of its component Chd1. Histone acetylation by the SAGA complex leads to transcription activation. An important contribution has been made recently regarding the implication of histone arginine methylation in transcription. The activity of the Set 1 complex is regulated by the asymmetric dimethylation of H3 R2. The patterns of H3 R2me2a and H3K4me3 are mutually exclusive because H3R2 methylation can prevent Spp1, a component of the Set1 complex, from binding H3K4me3 (27).

Figure 4. Epigenetic activation of transcription in S. cerevisiae. The activator proteins recruit the Rad6-Bre1 complex, which is loaded onto the RNA polymerase II along with Set1, Set2, and probably Dot1. After the first round of transcription, the gene is marked by H3K4me, H3K36me, and H3K79me. Chd1, a component of the SAGA complex, recognizes the methyl-H3K4, which leads to the histone acetylation and the activation of transcription.
DNA Methylation
Cytosine methylation occurs in the context of the CpG dinucleotide sequence. The frequency of the CpG dinucleotide in the human genome is lower than expected. However, approximately half of the human gene-promoter regions contain CpG-rich regions with lengths of 0.5 to several Kb, known as “CpG islands.” These regions are methylated according to a developmental program to control gene expression. In contrast, the CpG dinucleotides, which are spread through the genome, usually are methylated (48).
The enzymes directly responsible for CpG methylation are the DNA methyltransferases (DNMTs) . DNMT1 exhibits a 5- to 30-fold preference for hemimethylated substrates. This property led to the identification of DNMT1 as the enzyme responsible for maintaining the methylation patterns following DNA replication. DNMT3a and DNMT3b were identified by EST database searches and were proposed as the enzymes responsible for de novo methylation. DNMTs originally were classified as either maintenance or de novo DNA methyltransferases, but several lines of evidence indicate that all three DNMTs cooperate and possess both these functions in vivo (49).
The cell reads the DNA methylation code by the methyl-CpG binding proteins (MBDs). This family of proteins consists of five members, MeCP2, MBD1, MBD2, MBD3, and MBD4. They target the transcriptional repressive chromatin machinery to hypermethylated CpG islands (50). The first finding connecting DNA methylation and chromatin modification involved MeCP2, which can repress the transcription of methylated DNA through the recruitment of a histone deacetylase complex, HDAC. Other evidences that connect DNA methylation with histone modifications are that DNMTs and MBDs recruit repressive HMTs and HDACs complexes (51).
CpG islands of tumor-suppressor genes are hypermethylated in cancer cells. Therefore, they recruit multiple repressors that lead to a characteristic histone modification pattern: the deacetylation of histones H3 and H4, the methylation of lysine 9 of histone H3, and the demethylation of lysine 4 of histone H3 (52). In addition, it has been shown that the polycomb protein EZH2 associates with DNMTs and that specific methylation of lysine 27 of histone H3 is required to establish DNA methylation in a subset of target genes (6).
DNA methylation in mammals is an epigenetic modification involved in a range of cellular functions and pathologies, including X chromosome inactivation, genomic imprinting, tissue-specific gene expression, cell differentiation, regulation of chromatin structure, carcinogenesis, and aging, and is indispensable for the survival of differentiated cells.
Chemical Tools and Techniques
Epigenetic modifications are key translators between genotypes and phenotypes. The goal of scientists is to understand the significance of each chromatin modification within a biologic process, genomic region, and given organism. The simplest scenario would be that every epigenetic mark corresponded to definable and predictable outcomes. However, many lines of investigation have shown that the function of these chromatin marks is more complex than previously thought and depends entirely on the biologic context.
We can study the function of epigenetic modifications from different points of view; this includes the investigation of the global epigenetic modifications of every cell type to generate its epigenome and the other components involved in epigenetic modifications, such as the histone-modifying enzymes, the proteins that read each modification, and the enzymatic activities or functional protein complexes that are recruited to this modification, as well as the downstream biologic effects. Below we will discuss the technology that may be used to analyze epigenetic modifications.
The epigenome is the map of epigenetic modifications of a given cell in terms of DNA methylation and histone modifications. Many techniques for studying epigenetic modifications at specific loci exist, and several of them have been adapted for large-scale analyses.
Histone modifications
The most powerful technique for studying histone modifications coupled with particular DNA sequences is chromatin im- munoprecipitation, ChIP, with antibodies against specific modification. The immunoprecipitated DNA is analyzed by PCR with specific primers to investigate the presence of a candidate DNA sequence. ChIP may be scaled up for global analyses with microarrays (ChiP-on-chip). Its use with genomic platforms has begun to yield extensive maps of histone modifications in model organisms such as Arabidopsis thaliana, yeast, Drosophila melanogaster, mice, and recently normal human cells, including stem cells (53). The ChIP-on-chip assay can be applied to any histone modification for which an effective antibody is available, although it has been used only with lysine acetylation and methylation. Some important histone patterns nevertheless have been established already. ChIP analyses are limited by their requirement for good-quality antibodies, and they need to be highly specific if they are to yield results that can be comparable across experiments. The majority of ChIP experiments still use polyclonal antibodies, but the generation and use of monoclonal antibodies, by which differences between antibody batches may be minimized, is an important goal.
Mass spectrometry is the most accurate technique for detecting global levels of histone modification. However, it requires great technical expertise and is difficult to apply across genomes. Acceptable data on global levels of histone modification currently can be obtained by combining other methods. For example, all histones (H3, H4, H2A, H2B, and H1) can be isolated by HPLC, and the corresponding eluted fractions can be analyzed by HPCE and liquid chromatography-electrospray mass spectrometry (LC-ES/MS). Specific modifications at each amino acid residue also can be characterized by using antibodies in western blots, immunostaining, and tandem mass spectrometry (MS/MS).
DNA methylation
The most accurate method for analyzing DNA methylation is the bisulfite treatment of DNA, which reproducibly changes unmethylated cytosines to uracil but leaves methylated cytosine unchanged. Any laboratory can study DNA methylation by using the bisulfite treatment combined with genomic sequencing or amplification by methylation-specific PCR. To detect minimal amounts of aberrant DNA methylation, quantitative PCR-based methods can be used, such as the bisulfite treatment in combination with MethyLight or pyrosequencing (54). Some other approaches are based on the uses of restriction enzymes that can distinguish between methylated and unmethylated recognition sites in genes of interest. This technology is less accurate because incomplete cutting of the restriction enzymes within the studied regions is a limitation. Both methods can be coupled with several genomic approaches for detecting DNA methylation patterns. Recently, a ChIP-on-chip-based method has been applied to DNA methylation, the methyl-DIP (54). The DNA that is immunoprecipitated with an antibody against 5-methylcytosine can be used as a probe for hybridization to genomic microarray platforms, which allows the rapid identification of multiple CpG sites and simplifies the analysis of the DNA methylome.
References
1. Fisher AG. Cellular identity and lineage choice. Nat. Rev. Immunol. 2002; 2:977-982.
2. Miller OJ, Schnedl W, Allen J, Erlanger BF. 5-Methylcytosine localised in mammalian constitutive heterochromatin. Nature 1974; 251:636-637.
3. Craig JM. Heterochromatin—many flavours, common themes. Bioessays 2005; 27:17-28.
4. Delaval K, Feil R. Epigenetic regulation of mammalian genomic imprinting. Curr. Opin. Genet. Dev. 2004; 14:188-195.
5. Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 2004; 303:644-649.
6. Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006; 6:846-856.
7. Papp B, Muller J. Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 2006; 20:2041-2054.
8. Morris CA, Moazed D. Centromere assembly and propagation. Cell 2007; 128:647-650.
9. Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genomewide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006; 20:1123-1136.
10. Shi Y. Taking epigenetics center stage. Cell. 2007; 128:639-640.
11. Allis CD, Jenuwein T, Reinberg D. In: Epigenetics. 2007. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. pp. 29.
12. Cavalli G, Paro R. The Drosophila Fab-7 chromosomal element conveys epigenetic inheritance during mitosis and meiosis. Cell 1998; 93:505-518.
13. Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41-45.
14. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 1964; 51:786-794.
15. Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr Biol. 1998; 8:96-108.
16. Galasinski SC, Resing KA, Goodrich JA, Ahn NG. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem. 2002; 277:19618-19626.
17. Schubeler D, Francastel C, Cimbora DM, Reik A, Martin DI, Groudine M. Nuclear localization and histone acetylation: a pathway for chromatin opening and transcriptional activation of the human beta-globin locus. Genes Dev. 2000; 14:940-950.
18. Margueron R, Trojer P, Reinberg, D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005 Apr; 15(2):163-76.
19. Sobel RE, Cook RG, Perry CA, Annunziato AT, Allis CD. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:1237-1241.
20. Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311:844-847.
21. Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006; 20:1256-1261.
22. Kizer KO, Phatnani HP, Shibat Y, Hall H, Greanleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell Biol. 2005; 25:3305-3316.
23. Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002; 12:1052-1058.
24. Volkel P, Angrand PO. The control of histone lysine methylation in epigenetic regulation. Biochimie 2007; 89:1-20.
25. El Messaoudi S, Fabbrizio E, Rodriguez C, Chuchana Fauquier L, Cheng D, Theillet C, Vandel L, Bedford MT, Sardet C. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13351-13356.
26. Pal S, Sif S. Interplay between chromatin remodelers and protein arginine methyltransferase s. J. Cell Physiol. 2007; 213:306-315.
27. Kirmizis A, Santos-Rosa H, Penkett CJ, Singer MA, Vermeulen M, Mann M, Bahler J, Green RD, Kouzarides T. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature. 2007; 449:928-932.
28. Guccione E, Bassi C, Casadio F, Martinato F, Cesaroni M, Schuchlautz H, Luscher B, Amati B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007; 449:933-937.
29. Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 1999; 97:99-109.
30. Dai J, Sultan S, Taylor SS, Higgins JM. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005; 19:472-488.
31. Ajiro K. Histone H2B phosphorylation in mammalian apoptotic cells. An association with DNA fragmentation. J. Biol. Chem. 2000; 275:439-443.
32. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998; 273:5858-5868.
33. van Attikum H, Gasser S. The histone code at DNA breaks: a guide to repair? Nat. Rev. Mol. Cell. Biol. 2005; 6:757-765.
34. Clayton AL, Rose S, Barratt MJ, Mahadevan LC. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000; 19:3714-3726.
35. Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene 2007; 26:5528-5540.
36. Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007; 26:5310-5318.
37. Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell. Biol. 2005; 6:838-849.
38. Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006; 7:715-727.
39. Benevolenskaya EV. Histone H3K4 demethylases are essential in development and differentiation. Biochem. Cell Biol. 2007; 85:435-443.
40. Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development.Nature 2007; 449:731-734.
41. Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007; 318:447-450.
42. Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science 2007; 318:444-447.
43. de la Cruz X, Lois S, Sanchez-Molina S, Martinez-Balbas MA. Do protein motifs read the histone code? Bioessays. 2005; 27:164-175.
44. Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004; 18:1251-1262.
45. Grewal SI, Jia S. Heterochromatin revisited. Nat. Rev. Genet. 2007; 8:35-46.
46. Grewal SI, Moazed, D. Heterochromatin and epigenetic control of gene expression. Science. 2003; Aug 8;301 (5634):798-802.
47. Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003; 11:709-719.
48. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006; 31:89-97.
49. Kim GD, Ni J, Kelesoglu N, Roberts RJ, Pradhan S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002; 21:4183-4195.
50. Ballestar E, Esteller M. Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem. Cell. Biol. 2005; 83:374-384.
51. Esteller M. Epigenetics provides a new generation of oncogenes and tumour-suppressor genes. Br. J. Cancer 2007; (suppl 96):R26-R30.
52. Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J, Cigudosa JC, Huang TH, Esteller M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003; 22:6335-6345.
53. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007; 128:669-681.
54. Esteller M. Cancer epigenomics: DNA methylomes and histone- modification maps. Nat. Rev. Genet. 2007; 8:286-298.
Further Reading
Allis CD, Jenuwein T, Reinberg D. Epigenetics. 2007. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Chromatin structure and function, http://www.chromatin.us.
Ekwall K. Epigenetic control of centromere behavior. Annu. Rev. Genet. In Press.
Epigenetics and chromatin organization. Cell 2007; 128(4). Entire issue.
HEP, the Human Epigenome Project, http://www.epgenome.org.
Insight: Epigenetics. Nature 2007; 447(7143). Entire issue.
MethDB, Human DNA Methylation Database, http://www.methdb.de/.
Reviews on chromatin acetylation. Oncogene 2007; 26(37). Entire issue.
See Also
AdoMet-Dependent Methyltransferases, Chemistry of
DNA, Covalent Modifications of
Histone Acetyltransferases, Selective Inhibitors of
Post-Translational Modifications to Regulate Protein Function