CHEMICAL BIOLOGY
Chemistry of Flavoenzymes
Willem J. H. van Berkel, Laboratory of Biochemistry, Wageningen University, Wageningen, The Netherlands
doi: 10.1002/9780470048672.wecb168
Flavoenzymes are omnipresent in nature and are involved in many cellular processes. Flavoenzymes typically contain the vitamin B2 derivatives FAD and FMN as a redox-active prosthetic group. By varying the protein environment around the isoalloxazine ring of the flavin, evolution has created a great diversity of flavoprotein active sites and catalytic machineries. Most flavoenzymes perform one- or two-electron redox reactions and belong to the following groups: Flavoprotein reductases primarily use NAD(P)H as electron donor and pass these electrons to a protein substrate or another electron acceptor. Flavoprotein dehydrogenases oxidize organic substrates and mainly use quinones and electron transfer proteins as electron acceptors. Flavoprotein disulfide oxidoreductases contain active-site thiols. They use a dithiol substrate and NAD+ to form a disulfide product and NADH or act in the other direction yielding NAD(P)+ and a reduced (dithiol) product. Some flavoprotein (di)thiol oxidoreductases stabilize a reactive and reversibly oxidized cysteine in their active site. Flavoprotein oxidases catalyze the conversion of a substrate single bond to a double bond. The reduced flavin generated during this reaction is reoxidized by molecular oxygen to form hydrogen peroxide. Flavoprotein monooxygenases mainly use NAD(P)H as an electron donor and insert one atom of molecular oxygen into their substrates. By doing so, they act in different biological processes, ranging from lignin degradation and detoxification to the biosynthesis of polyketides and plant hormones.
Flavoenzymes are widespread in nature and are involved in many different chemical reactions. Flavoenzymes contain a flavin mononucleotide (FMN) or more often a flavin adenine dinucleotide (FAD) as redox-active prosthetic group. Both cofactors are synthesized from riboflavin (vitamin B2) by microorganisms and plants. Most flavoenzymes bind the flavin cofactor in a noncovalent mode (1). In about 10% of all flavoenzymes, the isoalloxazine ring of the flavin is covalently linked to the polypeptide chain (2, 3). Covalent binding increases the redox potential of the flavin and its oxidation power, but it may also be beneficial for protein stability, especially in flavin-deficient environments.
Flavoenzymes constitute about 2% of all biological catalysts and are classified in several ways. One classification is based on EC number (enzyme nomenclature) and refers to the type of reaction catalyzed. More sophisticated classifications concern the inclusion of sequence, fold, and function. Historically, a distinction is made between “simple” and “complex” flavoenzymes (4). The latter proteins contain besides flavin other cofactors like heme, tetrahydrobiopterin, and metal ions.
The catalytic cycle of each flavoenzyme consists of two distinct processes, the acceptance of redox equivalents from a substrate and the transfer of these equivalents to an acceptor. Accordingly, the catalyzed reactions consist of two half-reactions: a reductive half-reaction in which the flavin is reduced and an oxidative half-reaction, in which the reduced flavin is reoxidized. This review summarizes the chemistry of “simple” flavoprotein reductases, dehydrogenases, (di)thiol oxidoreductases, oxidases, and monooxygenases (Table 1) (5-40) This grouping provides a good appreciation about what type of common mechanisms can be distinguished and what type of substrates can be converted. Information on the chemistry of “complex” flavoenzymes can be found in the Further Reading section.
Biological Background
The intrinsic chemical properties of the isoalloxazine nucleus, as modulated by the protein environment, are at the heart of the success of flavoenzymes in nature. The chemical versatility of the flavin cofactor is used in a wide variety of biological processes that range from energy production, light emission, protein folding, and neural development to detoxification, apoptosis, chromatin remodeling, and DNA repair (41). With the developments in molecular life sciences, it is expected that many more functions of flavoenzymes will develop. The biological background of certain flavoprotein reductases, dehydrogenases, (di)thiol oxidoreductases, oxidases, and monooxygenases is summarized below.
Table 1. Examples of “simple” flavoenzymes
|
Enzyme |
EC number |
References |
|
Reductases |
|
|
|
NAD(P)H:quinone reductase |
1.6.5.2 |
(5) |
|
Flavin reductase |
1.5.1.30 |
(6, 7) |
|
Ferredoxin NADP+ reductase |
1.18.1.2 |
(8, 9) |
|
Dehydrogenases |
|
|
|
Acyl-CoA dehydrogenase |
1.3.99.3 |
(10-12) |
|
L-galactono-1,4-lactone dehydrogenase |
1.3.2.3 |
(13) |
|
Disulfide oxidoreductases |
|
|
|
Dihydrolipoamide dehydrogenase |
1.8.1.4 |
(14) |
|
Glutathione reductase |
1.6.4.2 |
(15, 16) |
|
Thioredoxin reductase |
1.6.4.5 |
(15, 17, 18) |
|
Sulfenic acid oxidoreductases |
|
|
|
NADH peroxidase |
1.11.1.1 |
(16, 19) |
|
NADH oxidase |
1.6.99.x |
(16, 19) |
|
Oxidases |
|
|
|
D-amino acid oxidase |
1.4.3.3 |
(20, 21) |
|
Monoamine oxidase |
1.4.3.4 |
(22, 23) |
|
Nitroalkane oxidase |
1.7.3.1 |
(24) |
|
Vanillyl-alcohol oxidase |
1.1.3.38 |
(3, 25, 26) |
|
Glucose oxidase |
1.1.3.4 |
(27) |
|
Cholesterol oxidase |
1.1.3.6 |
(12, 28, 29) |
|
Acyl-CoA oxidase |
1.3.3.6 |
(30) |
|
Monooxygenases |
|
|
|
p-Hydroxybenzoate 3-hydroxylase |
1.14.13.2 |
(31-34) |
|
Cyclohexanone monooxygenase |
1.14.13.22 |
(35, 36) |
|
Phenylacetone monooxygenase |
1.14.13.92 |
(37) |
|
Flavin-containing monooxygenase |
1.14.13.8 |
(38) |
|
Tryptophan 7-halogenase |
1.14.13.x |
(39, 40) |
Reductases
Flavoprotein reductases have many important cellular functions. NADH:cytochrome b5 reductase (EC 1.6.2.2) is a crucial housekeeping enzyme that controls the level of iron in the blood. Microsomal NADPH:cytochrome P450 reductase (EC 1.6.2.4) assists in hepatic drug metabolism by transferring electrons to many cytochrome P450 isoforms. Cytosolic NAD(P)H quinone reductase (NQO1; EC 1.6.5.2) protects cells from oxidative stress by catalyzing the reduction of exogenous and endogenous quinones to the corresponding hydroquinones. Recent studies suggest that mammalian quinone reductases also control the lifespan of transcription factors, such as p53, and hence participate in the development of apoptosis and cell transformation (5). Flavin reductases (EC 1.5.1.30) are widespread in microorganisms and use riboflavin, FMN, or FAD as substrate (6). The reduced flavin product is used by other enzymes for various purposes like for instance the emission of light (7). Plant-type ferredoxin NADP+ reductase (FNR; EC 1.18.1.2) acts in the reverse sense. Being involved in carbon fixation this enzyme receives electrons one at a time from ferredoxin and then transmits them in a two-electron step to NADP+ (8).
Dehydrogenases
Flavoprotein dehydrogenases are also widespread. Many of them occur in mitochondria where they are involved in energy production and the biosynthesis of essential nutrients. Disfunction of these enzymes may result in oxidative stress and neurobehavioral deficits. Acyl-CoA dehydrogenases (EC 1.3.99.3) play a crucial role in the mitochondrial P-oxidation of fatty acids (10). The reduced forms of these enzymes are reoxidized in two one-electron steps by ETF (electron transferring flavoprotein) (42). Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is a widely occurring inherited genetic disorder. During fasting or illness, it results in continued glucose consumption and strongly reduced ketone body production. L-galactono-1,4-lactone dehydrogenase (GALDH; EC 1.3.2.3) catalyzes the terminal step of vitamin C biosynthesis in plants (13). The mitochondrial enzyme uses cytochrome c as electron acceptor. GALDH homologs in animals (L-gulono-1,4-lactone oxidase), yeast (D-arabinono-1,4-lactone oxidase), and fungi (D-gluconolactone oxidase) use molecular oxygen as electron acceptor and are involved in the synthesis of L-ascorbate or its analogs D-erythorbate and D-erythroascorbate (3).
Disulfide oxidoreductases
Flavoprotein disulfide oxidoreductases are crucial metabolic and detoxification enzymes. Dihydrolipoamide dehydrogenase (EC 1.8.1.4) catalyzes the NAD+ dependent oxidation of dihydrolipoyl groups, which are covalently attached to the lipoyl domain of the acyltransferase components of the mitochondrial a-ketoacid dehydrogenase and glycine decarboxylase multienzyme complexes. Mutations to this homodimeric flavoprotein cause the often-fatal human disease known as E3 deficiency (14). Glutathione reductase (EC 1.6.4.2) and thioredoxin reductase (TrxR; EC 1.6.4.5) supply cells with high levels of dithiols, which are essential in keeping other thiols reduced (15). In red blood cells, up to 10% of the glucose consumption may be used for the production of reduced glutathione. The TrxR/thioredoxin system is involved in important physiological functions, such as cell growth, inflammation reactions, and apoptosis and regulation.
NADH peroxidase (Npx; EC 1.11.1.1) and NADH oxidase (Nox; EC 1.6.99.x) are disulfide oxidoreductases-related enzymes that contain a single redox-active cysteine (16). They supply strictly fermentative bacteria with NAD+ for glycolysis and play an important role in redox signaling in response to oxidative and nitrosative stress (19).
Oxidases
Flavoprotein oxidases are ubiquitous enzymes involved in the biosynthesis and biodegradation of a huge variety of compounds. D-amino acid oxidase (DAAO; EC 1.4.3.3) is the prototype amino acid oxidase. This peroxisomal enzyme is spread from yeasts to humans. Mammalian DAAO has been connected to the brain D-serine metabolism and to the regulation of glutamatergic neurotransmission (20). The outer mitochondrial membrane monoamine oxidase (MAO; EC 1.4.3.4) is perhaps the most well-known amine oxidase. In addition to the oxidation of neurotransmitters, such as dopamine and serotonin, this enzyme also oxidizes ingested amines such as phenethylamine and tyramine to prevent their functioning as false neurotransmitters. The human isoforms MAO-A and MAO-B are involved in many diseases and are important targets for antidepressant and neuroprotective drugs (22). Acyl-CoA oxidases (EC 1.3.3.6) are acyl-CoA dehydrogenase homologs involved in peroxisomal fatty acid breakdown in plants (30).
Monooxygenases
Flavoprotein monooxygenases are widely found in microorganisms and plants, and some are present in mammalian species (43). Aromatic hydroxylases like p-hydroxybenzoate 3-hydroxylase (PHBH; EC 1.14.13.2) are involved in lignin degradation. Baeyer-Villiger monooxygenases like cyclohexanone monooxygenase (EC 1.14.13.22) participate in microbial catabolic pathways by converting ketones (or aldehydes) into esters or lactones (35). Flavin-containing monooxygenases (EC 1.14.13.8) oxidize nitrogen-containing compounds and primarily are found in mammals and plants. The mammalian isoforms assist in the detoxification of drugs and other xenobiotics, whereas the plant enzymes are involved in the biosynthesis of auxin, the metabolism of glucosinolates, and in pathogen defense (38). Flavoprotein halogenases play an important role in the biosynthetic pathways of antibiotics, antitumor agents, and other natural products (39).
Chemistry
The isoalloxazine moiety of the flavin cofactor forms the catalytic heart of a flavoenzyme. It can undergo one- and two-electron redox transitions and form covalent adducts with substrates and protein residues. The redox properties of the flavin cofactor are modulated by the protein environment. In free flavin, the one-electron reduced state is thermodynamically unstable. Flavoenzymes, however, can stabilize the neutral or anionic semiquinone state (Fig. 1c) (44), and they can pass the electrons one at a time to other redox centers.
The redox states of the flavin cofactor in a purified flavoenzyme can be conveniently studied by optical spectroscopy (see also Flavoprotein Protocols article). Oxidized (yellow) flavin has characteristic absorption maxima around 375 and 450 nm (Fig. 1b and 1c). The anionic (red) and neutral (blue) semiquinone show typical absorption maxima around 370 nm and 580 nm, respectively (Fig. 1b and 1c). During two-electron reduction to the (anionic) hydroquinone state, the flavin turns pale, and the absorption at 450 nm almost completely disappears (Fig. 1b and 1c). The optical properties of the flavin can be influenced through the binding of ligands (substrates, coenzymes, inhibitors) or the interaction with certain amino acid residues. In many cases, these interactions result in so-called charge-transfer complexes that give the protein a peculiar color.
The catalytic cycle of each flavoenzyme consists of a reductive half-reaction, in which the flavin is reduced, and an oxidative half-reaction, in which the reduced flavin is reoxidized. The reduction and oxidation steps are in many cases irreversible, which enables the direct characterization of reaction intermediates (see “See Also” section and the Further Reading List).

Figure 1. (a) Redox states of the flavin cofactor. Flavoenzymes generally stabilize the anionic hydroquinone state (pKa free reduced flavin = 6.7). (b) Oxidized (—), anionic semiquinone (∙∙∙), and hydroquinone (-∙-) forms of the FAD cofactor of Arabidopsis thaliana GALDH (adapted from Reference 13). (c) Oxidized (—), neutral semiquinone (∙∙∙), and hydroquinone (-∙-) forms of the FMN cofactor of Bacillus subtilis flavodoxin YkuP (adapted from Reference 44).
Reductases
Flavoprotein reductases primarily use NAD(P)H (AH2) as electron donor:
![]()
and pass these electrons to a protein substrate or another electron acceptor (B) in two single-electron steps:

or, alternatively, in one two-electron step:
![]()
Several of the reductases mentioned here belong to the same structural family (the FNR family), and they are mechanistically related to each other (9). A two-electron reduction of the flavin by NAD(P)H in these enzymes typically involves the transient formation of an oxidized flavin-reduced pyridine nucleotide charge-transfer complex, which is followed by hydride transfer. After reduction, the flavin can transfer its electrons to different redox partners. With NADH:cytochrome b5 reductase, this transfer occurs in separate single-electron transfer steps. With NADPH: cytochrome P450 reductase, an enzyme containing which a “complex” flavoenzyme that contains two flavins, one electron is first intramolecularly transferred from FAD to FMN, before the reaction with cytochrome P450 takes place. With FNR, NADP+ first has to bind to the oxidized form, before the very fast one-electron transfer from the specifically interacting reduced ferredoxin (Fdred) occurs (8). Subsequent dissociation of the oxidized ferredoxin (Fdox) is rate-limiting in catalysis. The enzyme semiquinone-NADP+ complex then reacts with another reduced ferredoxin molecule to yield the flavin hydroquinone state. In the final steps of the catalytic cycle, the NADP+ is reduced and the NADPH dissociates:

NQO1 is a homodimer with a flavodoxin fold (5). This enzyme does not stabilize the semiquinone state. The obligate two-electron transfer mechanism prevents the generation of quinone radicals and redox cycling, which would result in oxidative stress. The NADPH and quinone substrates occupy the same site, consistent with the observed ping-pong bi-bi mechanism. NQO1 is inhibited by many (poly)aromatic compounds including the anticoagulant dicoumarol and the phytoalexin resveratrol (5).
Dehydrogenases
Flavoprotein dehydrogenases oxidize a large variety of organic substrates (AH2) and mainly use quinones and electron transfer proteins (B) as electron acceptors (see above scheme for reductases). Substrate dehydrogenation through flavin attack may either occur via a hydride transfer, carbanion, or radical mechanism (11). Determining the reaction mechanism of a given flavoprotein dehydrogenase (or oxidase) is a key step in its characterization and often rather controversial, especially because the mechanism may be dependent on the type of (model) substrate used.
MCAD is one of the best studied flavoprotein dehydrogenases (10). In this enzyme, the pro-R α-hydrogen of the acyl-CoA thioester is removed by the catalytic base Glu376 and the pro-R β-hydrogen of the substrate is transferred directly to flavin N5 as a hydride (11) (Fig. 2a). MCAD is inactivated by a range of acyl-CoA derivatives. One such compound is methylenecyclopropylacetyl-CoA which acts as a suicide inhibitor by forming a covalent adduct with flavin N5 (see Further Reading for more information).
GALDH shows a high enantio-preference for L-galactono-1,4-lactone (13). Reoxidation of the two-electron reduced enzyme by cytochrome c occurs in two single-electron steps and involves the intermediate formation of the red anionic flavin semiquinone (Fig. 1b). Related aldonolactone oxidoreductases act as true oxidases, which suggests that they provide a better access of molecular oxygen to the active site.

Figure 2. (a) Proposed hydride transfer mechanism for substrate oxidation in MCAD. (b) Proposed polar nucleophilic mechanism for the reductive half-reaction in MAO.
Disulfide oxidoreductases
FAD-dependent disulfide oxidoreductases contain active-site thiols. They use a dithiol substrate and NAD+ to form a disulfide product and NADH or act in the reverse direction yielding NAD(P)+ and a reduced (dithiol) product.
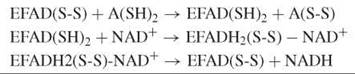
Disulfide oxidoreductases are structurally related homodimers with residues from both subunits participating in the catalysis of the two active sites (see the Further Reading for more information). The reductive half-reaction of lipoamide dehydrogenase involves the reduction of the active-site disulfide by the dihydrolipoamide substrate. During this reaction, the enzyme turns red, and a typical absorbance develops around 530 nm because of the formation of a charge-transfer species between the oxidized flavin and the nearby thiolate. The intensity of the charge-transfer absorption band is strongly pH dependent as influenced by the pK a-modulating properties of a C-terminal histidine, which acts as an acid-base catalyst. Binding of NAD+ then shifts the reducing equivalents from the disulfide to the flavin because of an increase of the redox potential of the FAD. The catalytic cycle is terminated by flavin reoxidation and dissociation of NADH.
Thioredoxin reductase (TrxR) acts in the reverse direction and shows a somewhat different mechanism, which is dependent on the protein source. Prokaryotes, plants, and lower eukaryotes contain a 35-kDa TrxR with one redox-active disulfide. Higher eukaryotes produce a 55-kDa TrxR that has either an additional redox-active disulfide or a selenenylsulfide in the flexible C-terminal part of the neighboring subunit (15). In low Mr TrxR, a large conformational change is required to move reducing equivalents from the apolar flavin site to the surface of the protein where the thioredoxin redox partner binds. In high Mr TrxR, this transfer is mediated by the second disulfide or selenylsulfide, and the conformational changes required are comparatively small (17).
Npx and Nox use a stable cysteine-sulfenate redox center, in concert with FAD, in catalysis (16). In Npx, the sulfenate form of the resting enzyme is reduced by NADH via the flavin, producing a thiolate:
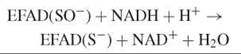
The thiolate is stabilized by a histidine, which facilitates the nucleophilic attack of the reduced enzyme-NADH complex by hydrogen peroxide:
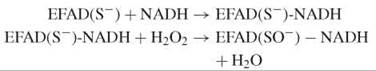
Again, NADH reduces the sulfenate via the flavin, which prepares the enzyme for the next cycle:
![]()
In Nox, NADH reduction of the thiolate form involves the stabilization of the four-electron reduced enzyme:
![]()
Subsequent reaction with molecular oxygen then yields the active-site sulfenate:
![]()
The Nox oxidizing substrate must be activated prior to its reaction with the active site thiolate. Kinetic studies with the Cys42Ser variant have indicated the formation of a flavin C4a-hydroperoxide as a primary oxygenated intermediate in reoxidation of the reduced enzyme-NAD+ complex (16). In the Cys42Ser variant, hydrogen peroxide is eliminated directly to give the oxidized enzyme. In wild-type Nox, Cys42 is in a favorable position for nucleophilic attack on the distal peroxyflavin oxygen, which yields the sulfenate and the flavin C4a-hydroxide in a monooxygenase type of reaction (vide infra). The flavin C4a-hydroxide eliminates water directly to give the oxidized enzyme.
Oxidases
Flavoprotein oxidases catalyze the conversion of a substrate single bond to a double bond. They differ from flavoprotein dehydrogenases in that the reduced flavin is reoxidized by molecular oxygen to form hydrogen peroxide.
![]()
The reaction of singlet-reduced flavin with triplet oxygen is spin forbidden and therefore involves the initial formation of a flavin semiquinone-superoxide anion radical pair (11). This caged radical pair can dissociate into oxygen radicals, or it can undergo a second electron transfer that produces hydrogen peroxide and oxidized flavin. Or, it can collapse to form a flavin-C4a-(hydro)peroxide covalent adduct. This adduct is the essential oxygenation species in flavoprotein monooxygenases (vide infra) but has never been detected in flavoprotein oxidases. The oxygen reactivity of flavoprotein oxidases can vary dramatically, and it is not entirely clear what determines this reactivity (45).
Flavoprotein oxidases come in many flavors. They can have different folds and topologies (46) and are active with many different substrates, which include (amino) acids, mono- and polyamines, nitroalkanes, aliphatic and aromatic alcohols, monosaccharides and oligosaccharides, thiols, thioesters, and so on. Flavoprotein oxidases obey either a ping-pong or ternary complex kinetic mechanism. In the latter case, the product (A) remains bound during the oxidative half-reaction. The rate-limiting step in catalytic turnover is often represented by the rate of flavin reduction or the rate of product release. The kinetic mechanism and also the rate-limiting step of catalysis may vary depending on the type of (model) substrate used.
DAAO is one of the most extensively studied flavoprotein oxidases. The homodimeric enzyme catalyzes the strictly stereospecific oxidative deamination of neutral and hydrophobic D-amino acids to give α-keto acids and ammonia (Fig. 3a). In the reductive half-reaction the D-amino acid substrate is converted to the imino acid product via hydride transfer (21). During the oxidative half-reaction, the imino acid is released and hydrolyzed. Mammalian and yeast DAAO share the same catalytic mechanism, but they differ in kinetic mechanism, catalytic efficiency, substrate specificity, and protein stability. The dimeric structures of the mammalian enzymes show a head-to-head mode of monomer-monomer interaction, which is different from the head-to-tail mode of dimerization observed in Rhodotorula gracilis DAAO (20). Benzoate is a potent competitive inhibitor of mammalian DAAO. Binding of this ligand strengthens the apoenzyme-flavin interaction and increases the conformational stability of the porcine enzyme.
MAO-A and MAO-B catalyze the oxidative deamination of aromatic amines to the corresponding aldehydes. Two active-site tyrosyl residues function in both isoforms as an “aromatic cage” (23). The kinetic mechanism of MAO is similar to that of DAAO (Fig. 4). The active sites of either MAO-A or MAO-B do not contain basic residues that could possibly function as a proton acceptor in the reductive half-reaction. However, the bent conformation of the isoalloxazine ring of the flavin might provide a clue as to how proton abstraction might occur (23). The strained conformation of the isoalloxazine moiety results in a higher electron density at N5 and lowered electron density at the C4a position of the flavin ring. This composition facilitates the nucleophilic attack of the basic substrate amine lone pair on the C4a position of the cofactor that results in a flavin-substrate adduct that would be isoelectronic with the reduced flavin ring (Fig. 2b). The N5 of the reduced flavin could constitute the strong base required to abstract the proton from the benzyl carbon of benzylamine substrates, and the “aromatic cage” might polarize the amine moiety of the substrate to make it more nucleophilic in accord with the proposed mechanism (23).
Nitroalkane oxidase (NAO; EC 1.7.3.1) from Fusarium oxysporum catalyzes the oxidation of neutral nitroalkanes to the corresponding aldehydes or ketones with the production of nitrite and hydrogen peroxide (24). The enzyme is evolutionary related to the acyl-CoA dehydrogenases and acyl-CoA oxidases, but it contains an aspartate (Asp402) instead of a glutamate as active site base. NAO obeys a ternary complex mechanism and is the only flavoprotein oxidase for which a carbanion has been established as an intermediate in catalysis. With nitroethane as a substrate, the rate-limiting step for flavin reduction is formation of the nitroethane anion. NAO prefers the neutral substrate. If the enzyme is incubated with a mixture of neutral and anionic nitroethane (pka = 8.0), then the enzyme is suicide inactivated through covalent flavin adduct formation with two substrate molecules (Fig. 5). At physiological pH, catalysis begins with proton abstraction from the neutral nitroalkane by Asp402. Nucleophilic attack of the nitroalkane anion on the flavin followed by nitrite loss would form an electrophilic cation (Fig. 5). Attack by hydroxide rather than a nitroalkane anion on the cation would form a species, which could eliminate the respective aldehyde to form reduced FAD (24).
Vanillyl-alcohol oxidase (VAO; EC 1.1.3.38) is the prototype of a family of flavoenzymes that favor the covalent binding of FAD (3). In addition to the oxidation of aromatic alcohols, the enzyme catalyzes demethylation, deamination, and hydroxylation reactions (25). The reaction with 4-methoxymethylphenol obeys a ternary complex mechanism (cf. Fig. 4). During binding to the oxidized enzyme, the substrate gets activated by deprotonation. In the rate-limiting reductive half-reaction, the substrate is converted to a quinone methide species via hydride transfer (Fig. 3b). During the oxidative half-reaction, the quinone methide is attacked by water in the active site with production of the aromatic aldehyde, methanol, and hydrogen peroxide. Studies with alternative substrates and site-directed mutants revealed that the attack by water is stereospecific and that the enantios- electivity of the enzyme can be reversed by transferring the catalytic base involved in water attack to the other site of the substrate-binding pocket (26).
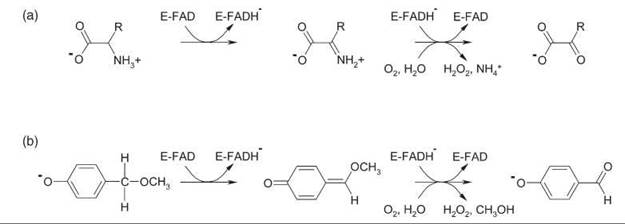
Figure 3. (a) Oxidative deamination of D-amino acids by DAAO. (b) Oxidative demethylation of 4-methoxymethylphenol by VAO.

Figure 4. Ternary complex kinetic mechanism of flavoprotein oxidases. Q: imine (DAAO and MAO); quinone methide (VAO). P1 + P2: a-keto-acid + ammonia (DAAO); aldehyde + ammonia (MAO); aldehyde + methanol (VAO).

Figure 5. Proposed carbanion mechanism for the reductive half-reaction of NAO. The lower path shows the off-pathway covalent adduct formed by attack of a second nitroalkane anion.
Monooxygenases
Flavoprotein monooxygenases mainly use NAD(P)H as electron donor and insert one atom of molecular oxygen into their substrates. Oxygen activation of flavoprotein monooxygenases involves the (transient) stabilization of a flavin C4a-(hydro) peroxide. This species performs either a nucleophilic or electrophilic attack on the substrate (Fig. 6). Oxygenation reactions catalyzed by flavoprotein monooxygenases include hydroxylations, epoxidations, Baeyer-Villiger oxidations, and sulfoxidations (43).
PHBH is the protype of the flavoprotein aromatic hydroxylases. Each subunit of this dimeric enzyme contains two active sites which, during catalysis, are alternately visited by the isoalloxazine ring of the FAD cofactor (31). Catalysis is initiated by reduction of the flavin in the exterior active site. The reduced flavin then moves to the interior active site where the reactions with oxygen occur. A similar conformational flexibility of the FAD cofactor has been observed in the crystal structures of phenol hydroxylase (EC 1.14.13.7) and 3-hydroxybenzoate 4-hydroxylase (EC 1.14.13.23). PHBH obeys the following kinetic mechanism:

Binding of 4-hydroxybenzoate (S) in the phenolate form facilitates flavin reduction by NADPH. After NADP+ release, the flavin hydroquinone reacts with molecular oxygen to yield the flavin C4a-hydroperoxide oxygenation species. Protonation of the distal oxygen of the peroxflavin facilitates the electrophilic attack on the nucleophilic carbon center of the substrate phenolate. After monooxygenation, the resulting hydroxyflavin is decomposed, and the 3,4-dihydroxybenzoate product (P) is released (Fig. 6). Studies from site-directed mutants have provided many insights in the process of substrate hydroxylation (32). In general, flavoprotein aromatic hydroxylases display a narrow substrate specificity and are very regioselective.
Baeyer-Villiger monooxygenases are another class of flavoprotein monooxygenases. These enzymes typically depend on NADPH as an electron donor and catalyze a relatively broad range of asymmetric oxygenation reactions with high enantios-electivity or enantiotoposelectivity (35). The kinetic mechanism of Baeyer-Villiger monooxygenases differs from that of the aromatic hydroxylases:

Here, NADP+ stays bound throughout the entire reaction cycle. Furthermore, Baeyer-Villiger monooxygenases usually promote the deprotonation of the flavin C4a-peroxide (Fig. 6), which thereby facilitates nucleophilic substitution reactions (36). Structural and kinetic studies on phenylacetone monooxygenase have revealed the importance of a conserved arginine in the reactivity with organic substrates (37).
Two-protein component flavoprotein monooxygenases generally consist of a reductase component and an oxygenase component. In addition to hydroxylation and epoxidation reactions, these enzymes can also catalyze desulfurization, halogenation, and light-emission reactions (43). Oxidative halogenation by tryptophan 7-halogenase has been proposed to involve the formation of hypochlorous acid through decomposition of the flavin C4a-hydroperoxide by chloride ion. Structural analysis suggests that the resulting hypochlorous acid is guided through a tunnel inside the enzyme to the tryptophan-binding pocket where it is activated to participate in the regioselective halogenation of the substrate (40).
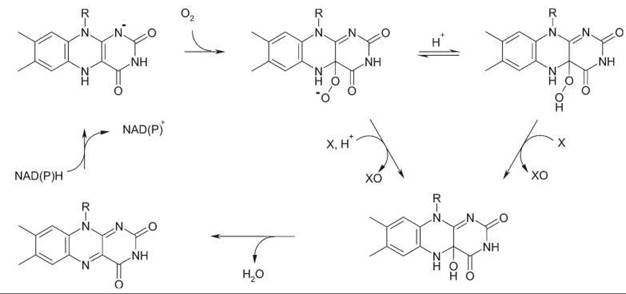
Figure 6. General mechanism for flavoprotein monooxygenases. With Baeyer-Villiger monooxygenases (nucleophilic oxygenation), NADP+ stays bound during the entire reaction cycle.
Chemical Tools and Techniques
Understanding the action mechanism of flavoenzymes heavily relies on the combination of different chemical tools and techniques. First, it is of utmost importance to have a pure and stable (recombinant) protein. Size exclusion chromatography will provide information about the enzyme quaternary structure, and mass spectrometry can establish posttranslational modifications. For a detailed insight into the protein structure, well-diffracting crystals are needed to determine the X-ray structure.
The properties of the flavin are exploited for flavoenzyme characterization. Absorbance spectroscopy (under anaerobisis, as needed) can be used for binding studies, redox titrations, and rapid-reaction kinetics (Note the relevant articles in the “See Also” section). Other useful techniques include fluorescence spectroscopy (at equilibrium or time-resolved, although in most cases the flavin fluorescence is quenched in the holoenzyme), EPR, and NMR. Several of these techniques have been developed early on (also) thanks to research on flavoproteins.
Preparation of the apoflavoprotein (1) and its reconstitution with 13C- and 15N-enriched flavins and subsequent NMR analysis yields information about the n-electron density of the atoms of the isoalloxazine ring in the different redox states (47). Reconstitution with chemically modified (“artificial”) flavins can be of help in substrate structure-activity relationship studies (23) and provide information about the solvent accessibility of the active site (48).
Site-directed mutagenesis can be used to establish the function of individual amino acid residues. This method also allows to introduce chemical probes (e.g., fluorophores) that may give insight into the dynamic features and folding properties of a flavoenzyme and its interaction with other protein partners (33, 34). In this context, it is important to stress that catalytically relevant conformational changes of flavoenzymes have been demonstrated that actually exploit the flavin itself as an excellent intrinsic spectroscopic probe (18, 31).
Bioinformatic tools are of increasing importance for the characterization of flavoenzymes. This finding holds for protein sequence and protein structural analysis as well as for gaining insight into the reactivity of the flavin cofactor by combined quantum mechanical and molecular mechanical (QM/MM) simulations (12).
Practical Applications and Future Research Directions
The available knowledge about the structural and mechanistic properties of flavoenzymes is extremely valuable for the discovery and characterization of new flavoenzymes and for the development of practical applications. Several flavoenzyme-inspired applications already exist but many more can be foreseen. Glucose oxidase (EC 1.1.3.4) (27) and cholesterol oxidase (EC 1.1.3.6) (28) are widely applied in diagnostics. Many flavoprotein oxidases and monooxygenases serve as biocatalysts for the production of fine chemicals and pharmaceuticals (41). DAAO is used for the enzymatic synthesis of the cephalosporin precursor 7-aminocephalosporanic acid and in gene therapy for tumor treatment (20). Other flavoenzymes, like e.g. MAO (22) and TrxR (15) serve as drug targets.
Flavoenzymes may be ideal model systems for the development of new tools to be made available for the study of enzymes in general. One example may be the use of cholesterol oxidase for single molecule enzymology, which exploits the flavin fluorescence changes during the catalytic cycle (29).
Sophisticated computational tools will facilitate the prediction of novel flavoenzyme functions from sequence, provide a better insight in enzyme and ligand dynamics, and improve methods for flavin-dependent biocatalyst design (41). Another challenge is to use the tools of chemical biology to study the functional properties of flavoenzymes in their natural environment, the living cell. In this article, only a selected group of “simple” flavoenzymes has been discussed. However, one should remember the “complex” flavoenzymes, and the flavin-dependent photoreceptors, and the emerging group of flavoenzymes that do not catalyze redox reactions (see Further Reading List).
References
1. Hefti MH, Vervoort J, van Berkel WJH. Deflavination and reconstitution of flavoproteins. Eur. J. Biochem. 2003; 270:4227-4242.
2. Mewies M, Mclntire WS, Scrutton NS. Covalent attachment of flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) to enzymes: the current state of affairs. Prot. Sci. 16. 1998; 7:7-20.
3. Leferink NGH, Heuts DPHM, Fraaije MW, van Berkel WJH. The growing VAO flavoprotein family. Arch. Biochem. Biophys. In press. 17.
4. Massey V, Hemmerich P. Flavin and pteridine monooxygenases. In: The Enzymes, 3rd edition. Boyer PD, ed. 1975. Academic Press, New York. pp. 191-252.
5. Deller S, Macheroux P, Sollner S. Flavin-dependent quinone 18. reductases. Cell. Mol. Life Sci. 2008; 65:141-160.
6. Kirchner U, Westphal AH, Muller R, van Berkel WJH. Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J. Biol. Chem. 2003; 278:47545-47553.
7. Nijvipakul S, Wongratana J, Suadee C, Entsch B, Ballou DP, Chaiyen P. LuxG is a functioning flavin reductase for bacterial luminescence. J. Bacteriol. 2008; 190:1531-1538.
8. Medina M, Gomez-Moreno C. Interaction of ferredoxin-NADP(+) reductase with its substrates: optimal interaction for efficient electron transfer. Photosynth. Res. 2004; 79:113-131.
9. Aliverti A, Pandini V, Pennati A, de Rosa M, Zanetti G. Structural and functional diversity of ferredoxin-NADP(+) reductases. Arch. Biochem. Biophys. In press.
10. Ghisla S, Thorpe C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur. J. Biochem. 2004; 271:494-508.
11. Ghisla S, Massey V. Mechanisms of flavoprotein-catalyzed reactions. Eur. J. Biochem. 1989; 181:1-17.
12. Bhattacharyya S, Stankovich MT, Truhlar DG, Gao J. Combined quantum mechanical and molecular mechanical simulations of one- and two-electron reduction potentials of flavin cofactor in water, medium-chain acyl-CoA dehydrogenase, and cholesterol oxidase. J. Phys. Chem. A. 2007; 111:5729-5742.
13. Leferink NGH, Van den Berg WAM, Van Berkel WJH. L-Galactono-γ-lactone dehydrogenase from Arabidopsis thaliana, a flavoprotein involved in vitamin C biosynthesis FEBS J. 2008; 275:713-726.
14. Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT. Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J. Mol. Biol. 2005; 350:543-552.
15. Huang HH, Arscott LD, Ballou DP, Williams CH Jr. Acid-base catalysis in the mechanism of thioredoxin reductase from Drosophila melanogaster. Biochemistry 2008; 47:1721-1731.
16. Claiborne A, Yeh JI, Mallett TC, Luba J, Crane IIIEJ, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 1999; 38:15407-15416.
17. Williams CH, Arscott LD, Muller S, Lennon BW, Ludwig ML, Wang PF, Veine DM, Becker K, Schirmer RH. Thioredoxin reductase two modes of catalysis have evolved. Eur. J. Biochem. 2000; 267:6110-6117.
18. van den Berg PAW, Mulrooney SB, Gobets B, van Stokkum IHM, van Hoek A, Williams CH, Jr., Visser AJWG. Exploring the conformational equilibrium of E. coli thioredoxin reductase: characterization of two catalytically important states by ultrafast flavin fluorescence spectroscopy. Protein Sci. 2001; 10:2037-2049.
19. Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 2004; 44:325-347.
20. Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G. Physiological functions of D-amino acid oxidases: from yeast to humans. Cell. Mol. Life Sci. 2007; 64:1373-1394.
21. Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Crystal structure of D-amino acid oxidase: a case of active site mirror-image convergent evolution with flavocytochrome b2. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:7496-7501.
22. Binda C, Wang J, Pisani L, Caccia C, Carotti A, Salvati P, Edmondson DE, Mattevi A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J. Med. Chem. 2007; 50:5848-5852.
23. Edmondson DE, Binda C, Mattevi A. Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch. Biochem. Biophys. 2007; 464:269-276.
24. Fitzpatrick PF, Orville AM, Nagpal A, Valley MP. Nitroalkane oxidase, a carbanion-forming flavoprotein homologous to acyl-CoA dehydrogenase. Arch. Biochem. Biophys. 2005; 433:157-165.
25. Fraaije MW, van Berkel WJH. Catalytic mechanism of the oxidative demethylation of 4-(methoxymethyl)phenol by vanillyl-alcohol oxidase. Evidence for formation of a p-quinone methide intermediate. J. Biol. Chem. 1997; 272:18111-18116.
26. van den Heuvel RHH, Fraaije MW, Ferrer M, Mattevi A, van Berkel WJH. Inversion of stereospecificity of vanillyl-alcohol oxidase. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:9455-9460.
27. van Hellemond EW, Leferink NGH, Heuts DPHM, Fraaije MW, van Berkel WJH. Occurrence and biocatalytic potential of carbohydrate oxidases. Adv. Appl. Microbiol. 2006; 60:17-54.
28. Srisawasdi P, Chaichanajarernkul U, Teerakranjana N, Kroll MH. Implementation of cellulomonas cholesterol oxidase for total serum cholesterol determination by the endpoint method. J Clin Lab Anal. 2008; 22:50-58.
29. Lu HP, Xun L, Xie XS. Single-molecule enzymatic dynamics. Science 1998; 282:1877-f1882.
30. Arent S, Pye VE, Henriksen A. Structure and function of plant acyl-CoA oxidases. Plant Physiol. Biochem. 2008; 46:292-301.
31. Entsch B, van Berkel WJH. Structure and mechanism of parahydroxybenzoate hydroxylase. FASEB J. 1995; 9:476-483.
32. Entsch B, Cole LJ, Ballou DP. Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Arch. Biochem. Biophys. 2005; 433:297-311.
33. Westphal AH, Matorin A, Hink MA, Borst JW, van Berkel WJH, Visser AJWG. Real-time enzyme dynamics illustrated with fluorescence spectroscopy of p-hydroxybenzoate hydroxylase. J. Biol. Chem. 2006; 281:11074-11081.
34. Kudryashova EV, Visser AJWG, van Berkel WJH. Monomer formation and function of p-hydroxybenzoate hydroxylase in reverse micelles and in dimethylsulfoxide/water mixtures. Chem- BioChem. 2008; 9:413-419.
35. Kamerbeek NM, Jansen DB, Van Berkel WJH, Fraaije MW. Baeyer-Villiger monooxygenases, an emerging family of flavin-dependent biocatalysis. Adv. Synth. Catal. 2003; 345:667-678.
36. Sheng D, Ballou DP, Massey V. Mechanistic studies of cyclohexanone monooxygenase: chemical properties of intermediates involved in catalysis. Biochemistry. 2001; 40:11156-11167.
37. Torres DE Pazmino, Baas BJ, Janssen DB, Fraaije MW. Kinetic mechanism of phenylacetone monooxygenase from Thermobifida fusca. Biochemistry 2008; 47:4082-4093.
38. Schlaich NL. Flavin-containing monooxygenases in plants: looking beyond detox. Trends Plant Sci. 2007; 12:412-418.
39. van Pee KH, Dong C, Flecks S, Naismith J, Patallo EP, Wage T. Biological halogenation has moved far beyond haloperoxidases. Adv. Appl. Microbiol. 2006; 59:127-157.
40. Dong C, Flecks S, Unversucht S, Haupt C, van Pee KH, Naismith JH. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005; 309:2216- 2219.
41. Joosten V, van Berkel WJH. Flavoenzymes. Curr. Opin. Chem. Biol. 2007; 11:195-202.
42. Toogood HS, Leys D, Scrutton NS. Dynamics driving function: new insights from electron transferring flavoproteins and partner complexes. FEBS J. 2007; 274:5481-5504.
43. van Berkel WJH, Kamerbeek NM, Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006; 124:670-689.
44. Lawson RJ, von Wachenfeldt C, Haq I, Perkins J, Munro AW. Expression and characterization of the two flavodoxin proteins of Bacillus subtilis, YkuN and YkuP: biophysical properties and interactions with cytochrome P450. Biochemistry 2004; 43:12390-12409.
45. Mattevi A. To be or not to be an oxidase: challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci. 2006; 31:276- 283.
46. Fraaije MW, Mattevi A. Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem Sci. 2000; 25:126-132.
47. Macheroux P, Ghisla S, Sanner C, Ruterjans H, Muller F. Reduced flavin: NMR investigation of N5-H exchange mechanism, estimation of ionisation constants and assessment of properties as biological catalyst. BMC Biochem. 2005; 6:26.
48. Ghisla S, Massey V. New flavins for old: artificial flavins as active site probes of flavoproteins. Biochem. J. 1986; 239:1-12.
Further Reading
Chapman SK, Reid GA, eds. Flavoprotein protocols. In Methods in Molecular Biology, Volume 131. 1999. Humana Press, Totowa, NJ.
Muller F, ed. Chemistry and Biochemistry of Flavoenzymes, Volume I. 1991. CRC Press, Boca Raton, FL.
Muller F, ed. Chemistry and Biochemistry of Flavoenzymes, Volume II. 1991. CRC Press, Boca Raton, FL.
Muller F, ed. Chemistry and Biochemistry of Flavoenzymes, Volume III. 1992. CRC Press, Boca Raton, FL.
Nishino T, Miura R, Kiyoshi F, and Tanokura M, eds. Flavins and Flavoproteins, Volume XV. 2005. ArchiTect Inc., Tokyo.
Palfey BA, Massey V. Flavin-dependent enzymes. In Comprehensive Biochemical Catalysis. Sinnott M, ed. 1998. Academic Press, New York, pp. 83-154.
See Also
NAD(P) Dependent Dehydrogenases
Oxygen-Activating Enzymes, Chemistry of
Transient State Enzyme Kinetics