CHEMICAL BIOLOGY
Fluorescent Labeling and Fluorescent Spectroscopy: Overview of Applications in Chemical Biology
Kim E. Sapsford, George Mason University, Manassas, Virginia and the Center for Bio/Molecular Science and Engineering Code 6900, U.S. Naval Research Laboratory, Washington, D.C.
Lorenzo Berti, National Research Center on NanoStructures and BioSystems at Surfaces, Modena, Italy
Igor L. Medintz, Center for Bio/Molecular Science and Engineering Code 6900, U.S. Naval Research Laboratory, Washington, D.C.
doi: 10.1002/9780470048672.wecb174
Fluorescence spectroscopy in all of its variations can be considered among the most powerful types of analysis available to chemical biology. However, to be useful almost all applications are dependent on optimal labeling of biomolecules with a fluorophore and on the appropriate choice of analytical technique. In this article, we examine the applications and contributions of fluorescent spectroscopy to chemical biology in three inter-related sections. We first examine the properties of the common fluorophores available from many disparate structural and functional classes, which includes a discussion of their individual benefits and liabilities in the context of their application. The available conjugation chemistries used to attach fluorophores to myriad biomolecules are next reviewed. As each class of biomolecule differs in both structure and function, the focus here is on strategies for the specific labeling of different functional groups. Last, some major types of fluorescent spectroscopy and the associated biologic questions and analysis that can be addressed with them are covered briefly.
Fluorescence can be defined as the emission of a photon from an excited singlet state electron returning to the ground state orbital of a molecule where it is paired with, but of opposite spin to, the second ground-state electron (1). Fluorescence, although it typically originates from molecules containing multiple double-bonded aromatic groups, can also be emitted from many structurally diverse substances. The power of fluorescent analysis results from the ability to label myriad biomolecules with fluorophores (or to use their intrinsic fluorescence), discretely excite and visualize them in a heterogeneous sample, and then monitor their function either in vitro or in vivo with a variety of different techniques. For the purpose of this review we define chemical biology as “applying the tools of chemistry to the understanding of biological problems” and focus in this article on the diverse classes of available fluorophores, their labeling chemistries, the predominant fluorescent techniques in use, and some biologic questions that can be answered by their application. As fluorescence is a diverse and complex discipline, an in-depth description of each aspect is beyond the current scope; the focus here is more a generalized overview of fluorophore structure/function, fluorescent applications and important considerations within a chemo-biologic context. The continuing challenge in this field remains in labeling the biologic molecule(s), both in vivo and in vitro, in a specific manner such that useful data can be derived from the configuration. Fortunately, many fluorophores are available commercially along with affordable instrumentation (2). Interestingly, the most popular application of fluorescent labeling is in microscopy, for example, antibody labeling or in situ hybridization, and the many facets of this particular technique are discussed in other related articles. Additionally, myriad other prominent analytical techniques rely on fluorescent detection, including fluorescence activated cell sorting (FACS), real-time polymerase chain reaction (PCR), and various microarray analyses, all of which are covered to some extent in other articles. For the interested reader, Lakowicz’s Principles of Fluorescence Spectroscopy can be considered the primary go-to reference for almost all questions on fluorescent analysis, including history, theory, basic and advanced concepts, techniques, fluorophores, instrumentation, and applications (1). Although primarily a catalog of fluorophores, Haughland’s The Handbook, A Guide to Fluorescent Probes and Labeling Technologies is another excellent resource (available from Invitrogen, Carlsbad, CA) (2).
Properties of Common Fluorophores
Fluorophores come in a huge diversity of structures based on the materials they are derived from and can be divided into three primary classes: organic, inorganic, and biologically derived materials. Fluorophores manifest many intrinsic physical properties that are exploited in the various experimental formats, which includes their absorption/emission profiles (spanning the UV-to-near-infrared (IR) regions of the electromagnetic spectrum), varying fluorescent lifetimes, stokes shifts and quantum yields, and in some instances their sensitivity to their local environment. The exact choice of fluorescent probe for a particular application obviously depends on several factors, including the nature of the system under investigation, the question to be addressed, the pertinent biolabeling chemistries, and the available analytical instrumentation.
Biologic materials
Amino acid fluorophores
Biologic materials are a class of fluorophores that can range from amino acids and enzymatic cofactors to fluorescent proteins. As their fluorescence originates from their structures, they are sometimes referred to as intrinsic fluorophores (1). The aromatic amino acids, tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe) are the simplest structural class of these fluorophores and are ubiquitous in most naturally occurring proteins (see Fig. 1a). The UV absorbance at 280 nm, which is commonly used for protein quantitation, and the resulting emission at 340-360 nm originates mostly from the indole ring of the tryptophan residue (1, 3). Tryptophan emission also reflects the polarity of its local solvent environment and can be sensitive to the binding of substrates/ligands, protein-protein association interactions, protein denaturation, and global conformational changes in structure (1, 4-6). These interactions can be used for protein characterization and sometimes can form the basis of certain types of biosensing (see below). Because of its UV emission, tryptophan has also been used as a donor in various fluorescence resonance energy transfer (FRET) studies examining intraprotein distances and conformation changes (3). Phenylalanine has a low quantum yield, and tyrosine residues are less prevalent in proteins and have been far less used for fluorescence.
The advent of recombinant DNA technology has enabled researchers to introduce these three residues into any desired site(s) within a cloned protein for potential fluorescent usage. It is particularly advantageous for FRET configurations in which optimal placement of a donor fluorophore is desired (2). Although it is an advantage in some cases, the widespread occurrence of these residues in almost all natural protein structures can also be considered a disadvantage, especially in applications in which observing a specific protein in a heterogeneous environment is desired. Another potential disadvantage includes excitation confined to the UV region, which can result in high background signals from scatter and autofluorescence within cells or other biomolecules in a sample matrix.
Fluorescent cofactors
Enzymatic cofactors, such as nicotinamide adenine dinucleotide (NADH), nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and pyridoxal phosphate, are fluorescent and commonly found associated with various proteins where they are responsible for electron transport (see Fig. 1b and Table 1). NADH and NADPH in the oxidized form are nonfluorescent, whereas conversely the flavins, FAD and FMN, are fluorescent only in the oxidized form. Both NADH and FAD fluorescence is quenched by the adenine found within their cofactor structures, whereas NADH-based cofactors generally remain fluorescent when interacting with protein structures. The fluorescence of these cofactors is often used to study the cofactors’ interaction with proteins as well as with related enzymatic kinetics (1, 9-12). However, their complex fluorescent characteristics have not led to widespread applications beyond their own intrinsic function.

Figure 1. Chemical structure of (a) the fluorescent amino acids and (b) various fluorescent cofactors, which are commonly found in biologic species. (Modified from Reference 3.)
Table 1. Physical properties of selected fluorescent probes
|
Species |
Approximate molecular weight (Da) |
λabs max (nm) |
λem max (nm) |
Quantum yield1 |
Lifetime (ns) |
|
Biologic Materials |
|
|
|
|
|
|
Tryptophan |
204 |
295 |
353 |
0.13 |
3.1 |
|
NADH |
709 |
340 |
460 |
|
0.4 |
|
FAD |
829 |
450 |
525 |
|
2.3 |
|
BFP |
25-30,000 |
380 |
440 |
0.17 |
|
|
GFP |
25-30,000 |
396,475 |
508 |
0.79 |
3.2 |
|
R-phycoerythrin |
240,000 |
495,536 |
576 |
0.84 |
|
|
Organic Materials |
|
565 |
|
|
|
|
Fluorescein |
332 |
490 |
514 |
0.95 |
4.0 |
|
FITC |
389 |
495 |
525 |
|
4.1 |
|
TAMRA |
430 |
555 |
580 |
|
|
|
ROX |
534 |
580 |
605 |
|
|
|
Texas Red |
817* |
595 |
615 |
|
4.2 |
|
Cy3 |
766* |
550 |
570 |
0.15 |
<0.3 |
|
Cy5 |
792* |
649 |
670 |
0.28 |
1.0 |
|
Alexa Fluor 488 |
643 |
495 |
519 |
|
4.1 |
|
Alexa Fluor 555 |
1250 |
555 |
565 |
|
— |
|
Alexa Fluor 647 |
1250 |
650 |
668 |
|
1.0 |
|
Inorganic Materials |
|
|
|
|
|
|
LanthaScreen Tb |
915 |
343 |
495,545,570 |
|
μs-ms |
|
Ru chelate2 |
940 |
445 |
615 |
|
500 |
|
550 nm CdSe/ZnS QDs |
>100 kd |
UV < 550 nm |
550 |
>0.20 |
5-10 |
Drawn from References 1, 3, 7, and 8.
*NHS Ester form.
1 Quantum yield added where available; the quantum yield and fluorescent lifetimes can be highly dependent on the local environment. A fluorophore free in solution may have a different quantum yield than the same fluorophore attached to a protein, which in turn also depends on the extent of protein-to-fluorophore labeling (1-3).
2 Sigma-Aldrich Product # 71603 Ru(bpy)2(phen-5-isothiocyanate) (PF6)2.
Fluorescent proteins
Fluorescent proteins (FPs) are a class of fluorescent probes of which green fluorescent protein (GFP) from the jellyfish Aequorea victoria (see Fig. 2) is probably the best characterized and can be considered the prototype (13-16). Their utility is derived primarily from the ability to append genetically and to coexpress these proteins as a chimeric fusion with a desired target in a wide range of host cells, including bacteria, lower eukaryotes such as yeast, almost all higher eukaryotic cells, and even within transgenic animals (17). This process makes FPs, in general, a very powerful chemical biology tool for many applications, including in vivo labeling of specific proteins in cells as well as monitoring of intercellular signaling and intracellular processes (18-25). Beyond basic research, many FPs are used in a variety of applications related to drug discovery. The field has progressed rapidly to the point where now several GFP variants exist, such as blue, cyan, yellow, and various dimeric (Ds) and monomeric (m) red fluorescent proteins (BFP, CFP, YFP, DsRed, mRFP) (26-28). Continued cloning from many different species, including coral (29), in conjunction with functional optimization has led to a variety of commercially available FPs with emission maxima that range across the spectrum from 470 nm to 630 nm (Fig. 2). Tsien’s monograph on the green fluorescent protein is perhaps the best source on many important photophysical considerations when using all fluorescent proteins (14). Issues of relevance when using FPs include size; maturation time; absorption and emission shifts; sensitivity to pH, O2, temperature, and certain ions; obligatory dimerization or oligomerization; optimal placement; and photobleaching (21, 26, 30). Another important consideration, when appending such large fluorescent probes onto a protein of interest, is verifying that the primary function/activity of the endogenous parent protein is not perturbed in any way (20, 21). Rationally designed FRET sensing in live cells to monitor signaling events would not have been possible without the ability to append genetically these FPs to species of interest, with some commonly used FRET FP pairs, including GFP/BFP and CFP/YFP; see below (3, 13-18).
Phycobiliproteins, such as phycocyanin and phycoerythrin, are members of a family of fluorescent accessory, nonchlorophyll-based pigments found in cyanobacteria and eukaryotic algae. The phycobiliproteins have characteristic broad absorption profiles spanning 450-600 nm, emissions ranging 570-660 nm, and small stokes shifts; see Fig. 3. The major structural subunits, phycoerythrobilin (PEB) or phycocyanobilin (PCB), have multiple covalently attached bilin chromophores (open chain tetrapyrol, between 6 and 34) per moiety, which result both in the large molecular weight (~10 times that of GFP) and a remarkable extinction coefficient (~100 times that of GFP). The strong brightness and photostability of these fluorophores have led to their wide use as a very sensitive probe (31, 32). Although appearing to succumb to classic photobleaching under constant illumination, the loss of specific fluorescence of some phycobiliproteins, such as B-phycoerythrin (B-PE). may actually be from the exciting of more than one chromophore per protein that can annihilate the excited state and decrease the quantum yield (1, 33). These FPs are currently available from several commercial sources (Martek Biosensciences Corporation, Columbia, MD; Invitrogen; Cyanotech Corporation, Kailua-Kona, HI; and Europa Bioproducts Ltd., Brussels, Belgium) and are commonly conjugated to antibodies or steptravidin for use in a variety of immunoassay and other detection formats.
Phytochromes are a family of bilin binding proteins that function as photoreceptors, regulating many physiologic processes in plants, cyanobacteria, and other microorganisms (34, 35). Unlike their light harvesting phycobiliprotein counterparts, native phytochromes binding their natural bilins (phytochromobilin or phycocyanobilin) result in a nonfluorescent conjugate caused by deactivation by an efficient double-bond isomerization. If, however, the apophytochrome is allowed to bind phycoerythrobilin (PEB; found naturally in phycobiliproteins), a highly fluorescent protein results, which is referred to as a phytofluor (36). These proteins have large extension coefficients, high quantum yields (0.7), and good photostability and pH stability. Highly fluorescent proteins can also result from site-directed mutagenesis of the apoprotein while maintaining the natural bilin (37). In contrast to phycobiliproteins, phytochrome-based FPs have not found widespread application as fluorescent probes principally because in their natural form, they are not fluorescent and only become so through either genetic mutation of the bilin binding site or insertion of a non-native bilin.

Figure 2. (a) Ribbon structure of the GFP and (b) fluorescence emission profiles from various representative fluorescent proteins. (Modified from Reference 3.)

Figure 3. Absorption and emission profile of the phycobiliprotein B-phycoerythrin (B-PE), which is a multi-subunit multi-chromophore fluorescent protein with exceptional absorption and emission properties.
Enzyme-generated luminescence
Chemiluminescence and bioluminescence are unique processes in the sense that the excited state species is generated enzymatically through a chemical reaction rather than photophysically. Although closely related, bioluminescence (BL) is considered a natural phenomenon found in certain beetles, bacteria, and marine species, whereas chemiluminescence (CL) originates from reactions with synthetic substrates. Both processes result from an enzymatically catalyzed reaction that generates an excited state chemical product that decays to produce light emission, generally between 400 nm and 550 nm (3). Although a variety of BL and CL enzymes/substrates combinations are available from which to choose, the firefly Luciferase/Luciferin pair is the most commonly exploited BL reporter system and Horseradish peroxidase (HRP) is the most popular CL system (3). BL and CL are dark field techniques, which means they do not require an excitation source, therefore, reducing the background fluorescence and greatly improving the sensitivity and potential limits of detection (LODs) when used for detection. BL/CL have been applied widely, including in microarrays and nanoarrays, in in vivo imaging ranging from whole animals down to single cells, in numerous biosensors, and as tracers in immunoassays such as enzyme-linked immunosorbent assays (ELISAs) (38-40). The only major disadvantages of using BL and CL are that multiple wash and reagent addition steps are often required in the “development” of the signal, which can make the analysis time longer, and the user typically has a limited time in which to collect the generated signal as the local substrate is consumed rapidly. Therefore, when multiple samples are run simultaneously in microtiter plates or microarray formats for high-throughput analysis, luminescent imaging techniques are preferred over the more traditional plate readers (41).
Organic materials
Standard organic fluorophores
Organic dye materials represent the largest and best characterized class of probes used in all manner of fluorescent analysis. As an overall class, these dyes are used in almost all areas of biotechnology, including biosensing, cellular imaging, clinical immunofluorescence, and in DNA/protein microarrays (42-45). Several major structural classes of organic fluorophore span the UV-to-near-IR spectrum; see Fig. 4. UV dyes are typically pyrene-based, naphthalene-based, and coumarin-based structures, whereas the Vis/near-IR dyes include a variety of fluorescein-based, rhodamine-based, and cyanine-based derivatives; see Fig. 5. Fluorescein dyes are extremely popular because they have good quantum yields, are relatively cheap, water soluble and readily bioconjugated, and easily excited using a standard argon-ion laser (488 nm). However, fluorescein has a high rate of photobleaching, is sensitive to pH (sometimes considered an advantage; see below), and can self-quench at high degrees of substitution onto biomolecules. Various alternatives are available such as Oregon Green dyes (Invitrogen, Carlsbad, CA), which are fluorinated fluorescein analogs; AlexaFluor dyes [Invitrogen; (46)]; Cy dyes (GE Healthcare, Buckinghamshire, United Kingdom); and BIODIPY dyes (Invitrogen), all of which claim to alleviate some of these issues. For some redder dyes, overlabeling can induce protein precipitation because of their low solubility in aqueous environments (47). Also, several organic-based dyes are used as fluorescent stains for visualizing cell membranes, proteins, and DNA in cells or separation gels (1, 2, 48). These dyes also include fluorescent dyes attached to lipids that allow membrane labeling and intercalating probes like ethidium bromide, which are typically weakly fluorescent until bound to DNA (1, 2, 48). In general, organic dyes have several issues that have to be considered before use; these include broad absorption/emission profiles with small stokes shifts (which can be problematic for FRET-based applications or multiplexing), solubility issues, and susceptibility to environmental influences. Cumulatively their many advantages, which include extensive characterization in the literature, their wide availability from several commercial sources, relative cost, ease of use, and many different available bioconjugation chemistries, often outweigh the above liabilities and mean that they are usually the first choice in most experiments. Haughland’s handbook is an excellent reference on many aspects of most commercial organic fluorophores (2).

Figure 4. Examples of commercially available fluorophore families. (Modified from Reference 3.) Absorbance and emission maxima along with spectral regions covered by a particular dye family are highlighted. The major suppliers are as follows: Molecular Probes, Inc. (Eugene, OR; Fluorescein, rhodamine-TMR, TAMRA and ROX, AlexaFluor, BODIPY, Oregon Green, and Texas red), GE Healthcare (Cy dyes) AnaSpec, Inc. (San Jose, CA; HiLyte Fluors), ATTO-TEC GmbH (Siegen, Germany; ATTO dyes), Molecular Biotechnology (DY dyes). and Pierce Biotechnology, Inc. (Rockford, IL; DyLight 547 and 647 dyes).

Figure 5. Chemical structures of common UV/Vis fluorescent dyes. Typical groups at the R position include CO2-, SO3-, OH, OCH3, CH3, and NO2; Rx marks the typical position of the bioconjugation linker. (Modified from Reference 3.)
Environmentally sensitive fluorophores
Environmentally sensitive fluorophores that exhibit some change in their absorption or emission properties as a function of their environment are often referred to as indicator probes (1, 3). Such dyes may be sensitive to changes in pH, ionic strength, ionic type, oxygen, solvation, or polarity. Although many dyes will exhibit some sensitivity to perturbation of their local environment, fluorescein is probably the best known; with sensitivity to pH, see Fig. 6. Again Haughland’s handbook is an excellent reference for organic dye probes optimized for environmental sensing (2). Many new indicator probes, which are sensitive to neutral and ionic molecules as well as to oxygen reactive species, have been reviewed recently in the literature (49, 50). These indicator dyes have applications for chemical sensing both in vitro and in vivo. Important considerations when loading cells with these indicator probes include whether the probe is as follows: toxic to the cell, and at what concentration, and where the dye will likely be sequestered during loading into the cell, such as within the organelles.

Figure 6. (a) Normalized absorption and emission (b) changes of fluorescein as a function of its ionization state (c). The monocationic and dicationic species are the more fluorescent. (Modified from Reference 3.)
Fluorescent polymeric microspheres and nanospheres
One photophysical limitation of organic fluorescent dyes is the tendency toward self-quenching that occurs when attempting high conjugation ratios to improve both sensitivity and LODs. Researchers have overcome this limitation by functionalizing polymeric microspheres and nanoparticles with multiple fluo- rophores (100s-1000s) that result in highly fluorescent particles. This strategy also allows one to label biomolecules with dyes that would otherwise lack reactive groups for bioconjugation or are inherently insoluble in an aqueous environment (3). Fluorescently functionalized microspheres and nanospheres are available commercially from several sources, spanning the UV-to-IR, in a variety of sizes ranging from 20 nm to 5 pm (3). Fluorescent microspheres are also commonly labeled with primary or secondary antibodies and are used as solid supports in many sandwich immunoassays, where they take full advantage of solution-based kinetics (51-53). Alternatively, microspheres can be obtained with carboxyl, amine or other surface-displayed functional groups for chemical conjugation to biologic molecules (3). The recently developed FloDots, which are functionalized with either antibodies or DNA, have demonstrated applications in several areas, including bioimaging, cell detection, gene detection, and protein arrays (54). Similar sol-gelderived silica nanoparticles have also been investigated (55). Coded microspheres, which are internally labeled with specific concentrations of two fluorescent dyes, are an integral part of the Luminex flow cytometry technology and have been used in many biomedical research and diagnostic applications (56, 57). Environmentally sensitive dyes have been immobilized in various cross-linked polymers to produce PEBBLE (Probes Encapsulated by Biologically Localized Embedding) nanosensors for several ionic and neutral species. When delivered into cells, these PEBBLE nanosensors are used to image and monitor the presence of intracellular chemical species (58, 59). In general, the liability of working with a microsphere that is far larger than an organic or intrinsic fluorophore can be offset greatly by the increased sensitivity and stability.
Inorganic materials
Metal chelates and long-lifetime fluorophores
Long-lifetime dyes typically consist of either the luminescent lanthanides or the ruthenium-metal chelates. The principle advantage of working with long-lifetime dyes over conventional fluorophores originates from the ability to gate out, through time-resolved measurements, any background fluorescence originating from matrix components in the sample (such as autofluorescence and scattering), thus greatly improving the sensitivity of detection. Of the four lanthanides, terbium (Tb), europium (Eu), samarium (Sm), and dysprosium (Dy), that emit in the visible region, Tb and Eu are the most commonly used probes (60-63). In particular, chelate ligands of the lanthanide ions are typically used for biophysical applications. The chelates are designed to 1) tightly bind the lanthanide ion, imparting high thermodynamic and photochemical stability; 2) position a sensitizing chromophore in close proximity to the ion; and 3) contain a reactive group allowing bioconjugation (see Fig. 7). Because of their relatively low extinction coefficients (~1M-1 cm-1), lanthanide ions are almost impossible to excite directly so the sensitizing chromophore is an essential part of the lanthanide chelate probe as it functions as the initial light-harvesting antenna. Lanthanide probes are characterized by unique, sharp emission profiles (see Fig. 7), which originate from both magnetic and electric dipole transitions, along with large stokes shift (excitation 343 nm, emission > 500 nm). The fluorescent lifetimes of these materials range from μs to ms, and this allows lanthanide probes to be used in luminescence resonance energy transfer (LRET) applications along with various clinical bioassays (60, 61, 63-65). Commercial sources for lanthanide probes include CIS-Bio International (Yvette, France) and Packard Instrument Company (Downers Grove, IL; cryptate-based probes), PerkinElmer, Inc. (Waltham, MA; LANCE, a pyridine-based system), Invitrogen (LanthaScreen; polyaminocarboxylate chelates, coupled with a CS124 sensitizer), and GE Healthcare who market a europium (TMT) chelate donor in the isothiocyanate form for bioconjugation to amines. Also, an increasing number of reports appear in the literature of lanthanide-doped nanoparticles being used as biolabels where their benefits can be compounded for exploitation (66-68).
Long-lifetime ruthenium metal-chelate complexes were originally used by Lakowicz as anisotropy labels for measuring the rotational dynamics of proteins (69, 70). These transition metal complexes typically contain one or more bi-dentate or tri-dentate ligands and use metal-to-ligand charge transfer processes; see Fig. 8 (1, 71). Sigma-Aldrich Corporation (St. Louis, MO) offers a series of reactive ruthenium complexes for biolabeling that are derived from Lakowicz’s work. They are characterized by relatively small extinction coefficients (14,500 M-1 cm-1) and low quantum yields (0.05), but they possess long lifetimes, typically 500 ns, coupled with a fairly large stokes shift. As with the lanthanide probes, several applications have used the ruthenium complex as a donor in LRET-based studies. The major advantage of these transition metal complexes is again the ability to gate out any background signal and direct excitation of the acceptor dye, which leads to significantly improved sensitivity (72).

Figure 7. Luminescent lanthanides. (Modified from Reference 3.) (a) Structure of the LanthaScreen Tb probe from Invitrogen. The bioconjugation group is typically a NHS ester, isothiocyanate, or maleimide group. (b) The unique sharp emission profile of the LanthaScreen Tb probe λex = 343 nm.
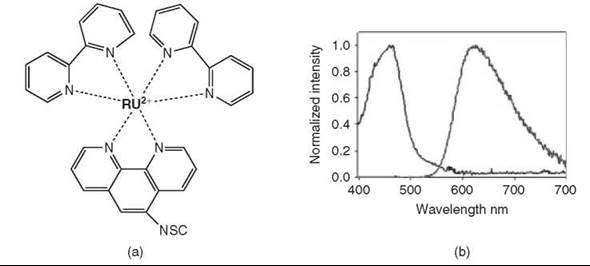
Figure 8. Transition metal chelates. (Modified from Reference 3.) (a) Structure of the commercially available ruthenium complex, Ru(bpy)2(phen-5-isothiocyanate)(PF6)2. (b) The resulting absorption and emission profiles of the ruthenium complex.
Fluorescent metal nanoparticles
Atomic clusters consisting of a few noble-metal atoms have demonstrated intense fluorescence emission as well as some interesting nonlinear optical properties. Gold (73), silver (74, 75), and copper (76) nanoparticles have both size-dependent and shape-dependent absorption and luminescent profiles, with the resulting emission exhibiting strong resistance to photobleaching. The potential benefit here would be the extremely small size; however, these nanoparticles are currently not available commercially and to date have found limited use as fluorescent labels, mostly because of complex synthesis and stabilization along with lack of bioconjugation routes.
Semiconducting nanoparticles: quantum dots
The II-VI group of semiconductor nanocrystal materials or quantum dots (QDs), namely CdSe and CdS, are extremely well characterized in the literature and have proven their applicability as fluorescent probes in a wide range of biologic applications, including FRET-based investigations (3, 77), biosensing (78), drug discovery (23), cancer diagnostics and investigative treatments (79, 81), and many other areas of biomedical research (82, 83). QDs or Qdots have several unique properties that make them ideal fluorescent probes compared with organic dyes: 1) high quantum yields and molar extinction coefficients >10 to 100 times those of organic dyes that steadily increase toward the UV; 2) high resistance to photobleaching and chemical degradation; 3) broad absorption coupled with narrow emission profiles (see Fig. 9b); 4) large stokes shifts, >200 nm for the red-emitting QDs; and most importantly, 5) size-tunable photoluminescence; see Fig. 9a (83, 88). Although QDs have been synthesized from many binary and ternary semiconductors, the most commonly used are a CdSe QD core overcoated with a ZnS shell, which acts to passivate and protect the core (83, 89). Critical to the development of QD biotechnology has been the ability to make these inherently insoluble materials biocompatible through the use of bifunctional ligands (83, 87). QDs are available commercially from Invitrogen and Evident Technologies, Inc. (Troy, NY); they are typically overcoated with a protecting layer and then precoated with avidin or other proteins that facilitate bioconjugation. Several detailed discussions have occurred on the synthesis of QD core-shell materials (89, 92). Many applications specifically aim to minimize the large protecting outer layers, which is often desired in FRET-based applications where acceptor distance from the QD donor core can be an issue (77, 93).
Silicon and more recently germanium semiconductor-based nanocrystals have also demonstrated intense fluorescence emission (94-98). Si-based nanoparticles have found some applications as biolabels (98, 99), whereas Si-based and Ge-based nanoparticles are still in the early stages of development. However, all are characterized by immature synthesis and stabilization that has limited their biologic use to date. That said, they still have a promising future as optical probes that potentially may be a nontoxic alternative to semiconductor materials.

Figure 9. Semiconductor QDs. (a) Image showing the effect of CdSe core size on the wavelength of emission. Note that all QDs were excited using a UV light source (λex = 365 nm). (b) The absorption of 510-nm QDs and emission profiles of six different QDs (in buffer) used in several assays. (Modified from Reference 83.)
Fluorophore Bioconjugation Chemistries
To exploit analytical fluorescence in biomolecular assays fully, it is often necessary to label target biomolecules by introducing a fluorophore, usually through a chemical reaction. Ideally, the reaction should take place in aqueous conditions; be fast, high yielding, and nondestructive; target reactive groups already present on the biomolecule; and allow easy purification of product from unreacted dye. Many reactions have been developed that fit some or most of the above criteria, and this section is only intended to give the reader a general outline of the common/practical chemical methods of achieving fluorescent labeling. Several excellent reviews (100) and books (3, 101, 102) cover this extensive topic exhaustively, and the reader is referred to these and to the current literature in the field of bioconjugation for additional insight.
Synthetic considerations
Labeling biomolecules with fluorophores can be challenging because of the differences in the nature of the two reactants and the small quantities of reagents employed (pmol to pmol). Because of their widespread incorporation in countless assays, many fluorophores are now available commercially in reactive form, targeting chemical groups already present in biomolecules, with amines and thiols being by far the most common targets (2). Although sometimes expensive, reactive fluorophores are the most convenient to use as a stock solution can be prepared and the conjugation reaction can be performed by adding a known concentration of fluorophore to the biomolecule dissolved in the appropriate buffer. Fluorophores are often hydrophobic molecules with limited water solubility, whereas biomolecules are soluble in water at a wide pH range. To facilitate the reaction between these disparate entities, a large excess of dye is usually employed. As a result, a common problem often encountered in fluorescent labeling of biomolecules can be the presence of a heterogeneous reaction mixture: soluble biomolecule and solid dye in two immiscible phases. One way to address partially this issue consists of adding small quantities (usually between 20% and 50% of the total reaction volume) of a carrier solvent, such as dimethylsulfoxide (DMSO) or N,N-dimethylformamide (DMF). The carrier solvent facilitates the solubilization of some hydrophobic dye in the water medium and diminishes the problems associated with the formation of aggregates. Labeling reactions usually take place under some moderate-to-vigorous stirring, and if the desired substitution level is not achieved in a reasonable reaction time (usually 1-2 hours), repeating the conjugation is preferred over extended reaction times as many reagents tend to hydrolyze over time. Because of the necessity of lowering background fluorescence in the final assay, purification of the fluorescent adduct from the unreacted fluorophore is necessary and sometimes complex. The large excess of fluorophore employed in the labeling reaction can compound purification of the labeled adduct, and several rounds of purification using different techniques may be necessary. Centrifugation and precipitation are the first steps that can help in removing suspended unreacted fluorophore and in facilitating additional purification steps. Gel-permeation, reverse-phase high-performance liquid chromatography, electrophoresis, dialysis, and combinations thereof are the techniques more commonly employed to achieve a high-purity labeled product. Depending on application, site-specific labeling may be a preferred strategy as opposed to nonspecific or generalized labeling of a common functional group, for example, a single-labeled protein-thiol versus multiple lysine-amines. Alysines are abundant on most proteins, there is also the possibility of a distribution of labeled-adducts expressing various degrees of functionalization. Furthermore, lysine residues may be the key to subsequent function, and labeling can interfere with this process.
Polypeptides and nucleic acids as biomolecular targets
Proteins, antibodies, and enzymes consist of long polypeptide sequences folded in ways that favor the positioning of hydrophobic residues at their interior, whereas more hydrophilic or easily ionizable residues are at the periphery. Water-exposed residues are therefore important and available targets for bioconjugation, most notably the carboxyl group of aspartic (103) and glutamic acid (104), the primary amine of lysine and arginine (105, 106), the secondary amine of histidine (107, 108), the thiol of cysteine (109, 110), and the phenolic hydroxyl of tyrosine (111). All behave as nucleophiles under appropriate conditions, such as specific pH ranges, and their reactivity can be exploited for the attachment of several reactive probes (112, 113). Such target-expressing residues can also be introduced recombinantly at well-defined specific sites in proteins (114, 115). Besides the presence of these residues, it is also possible to introduce alternative functional groups, or to interconvert existing ones, through the use of versatile and readily available bifunctional reagents (116). For example, 2-bromoethylamine can be used to transform thiol-containing amino acids (cysteine) into primary amines, whereas N-succinimidyl-s-acetylthioacetate (SATA) reacts with amine containing amino acids to form a protected sulfhydryl group, which can be deprotected readily using hydroxylamine to a free thiol (101). The use of bifunctional reagents provides versatility to the bioconjugation chemist, extending the repertoire of reactions and functional groups available and permitting the fine-tuning of other factors, such as the spacing or the solubility of the fluorophore.
Nucleic acids do not display the same promiscuous chemical reactivity of proteins. Instead, individual synthetic nucleotides can display a unique functional group that can be exploited even more for direct attachment of probes (117). The simplest method of labeling DNA uses high-affinity bis-intercalating dyes such as ethidium bromide, acridine, and thiazole orange monomers and dimers (118). These dyes, which have almost no emission in water, display fluorescent enhancements of 30-1000-fold after binding to DNA and remain bound even during electrophoresis, hence, their use as gel stains. Different DNA samples can be prestained with differentially emissive dyes before electrophoresis, then mixed, and finally coanalyzed (119). Perhaps the biggest liability of these dyes is their toxicity, which necessitates careful handling and disposal. The 5’-backbone phosphates can be targets for modification through carbodiimide-mediated formation of a phosphoramidate (120, 121). Such reactions, however, are not easy to control, and the expertise required makes this type of approach not very common. Two other techniques are usually preferred for tagging DNA with a fluorescent probe: 1) direct enzymatic labeling and 2) postsynthetic modification. Direct enzymatic labeling usually uses some type of enzymatic introduction of modified ddNTPs (122, 128), which results in the generation of an oligonucleotide sequence that contains the probe or an appropriate functional moiety such as an amine to which a probe can be attached. Several enzymatic techniques are available, including PCR, nick-translation, random-primed labeling, and 3 tailing with terminal transferase, which are all methods reviewed in References 129 and 130. Issues of this enzymatic insertion include the limited types of modified nucleotides that can be introduced, as the enzymes employed can be very selective when it comes to incorporating non-natural substrates, cost, and the necessity of long incubations in some cases. Postsynthetic modification of nucleotides usually consists of the automated synthesis of a short DNA fragment incorporating a specific chemical moiety—usually an amine or a thiol—within or at the termini of the sequence (117, 130-132). The functionalized oligo is then tagged with the fluorescent probe, which exploits reactions very similar to those employed for the proteins above or listed in Table 2. Alternatively, a fluorophore can be introduced directly through automated synthesis whenever its phosphoramidate building block is available, provided that the dye is stable to the synthetic and resin-cleavage conditions.* This, however, can be a costly approach because of the use of specialized synthetic building blocks and modified chemistries. Small oligonucleotides (i.e., DNA, RNA, and PNA) that have been dye-labeled during synthesis are commonly used in techniques such as quantitative PCR, in situ hybridization, and array labeling. Most commercial DNA vendors offer a variety of dye choices and modification sites as part of their services, and again, cost, the need for postsynthetic purification, and some limitations on dyes choices may be an issue. The alternative choice is a derivative of the previous one and simply involves synthesizing oligonucleotides with unique modifications, i.e., amines or thiols, and then custom labeling them with appropriate reactive dyes as desired (3, 171). The last method is an interesting hybrid technology where enzymes such as peroxidases are attached to the oligonucleotide and then used to generate light via enhanced chemiluminescence (ECL) (172, 173). In this case, biotinylated oligonucleotides are exposed to streptavidin-enzyme conjugates for specific and localized labeling. The enzymes then generate light when exposed to the substrate, which is captured on film or by a detection panel. This last method has become a sensitive and attractive alternative to the use of radioactivity for classic techniques such as Southern analysis.
Table 2. Selected biologic functional groups and their corresponding target chemistries
|
Target |
Reactive group |
Product |
References |
|
Free Thiol |
Maleimide |
Thioether |
(133-138) |
|
|
Haloacetyl/Alkyl Halide |
Thioether |
(139-141) |
|
|
Arylating agents Aziridine |
Thioether |
(142) (143) |
|
|
Acryloyl derivatives |
Thioether |
(144, 145) |
|
|
Pyridyl disulfides, 5-thio-2-nitrobenzoic (TNB) acid |
Mixed disulfide |
(14, 147) |
|
|
Hydrazine |
Hydrazone |
(148) |
|
Aldehyde/Ketone |
Amines |
Schiff’s base (imine)* |
(149,150) |
|
Free Amine |
N-hydroxysuccinimide ester (NHS) |
Amide |
(151, 152) |
|
|
Isocyanates, Isothiocyanates |
Urea, Thiourea |
(153-155) |
|
|
Acyl azides |
Amide |
(101) |
|
|
Sulfonyl chlorides |
Sulfonamide |
(156) |
|
|
Aldehydes, Glioxals |
Imine, secondary amine1 |
(157, 158) |
|
|
Epoxides, Oxiranes |
Secondary amines |
(101) |
|
|
Carbonates |
Carbamate |
(101) |
|
|
Arylating agents |
Arylamine |
(101) |
|
|
Imidoesters |
Amidine |
(159, 160) |
|
|
Carbodiimides, Anhydrides |
Amine2 |
(120, 121, 161) |
|
|
Diazoalkanes, Diazoacetyl |
Ester |
(162, 163) |
|
Carboxylate |
Carbonyldiimidazole, Carbodiimides |
Amides2 |
(159, 164) |
|
|
Epoxides |
Ether |
(101) |
|
Hydroxyl |
Cabonyldiimidazole, N,N’-disuccinimidyl carbonate, N-hydrosuccinimidyl chloroformate |
Carbamate or Urethane2
Ether |
(165, 167) |
|
|
Alkyl halogens Isocyanates |
carbamate |
(168, 169) (170) |
*Might be followed by reducing amination to form a stable product.
1 After reduction.
2 Via reactive intermediate.
Type of linkages
Table 2 presents a list of the most common target groups used for fluorescent biolabeling along with their cognate reactive partner and the type of product formed. Amines and thiols are by far the most commonly exploited target functional groups as they are both good nucleophiles, readily available and/or easily introduced in biomolecules. Figure 10 shows the most common reactions for targeting amines. Isothiocyanate (ITC) (153-155) and succinimidyl ester (OSu) (151, 152) derivatives are the most common amine-reactive groups readily forming, respectively, stable thiourea and amide linkages (Fig. 10a, 10b). ITCs, however, present long-term storage stability problems, which makes the OSu derivatives more popular. Nonetheless, certain types of reactive ITC probes, such as the fluorescein (FITC) and tetramethylrhodamine (TRITC) derivatives, are still used widely. OSu derivatives are extremely common, and many of them are available commercially (3). OSu derivatives can also be prepared easily starting from a carboxylic derivative and N-Br-Succinimide as reagent. Less common, but still viable, reactive partners for amines are sulfonyl chlorides, unstable in aqueous (156), which leads to more stable sulfonamides (Fig. 1c) and aldehydes (157) yielding Schiff bases (imines) (Fig. 1d). Carboxylic acids can also be reactive toward amines, ultimately forming amide bonds, but they require prior activation usually achieved by employing a water-soluble carbodiimide reagent (Fig. 1e) (159, 164).
Maleimides (133-138), iodoacetamides, and alkyl halide derivatives (133, 139-141) are the most common thiol-reactive groups (Fig. 11a, 11b). All of these reagents readily react when a free thiol is present to give stable thioethers. Disulfide exchange of a free thiol with an activated piridyldisulfide is another common and effective reaction (146, 147, 171, 174) (Fig. 11c), but the resulting disulfide containing product may not be stable under reducing conditions or in the presence of other free thiols.
Other chemical groups such as aldehydes, ketones, and alcohols are also exploitable targets for bioconjugation. The reactivity of alcohols in water is very low, and usually intermediate steps are involved to transform the hydroxyl into a more reactive or more easily exchangable group. The resulting multistep scheme, although not an alternative to more readily labeled groups, is an option when other reactive groups, such as carbohydrates, are not present (175). Aldehydes and ketones react with amines to give imines (149, 150). However, both groups are not commonly found and often have to be generated by oxidation of the corresponding alcohol or vicinal diol. Hydrazines are also very good reactive partners for ketones and, to some extent, aldehydes, which yield hydrazones (148). The hydrazones can be stabilized even more by reduction yielding an irreversible product. Interestingly, these same groups are also common targets for modifying sugars and carbohydrates using some of the same chemistry. However, sugar modification is a less well-developed chemistry in general and thus less prevalent, although strong research in this area continues (2, 101).
Another commonly exploited labeling method takes advantage of the very strong interaction between the protein Avidin (or Streptavidin) (176) and its natural ligand biotin, as reviewed in Reference 2. This labeling scheme usually starts with the biotinylation of the target biomolecule by employing one of many commercially available biotinylating reagents. The biotinylation chemistries available are essentially similar to the ones already mentioned for the fluorphores above. After biotinylation, fluorescently labeled Avidin or Streptavidin is added, which results in strong binding to the biotin and forms a basically irreversible linkage that has found use in countless biologic applications (177, 178).
Beyond the above chemical methods for the introduction of fluorophores, several emerging technologies target in vivo fluorescent labeling for applications where the probe has to be located inside a target cell. Fluorescent proteins such as the GFP can be appended onto the target protein by recombinant techniques resulting in the coexpression of fluorescent protein chimeras (14, 179). FLASH technology can be used in the specific in vivo tagging of proteins expressing a Cys-Cys-Pro-Gly-Cys-Cys sequence by employing a cell-permeable dye that becomes fluorescent only during labeling (180-182). The HaloTag method consists of a fusion protein with a dehalogenase domain that conjugates a fluorescent ligand through chloride substitution (183). Also, the SNAP method allows both in vivo or solution labeling of proteins by using a modified alkylguanine-DNA alkyltransferase reacting with a p-benzylguanine modified fluorophore to form a thioether bond (3).
A recent and growing conjugation methodology worth mentioning is the implementation of click chemistry to bioconjugation. Click chemistry is a fairly generic term referring to a certain class of quick, high-yielding reactions. For bioconjugation, this term usually indicates the Cu(I) catalyzed 1,3-dipolar (Huisgens) cycloaddition between an alkyne and an azide to give a 1,2,3-triazole as the product. This reaction proceeds very rapidly and is compatible with both aqueous chemistry and a variety of functional groups, which makes it an excellent candidate for future development and applications (184, 185). However, the current drawback to this chemistry is the unavailability of widely applicable commercial kits for introducing the necessary alkyne and azide cognate precursors onto both target and probe. However, the Click-iT kit (Invitrogen) targeting glycoproteins probably represents the first of many applications to come that will use this exciting chemistry.

Figure 10. Schematic representation of the most common reactions for labeling an amine: (a) reaction with isothiocyanate to give a thiourea; (b) reaction with a Succinimidil ester to give an amide; (c) reaction with a sulfonyl chloride to give a sulfonamide; (d) reaction with an aldehyde to give an imine (Schiff's base); and (e) reaction with a carbodiimide-activated carboxylic acid to give an amide.

Figure 11. Schematic representation of the most common reactions for labeling a thiol: (a) reaction with an alkyl halide to give a thioether; (b) reaction with a maleimide to give a thioether; and (c) reaction with an activated piridyl-disulfide to give a mixed-disulfide through disulfide interchange.
________________________
*Glen Research (Sterling, VA; www.glenres.com) and TriLink BioTechnologies (San Diego, CA; www.trilink.com) offer a variety of fluorescent phosphoramidates for automated DNA synthesis.
Selected Fluorescent Techniques
Fluorescence anisotropy
The extent of the polarized emission from excited state fluorophores in a solution can be described in terms of their anisotropy (r), and measuring this can provide insight into the angular displacement of the biomolecule(s) to which the fluorophores are attached (1). Within a homogeneous solution, the ground-state fluorophores are all oriented randomly. However, when exposed to a polarized excitation source, the molecules with their absorption transition moments oriented along the electronic vector component of the light will be excited preferentially (1), which means the excited population is not random in orientation, but their transition moments are all oriented. For chemical biology, the factors that affect the rotational correlation time, or anisotropy, can then be investigated, including biomolecule-biomolecule interactions, modifications, or denaturation. Anisotropy is also a powerful tool for measuring viscosity in select bioenvironments such as membranes and lipid composition. Again, the interested reader is referred to Lakowicz’s text for more detailed reading on all facets of this subject (1).
Anisotropy, which is sometimes used interchangeably with polarization, although the former is preferred, is a dimensionless quantity that is independent of sample intensity. A fluorometer and appropriate polarizing filters (parallel and perpendicular) are the simplest instrumental setup that can be applied (1). Measuring the steady-state fluorescence anisotropy provides data on only the average anisotropy decay and the interpretation can be complex. Far more information is gained from measuring the time-resolved anisotropy; however, the equipment and analysis required is more complex. Direct monitoring of fluorescent anisotropy provided insights into the binding modes of the G-protein-coupled type A and B cholecystokinin receptors (186). These receptors, which share homology to rhodopsin and P-adrenergic receptors, have important regulatory functions in certain hormonal responses. Monitoring changes in anisotropy of differentially labeled probes while interacting with the receptors confirmed that each type uses a distinct mode of high affinity binding. The structure of UreG, which is an essential Bacillus pasteurii protein required for the in vivo activation of the enzyme urease, has also been probed with both steady-state and time-resolved anisotropy analysis (187). Although this protein behaves as an intrinsic unstructured dimer, the conformation it assumes is unknown, for example, fully folded, molten globule, or random coil. Direct analysis of steady-state anisotropy and intrinsic tryptophan fluorescence wavelength shift allowed monitoring of transitions between native and unfolded states upon increasing concentrations of a denaturant (see Fig. 12). Furthermore, the hydrodynamic parameters obtained by time-resolved fluorescence anisotropy in the presence of a denaturant confirmed the existence of a stable but disordered dimer formed at a unique cysteine residue. This key bond acts to stabilize the dimer under native conditions.

Figure 12. Conformational transition of BpUreG as revealed by steady-state fluorescence signals. (a) Steady-state emission spectra of BpUreG at 24° C at increasing concentrations of GuHCl (from 0 M to 3 M, incubation time of 10 min). (b) Changes in emission max (black circles) and steady-state anisotropy (clear squares) as a function of denaturant concentration. The solid lines represent the fits by a nonlinear least-squares method of the experimental data. (Reprinted from Reference 187 with permission of the ACS.)
DNA and fluorescence
Fluorescent labeling and spectroscopic analysis have had a profound impact on two areas: research into DNA structure/function and myriad diagnostic and sequencing applications. For the former area, the focus is to understand complex nucleic acid chemistry and more recently to exploit the inherent self-assembled structures for creating precisely formed nanoscale architectures (188, 189). For the latter area, the focus has been on all aspects of genomic analysis from clinical/genetic diagnostics to the creation of the genomic databases (190). Although this field is relatively young, with fluorescent DNA sequencing described just ~20 years ago (191), commercial applications have driven progress and the completion of the Human Genome map (192); many focused reviews are available in this area (3, 193-195).
Fluorescent sensing
Fluorescent sensing can provide a powerful tool to the chemical biologist especially for in vivo applications. One version of this analysis originates almost exclusively from the environmental sensitivity of selected fluorophores discussed earlier (1-3). Fluorescein, for example, has been exploited for intracellular pH monitoring (see Fig. 6). Intracellular calcium-sensing techniques are a powerful and widely used technology with many applications in neuroscience and in cardiovascular and signal transduction research. It has also proven useful recently in monitoring receptors associated with odor detection (196). This particular technique is discussed extensively in other articles; see, for example, the article “Calcium Signaling.” The ability of membrane localized receptors to “sense” and transduct an odorant was monitored by their ability to elicit a coupled-intracellular calcium transient. Invitrogen also offers a variety of commercial probes targeting diverse ions as modified esters for intracellular delivery and Haughland’s handbook is an excellent reference on this subject (2). This type of fluorescent sensing is well developed but may be limited by the lack of multiplexing capability. That is, only one or two of these dyes can be used simultaneously because of their broad absorption/emission profiles.
Fluorescent sensing has been exploited even more by implanting environmentally sensitive fluorophores into select proteins to create a variety of in vitro and in vivo sensors. The proteins provide biologic specificity for target recognition, whereas the fluorophores provide signal transduction. This type of sensing relies on some change in the proteins structure during binding, which in turn causes a change in the local environment of the fluorescent dyes and, thus, its photophysical state. The superfamily of bacterial periplasmic binding proteins has provided an excellent source of sensing proteins for diverse analytes from which to begin designing such sensors (197), but long-term fluorophore instability has remained an issue and may necessitate preparation of fresh sensors for each experiment. The prototype for this sensor design has been the maltose binding protein (198), and many different maltose sensors have been assembled to test a variety of signal transduction modalities, including a variety of surface-tethered versions with complex kinetic functions; see Fig. 13 (198, 199). Hellinga and Frommer (197, 200) have been at the forefront of this field where they have applied computational design to identify critical sensing sites within a variety of natural and de novo rationally designed proteins.

Figure 13. (a) Schematic of a surface-tethered maltose biosensor with complex binding kinetics. The modular arm consists of DNA, a dye, and terminates in the maltose analog beta-cyclodextran (β-CD). The maltose binding protein (MBP) is site-specifically dye labeled, and both it and the β-CD are assembled onto a Neutravidin (NA) functionalized surface using biotin (b). For MBP, this uses a biotin-nickel nitroloacetic acid (Ni-NTA) intermediary to bind the MBP's 5-histidine sequence, 5-HIS. MBP binding of the β-CD-dye-DNA arm assembles the final sensor by bringing dyes 1 and 2 into proximity, which allows FRET or FRET quenching. Maltose displaces the β-CD disrupting FRET in a concentration-dependent manner. The addition of second modulator DNA that hybridizes to the arm can alter and extend the binding kinetics. (b) Representative binding curve and approximate binding constant (Kapp) for titrating the sensor against maltose. (Reprinted from Reference 199 with permission of the ACS.)
Energy transfer
As FRET is a significant analytical technique for a variety of in vivo and in vitro biosensing configurations, the concepts and applications will be discussed in other articles. However, FRET is heavily dependent on optimal placement of two or more fluorophores either within a single biomolecule or two cognate biologic entities (1, 3, 201). As such, discussion is warranted on some relevant issues. The first is choice of fluorophores with appropriate spectral overlap and their placement such that enough proximity exists between the fluorophores for efficient FRET (1, 3, 201). Depending on the structure and the available functional groups, site-specific sequential or orthogonal labeling of a single biomolecule with multiple fluorophores is still challenging. DNA and other oligonucleotides are synthesized readily with both site-specific amine- or thiol-functions for facile labeling with appropriately reactive dyes (2, 101). Single cysteines can be introduced recombinantly into specific protein sites; however, the presence of other cysteines can cause “thiol-scrambling” of the structure and subsequent loss of function (3). Hellinga and colleagues (202) have described recently a method for the sequential labeling of multiple cysteines within a single protein to address this issue specifically. Primary amines are ubiquitous to proteins, and thus, fluorophores targeting these functions tend to be nonspecific. The same considerations for thiols and amines usually apply to labeling synthetic peptides.
Proteins can also be engineered to express a variety of fluorescent proteins appended at different points within the structure. Frommer and colleagues (203, 204) have designed advanced sensors that couple two fluorescent proteins at select sites within sensing proteins and ligand binding is signaled by changes in their FRET efficiency. Figure 14 is an example of a cell-surface expressed version of such a sensor that detects glutamate release from neurons (205). The recently developed FLASH, HaloTag, and SNAP tag techniques offer alternative methods for labeling specific recombinant sequences in vivo with proprietary reactive dyes (3). The use of two disparate fluorophore classes as donors/acceptors in FRET configurations is steadily growing as exemplified by quantum dot—dye or fluorescent protein—dye pairings (3, 7). Interestingly, although not fluorophores per se, fluorescent quenchers and gold nanoparticles have found utility as acceptors in myriad FRET applications (3). In vivo sensing of proteases and second messengers with FRET-based fluorescent protein sensors is an important and related area covered in several other chapters.

Figure 14. Cell-surface glutamate nanosensor. (a) Model of the glutamate sensor. The two lobes of the protein are shown in green with glutamate in red in the central binding pocket. Enhanced cyan-fluorescent protein (ECFP) and Venus, a yellow fluorescent protein, are fused to one lobe. Binding of glutamate causes the two lobes to move relative to each other, which alters the energy transfer efficiency between the two fluorescent proteins. (b) Image of hippocampal neurons expressing the glutamate sensor with highest concentration at the plasma membrane. (Provided by W. Frommer and reproduced from Reference 205 with permission and Copyright National Academy of Sciences, USA.)
Single-molecule analysis
In theory, single-molecule analysis (SMD) represents the ultimate analysis as it can provide the highest sensitivity while providing stochastic sensing. In an ideal experiment, fluorescent signal transduction is suited particularly to this analysis as a lone fluorophore can emit photons against a dark background. Practice has proven far more complex, however, and this remains a technically challenging and still nascent technique (1). The two confounding issues remain sample immobilization coupled with optical detection configuration. Almost all SMD is dependent on microscopy and the 1) immobilization of biomolecules by surface tethering or fixation within a network, or 2) monitoring and averaging of a continuous sample of individual molecules to overcome any photobleaching effects. The required instrumentation is still some combination of confocal imaging, cooled avalanche photodiode (APD) detector, and total internal reflection or similar microscope, although continuous technological innovations have made these simpler and more affordable. SMD has been applied recently to detecting the cystic fibrosis transmembrane conductance regulator (CFTR) localized to the erythrocyte plasma membrane (206). In this case, a novel experimental approach that combined atomic force microscopy with quantum-dot-labeled anti-CFTR antibodies was employed to detect individual CFTR molecules. The results suggested that quantification of CFTR in a blood sample could be useful in the diagnosis of CFTR-related diseases. An important issue for every SMD experiment is whether immobilizing the analyte effects its function, and thus, monitoring in this state may reflect “unrealistic” data. The benefits of SMD include bypassing ensemble averaging completely, taking measurements from fixed “quantities” of analyte, and the ability to monitor intermediary reaction states that can be “masked” in an ensemble. For all pertinent issues, the reader is referred to References 1, 207, and 208 for a comparison of ensemble versus single-molecule FRET studies. The interested reader is referred to other pertinent articles on SMD of various analytes.
Fluorescence correlation spectroscopy
Fluorescence correlation spectroscopy (FCS) is a technique that allows monitoring of single molecules but does not necessitate surface immobilization as above. At its most basic level, this process is a monitoring of fluorophore translational diffusion into/out of a minute volume defined by a focused laser and imaged with a confocal aperture (1). Diffusion continuously replenishes the analyzed molecules and their short transit times through the laser focal point to mitigate any photobleaching issues. The fluorescent bursts are collected and analyzed with the Stokes-Einstein equations to correlate their diffusion coefficients and thus provide insight into their size, rotational properties, concentration, and so on. FCS has found extensive applications in biochemical reaction monitoring and kinetic analysis. FCS has been applied recently to investigating the hydrodynamic sizes of monomeric polyglutamine (209). The data suggest that the monomeric polyglutamine ensemble is made up of a heterogeneous collection of collapsed structures despite the absence of hydrophobic residues. Understanding this process has important implications for diseases where polyglutamine structures aggregate, such as the molecular mechanism behind Huntington’s disease, which is caused by polyglutamine stretches in the Huntington’s protein. The benefits of this technique include the ability to analyze very small sample concentrations/volumes and to bypass photobleaching issues that may originate from continuous sample excitation. For information on theory, analysis, applications, and instrumentation, the reader is referred to References 1 and 210.
Summary
A strong case can be made that fluorescent analysis in all its myriad variations is the most powerful tool available to the chemical biologist. This article has attempted to provide an overview of this field from the prospective of the fluorophores. The authors realize that we have left out far more than we have included, and beyond our apologies for this, we hope that this article will serve rather to stimulate more reading and experimentation.
References
1. Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd edition. 2006. Springer, New York.
2. Sapsford KE, Berti L, Medintz IL. Materials for fluorescence resonance energy transfer: beyond traditional ‘dye to dye’ combinations. Angewandte Chemie-Internat. Ed. 2006; 45:4562-4588.
3. Haughland RP. The Handbook. A Guide to Fluorescent Probes and Labeling Technologies. 10th edition. 2005. Invitrogen, San Diego CA.
4. Lakowicz JR. On spectral relaxation in proteins. Photochem. Photobiol. 2000; 72:421-437.
5. Buehler C, Dreessen J, Mueller K, So PTC, Schilb A, Hassiepen U, Stoeckli KA, Auer M. Multi-photon excitation of intrinsic protein fluorescence and its application to pharmaceutical drug screening. Assay Drug Devel. Technol. 2005; 3:155-167.
6. Kamal JKA, Zhao L, Zewail AH. Ultrafast hydration dynamics in protein unfolding: Human serum albumin. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13411-13416.
7. Clapp AR, Medintz IL, Mauro JM, Fisher BR, Bawendi MG, Mattoussi H. Fluorescence resonance energy transfer between quantum dot donors and dye-labeled protein acceptors. J. Am. Chem. Soc. 2004; 126:301-310.
8. Medintz IL, Clapp AR, Mattoussi H, Goldman ER, Fisher B, Mauro JM. Self-assembled nanoscale biosensors based on quantum dot FRET donors. Nature Materials 2003; 2:630-638.
9. Agranovski V, Ristovski ZD, Ayoko GA, Morawska L. Performance evaluation of the UVAPS in measuring biological aerosols: fluorescence spectra from NAD(P)H coenzymes and riboflavin. Aerosol Sci. Technol. 2004; 38:354-364.
10. Galban J, Andreu Y, Sierra JF, de Marcos S, Castillo JR. Intrinsic fluorescence of enzymes and fluorescence of chemically modified enzymes for analytical purposes: a review. Luminescence 2001; 16:199-210.
11. Wolfbeis OS. Fiber optic biosensing based on molecular recognition. Sensors Actuators B-Chem. 1991; 5:1-6.
12. Zhao HW, Ge M, Zhang ZX, Wang WF, Wu GZ. Spectroscopic studies on the interaction between riboflavin and albumins. Spectrochim. Acta Part A-Molec. Biomolec. Spectros. 2006; 65:811-817.
13. Wachter RM. The family of GFP-like proteins: structure, function, photophysics and biosensor applications. Introduction and perspective. Photochem. Photobiol. 2006; 82:339-344.
14. Tsien RY. The green fluorescent protein. Ann. Rev. Biochem. 1998; 67:509-544.
15. Stewart CN. Go with the glow: fluorescent proteins to light transgenic organisms. Trends Biotechnol. 2006; 24:155-162.
16. Schmid JA, Neumeier H. Evolutions in science triggered by green fluorescent protein (GFP). ChemBioChem 2005; 6:1149-1156.
17. Chen H, Yang J, Wang YX, Rang Q, Xu H, Song HY. Development of a transgenic zebrafish in which the expression of EGFP is driven by vtg1 promoter. Progress Biochem. Biophys. 2006; 33:965-970.
18. Hanton SL, Brandizzi F. Fluorescent proteins as markers in the plant secretory pathway. Micros. Res. Technique 2006; 69:152- 159.
19. Du W, Wang Y, Luo QM, Liu BF. Optical molecular imaging for systems biology: from molecule to organism. Analyt. Bioanalyt. Chem. 2006; 386:444-457.
20. Day RN, Schaufele F. Imaging molecular interactions in living cells. Molecul. Endocrinol. 2005; 19:1675-1686.
21. Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. Review—The fluorescent toolbox for assessing protein location and function. Science 2006; 312:217-224.
22. Knopfel T, Diez-Garcia J, Akemann W. Optical probing of neuronal circuit dynamics: genetically encoded versus classical fluorescent sensors. Trends Neurosci. 2006; 29:160-166.
23. Lang P, Yeow K, Nichols A, Scheer A. Cellular imaging in drug discovery. Nature Rev. Drug Discov. 2006; 5:343-356.
24. Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 2004; 279:22284-22293.
25. Tsien RY. Imagining imaging’s future. Nature Cell Biol. 2003 (Suppl): SS16-SS21.
26. Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nature Methods 2005; 2:905-909.
27. Baird GS, Zacharias DA, Tsien RY. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:11984-11989.
28. Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:7877-7882.
29. Matz MV, Marshall NJ, Vorobyev M. Symposium-in-print: Green fluorescent protein and homologs. Photochem. Photobiol. 2006; 82:345-350.
30. Shu XK, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Novel chromophores and buried charges control color in mFruits. Biochem. 2006; 45:9639-9647.
31. Glazer AN. Phycobiliproteins—a family of valuable, widely used fluorophores. J. Appl. Phycol. 1994; 6:105-112.
32. Glazer AN. Light harvesting by phycobilisomes. Ann. Rev. Biophys. Biophys. Chem. 1985; 14:47-77.
33. Holzwarth AR, Wendler J, Suter GW. Studies on chromophore coupling in isolated phycobiliproteins. 2. Picosecond energy- transfer kinetics and time-resolved fluorescence-spectra of C- phycocyanin from synechococcus-6301 as a function of the aggregation state. Biophys. J. 1987; 51:1-12.
34. Smith H. Phytochromes and light signal perception by plants—an emerging synthesis. Nature 2000; 407:585-591.
35. Rockwell NC, Su YS, Lagarias JC. Phytochrome structure and signaling mechanisms. Ann. Rev. Plant Biol. 2006; 57:837-858.
36. Murphy JT, Lagarias JC. The phytofluors: a new class of fluorescent protein probes. Curr. Biol. 1997; 7:870-876.
37. Fischer AJ, Rockwell NC, Jang AY, Ernst LA, Waggoner AS, Duan Y, Lei HX, Lagarias JC. Multiple roles of a conserved GAF domain tyrosine residue in cyanobacterial and plant phytochromes. Biochem. 2005; 44:15203-15215.
38. Dodeigne C, Thunus L, Lejeune R. Chemiluminescence as a diagnostic tool. A review. Talanta 2000; 51:415-439.
39. Roda A, Pasini P, Mirasoli M, Michelini E, Guardigli M. Biotechnological applications of bioluminescence and chemiluminescence. Trends Biotechnol. 2004; 22:295-303.
40. Michelini E, Guardigli M, Magliulo M, Mirasoli M, Roda A, Simoni P, Baraldini M. Bioluminescent biosensors based on genetically engineered living cells in environmental and food analysis. Analytical Lett. 2006; 39:1503-1515.
41. Roda A, Guardigli M, Pasini P, Mirasoli M, Michelini E, Musiani M. Bio- and chemiluminescence imaging in analytical chemistry. Analyt. Chim. Acta 2005; 541:25-36.
42. Waggoner A. Fluorescent labels for proteomics and genomics. Curr. Opin. Chem. Biol. 2006; 10:62-66.
43. Taitt CR, Anderson GP, Ligler FS. Evanescent wave fluorescence biosensors. Biosensors Bioelectr. 2005; 20:2470-2487.
44. Schaferling M, Nagl S. Optical technologies for the read out and quality control of DNA and protein microarrays. Analyt. Bioanalyt. Chem. 2006; 385:500-517.
45. Nagl S, Schaeferling M, Wolfbeis OS. Fluorescence analysis in microarray technology. Microchim. Acta 2005; 151:1-21.
46. Panchuck-Voloshina N, Haugland RP, Bishop-Stewart J, Bhalgat MK, Millard PJ, Mao F, Leung WY, Haugland RP. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J. Histochem. Cytochem. 1999; 47:1179-88.
47. Buschmann V, Weston KD, Sauer M. Spectroscopic study and evaluation of red-absorbing fluorescent dyes. Bioconjug. Chem. 2003; 14:195-204.
48. Miller I, Crawford J, Gianazza E. Protein stains for proteornic applications: which, when, why? Proteomics 2006;6:5385-5408.
49. Mohr GJ. New chromogenic and fluorogenic reagents and sensors for neutral and ionic analytes based on covalent bond formation—a review of recent developments. Analyt. Bioanalyt. Chem. 2006; 386:1201-1214.
50. Soh N. Recent advances in fluorescent probes for the detection of reactive oxygen species. Analyt. Bioanalyt. Chem. 2006; 386:532-543.
51. Pataki J, Szabo M, Lantos E, Szekvolgyi L, Molnar M, Hegedus E, Bacso Z, Kappelmayer J, Lustyik G, Szabo G. Biological microbeads for flow-cytometric immunoassays, enzyme titrations, and quantitative PCR. Cytometry Part A 2005; 68A:45-52.
52. Martins TB, Augustine NH, Hill HR. Development of a multiplexed fluorescent immunoassay for the quantitation of antibody responses to group A streptococci. J. Immunol. Methods 2006; 316:97-106.
53. Wang C, Zhang Y. A microsphere coupled micropatterning method for cytokine detection. Sensors and Actuators B-Chemical 2006; 120:125-129.
54. Yao G, Wang L, Wu YR, Smith J, Xu JS, Zhao WJ, Lee EJ, Tan WH. FloDots: luminescent nanoparticles. Analyt. Bioanalyt. Chem. 2006; 385:518-524.
55. Burns A, Ow H, Wiesner U. Fluorescent core-shell silica nanoparticles: towards “Lab on a Particle” architectures for nanobiotechnology. Chem. Soc. Rev. 2006; 35:1028-1042.
56. Hindson BJ, Makarewicz AJ, Setlur US, Henderer BD, McBride MT, Dzenitis JM. APDS: the autonomous pathogen detection system. Biosensors Bioelectron. 2005; 20:1925-1931.
57. Wilson R, Cossins AR, Spiller DG. Encoded microcarriers for high-throughput multiplexed detection. Angewandte Chemie-Internat. Ed. 2006; 45:6104-6117.
58. Buck SM, Koo YEL, Park E, Xu H, Philbert MA, Brasuel MA, Kopelman R. Optochemical nanosensor PEBBLEs: photonic explorers for bioanalysis with biologically localized embedding. Curr. Opin. Chem. Biol. 2004; 8:540-546.
59. Sumner JP, Westerberg NM, Stoddard AK, Fierke CA, Kopelman R. Cu+- and Cu2+-sensitive PEBBLE fluorescent nanosensors using DsRed as the recognition element. Sensors Actuators B-Chem. 2006; 113:760-767.
60. Selvin PR. Principles and biophysical applications of lanthanide-based probes. Ann. Rev. Biophys. Biomolec. Struct. 2002; 31:275-302.
61. Bunzli JCG, Piguet C. Taking advantage of luminescent lanthanide ions. Chem. Soc. Rev. 2005; 34:1048-1077.
62. Hemmila I, Laitala V. Progress in lanthanides as luminescent probes. J. Fluores. 2005; 15:529-542.
63. Yuan JL, Wang GL. Lanthanide-based luminescence probes and time-resolved luminescence bioassays. Trac-Trends Analyt. Chem. 2006; 25:490-500.
64. Posson DJ, Ge PH, Miller C, Bezanilla F, Selvin PR. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature 2005; 436:848-851.
65. Laitala V, Hemmila L. Homogeneous assay based on low quantum yield Sm(III)-donor and anti-Stokes’ shift time-resolved fluorescence resonance energy-transfer measurement. Analyt. Chim. Acta 2005; 551:73-78.
66. Sivakumar S, Diamente PR, van Veggel FC. Silica-coated Ln (3+)-doped LaF3 nanoparticles as robust down- and upconvert- ing biolabels. Chem. Euro. J. 2006; 12:5878-5884.
67. Dosev D, Nichkova M, Liu MZ, Guo B, Liu GY, Hammock BD, Kennedy IM. Application of luminescent Eu: Gd2O3 nanoparticles to the visualization of protein micropatterns. J. Biomed. Optics 2005; 10:064006.
68. Casanova D, Giaume D, Gacoin T, Boilot JP, Alexandrou A. Single lanthanide-doped oxide nanoparticles as donors in fluorescence resonance energy transfer experiments. J. Phys. Chem. 2006; 110:19264-19270.
69. Terpetschnig E, Szmacinski H, Lakowicz JR. Long-lifetime metal-ligand complexes as probes in biophysics and clinical chemistry. Methods Enzymol. Fluores. Spectros. 1997; 278:295- 321.
70. Szmacinski H, Castellano FN, Terpetschnig E, Dattelbaum JD, Lakowicz JR, Meyer GJ. Long-lifetime Ru(II) complexes for the measurement of high molecular weight protein hydrodynamics. Biochim. Biophys. Acta-Protein Structure and Molec. Enzymol. 1998; 1383:151-159.
71. Medlycott EA, Hanan GS. Synthesis and properties of mono- and oligo-nuclear Ru(II) complexes of tridentate ligands: the quest for long-lived excited states at room temperature. Coordinat. Chem. Rev. 2006; 250:1763-1782.
72. Kang JS, Piszczek G, Lakowicz JR. Enhanced emission induced by FRET from a long-lifetime, low quantum yield donor to a long-wavelength, high quantum yield acceptor. J. Fluores. 2002; 12:97-103.
73. Zheng J, Zhang CW, Dickson RM. Highly fluorescent, water-soluble, size-tunable gold quantum dots. Phys. Rev. Lett. 2004; 93.
74. Lee TH, Gonzalez JI, Zheng J, Dickson RM. Single-molecule optoelectronics. Accounts Chem. Res. 2005; 38:534-541.
75. Peyser LA, Vinson AE, Bartko AP, Dickson RM. Photoactivated fluorescence from individual silver nanoclusters. Science 2001; 291:103-106.
76. Ganeev RA, Ryasnyansky AI, Stepanov AL, Usmanov T. Nonlinear optical response of silver and copper nanoparticles in the near-ultraviolet spectral range. Phys. Solid State 2004; 46:351- 356.
77. Clapp AR, Medintz IL, Mattoussi H. Forster resonance energy transfer investigations using quantum-dot fluorophores. ChemPhysChem 2006; 7:47-57.
78. Sapsford KE, Pons T, Medintz IL, Mattoussi H. Biosensing with luminescent semiconductor quantum dots. Sensors 2006; 6:925-953.
79. Cuenca AG, Jiang HB, Hochwald SN, Delano M, Cance WG, Grobmyer SR. Emerging implications of nanotechnology on cancer diagnostics and therapeutics. Cancer 2006; 107:459-466.
80. Samia ACS, Dayal S, Burda C. Quantum dot-based energy transfer: Perspectives and potential for applications in photodynamic therapy. Photochem. Photobiol. 2006; 82:617-625.
81. Weng JF, Ren JC. Luminescent quantum dots: A very attractive and promising tool in biomedicine. Curr. Med. Chem. 2006; 13:897-909.
82. Klostranec JM, Chan WCW. Quantum dots in biological and biomedical research: recent progress and present challenges. Adv. Mat. 2006; 18:1953-1964.
83. Medintz I, Uyeda H, Goldman E, Mattoussi H. Quantum dot bioconjugates for imaging, labeling and sensing. Nature Mat. 2005; 435-446.
84. Chan WCW, Nie S. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science 1998; 281:2016-2018.
85. Bruchez M Jr, Moronne M, Gin P, Weiss S, Alivisatos AP. Semiconductor nanocrystals as fluorescent biological labels. Science 1998; 281:2013-2016.
86. Murphy CJ. Optical sensing with quantum dots. Analytical Chem. 2002; 74:520A-526A.
87. Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 2005; 307:538-544.
88. Pinaud F, Michalet X, Bentolila LA, Tsay JM, Doose S, Li JJ, Iyer G, Weiss S. Advances in fluorescence imaging with quantum dot bio-probes. Biomaterials 2006; 27:1679-1687.
89. Dabbousi BO, RodriguezViejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. (CdSe)ZnS core-shell quantum dots: Synthesis and characterization of a size series of highly luminescent nanocrystallites. J. Phys. Chem. B 1997; 101:9463-9475.
90. Mattoussi H, Kuno K, Goldman ER, Anderson GP, Mauro JM. Colloidal semiconductor quantum dot conjugates in biosensing. In Optical Biosensors: Present and Future. Ligler FS, Rowe Tait CA, eds. 2002. Elsevier, San Diego, CA. pp. 537-569.
91. Peng ZA, Peng X. Formation of high-quality CdTe, CdSe, and CdS nanocrystals using CdO as precursor. J. Am. Chem. Soc. 2001; 123:183-184.
92. Qu L, Peng ZA, Peng X. Alternative routes toward high quality CdSe nanocrystals. Nano Lett. 2001; 1:333-337.
93. Medintz IL, Clapp AR, Melinger JS, Deschamps JR, Mat- toussi H. A reagentless biosensing assembly based on quantum dot donor Forster resonance energy transfer. Adv. Mat. 2005; 17:2450-2455.
94. Belomoin G, Therrien J, Smith A, Rao S, Twesten R, Chaieb S, Nayfeh MH, Wagner L, Mitas L. Observation of a magic discrete family of ultrabright Si nanoparticles. Appl. Phys. Lett. 2002; 80:841-843.
95. Kapaklis V, Politis C, Poulopoulos P, Schweiss P. Photoluminescence from silicon nanoparticles prepared from bulk amorphous silicon monoxide by the disproportionation reaction. Appl. Phys. Lett. 2005; 87.
96. Warner JH, Tilley RD. Synthesis of water-soluble photoluminescent germanium nanocrystals. Nanotechnol. 2006; 17:3745-3749.
97. Warner J, Hoshino A, Yamamoto K, Tilley R. Water-soluble photoluminescent silicon quantum dots. Angewandte Chemie-Internat. Ed. 2005; 44:2-6.
98. Tilley RD, Yamamoto K. The microemulsion synthesis of hydrophobic and hydrophilic silicon nanocrystals. Adv. Mat. 2006; 18:2053-2056.
99. Wang L, Reipa V, Blasic J. Silicon nanoparticles as a luminescent label to DNA. Bioconj. Chem. 2004; 15:409-412.
100. Kwon OH, Ito Y. Bioconjugation for enzyme technology. Biotechnol. Genetic Engineer. Rev. 2001; 18:237-263.
101. Hermanson GT. Bioconjugate Techniques. 1996. Academic Press, San Diego, CA.
102. Wong SS. Chemistry of Protein Conjugation and Cross-Linking. 1991. CRC Press, Boca Raton, FL.
103. Song AI, Rana TM. Synthesis of an amino acid analogue to incorporate p-aminobenzyl-EDTA in peptides. Bioconj. Chem. 1997; 8:249-252.
104. Wen X, Jackson EF, Price RE, Kim EE, Wu Q, Wallace S, Charnsangavej C, Gelovani JG, Li C. Synthesis and characterization of poly (L-glutamic acid) gadolinium chelate: a new biodegradable MRI contrast agent. Bioconj. Chem. 2004; 15:1408-1415.
105. Parameswaran KN, Velasco PT, Wilson J, Lorand L. Labeling of epsilon-lysine cross-linking sites in proteins with peptide- substrates of Factor-Xiiia and transglutaminase. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:8472-8475.
106. Sakal E, Gertler A, Shechter Y. Biological-activity of a fluorescein human growth-hormone derivative prepared by specific covalentlabeling oflysine-70. Biochemistry 1991; 30:8899-8904.
107. Li XH, Ma HM, Dong SY, Duan XJ, Liang SC. Selective labeling of histidine by a designed fluorescein-based probe. Talanta 2004; 62:367-371.
108. Li XH, Ma HM, Nie LH, Sun M, Xiong SX. A novel fluorescent probe for selective labeling of histidine. Analytica Chimica Acta 2004; 515:255-260.
109. Griep MA, Mesman TN. Fluorescent labeling of cysteine-39 on Escherichia-coli primase places the dye near an active-site. Bioconj. Chem. 1995; 6:673-682.
110. Schuler B, Pannell LK. Specific labeling of polypeptides at amino-terminal cysteine residues using Cy5-benzyl thioester. Bioconj. Chem. 2002; 13:1039-1043.
111. Toyo’oka T, Mantani T, Kato M. Reversible labeling of tyrosine residue in peptide using 4-fluoro-7-nitro-2,1,3-benzoxadiazole and N-acetyl-L-cysteine. Analyt. Sci. 2003; 19:341-346.
112. Brinkley M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconj. Chem. 1992; 3:2-13.
113. Waggoner A. Covalent labeling of proteins and nucleic-acids with fluorophores. In Biochemical Spectroscopy. Methods Enzymol. 1995; 246:362-373.
114. De Lorimier RM, Smith JJ, Dwyer MA, Looger LL, Sali KM, Paavola CD, Rizk SS, Sadigov S, Conrad DW, Loew L, Hellinga HW. Construction of a fluorescent biosensor family. Protein Sci. 2002; 11:2655.
115. Gentle IE, De Souza DP, Baca M. Direct production of proteins with N-terminal cysteine for site-specific conjugation. Bioconj. Chem. 2004; 15:658-663.
116. Mattson G, Conklin E, Desai S, Nielander G, Savage MD, Morgensen SA. practical approach to crosslink. Molec. Biol. Reports 1993; 17:167-183.
117. Davies MJ, Shah A, Bruce IJ. Synthesis of fluorescently labelled oligonucleotides and nucleic acids. Chem. Soc. Rev. 2000; 29:97-107.
118. Benson SC, Mathies RA, Glazer AN. Heterodimeric DNA-binding dyes designed for energy-transfer—stability and applications of the DNA complexes. Nucleic Acids Res. 1993; 21:5720- 5726.
119. Shi YN, Simpson PC, Scherer JR, Wexler D, Skibola C, Smith MT, Mathies RA. Radial capillary array electrophoresis microplate and scanner for high-performance nucleic acid analysis. Analyt. Chem. 1999; 71:5354-5361.
120. Chu BCF, Kramer FR, Orgel LE. Synthesis of an amplifiable reporter RNA for bioassays. Nucleic Acids Res. 1986; 14:5591.
121. Ghosh SS, Kao PM, McCue AW, Chapelle HL. Use of maleimide-thiol coupling chemistry for efficient syntheses of oligonucleotide-enzyme conjugate hybridization probes. Bioconj. Chem. 1990; 1:71.
122. Temsamani J, Agrawal S. Enzymatic labeling of nucleic acids. Molec. Biotechnol. 1996; 5:223-232.
123. Cruickshank KA. Quantitation of fluorescent nucleotide incorporation by capillary gel electrophoresis and laser-induced fluorescence detection. Analyt. Biochem. 1999; 269:21.
124. Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Analyt. Biochem. 1983; 132:6.
125. Langer PR, Waldrop AA, Ward DC. Enzymatic-synthesis of biotin-labeled polynucleotides—novel nucleic-acid affinity probes. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:6633.
126. Kumar A, Tchen P, Roullet F, Cohen J. Nonradioactive labeling of synthetic oligonucletide probes with terminal deoxynucleotidyl transferase. Analyt. Biochem. 1988; 169:376-382.
127. Guerra CE. Analysis of oligonucleotide microarrays by 3 end labeling using fluorescent nucleotides and terminal transferase. Biotechniques 2006; 41:53-56.
128. Medintz IL, Wong WW, Mathies RA. Fluorescence labeling methods for microchannel plate capillary electrophoresis DNA sizing. J. Capillary Electrophor. Microchip Technol. 2002; 7:43-49.
129. Kricka LJ. Nonisotopic DNA Probe Techniques. 1992. Academic Press, New York.
130. Richter A, Hentze S, Ansorge W, Hentze MW, Muckenthaler M. Comparison of fluorescent tag DNA labeling methods used for expression analysis by DNA microarrays. Biotechniques 2002; 33:620-630.
131. Skrzypczynski Z, Wayland S. New reagents for the introduction of reactive functional groups into chemically synthesized DNA probes. Bioconj. Chem. 2003; 14:642-652.
132. Proudnikov D, Mirzabekov A. Chemical methods of DNA and RNA fluorescent labeling. Nucleic Acids Res. 1996; 24:4535-4542.
133. Schelte P, Boeckler C, Frisch B, Schuber F. Differential reactivity of maleimide and bromoacetyl functions with thiols: application to the preparation of liposomal diepitope constructs. Bioconj. Chem. 2000; 11:118-123.
134. Reddy PY, Kondo S, Fujita S, Toru T. Efficient synthesis of fluorophore-linked maleimide derivatives. Synthesis-Stuttgart 1998; 999-1002.
135. Partis MD, Griffiths DG, Roberts GC, Beechey RB. Cross-linking of protein by omega-maleimido alkanoyl N-hydroxysuccimimido esters. J. Protein Chem. 1983; 2:263-277.
136. Gorin G, Matic PA, Doughty G. Kinetics of reaction of N-ethylmaleimide with cysteine and some congeners. Arch. Biochem. Biophys. 1966; 115:593.
137. Smyth DG, Blumenfeld OO, Konigsberg W. Reactions of N-ethylmaleimide with peptides + amino acids. Biochem. J. 1964; 91:589.
138. Heitz JR, Anderson CD, Anderson BM. Inactivation of yeast alcohol dyhydrogenase by N-alkylmaleimides. Arch. Biochem. Biophys. 1968; 127:627.
139. Ramseier U, Chang JY. Modification of cysteine residues with N-methyl iodoacetamide. Analyt. Biochem. 1994; 221:231-233.
140. Bernhard WR. Differential modification of metallothionein with iodoacetamide. Methods Enzymol. 1991; 205:426-433.
141. Cole RD, Stein WH, More S. Cysteine content of human hemogolbin. J. Biol. Chem. 1958; 233:1359-1363.
142. Eisen HN, Belman S, Carsten ME. The reaction of 2,4-dinitro-benzenesulfonic acid with free amino groups of proteins. J. Am. Chem. Soc. 1953; 75:4583-4585.
143. Grossman SH, Pyle J, Steiner RJ. Kinetic evidence for active monomers during the reassembly of denatured creatine-kinase. Biochemistry 1981: 21:6122-6128.
144. Yem AW, Epps DE, Mathews WR, Guido DM, Richard KA, Staite ND, Deibel MR. Jr Site-specific chemical modification of interleukin-1-Beta by acrylodan at cysteine-8 and lysine-103. J. Biol. Chem. 1992; 267:3122-3128.
145. Epps DE, Yem AW, Fisher JF, McGee JE, Paslay JW, Deibel MR Jr. Spectral characterization of environment-sensitive adducts of interleukin- 1-beta. J. Biol. Chem. 1992; 267:3129-3135.
146. Ellman GL. A colorimeteric method for determining low concentrations of mercaptans. Arch. Biochem. Biophys. 1958; 74:443-450.
147. King P, Li Y, Kochoumian L. Preparation of protein conjugates via intermoleculr disulfide bond formation. Biochemistry 1978; 17:1499-1506.
148. Chang YS, Jeong JM, Lee YS, Kim HW, Rai GB, Lee SJ, Lee DS, Chung JK, Lee MC. Preparation of F-18-human serum albumin: A simple and efficient protein labeling method with F-18 using a hydrazone-formation method. Bioconj. Chem. 2005; 16:1329-1333.
149. Rice KG. Preparation of fluorescent glycoconjugates for energy- transfer studies. Methods Enzymol. 1994; 247:30-43.
150. Amir D, Haas E. Estimation of intramolecular distance distributions in bovine pancreatic trypsin-inhibitor by site-specific labeling and nonradiative excitation energy-transfer measurements. Biochemistry 1987; 26:2162-2175.
151. Cuatrecasas P, Parikh I. Adsorbents for affinity chromatography-use of N-hydroxysuccimide esters of agarose. Biochemistry 1972; 11:2291.
152. Staros JV. N-hydroxysulfosuccinimide active esters-bis (N-hydroxysulfuccinimide esters of 2-dicarboxylic-acids are hydrophilic, membrane-impermeant, protein cross-linkers. Biochemistry 1982; 21:3950-3955.
153. Rifai A, Wong SS. Preparation of phosphorylcholine-conjugated antigens. J. Immunol. Methods 1986; 94:25-30.
154. Annunziato ME, Patel US, Ranade M, Palumbo PS. P-maleimidophenyl isocyanate—A novel heterobifunctional linker for hydroxyl to thiol coupling. Bioconjugate Chemistry 1993; 4:212-218.
155. Banks PR, Paquette DM. Comparison of 3 common amine reactive flourescent probes for conjugation to biomolecules by capillary zone electrophoresis. Bioconj. Chem. 1995; 6:447-458.
156. Lefevre C, Kang HC, Haugland RP, Malekzadeh N, Arttamangkul S, Haugland RP. Texas Red-X and rhodamine Red-X, new derivatives of sulforhodamine 101 and lissamine rhodamine B with improved labeling and fluorescence properties. Bioconj. Chem. 1996; 7:482-489.
157. Liu J, Hsieh YZ, Wiesler D, Novotny M. Design of 3-(4-carboxy-benzoyl)-2-quinolinecarboxyaldehyde as a reagent for ultrasensitive determination of primary amines by capillary electrophoresis using laser fluorescence detection. Analyt. Chem. 1991; 63:408-412.
158. Beale SC, Hsieh Y-Z, Wiesler D, Novotny M. Application of 3-(2-furolyl)quinoline-2-carbaldehyde as a fluorogenic reagent for the analysis of primary amines by liquid-chromatography with laser-induced fluorescence detection. J. Chromatog. A 1990; 499:579-587.
159. Nakajima N, Ikada Y. Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media. Bioconj. Chem. 1995; 6:123-130.
160. Wilbur DS. Radiohalogenation of proteins-an overview of radionuclides, labeling methods, and reagents for conjugate labeling. Bioconj. Chem. 1992; 3:433-470.
161. Hoare DG, Koshland DE. A procedure for selective modification of carboxyl groups in proteins. J. Am. Chem. Soc. 1966; 88:2057.
162. DeMar JC, Disher RM, Wensel TG. HPLC analysis of protein- linked fatty acids using fluorescence detection of 4-(diazomethyl)-7-diethyl aminocoumarin derivatives. FASEB J. 1992; 61:A81.
163. Ito K, Maruyama J. Studies on stable diazoalkanes as potential fluorogenic reagents. 1,7-substituted 4-diazomethylcoumarins. Chem. Pharmaceut. Bull. 1983; 31:3014-3023.
164. Paul R, Anderson GW. N,N’-carbonyldiimidazole, a new peptide forming reagent. J. Am. Chem. Soc. 1960;82:4596-4600.
165. Bethell GS, Ayers JS, Hancock WS, Hearn MTW. Novel method of activation of cross-linked agaroses with 1,1-carbonyldiimidazole which gives a matrix for affinity chromatography devoid of additional charged groups. J. Biol. Chem. 1979; 254:2572-2574.
166. Hearn MTW, Bethell GS, Ayers JS, Hancock WS. Application of 1,1-carbonyldiimidazole-activated agarose for the purification of proteins. 2. Use of an activated matrix devoid of additional charged groups for the purification of thyroid proteins. J. Chromatog. 1979; 185:463-470.
167. Beauchamp CO, Gonias SL, Menapace DP, Pizzo SV. A new procedure for the synthesis of polyethylene glycol-protein adducts—effects on function, receptor recognition, and clearance of superoxide-dismutase, lactoferrin, and alpha-2-macroglobulin. Analyt. Biochem. 1983; 131:25-33.
168. Brunswick M, Finkelman FD, Highet PF, Inman JK, Dintzis HM, Mond JJ. Picogram quantitites of anit-Ig antibodies coupled to dextran induce B-cell proliferation. J. Immunol. 1988; 140:3364- 3372.
169. Noguchi A, Takahashi T, Yamaguchi T, Kitamura K, Takakura Y, Hashida M, Sezaki H. Preparation and properties of the immuno-conjugate composed of anti-human colon cancer monoclonal-antibody and mitomycin C-dextran conjugate. Bioconj. Chem. 1992; 3:132-137.
170. Takadate A, Irikura M, Suehiro T, Fujino H, Goya S. New labeling reagents for alcohols in fluorescence high-performance liquid chromatography. Chem. Pharmaceut. Bull. 1985;33:1164-1169.
171. Berti L, Xie J, Medintz IL, Glazer AN, Mathies RA. Energy transfer cassettes for facile labeling of sequencing and PCR primers. Analyt. Biochem. 2001; 292:188-197.
172. Kricka LJ. Stains, labels and detection strategies for nucleic acids assays. Annals Clinical Biochem. 2002; 39:114-129.
173. Kricka LJ. Clinical applications of chemiluminescence. Analyt. Chim. Acta 2003; 500:279-286.
174. Na DH, Woo BH, Lee KC. Quantitative analysis of derivatized proteins prepared with pyridyl disulfide-containing cross-linkers by high-performance liquid chromatography. Bioconj. Chem. 1999; 10:306-310.
175. Fukase K, Nakayama H, Kurosawa M, Ikegaki T, Kanoh T, Hase S, Kusumoto S. Functional fluorescence labeling of carbohydrates and its use for preparation of neoglycoconjugates. J. Carbohydrate Chem. 1994; 13:715-736.
176. Gonzalez M, Bagatolli LA, Echabe I, Arrondo JLR, Argarana CE, Cantor CR, Fidelio GD. Interaction of biotin with streptavidin—Thermostability and conformational changes upon binding. J. Biol. Chem. 1997; 272:11288-11294.
177. Gruber HJ, Marek M, Schindler H, Kaiser K. Biotin-fluorophore conjugates with poly(ethylene glycol) spacers retain intense fluorescence after binding to avidin and streptavidin. Bioconj. Chem. 1997; 8:552-559.
178. Kakabakos SE, Khosravi MJ. Direct time-resolved fluorescence immunoassay of progesterone in serum involving the biotin streptavidin system and the immobilized-antibody approach. Clin. Chem. 1992; 38:725-730.
179. Miyawaki A, Sawano A, Kogure T. Lighting up cells: labelling proteins with fluorophores. Nature Cell Biol. 2003; (Suppl):S1-S7.
180. Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998; 281:269.
181. Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. New biarsenical Ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J. Am. Chem. Soc. 2002; 124:6063-6067.
182. Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Eisman MH, Lohse MJ. A FlAsH-based FRET approach to determine G protein— coupled receptor activation in living cells. Nature Methods 2005; 2:171-176.
183. Los GV, Darzins A, Karassina N, Zimprich C, Learish R, McDougall MG, Encell LP, Friedman-Ohana R, Wood M, Vidugiris G, Zimmerman K, Otto P, Klaubert DH, Wood KV. HaloTag Interchangeable labeling technology for cell imaging and protein capture. Cell Notes Promega 2005; 11:2-6.
184. Brennan JL, Hatzakis NS, Tshikhudo TR, Dirvianskyte N, Razumas V, Patkar S, Vind J, Svendsen A, Nolte RJM., Rowan AE, Brust M. Bionanoconjugation via click chemistry: The creation of functional hybrids of lipases and gold nanoparticles. Bioconj. Chem. 2006; 17:1373-1375.
185. Gierlich J, Burley P, Gramlich ME, Hammond DM, Carell T. Click chemistry as a reliable method for the high-density postsynthetic functionalization of alkyne-modified DNA. Organic Letters 2006; 8:3639-3642.
186. Harikumar KG, Pinon DI, Miller LJ. Fluorescent indicators distributed throughout the pharmacophore of cholecystokinin provide insights into distinct modes of binding and activation of type A and B cholecystokinin receptors. J. Biol. Chem. 2006; 281:27072-27080.
187. Neyroz P, Zambelli B, Ciurli S. Intrinsically disordered structure of Bacillus pasteurii UreG as revealed by steady-state and time-resolved fluorescence spectroscopy. Biochemistry 2006; 45: 8918-8930.
188. Lin CX, Liu Y, Rinker S, Yan H. DNA tile based self-assembly: Building complex nanoarchitectures. ChemPhysChem 2006; 7:1641-1647.
189. Bando T, Sugiyama H. The chemical biology that controls DNA function and structure. J. Synthetic Organic Chem. Japan 2005; 63:1016-1027.
190. Bai XP, Edwards J, Ju JY. Molecular engineering approaches for DNA sequencing and analysis. Expert Rev. Molec. Diagnos. 2005; 5:797-808.
191. Prober JM, Trainor GL, Dam RJ, Hobbs FW, Robertson CW, Zagursky RJ, Cocuzza AJ, Jensen MA, Baumeister K. A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science 1987; 38:336-341.
192. Kheterpal I, Mathies RA. Capillary array electrophoresis DNA sequencing. Analyt. Chem. 1999; 71:31A-37A.
193. Rebrikov DV, Trofimov DY. Real-time PCR: a review of approaches to data analysis. Appl. Biochem. Microbiol. 2006; 42:455-463.
194. Park HG, Song JY, Park KH, Kim MH. Fluorescence-based assay formats and signal amplification strategies for DNA microarray analysis. Chem. Engineer. Sci. 2006; 61:954-965.
195. Didenko VV. DNA probes using fluorescence resonance energy transfer (FRET): designs and applications. Biotechniques 2002; 32:1012.
196. Silver WL, Clapp TR, Stone LM, Kinnamon SC. TRPV1 receptors and nasal trigeminal chemesthesis. Chem. Senses 2006; 31:807-812.
197. Dwyer MA, Hellinga HW. Periplasmic binding proteins: a versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 2004; 14:495-504.
198. Medintz IL, Deschamps JR. Maltose-binding protein: a versatile platform for prototyping biosensing. Curr. Opin. Biotechnol. 2006; 17:17-27.
199. Medintz IL, Anderson GP, Lassman ME, Goldman ER, Bettencourt LA, Mauro JM. General strategy for biosensor design and construction employing multifunctional surface-tethered components. Analyt. Chem. 2004; 76:5620-5629.
200. Fehr M, Ehrhardt DW, Lalonde S, Frommer WB. Minimally invasive dynamic imaging of ions and metabolites in living cells. Curr. Opin. Plant Biol. 2004; 7:345-351.
201. Sapsford KE, Berti L, Medintz IL. Fluorescence resonance energy transfer—concepts, applications and advances. Minerva Biotecnologi. 2004; 16:247-273.
202. Smith JJ, Conrad DW, Cuneo MJ, Hellinga HW. Orthogonal site-specific protein modification by engineering reversible thiol protection mechanisms. Protein Sci. 2005; 14:64-73.
203. Medintz IL. Recent progress in developing FRET-based intracellular sensors for detection of small molecule nutrients and ligands. Trends Biotechnol. 2006; 24:539-542.
204. Fehr M, Frommer WB, Lalonde S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:9846-9851.
205. Okumoto S, Looger LL, Micheva KD, Reimer RJ, Smith SJ, Frommer WB. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:8740-8745.
206. Lange T, Jungmann P, Haberle J, Falk S, Duebbers A, Bruns R, Ebner A, Hinterdorfer P, Oberleithner H, Schillers H. Reduced number of CFTR molecules in erythrocyte plasma membrane of cystic fibrosis patients. Molec. Membrane Biol. 2006; 23:317-323.
207. Weiss S. Fluorescence spectroscopy of single molecules. Science 1999; 283:1676-1683.
208. Pons T, Medintz IL, Wang X, English DS, Mattoussi H. Solution-phase single quantum dot fluorescence resonant energy transfer sensing. J. Am. Chem. Soc. 2006; 128:15324-15331.
209. Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:16764-16769.
210. Rigler R, Elson ES. Fluorescence correlation spectroscopy: theory and applications. 2001. Springer, New York.
Further Reading
Fluorophore spectra:
http://probes.invitrogen.com/resources/spectraviewer/.
http://microscopy.biorad.com/fluorescence/fluorophoreDatab.htm.
Pierce linker selection guides:
http://www.piercenet.com/products/browse.cfm?fldID=78C0D455-A2D3-11D5-9E2A-00508BD9167A.
See Also
Chemical Labeling: Overview of Applications in Chemical Biology
Chemiluminescence and Bioluminescence Techniques
Fluorescence in Living Systems: Overview of Applications in Chemical Biology
Fluorescence Lifetime and Anisotropy Measurements: Proteins
Fluorescence Resonance Energy Transfer: Proteins
Fluorescence Techniques: Lipids