CHEMICAL BIOLOGY
Glycosylation of Proteins in the Golgi Apparatus
Marguerite M. Desko and Jennifer J. Kohler, Stanford University, Stanford, California,
doi: 10.1002/9780470048672.wecb212
Oligosaccharides are essential for interactions of cells with their environments. These complex carbohydrates are often found covalently attached to proteins embedded in eukaryotic cell membranes. Protein glycosylation is heterogeneous;this heterogeneity stems from the biosynthesis of these polymers. As proteins destined for secretion or cell-surface presentation traffic through the endoplasmic reticulum and the Golgi apparatus, they are modified with sugars in a stepwise fashion by enzymes called glycosyltransferases. The differential expression of these enzymes leads to a multiplicity of specific oligosaccharides both among and within cells because not all cells contain all enzymes and because not all substrate proteins will encounter every enzyme. Although myriad oligosaccharides are found attached to proteins, most of these diverse structures can be grouped into several classes of glycans. In this article, we will discuss some of the most common forms of Golgi protein glycosylation: mucin-type O-linked glycosylation, N-linked glycosylation, and the formation of glycosaminoglycans. In addition, we will briefly consider some less common, but essential, forms of glycosylation.
The Organelles of the Secretory Pathway form an Assembly Line for Glycoprotein Biosynthesis
A large percentage of eukaryotic proteins have oligosaccharides covalently linked to certain amino acid side chains posttranslationally. These posttranslational modifications are essential for cell-cell recognition, cell-matrix interactions, and cell-pathogen recognition. The protein-linked oligosaccharides are known as glycans. Biosynthesis of glycans occurs in the secretory pathway of eukaryotic cells.
Cellular glycans are biosynthesized in eukaryotic cells by sugar-transfer enzymes called glycosyltransferases. These enzymes reside in the secretory pathway, which comprises the endoplasmic reticulum (ER) and the Golgi apparatus. Glycosyltransferases transfer sugars from activated nucleotide sugar donors to a protein or nascent oligosaccharide substrate. Most Golgi-resident glycosyltransferases are type II membrane proteins, which are characterized by a short N-terminal cytoplasmic tail and a single-pass transmembrane region, followed by a variable-length stem region and catalytic domain, both of which extend into the Golgi lumen. Glycosyltransferase enzymes are localized to different parts of the Golgi; enzyme localization influences the order in which substrate proteins encounter different glycosyltransferases and thus the order and arrangement of sugar attachment. The cytoplasmic tail, transmembrane domain, and stem region have all been implicated in controlling localization of glycosyltransferases within the Golgi.
Nucleotide sugar donors, such as UDP-GalNAc, UDP-GlcNAc, GDP-fucose, and CMP-sialic acid, serve as substrates for the glycosyltransferases and are the source of the sugars that are added to substrate proteins (Table 1). Nucleotide sugar donors are synthesized in the cytoplasm and imported into the secretory pathway by membrane-resident transporters (1).
Glycan biosynthesis, unlike DNA, RNA, or protein biosynthesis, is not template directed. Rather, the secretory pathway functions as an assembly line that substrate proteins traffic through on their way to the cell surface. Substrate proteins enter the secretory pathway in the ER. As the proteins pass through the ER and the Golgi, they are modified by enzymes residing in these compartments. Glycosyltransferases that add sugars directly to the protein tend to be found earlier in the secretory pathway, particularly in the ER and the cis-Golgi. Other enzymes are responsible for further glycan modification. These elaborating enzymes tend to be localized in later Golgi compartments (the medial and trans-Golgi) or in the trans-Golgi network (TGN) (Fig. 1). The exact modifications that occur depend on two factors: the availability of activated sugar donor and the order in which the substrate protein encounters glycosyltransferases.
The nontemplated nature of glycan biosynthesis means that not every substrate protein encounters every glycosyltransferase in the secretory pathway; thus, glycosylation reactions are not quantitative. These nonquantitative yields lead to production of a heterogeneous collection of oligosaccharides. Furthermore, glycosyltransferases with similar substrate specificity and localization can compete for substrate molecules, leading to the formation of different glycan products.
Here, we delineate the biosynthetic pathways of the most common types of protein glycosylation occurring in the secretory pathway: mucin-type O-linked glycosylation, N-linked glycosylation, and the formation of glycosaminoglycans. In addition, we will briefly visit the biosynthesis of some less-common varieties of protein glycosylation.
Table 1. Abbreviations and symbols used to indicate common monosaccharides.
|
Common abbreviations and symbols |
|
|
Glucose (Glc) |
|
|
Mannose (Man) |
|
|
Galactose (Gal) |
|
|
Fucose (Fuc) |
|
|
Sialic acid (Sia) |
|
|
N-acetylglucosamine (GlcNAc) |
|
|
N-acetylgalactosamine (GaINac) |
|
|
Glucuronic acid (GIcA) |
|
|
Iduronic acid (IdoA) |

Figure 1. Cartoon of the secretory pathway, indicating the ER and the subcompartments of the Golgi: (a) c/s-Golgi, (b) medial Golgi, (c) trans-Golgi, and (d) trans-Golgi network (TGN). As proteins traffic from the ER through these compartments, they encounter glycosyltransferases in a specific order; this order determines the structure of the final oligosaccharide.
Secretory Pathway Enzymes Perform the Chemistry of Glycosylation
Mucin-Type O-Linked Glycosylation
Glycans can be attached via a glycosidic bond to the hydroxyl group of serine or threonine in a polypeptide. In these cases, the glycosidic bonds connect a sugar to an oxygen in the polypeptide; therefore, this type of glycosylation is termed O-linked. In mammals, the most common type of O-linked glycosylation is the mucin-type, in which N-acetylgalactosamine (GalNAc) is a-linked to either serine or threonine and is subsequently modified by additional sugars (Fig. 2) 2. The name mucin-type derives from the initial isolation of glycoproteins containing these sugar moieties from mucus. Mucins are secreted by many types of tissues, including the linings of the digestive tract and airways 3.
Mucin-type O-linked glycans are synthesized in the Golgi apparatus of eukaryotic cells. Synthesis is stepwise, with individual Golgi-resident glycosyltransferases transferring sugars one at a time to the growing glycoprotein. A wide variety of final glycan structures is possible. The heterogeneity depends on the set of glycosyltransferases that are present in a particular cell and the order in which substrate polypeptide encounters its modifying enzymes. The sequence of steps required for biosynthesis of mucin-type glycans can loosely be divided into the following types of sugar transfer events: initiation, core formation, elongation/branching, and termination.
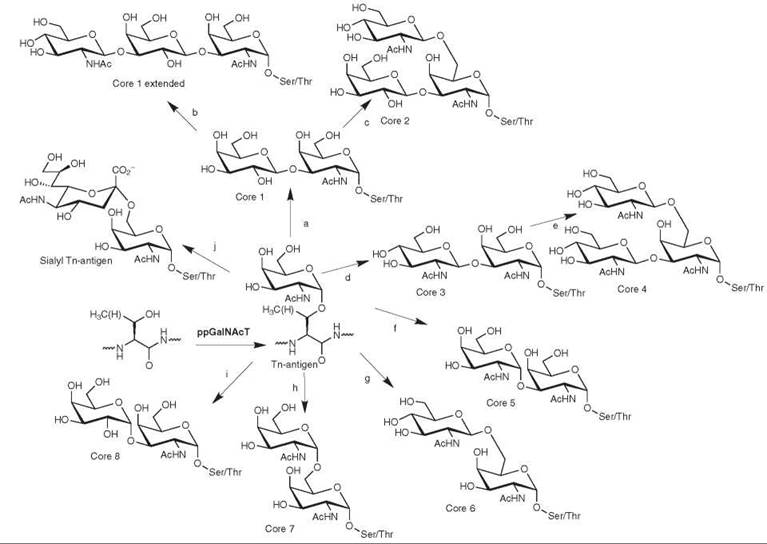
Figure 2. Biosynthesis and structure of mucin-type O-linked glycans. O-linked glycosylation is initiated by the addition of GalNAc to serine or theonine of a polypeptide. This sugar can be further elaborated at the C3 or C6 positions to form the core O-linked glycan structures. The enzymes that form these linkage are: (a) β3 GalT; (b) β3GlcNAcT-3; (c) Core 2 β6-GlcNAcT; (d) Core 3 β3-GlcNAcT; (e) M-type Core 2 [36-GlcNAcT; (f) α1-3 GalNAcT; (g) β1-6 GlcNAcT ; (h) α1-6 GalNAc ; (i) α1-3GalT; and (j) α2-6 SiaT.
Initiation: The ppGalNAcTs
Polypeptide α-GalNAc-transferases (ppGalNAcTs) initiate biosynthesis of mucin-type O-linked glycans by transferring GalNAc from UDP-GalNAc to an acceptor protein. Sequence analysis has revealed approximately 24 mammalian genes with high homology to known ppGalNAcTs. So far, 21 of these genes have been demonstrated to encode proteins that exhibit ppGalNAcT activity. Within the large ppGalNAcT family, individual isoforms display discrete patterns of tissue- and stage-specific expression (4), suggesting that they serve distinct developmental roles. However, there is some functional redundancy among family members as evidenced by the absence of discernable phenotypes in mice harboring targeted disruptions of individual ppGalNAcT genes.
Subcellular localization studies have been completed for some, but not all, of the ppGalNAcTs. Those enzymes that have been examined localize to positions within the Golgi; however, the exact distributions are isoform and cell-type specific. For example, α-GalNAcT1 (5) is localized to the cis-Golgi in porcine submaxillary gland, although it is distributed throughout the Golgi in other types of tissue. Other ppGalNAcT isoforms localize to the medial and trans-Golgi compartments. Examples include human ppGalNAc-T2 and -T3, which localize to the trans-Golgi in HeLa cells (6). The localization of the initiating ppGalNAcT determines which other enzymes the substrate protein can encounter as it completes its transit through the secretory pathway. Therefore, the localization of the initiating ppGalNAcT affects the final structure: If the ppGalNAcT is localized relatively late in the Golgi, fewer additional modifications are possible.
Despite the fact that only certain serine and threonine residues found in secreted proteins are glycosylated, no consensus sequence for mucin-type O-linked glycosylation has been discovered (7). Analysis of glycosylated sequences reveals a preference for prolines and acidic amino acids near the glycosylation site (8), and the secondary structure of the substrate also appears to play a role (9). Among the ppGalNAcT family members, different isoforms have overlapping but distinct substrate preferences (10). Notably, many ppGalNAcTs prefer to transfer GalNAc to glycopeptide substrates that already have GalNAc installed at other serines or threonines (11-13).
Core Formation
Addition of a single GalNAc to serine or threonine constitutes a structure called the Tn-antigen, often observed on tumor cells. However, in most cases, one or more additional monosaccharides are attached to the 3 or 6 positions of the initiating GalNAc, forming the core O -linked structures (Fig. 2), eight of which have been characterized. Synthesis of core structures is developmentally regulated; for example, mouse core 2 GlcNAcT is highly expressed early in gestation, but later it is expressed only in tissues that produce mucins and cartilage 14.
Core 1, the most common mucin-type O-linked core structure, is formed by the action of β1-3 galactosyltransferase (β3GalT), which adds galactose to form Galβ1-3GalNAc (Fig. 2a). P3GalT is expressed in most mammalian cells. The substrate specificity of this enzyme, like others involved in O-linked core synthesis, is influenced by the amino acid sequences of substrate proteins and their glycosylation patterns.
An alternative to core 1-based glycans is the core 3 structure; the 3 position of the initiating GalNAc can be modified by the core 3 β3-GlcNAc-transferase (Fig. 2d), yielding the GlcNAcβ1-3GalNAc structure. The core 3 structure is not as ubiquitous as core 1 and has been found only in mucin-secreting tissues.
The core 2 and 4 structures are synthesized by the addition of GlcNAc to the 6 position of GalNAc in the core 1 (Fig. 2c) and core 3 structures (Fig. 2e), respectively. These structures are produced by a family of β1-6GlcNAc-transferases that catalyzes the formation of the GlcNAcβ1-6GalNAc linkage. At least three β6GlcNAcT isoforms exist; they vary in their preference for core 1 or core 3 substrates (15). For example, the L-type (leukocyte) core 2 β6GlcNAcT accepts only the core 1 substrate, producing core 2 antigen. The M-type (mucin) isoform, expressed in mucin-secreting cell types, exhibits broader substrate scope and is capable of synthesizing both core 2 and core 4 structures, as well as the I antigen (discussed below) (16). Core 2 β6GlcNAcT localizes to the cis- and medial Golgi (17).
The addition of GlcNAc to core 1 by β1-3 GlcNAc-transferase-3 (β3GlcNAcT-3) results in the formation of extended core 1 structures (Fig. 2b). These glycans are frequently modified with additional sugars to form sialyl Lewis x or 6-sulfo sialyl Lewis x; this elaboration is essential to leukocyte rolling and L-selectin binding. Extended core 1 oligosaccharides may be further elaborated by the addition of GlcNAc to the 6 position of GalNAc, forming biantennary glycans containing core 2(18).
Addition of GlcNAc in a β1-6 linkage to Tn antigen generates the putative core 6 structure (Fig. 2g). Core 6 structures have been reported to occur in human ovarian tissue, but an enzyme responsible for core 6 formation has not been identified (19).
Less common core structures are formed when the initiating GalNAc is modified by the addition of a-linked sugars at either the 3 or 6 positions. Core 5 is formed by the addition of a second GalNAc in an a 1-3 linkage to the first (Fig. 2f) (19-21). This enzymatic activity has been detected in biological samples, but the responsible gene has not been identified. The core 7 (GalNAca1-6GalNAc) (Fig. 2h) (22) and core 8 (Gala1-3GalNAc) (Fig. 2i) structures (23) are produced in restricted tissues and the responsible genes are not yet known.
Branching and Elongation
A common motif found in larger O-linked glycans is poly-N-acetyllactosamine (-Galβ1-4GlcNAcβ1-3-)n, also called poly-LacNAc or the type 2 backbone (Fig. 3a). PolyLacNAc repeats are commonly found on fetal erythrocytes, where they constitute the blood group i antigen. PolyLacNAc chains of varying length are synthesized by the alternate action of two enzymes, a β1-4galactosyltransferase (β4-GalT) and a β1-3GlcNAc-transferase (β1-3 GlcNAcT). A family of at least five β4-GalTs has been discovered and at least eight β1-3 GlcNAcTs have been identified; these biosynthetic enzymes are widely expressed. The most well-characterized of the β1-3 GlcNAcTs is known as iGnT because of its role in synthesis of the i antigen (24).
PolyLacNAc can be extended from sugars attached to either the 3 or 6 position of the initiating GalNAc; however, addition to the 6 (upper) branch is more common (Fig. 3c). Additional branching can be introduced into the LacNAc backbone by the action of β1-6 GlcNAc-transferases, including the previously mentioned core 2 β1-6GlcNAcT M isoform. This type of branched structure, the I antigen, is commonly found on adult erythrocytes. Members of the β6GlcNAc-transferase family that produce branched structures are known as IGnTs. Synthesis of branched structures is developmentally regulated, and members of IGnT enzyme family have differing substrate specificities. Some IGnTs select for terminal Gal whereas others modify internal Gal residues.
The type 1 (-Galβ1-3GlcNAcβ1-3-)n polymer, also called lacto-A-biose, is a related but less common, backbone motif (Fig. 3b). Lacto-A-biose is synthesized by a β1-3galactosyl-transferase (β3-GalT) and a β1-3GlcNAc-transferase (β3- GlcNAcT).

Figure 3. Common glycan modifications. Once the core structure is assembled, mucin-type O-linked glycans are elongated by the addition of repeating polymers of (a) LacNAc or (b) lacto-N-biose. (c) Branching occurs by the addition of GlcNAc to the 6 position of GalNAc in these polymers forming the ''I antigen.'' Fucosylation, sialylation, and sulfation are terminal modifications. (d) α1,2 fucosylation; (e) α1,3 fucosylation; (f) α1,4 fucosylation; (g) α2,3 sialylation (*) and α2,6 sialylation (**) of terminal galactose; (h) α2,3 sialylation of GalNAc; (i) 3-O-sulfation; and (j) 6-O-sulfation.
Further Modification and Termination
Mucin-type O-linked glycans are decorated with a variety of capping structures. Given their prominent location, these structures play critical roles in cell-cell and cell-matrix recognition events. The most common modifications are fucosylation, sialylation, and sulfation. Terminal sialic acids can also be further modified by O-acetylation at the 7 or 9 position.
Fucose can be added to either galactose or GlcNAc. α1-2 fucosyltransferases add fucose to galactose forming a structure (Fuca1-2Gal) known as the H antigen (Fig. 3d). The two known α1-2 fucosyltransferases (FucT1 and FucT2) localize to the trans-Golgi and fucosylate only terminal (i.e., nonreducing) galactose (25). The Fuca1-3GlcNAc linkage (Fig. 3e) is produced by a family of six known α1-3 fucosyltransferases (FucT3, FucT4, FucT5, FucT6, FucT7, and FucT9) (26). Some members of this family can distinguish between Galβ1,4GlcNAc and Siaα1-3Galβ1-4GlcNAc, whereas others can fucosylate both. The resultant structures (Galβ1-4[Fucα1-3]GlcNAc and Siaα2-3Galβ1-4[Fuca1-3]GlcNAc) are known as the Lewis x and the sialyl Lewis x antigens, respectively. Individual family members exhibit distinct localization patterns throughout the Golgi and TGN. Enzymes that localize earlier within the Golgi encounter more Galβ1,4GlcNAc substrate, whereas enzymes that localize later are more likely to fucosylate Siaα2-3Galβ1-4GlcNAc. FucT3 is also capable of adding fucose in α1-4 linkage to GlcNAc (Fig. 3f) and can act on both unsialylated and sialylated LacNAc (27).
Sialylation of mucin-type O-linked glycans is accomplished by two families of enzymes: α2-3 sialyltransferases and α2-6 sialyltransferases. The six known α2-3SiaTs transfer sialic acid to terminal galactose, which can be found in core 1 and core 2 structures, as well as at the ends of polyLacNAc chains. The resultant structure, Siaα2-3Gal (Fig. 3g), can only be further extended by the addition of α2-6 sialic acid to the Siaα2-3Gal (Fig. 3g). As a result of their localization to the medial and trans-Golgi, the α2-3SiaTs are able to compete with both branching and elongation and are responsible for the early termination of oligosaccharide chains (16). The α2-6SialTs exhibit varying degrees of discrimination among the following three substrates: unmodified GalNAc (Tn-antigen) (Fig. 3h), Galβ1-3GalNAc (core 1), and Sia1α2-3Galβ1-3GalNAc (28). One α2-6SiaT, ST6GalI, is known to sialylate terminal galactose found in polyLacNAc. As sialylation tends to be a terminal modification, most sialyltransferases localize to the later compartments of the secretory pathway. For example, the two rat isoforms of ST6GalI localize to the medial and trans-Golgi (29).
Another common modification of mucin-type O-linked glycans is sulfation. At least four sulfotransferases add sulfate to the 6 position of GlcNAc (30) (Fig. 3j), whereas several others sulfate the 3 position of galactose in core 1 and in polyLacNAc chains (Fig. 3i) (31, 32). Sulfation at the 6 position of GlcNAc interferes with branching by blocking the action of β6GlcNAc-transferases. Despite the fact that sulfation is often observed on internal GlcNAc residues, the GlcNAc-6-O-sulfotransferases all exhibit a strong preference for terminal GlcNAc substrates, suggesting that these enzymes sulfate GlcNAc after it is added to the oligosaccharide chain but before the glycan is extended (33). The sulfotransferases exhibit varying localization patterns throughout the Golgi, allowing sulfotransferases to intercept mucins at various stages of synthesis.
N-Linked Glycosylation
The best known form of protein glycosylation is the asparagine- or N-linked variety. These large, branched structures contain a conserved core structure that is produced in the majority of eukaryotes, including yeasts, plants, and mammals. Other eukaryotes, including protists, also produce related N-glycans, albeit with simplified cores (34). N-linked glycans are synthesized by an elaborate process that begins on the cytoplasmic face of the ER and continues in the lumens of the ER and the Golgi. In this section, we outline the steps involved in synthesis of the common oligosaccharide precursor, its transfer to polypeptide substrates, and subsequent processing and elaboration events that occur as the N-glycosylated protein completes its transit through the secretory pathway.
Assembly of the Dolichol Oligosaccharide Donor
The dolichol oligosaccharide donor (35) is composed of a lipid pyrophosphate that is attached to an oligosaccharide composed of 14 individual sugars: dolichol-P-P-GlcNAc2Man9Glc3. The dolichol donor is assembled by the action of the Alg family of glycosyltransferases, which add the sugars one by one (Fig. 4a-e) (36-38). Assembly of the dolichol donor begins on the cytoplasmic face of the ER membrane, where a series of Alg GlcNAc-transferases and mannosyltransferases converts dolichol phosphate to dolichol-P-P-GlcNAc2Man5 (Fig. 4A, a-e). At this stage, Rft1 functions as a “flippase” to transfer dolichol-P-P-GlcNAc2Man5 across the ER membrane (39), so that the carbohydrates are now in the lumen of the ER (Fig. 4A, f). Inside the ER, Alg mannosyltransferases and glucosyltransferses add additional monosaccharides to form dolichol-P-P-GlcNAc2Man9Glc3 (Fig. 4A, g-l).
Transfer of the Oligosaccharide to Polypeptides
The oligosaccharyltransferase (OT) transfers GlcNAc2Man9Glu3 from the dolichol donor to an asparagine (Asn) of a nascent glycoprotein (Fig. 4A, m). N-glycosylation occurs co-translationally, as the newly synthesized polypeptide enters the ER through the translocon (40, 41). The heteromeric OT complex is composed of at least nine different polypeptides, including the STT3 subunit that provides the active site (42-44). The oligosaccharide is transferred to asparagines within the minimal consensus sequence Asn-X-Thr/Ser, where X is any amino acid except proline (45). The amino acids surrounding the consensus sequence also affect whether a particular Asn is a substrate for glycosylation. In addition, Asn-X-Cys sequences are occasionally glycosylated. After the oligosaccharide is transferred to a substrate protein, it is processed by ER- and Golgi-resident glycosidases.

Figure 4. Biosynthesis and structure of N-linked glycans. (A) The ER-resident Alg glycosyltransferases are responsible for addition of the individual monosaccharides to assemble the dolichol oligosaccharide donor. Initial steps are accomplished by Alg7 (a), Alg13/14 (b), Alg1 (c), Alg2 (d), and Alg11 (e) on the cytosolic face of the ER. The oligosaccharide is ''flipped'' from the cytoplasm to the ER lumen by Rft1 (f). Inside the ER lumen, additional sugars are added by glycosyltransferases Alg3 (g), Alg9 (h), Alg12 (i), Alg6 (j), Alg8 (k), and Alg10 (l). The assembled dolichol donor is transferred to the nascent polypeptide by the oligosaccharyltransferase (m). Trimming of the dolichol donor is catalyzed by glucosidase I (n), glucosidase II (o), and a-mannosidase I (p). In the Golgi, additional trimming is performed by Golgi-resident a-mannosidases (q) and GlcNAc is added by GlcNAcT-I (r). In the synthesis of typical complex glycan, two more mannoses are removed by a-mannosidase II (s), followed by the addition of GlcNAc residues by GlcNAcT-II (t) and GlcNAcT-V (u) and galactose residues by a β4-GalT (v). Extension of polyLacNAc chains is performed by β1-3 GlcNAcTs (w) and β4-GalTs (x), whereas terminal capping structures are added by fucosyltransferases (y) and sialyltransferases (z). (B) Subtypes of N-linked glycans include (a) mannan-type glycan; (b and c) monoantennary hybrid glycans; (d) a biantennary complex glycan; (e) a triantennary complex glycan; (f) a tetraantennary complex glycan; and (g) an N-linked glycan with a ''bisecting GlcNAc.''
Exit from the ER
The ER-resident glucosidase I removes the terminal α1-2 linked glucose from the oligosaccharide (Fig. 4A, n). Subsequently, glucosidase II removes the α1-3 glucose-linked glucose and then, more slowly, removes the α1-3 mannose-linked glucose (Fig. 4A, o) (46, 47). The presence of this mannose-linked glucose is intimately associated with protein folding. If a protein is folded improperly, the mannose-linked glucose is reinstalled by a glucosyltransferase (UGGT) and the protein remains in the ER to complete its folding (48, 49). Once the protein is properly folded and all three glucoses removed, a-mannosidase I removes the terminal α1-2 linked mannose from the middle chain (50) (Fig. 4A, p). The properly folded protein with its remaining Man8GlcNAc2-Asn glycan is now able to exit the ER and traffic to the cis-Golgi.
Trimming in the Golgi
Proteins destined for the cell surface or secretion are processed by Golgi-resident a-mannosidases, which remove additional mannoses to produce Man5GlcNAc2-Asn (Fig. 4A, q). Man5GlcNAc2 is also referred to as high-mannose (51) (Fig. 4B, a), and it serves as the starting point for the synthesis of a variety of other N-glycan subtypes, described below.
An alternative processing pathway is used by proteins that will traffic to the lysosome. GlcNAc-phosphotransferase adds phosphate ester-linked GlcNAc residues to two mannoses in the oligosaccharide (52). A GlcNAcase then removes the GlcNAc sugars (53), revealing mannose-6-phosphate, which serves as a signal for the protein to be shuttled to the lysosome.
N-Glycan Subtypes
Despite the conservation of the core structure, the ways in which N-linked glycans are elaborated vary among organisms and cell types. In yeasts, Man5GlcNAc2 is elaborated by several manno- syltransferase to form large mannan-type structures. In muticellular organisms, there are 3N-glycan subtypes: high-mannose, hybrid, and complex. Insects and other invertebrates produce high-mannose and hybrid-type N-glycans, but not complex structures (54). In vertebrate organisms, MansGlcNAc2 can be modified to form hybrid and complex N-glycans. The central β1-4-linked mannose bears two mannoses, which are α1-3- and α1-6-linked. Addition of GlcNAc to the α1-3-linked mannose yields the hybrid structures (Fig. 4B, b-c), whereas complex structures have GlcNAc added to both the α1-3- and α1-6-linked mannoses (Fig. 4B, d-f). Hybrid and complex glycans can be described by the number of GlcNAc-containing branches, or antennae, they possess. Six different GlcNAc-transferases can initiate branch formation, generating various antennary structures (55).
Synthesis of both hybrid and complex N-glycans begins with the addition of GlcNAc β1-2 to the α1-3-linked mannose, forming a monoantennary hybrid glycan (Fig. 4A, r). GlcNAc addition is catalyzed by the GlcNAcT-I enzyme encoded by the Mgat1 gene (56). The resultant glycan is a substrate for the medial Golgi enzyme, α-mannosidase II, which removes two mannoses to generate GlcNAc1Man3GlcNAc2 (Fig. 4A, s). This same glycan can be produced by an alternative route: α-mannosidase-III catalyzed removal of two mannoses from the high-mannose glycan and subsequent addition of GlcNAc by GlcNAcT-I. Synthesis of biantennary hybrid glycan is also possible, through the action of GlcNAcT-IV, which adds a GlcNAc in a β1-4 linkage to the α1-3-linked mannose (57).
Complex glycans (Fig. 4B, d-f) are produced when GlcNAcT-II adds GlcNAc in a β1-2 linkage to α1-6-linked mannose (47). Once this linkage occurs, an additional antenna can be produced by GlcNAcT-V, which adds GlcNAc in a β1-6 linkage to the α1-6-linked mannose (Fig. 4B, e-f) (58). Finally, addition of GlcNAc in an β1-4 linkage to the α1-6-linked mannose is a rare modification catalyzed by GlcNAcT-VI.
Extension and Elaboration
Once the number of antennae is established, further extension is possible through addition of backbone polymers and terminal structures similar to those found on mucin-type glycans. LacNAc polymers can be added to any of the aforementioned GlcNAcs (Fig. 4A, w-x). Similarly, these polymers can be elaborated with fucose, sulfate, and sialic acid added in the same linkages described for mucin-type structures (Fig. 4A, y-z). Most fucosyltransferases and sulfotransferases, as well as the sialyltransferases that cap polyLacNAc, are capable of modifying both O-linked and N -linked glycans; some exhibit a preference for one or the other. These biases are likely a result of differences in enzyme localization.
Fucosylation of the Asn-linked GlcNAc is a common modification unique to N-linked glycans. In vertebrates, core FucT adds fucose in an α1-6 linkage (59), whereas in plants and invertebrates α1-3-linked fucose is observed (60).
Elaboration of N-linked glycans can be dramatically altered by GlcNAcT-III activity. GlcNAcT-III adds GlcNAc to the β1-4-linked mannose. This β1-4-linked GlcNAc is also referred to as the “bisecting GlcNAc” (Fig. 4B, g). Unlike other mannose-linked GlcNAc residues, this sugar cannot be further elaborated (61, 62). Furthermore, the addition of the bisecting GlcNAc leads to the appearance of “unprocessed hybrid” glycan by preventing mannose trimming by α-mannosidase II and subsequent elaboration.
Glycosaminoglycans and Proteoglycans
Glycosaminoglycans (GAGs) are defined by their composition; they are composed of long chains of repeating disaccharides. Alternating amino sugars (GlcNAc or GalNAc) and uronic acids (glucuronic acid or iduronic acid) comprise their disaccharide building blocks. The exact sugar composition and modifications to the sugars determine the classification of the GAG. Commonly occuring GAGs include heparin, heparan sulfate (HS), chondroitin sulfate (CS), and dermatan sulfate (DS). GAGs contain a common core tetrasaccharide linking them to a protein. Proteins or polypeptides with GAG chains attached are known as proteoglycans.
Biosynthesis of the Linkage Tetrasaccharide
Heparin, HS, CS, and DS share a common serine-linked core tetrasaccharide, GlcAβ1-3-Galβ1-3-Galβ1-4-Xyl-Ser. Biosynthesis of this core tetrasaccharide, known as the linkage tetrasac- charide, is initiated in the ER by the transfer of xylose to serine by a xylosyltransferase (63, 64) (Fig. 5). This enzyme prefers to transfer sugars to serines followed by glycine and flanked by one or more acidic residues (65). The xylosylated protein is then transported to the Golgi to undergo further modification by three Golgi resident enzymes. GalT-I adds galactose in a β1-4 linkage (66, 67). The resulting disaccharide is then elongated by GalT-II (β1-3GalT6), a β1-3 galactosyltransferase. Finally, GlcAT-I, a GlcA transferase specific for this trisaccharide precursor, adds β1-3 glucuronic acid to complete synthesis of the linkage tetrasaccharide (68-69).
Sugars in the linkage tetrasaccharide can be modified. Common modifications are phosphorylation of xylose at C2 (70, 71) and sulfation of the second galactose at C4 (72). The function of these modifications is not known; not all proteoglycans containing the linkage tetrasaccharide are modified (73, 74).
The Branching Point: Addition of GlcNAc or GalNAc
Once the synthesis of the linkage tetrasaccharide is complete, it can be further modified by one of two Golgi-resident glycosyltransferases. The enzyme α-GlcNAcT-I can add α1-4 linked GlcNAc, thereby initiating the heparin/HS biosynthetic pathway. In vitro activity assays indicate that α-GlcNAcT-I also appears to be capable of adding an α-linked GalNAc to cap the linkage tetrasaccharide, resulting in a pentasaccharide that cannot be further elongated (75); however, the biological relevance of this modification is not known. If the protein is not modified by α-GlcNAcT-I, β-GalNAcT II can add GalNAc β1,4 to the glucuronic acid, initiating the CS/DS pathway (76). The CS/DS pathway is suggested to be the default pathway for linkage tetrasaccharides not intercepted by α-GlcNAcT-I.
Heparin and HS
After the initiation of heparin/HS biosynthesis by the addition of GlcNAc to the linkage tetrasaccharide, polymerization of the disaccharide β1-4GlcA-α1-4GlcNAc begins (Fig. 5). The polymerization reaction is catalyzed by two bifunctional Golgi-resident glycosyltransferases, EXT1 and EXT2. Individually, these enzymes each exhibit GlcNAc- and glucuronic acid-transferase activity, but they are most active when they are physically associated with one another and localized to the Golgi (77).
As the (β1-4GlcA-α1-4GlcNAc)n polymer is produced, it is modified by a number of Golgi-resident enzymes that deacetylate, epimerize, and sulfate the growing GAG. The GlcNAc N-deacetylase/N-sulfotransferases (NDSTs) catalyze both the deacetylation and N-sulfation of GlcNAc in the repeating disaccharide. The four different NDST isozymes display different substrate selectivities and vary in their enzymatic activities (78-83). Glucuronic acid (GlcA) residues adjacent to N-sulfated GlcNAc can be epimerized to iduronic acid (IdoA) by GlcA-C5 epimerase. The newly formed IdoA can then be sulfated at the 2 position by the 2-O-sulfotransferase, which prevents reverse epimerization to GlcA. The epimerase and 2-O-sulfotransferase work in close concert and are known to physically associate with one another (84). Further O-sulfation of the GAG chain is accomplished by the 6-O-sulfotransferases (6-OSTs) and 3-O-sulfotransferases (3-OSTs). The three 6-OSTs that add sulfate to N-sulfated GlcN (deacetylated GlcNAc) (85) and the six 3-OSTs that sulfate the 3 position of the hexosamine (86) exhibit varying substrate specificities (87, 88).
Heparin and HS are formed from the same disaccharide repeat and differ only in their degrees of modification. Heparin is highly modified, containing more IdoA than GlcA and having many sites of N- and O-sulfation; the predominant carbohydrate motif is a repeating trisulfated IdoA(2-O-SO3)-GlcN-SO3 (6-O-SO3)- unit. HS is considerably more heterogeneous; modifications are confined to distinct regions of the oligosaccharide chain. HS can be rather large, reaching up to 70kDa, whereas heparin is generally only 10-12 kDa. Like other glycans, the structures of heparin and HS GAGs are ultimately determined by expression levels of Golgi-resident glycosyltransferases and sulfotransferases and the availability of activated substrate.
CS and DS
Linkage tetrasaccharides not modified by αGlcNAcT-I in the Golgi will be converted to CS or DS in the TGN. The oligosaccharide chain is elongated by the bifunctional CS synthase, which produces the (-β1-3GlcA-β1-4GalNAc-)n polymer (89-90) (Fig. 5).
As the oligosaccharide is elongated, it is modified by several sulfotransferases. At any point during these modifications, a C5-epimerase is able to convert GlcA to IdoA (91). The presence of IdoA indicates that the oligosaccharide is DS rather than CS (Fig. 5). Three GalNAc4-O-STs are involved in the transfer of sulfate to GalNAc in GlcA-rich regions of the oligosaccharide (92). The C5-epimerase works in conjunction with a DS-specific GalNAc4-O-ST (D4ST-1) (93). A 2-O-sulfotransferase known as CS/DS2ST catalyzes the addition of sulfate to both IdoA and GlcA (94). In addition, two different 6-O-sulfotransferases can produce chondroitin 6-sulfate. These two sulfotransferases have different substrate selectivities, with C6ST-II sulfating GalNAc(4-O-SO3) and with C6ST producing GlcA(2-O-SO3)-GalNAc(6-O-SO3). In addition, a DS-specific GalNAc6-O-ST (D6ST) sulfates C6 on GalNAc flanked by two IdoAs (95).
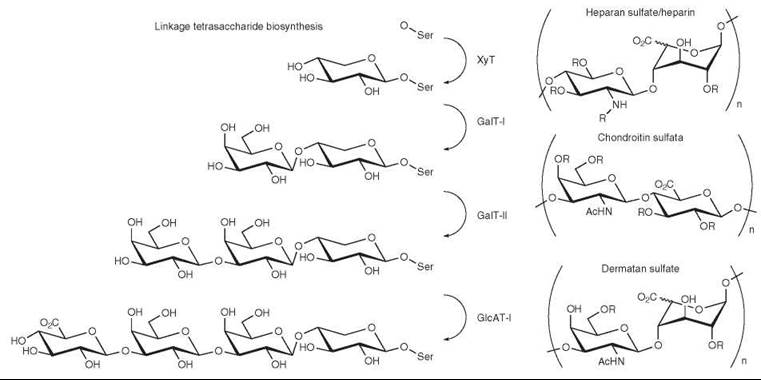
Figure 5. Biosynthesis and structure of glycosaminoglycans. Synthesis of the core linkage tetrasaccharide is accomplished by XylT, GalT-I, GalT-II, and GlcAT-I. The repeating disaccharides of heparin/HS, CS, and DS are shown.
Keratan Sulfate
Keratan sulfate (KS) is often grouped with the GAGs even though it does not meet the definition of a GAG. KS consists of polyLacNAc sulfated at C6 on both hexoses. Three distinct types of KS exist that are differentiated by the way the sulfo-polyLacNAc is linked to the protein; these types are designated KSI, KSII, and KSIII (96).
KSI biosynthesis is initiated in the same way as N-linked glycan biosynthesis, with the addition of an oligosaccharide to Asn from the dolichol donor in the ER. The oligosaccharide is trimmed to form the KSI linkage oligosaccharide, which is of the complex biantennary type (97). The linkage oligosaccharide can be modified on either the C6 branch or the C3 branch to form KS structures (98-100). The nonreducing terminus is usually very highly sulfated, whereas the sugars nearest the reducing end are not sulfated (101). KSI chains are frequently terminated with sialic acid and less frequently with GalNAc or Gal (97, 102).
KSII biosynthesis is initiated by the formation of mucin-type core 2 disaccharide (103). KSII is usually capped with sialic acid at either C3 or C6 of terminal GlcNAc. Sulfated GlcNAc in KSII is often modified with α1-3-linked fucose, although not within four sugars of the terminus (104).
KSIII is the least-characterized member of the KS family. This polyLacNAc polymer is extended from a serine-linked mannose and has been found in brain tissue (105).
Elongation of KS is not well understood. The activities of β4GalT-I, β3GnT, and iGnT have been suggested to fulfill this role, but none of these enzymes have been directly linked to KS biosynthesis (106-109).
Sulfation of KS is catalyzed by at least two sulfotransferases. KSGal6ST can sulfate terminal and internal galactoses of the elongating oligosaccharide (110); sulfation of terminal galactose is believed to block further KS polymerization (111). In contrast, GlcNAc6-O-sulfation occurs only on nonreducing GlcNAc and must be simultaneous with chain elongation. GlcNAc6ST-5 is the most likely candidate to provide the GlcNAc6-O-sulfation activity (112).
Other Types of Protein Glycosylation in the Golgi
O-fucose is an important modification that mediates cell-cell interactions and leads to intracellular signaling events. Fucose is linked to either serine or threonine found in the consensus sequence C2XXGGS/TC3. The best-characterized modification sites are in the EGF domain of Notch (113). O-Fuc is added by protein O-fucosyltransferase (O-FucT-I) and may be either a standalone modification or further extended. If extended, Fringe glycosyltransferase, a β1-3GlcNAcT, adds GlcNAc to fucose, followed by addition of galactose by β1-4GalT-I. The single LacNAc is terminated with α2-6 sialic acid, although this modification is not essential to Notch signaling.
Glycans can also be attached to proteins via an O-linked mannose. Mannose is added in the ER and can be further elaborated in the Golgi to form a tetrasaccharide, Siaα2-3Galβ1-4GlcNAcβ1-2Man-Ser/Thr (114). O-mannose glycans are essential to proper brain and muscle function and their absence is associated with muscular dystrophy.
Polysialic acid (PSA) is a linear homopolymer of α2-8- or α2-9-linked sialic acid attached to protein scaffolds including the neural cell adhesion molecule (NCAM). Two Golgi-resident polysialyltransferases, PST and STX, are responsible for its synthesis. Although PSA is widely distributed in embryonic tissues, it is normally found only in regenerating neural and muscle tissues in adults. Neo-expression of PSA is often associated with metastatic cancer (115).
Collagen is an important glycoprotein whose glycosylation does not fit into any of the categories previously described. In collagen, many prolines and lysines are posttranslationally hydroxylated and the hydroxyl-lysine residues further glycosylated. Hydroxyl-lysine may be modified by the presence of either a single galactose or Glcα1-2Gal (116).
Chemical Tools to Manipulate Cellular Glycosylation
Protein glycosylation affects a large number of biological interactions, including developmental processes, cancer metastasis, and host-pathogen interactions. Small molecule tools allow researchers to control glycosylation in vivo and determine the roles of oligosaccharides in biological processes. Chemical tools provide advantages over genetic approaches because they offer time-dependent, dose-dependent, and reversible control of glycosylation events.
Small molecules such as brefeldin A and nocodazole are molecules that disrupt the architecture of the Golgi apparatus, which results in a perturbation of the spatial organization of Golgi-resident proteins, including glycosyltransferases. Substrate proteins no longer encounter glycosyltransferases in the correct order, leading to gross changes in cellular glycans. Brefeldin A reversibly inhibits vesicle trafficking from the ER to the Golgi; nocodazole depolymerizes microtubules and arrests the cell cycle, leading to improper formation of the Golgi.
Competitive primers function as alternative substrates for elaborating glycosyltransferases. These small molecules have provided a facile way to interfere with mucin-type synthesis and lead to truncation of the O-linked glycans found on cellular substrates. The competitive primer α-benzyl-GalNAc (Fig. 6a) mimics the Tn antigen, whereas Galβ1-3GlcNAcβ-O-naphthalenemethanol (Fig. 6c) and Galβ1-4GlcNAcβ-O-naphthalenemethanol (Fig. 6b) are competitive with type 1 and type 2 backbone polymers.
Inhibition of the initiating ppGalNAcTs is an alternative strategy for obstructing mucin-type biosynthesis. A uridine-based library has yielded several competitive inhibitors of the ppGal-NAcTs, including 1-68A (Fig. 6d) and 2-68A (Fig. 6e) (117). These molecules have been used successfully in cellular and organ culture (118).
Chemical control of the biosynthesis of N-linked glycans has been facilitated by the availability of cell-permeable natural products that interfere with various steps in oligosaccharide processing. Tunicamycin (Fig. 6h) impedes the first step of dolichol oligosaccharide donor synthesis, preventing transfer of GlcNAc from UDP-GlcNAc to Dol-P. As a result, no N-linked glycans are transferred to polypeptides. Once the oligosaccharide is added to the protein, deoxynojirimycin (Fig. 6g), nojirimycin, and related derivatives can be used to prevent further processing: These molecules inhibit trimming by ER-resident glucosidase (119, 120). Another azasugar, 1-deoxymannojirimycin (Fig. 6f), is a mannose analog that is able to inhibit the ER a-mannosidase I, keeping the substrate protein from being transported from the ER to the Golgi (121). Swainsonine (Fig. 6i) functions in the Golgi as a reversible inhibitor of α-mannosidase II. Treatment with swainsonine blocks elaboration of the oligosaccharide, precluding modifications by the GlcNAc-transferases (122). As each of these molecules functions at an early stage in N -linked glycan biosynthesis, they have a global effect on N-linked glycosylation.
Competitive primers can also be used as alternate substrates for GAG biosynthesis. β-D-xylosides containing two fused aromatic rings intercept the galacotsyltransferases involved in linkage tetrasaccharide biosynthesis (123).

Figure 6. Chemical tools can be used to interfere with cellular glycosylation. Tools to interfere with mucin-type O-linked glycosylation include (a) α-benzyl GalNAc; (b) Galβ1-4GlcNAcβ-O-naphthalenemethanol; (c) Galβ1-3GlcNAcβ-O-naphthalenemethanol; (d) 1-68A; and (e) 2-68A. Molecules used to disrupt N-linked glycosylation at different steps include (f) 1-deoxymannojirimycin; (g) deoxynojirimycin; (h) tunicamycin; and (i) swainsonine.
Summary
The plethora of oligosaccharide structures synthesized by living organisms provides a tantalizing diversity of structures for chemists to explore. Although methods to analyze glycoconjugates are rapidly improving, we remain unable to predict or program cellular glycosylation events because of the challenges posed by the nontemplated nature of glycan biosynthesis. In the future, systems biology approaches may give a predictive understanding of the glycans produced by a given cell. Further exploration of these pathways will be enhanced by chemical biologists’ development of additional chemical tools with targeted, rather than global, effects on cellular glycosylation.
Acknowledgments
We thank Dr. Danielle H. Dube, Michelle R. Bond, Peter L. Lee, and Ethan J. Greenblatt for discussions and comments on the manuscript.
References
1. Abeijon C, Mandon EC, Hirschberg CB. Transporters of nucleotide sugars, nucleotide sulfate and ATP in the Golgi apparatus. Trends Biochem. Sci. 1997; 22(6):203-207.
2. Hanisch FG. O-glycosylation of the mucin type. Biol. Chem. 2001; 382(2):143-149.
3. Roussel P, Lamblin G, Lhermitte M, Houdret N, Lafitte JJ, Perini JM, Klein A, Scharfman A. The complexity of mucins. Biochimie 1988; 70(11):1471-1482.
4. Kingsley PD, Hagen KG, Maltby KM, Zara J, Tabak LA. Diverse spatial expression patterns of UDP-GalNAc: polypeptide N-acetylgalactosaminyltransferase family member mRNAs during mouse development. Glycobiology 2000; 10(12):1317-1323.
5. Roth J, Wang Y, Eckhardt AE, Hill RL. Subcellular localization of the UDP-N-acetyl-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase-mediated O- glycosylation reaction in the submaxillary gland. Proc. Natl. Acad. Sci. U.S.A. 1994; 91(19):8935-8939.
6. Rottger S, White J, Wandall HH, Olivo JC, Stark A, Bennett EP, Whitehouse C, Berger EG, Clausen H, Nilsson T. Localization of three human polypeptide GalNAc-transferases in HeLa cells suggests initiation of O-linked glycosylation throughout the Golgi apparatus. J. Cell Sci. 1998; 111(1):45-60.
7. Pratt MR, Hang HC, Ten Hagen KG, Rarick J, Gerken TA, Tabak LA, Bertozzi CR. Deconvoluting the functions of polypeptide N-α-acetylgalactosaminyltransferase family members by glycopeptide substrate profiling. Chem. Biol. 2004; 11(7):1009-1016.
8. Julenius K, Molgaard A, Gupta R, Brunak S. Prediction, conservation analysis, structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 2005; 15(2):153-164.
9. Wragg S, Hagen FK, Tabak LA. Kinetic analysis of a recombinant UDP-N-acetyl-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase. J. Biol. Chem. 1995; 270(28):16947-16954.
10. Elhammer AP, Kezdy FJ, Kurosaka A. The acceptor specificity of UDP-GalNAc: polypeptide N-acetylgalactosaminyltransferases. Glycoconj. J. 1999; 16(2):171-180.
11. Ten Hagen KG, Bedi GS, Tetaert D, Kingsley PD, Hagen FK, Balys MM, Beres TM, Degand P, Tabak LA. Cloning and characterization of a ninth member of the UDP-GalNAc: polypeptide N-acetylgalactosaminyltransferase family, ppGaNTase-T9. J. Biol. Chem. 2001; 276(20):17395-17404.
12. Ten Hagen KG, Tetaert D, Hagen FK, Richet C, Beres TM, Gagnon J, Balys MM, VanWuyckhuyse B, Bedi GS, Degand P, Tabak LA. Characterization of a UDP-GalNAc: polypeptide N-acetylgalactosaminyltransferase that displays glycopeptide N-acetylgalactosaminyltransferase activity. J. Biol. Chem. 1999; 274(39):27867-27874.
13. Bennett EP, Hassan H, Hollingsworth MA, Clausen H. A novel human UDP-N-acetyl-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase, GalNAc-T7, with specificity for partial GalNAc-glycosylated acceptor substrates. FEBS Lett. 1999; 460(2):226-230.
14. Granovsky M, Bielfeldt T, Peters S, Paulsen H, Meldal M, Brockhausen J, Brockhausen I. UDP galactose:glycoprotein-N-acetyl-D-galactosamine 3-p-D-galactosyltransferase activity synthesizing O-glycan core 1 is controlled by the amino acid sequence and glycosylation of glycopeptide substrates. Eur. J. Biochem. 1994; 221(3):1039-1046.
15. Dalziel M, Whitehouse C, McFarlane I, Brockhausen I, Gschmeissner S, Schwientek T, Clausen H, Burchell JM, Taylor-Papadimitriou J. The relative activities of the C2GnT1 and ST3Gal-I glycosyltransferases determine O-glycan structure and expression of a tumor-associated epitope on MUC1. J. Biol. Chem. 2001; 276(14):11007-11015.
16. Kuhns W, Rutz V, Paulsen H, Matta KL, Baker MA, Barner M, Granovsky M, Brockhausen I. Processing O-glycan core 1, Galβ1-3GalNAcα-R. Specificities of core 2, UDP-GlcNAc: Galβ1-3 GalNAc-R(GlcNAc to GalNAc)β6-N-acetylglucosaminyltransferase and CMP-sialic acid: Galβ1-3GalNAc-Rα3-sialyltransferase. Glycoconj. J. 1993; 10(5):381-394.
17. Skrincosky D, Kain R, El-Battari A, Exner M, Kerjaschki D, Fukuda M. Altered Golgi localization of core 2β-1,6-N-acetylglucosaminyltransferase leads to decreased synthesis of branched O-glycans. J. Biol. Chem. 1997; 272(36):22695-22702.
18. Mitoma J, Petryniak B, Hiraoka N, Yeh JC, Lowe JB, Fukuda M. Extended core 1 and core 2 branched O-glycans differentially modulate sialyl Lewis X-type L-selectin ligand activity. J. Biol. Chem. 2003; 278(11):9953-9961.
19. Hounsell EF, Lawson AM, Feeney J, Gooi HC, Pickering NJ, Stoll MS, Lui SC, Feizi T. Structural analysis of the O-glycosidically linked core region oligosaccharides of human meconium glycoproteins which express oncofetal antigens. Eur. J. Biochem. 1985; 148(2):367-377.
20. Wieruszeski JM, Michalski JC, Montreuil J, Strecker G, Peterkatalinic J, Egge H, Vanhalbeek H, Mutsaers JHGM, Vliegenthart JFG. Structure of the monosialyl oligosaccharides derived from salivary gland mucin glycoproteins of the Chinese swiftlet (genus Collocalia) Eur. J. Biol. Chem. 1987; 262(14):6650-6657.
21. Hanisch FG, Peter-Katalinic J. Structural studies on fetal mucins from human amniotic fluid. Core typing of short-chain O-linked glycans. Eur. J. Biochem. 1992; 205(2):527-535.
22. Chai WG, Hounsell EF, Cashmore GC, Rosankiewicz JR, Bauer CJ, Feeney J, Feizi T, Lawson AM. Neutral oligosaccharides of bovine submaxillary mucin. A combined mass spectrometry and 1H-NMR study. Eur. J. Biochem. 1992; 203(1-2):257-268.
23. Van Halbeek H, Breg J, Vliegenthart JF, Klein A, Lamblin G, Roussel P. Isolation and structural characterization of low-molecular-mass monosialyl oligosaccharides derived from respiratory-mucus glycoproteins of a patient suffering from bronchiectasis. Eur. J. Biochem. 1988; 177(2):443-460.
24. Ujita M, Fukuda M. Regulation of poly-N-acetyllactosamine biosynthesis in O-glycans. Trends Glycosci. Glycotech. 2001; 13(70):177-191.
25. Liu YH, Fujitani N, Koda Y, Kimura H. Distribution of H type 1 and of H type 2 antigens of ABO blood group in different cells of human submandibular gland. J. Histochem. Cytochem. 1998; 46(1):69-76.
26. Grabenhorst E, Nimtz M, Costa J, Conradt HS. In vivo specificity of human α1,3/4-fucosyltransferases III-VII in the biosynthesis of Lewis X and Sialyl Lewis X motifs on complex-type N-glycans. Coexpression studies from BHK-21 cells together with human beta-trace protein. J. Biol. Chem. 1998; 273(47):30985-30994.
27. Javaud C, Dupuy F, Maftah A, Julien R, Petit JM. The fucosyltransferase gene family: an amazing summary of the underlying mechanisms of gene evolution. Genetica 2003; 118(2-3):157-170.
28. Bergh ML, van den Eijnden DH. Aglycon specificity of fetal calf liver and ovine and porcine submaxillary gland α-N-acetylgalactosaminide α2-6-sialyltransferase. Eur. J. Biochem. 1983; 136(1):113-118.
29. Chen TL, Chen C, Bergeron NQ, Close BE, Bohrer TJ, Vertel BM, Colley KJ. The two rat α2,6-sialyltransferase (ST6Gal I) isoforms: evaluation of catalytic activity and intra-Golgi localization. Glycobiology 2003; 13(2):109-117.
30. Carter SR, Slomiany A, Gwozdzinski K, Liau YH, Slomiany BL. Enzymatic sulfation of mucus glycoprotein in gastric mucosa. Effect of ethanol. J. Biol. Chem. 1988; 263(24):11977-11984.
31. Kuhns W, Jain RK, Matta KL, Paulsen H, Baker MA, Geyer R, Brockhausen I. Characterization of a novel mucin sulphotransferase activity synthesizing sulphated O-glycan core 1,3-sulphate-Galβ1-3GalNAcα-R. Glycobiology 1995; 5(7):689-697.
32. Lo-Guidice JM, Perini JM, Lafitte JJ, Ducourouble MP, Roussel P, Lamblin G. Characterization of a sulfotransferase from human airways responsible for the 3-O-sulfation of terminal galactose in N-acetyllactosamine-containing mucin carbohydrate chains. J. Biol. Chem. 1995; 270(46):27544-27550.
33. Grunwell JR, Rath VL, Rasmussen J, Cabrilo Z, Bertozzi CR. Characterization and mutagenesis of Gal/GlcNAc-6-O-sulfotransferases. Biochemistry 2002; 41(52):15590-15600.
34. Southworth MW, Fuhrman JA, Robbins PW, Beauregard K, Perler FB. Gene cloning and production of active recombinant Brugia malayi microfilarial chitinase. Gene 1996; 177(1-2):55- 58.
35. Hsu AF, Baynes JW, Heath EC. Role of a dolichol oligosaccharide as an intermediate in glycoprotein biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 1974; 71(6):2391-2395.
36. Cipollo JF, Trimble RB. The accumulation of Man6GlcNAc2-PP-dolichol in the Saccharomyces cerevisiae δalg9 mutant reveals a regulatory role for the Alg3p α1,3-Man middle-arm addition in downstream oligosaccharide-lipid and glycoprotein glycan processing. J. Biol. Chem. 2000; 275(6):4267-4277.
37. Cipollo JF, Trimble RB. The Saccharomyces cerevisiae δalg12 mutant reveals aroleforthe middle-arm α1,2Man- and upper-arm α1,2Manα1,6Man-residues of Glc3Man9GlcNAc2-PP-Dol in regulating glycoprotein glycan processing in the endoplasmic reticulum and Golgi apparatus. Glycobiology 2002; 12(11):749-762.
38. Oriol R, Martinez-Duncker I, Chantret I, Mollicone R, Codogno, P. Common origin and evolution of glycosyltransferases using Dol-P-monosaccharides as donor substrate. Mol. Biol. Evol. 2002; 19(9):1451-1463.
39. Helenius J, Ng DTW, Marolda CL, Walter P, Valvano MA, Aebi M. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature 2002; 415(6870):447- 450.
40. Nikonov AV, Kreibich G. Organization of translocon complexes in ER membranes. Biochem. Soc. Trans. 2003; 31(6):1253-1256.
41. Chavan M, Yan AX, Lennarz WJ. Subunits of the translocon interact with components of the oligosaccharyl transferase complex. J. Biol. Chem. 2005; 280(24):22917-22924.
42. Yan Q, Lennarz WJ. Oligosaccharyltransferase: a complex multisubunit enzyme of the endoplasmic reticulum. Biochem. Bio- phys. Res. Comm. 1999; 266(3):684-689.
43. Karamyshev AL, Kelleher DJ, Gilmore R, Johnson AE, von Heijne G, Nilsson I. Mapping the interaction of the STT3 subunit of the oligosaccharyl transferase complex with nascent polypeptide chains. J. Biol. Chem. 2005; 280(49):40489-40493.
44. Nilsson I, Kelleher DJ, Miao YW, Shao YL, Kreibich G, Gilmore R, von Heijne G, Johnson AE. Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J. Cell Biol. 2003; 161(4):715-725.
45. Imperiali B, Hendrickson TL. Asparagine-linked glycosylation: specificity and function of oligosaccharyl transferase. Bioorg. Med. Chem. 1995; 3(12):1565-1578.
46. Bause E, Schweden J, Gross A, Orthen B. Purification and characterization of trimming glucosidase-I from pig liver. Eur. J. Biochem. 1989; 183(3):661-669.
47. Tulsiani DRP, Coleman VD, Touster O. Asparagine-linked glycoprotein biosynthesis in rat brain - identification of glucosidase-I, glucosidase-II, and an endomannosidase (glucosyl mannosidase). Arch. Biochem. Biophys. 1990; 277(1):114-121.
48. Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science 2001; 291(5512):2364-2369.
49. Helenius A, Aebi M. Roles of N -linked glycans in the endoplasmic reticulum. Ann. Rev. Biochem. 2004; 73:1019-1049.
50. Tulsiani DRP, Touster O. The purification and characterization of mannosidase Ia from rat liver Golgi membranes. J. Biol. Chem. 1988; 263(11):5408-5417.
51. Lubas WA, Spiro RG. Golgi endo-Alpha-D-mannosidase from rat liver, a novel N-linked carbohydrate unit processing enzyme. J. Biol. Chem. 1987; 262(8):3775-3781.
52. Bao M, Elmendorf BJ, Booth JL, Drake RR, Canfield WM. Bovine UDP-N-acetylglucosamine: lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. II. Enzymatic characterization and identification of the catalytic subunit. J. Biol. Chem. 1996; 271(49):31446-31451.
53. Lee JK, Pierce M. Purification and characterization of human serum N-acetylglucosamine-1-phosphodiester a-N-acetylglucosaminidase. Arch. Biochem. Biophys. 1995; 319(2):413-425.
54. Altmann F. N -glycosylation in insects revisited. Trends Glycosci. Glycotech. 1996; 8(40):101-114.
55. Kobata A. Structures and functions of the sugar chains of glycoproteins. Eur. J. Biochem. 1992; 209(2):483-501.
56. Tan J, Dagostaro GAF, Bendiak B, Reck F, Sarkar M, Squire JA, Leong P, Schachter H. The human UDP-N-acetylglucosamine-α-6-D-mannoside-β-1,2-N-acetylglucosaminyltransferase-II gene (Mgat2) - cloning of genomic DNA, localization to chromosome 14q21, expression in insect cells and purification of the recombinant protein. Eur. J. Biochem. 1995; 231(2):317-328.
57. Oguri S, Minowa MT, Ihara Y, Taniguchi N, Ikenaga H, Takeuchi M. Purification and characterization of UDP-N-acetylglucosamine: α1,3-D-mannoside β1,4-N-acetylglucosaminyltransferase (N-acetylglucosaminyltransferase-IV) from bovine small intestine. J. Biol. Chem. 1997; 272(36):22721-22727.
58. Shoreibah MG, Hindsgaul O, Pierce M. Purification and Characterization of rat kidney UDP-N-acetylglucosamine-α-6-D-mannoside β-1,6-N-acetylglucosaminyltransferase. J. Biol. Chem. 1992; 267(5):2920-2927.
59. Lin AI, Philipsberg GA, Haltiwanger RS. Core fucosylation of high-mannose-type oligosaccharides in GlcNAc transferase I-deficient (Lec1) CHO cells. Glycobiology 1994; 4(6):895-901.
60. Fabini G, Freilinger A, Altmann F, Wilson IBH. Identification of core α1,3-fucosylated glycans and cloning of the requisite fucosyltransferase cDNA from Drosophila melanogaster - potential basis of the neural anti-horseradish peroxidase epitope. J. Biol. Chem. 2001; 276(30):28058-28067.
61. Sasai K, Ikeda Y, Eguchi H, Tsuda T, Honke K, Taniguchi N. The action of N-acetylglucosaminyltransferase-V is prevented by the bisecting GlcNAc residue at the catalytic step. FEBS Lett. 2002; 522(1-3):151-155.
62. Ikeda Y, Taniguchi N. Enzymatic properties and biological functions of β1,4-N-acetylglucosaminyltransferase III. Trends Glycosci. Glycotech. 2001; 13(70):167-176.
63. Grebner EE, Hall CW, Neufeld EF. Glycosylation of serine residues by a uridine diphosphate-xylose-protein xylosyltransferase from mouse mastocytoma. Arch. Biochem. Biophys. 1966; 116(1-3):391-398.
64. Schwartz NB. Xylosylation - the first step in synthesis of proteoglycan. Trends Glycosci. Glycotech. 1995; 7(37):429-445.
65. Esko JD, Zhang LJ. Influence of core protein sequence on glycosaminoglycan assembly. Curr. Opin. Struct. Biol. 1996; 6(5):663-670.
66. Okajima T, Yoshida K, Kondo T, Furnkawa K. Human homolog of Caenorhabditis elegans Sqv-3 gene is galactosyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1999; 274(33):22915-22918.
67. Almeida R, Levery SB, Mandel U, Kresse H, Schwientek T, Bennett EP, Clausen H. Cloning and expression of a proteoglycan UDP-galactose:β-xyloseβ1,4-galactosyltransferase I - A seventh member of the human β4-galactosyltransferase gene family. J. Biol. Chem. 1999; 274(37):26165-26171.
68. Kitagawa H, Tone Y, Tamura J, Neumann KW, Ogawa T, Oka S, Kawasaki T, Sugahara K. Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1998; 273(12):6615-6618.
69. Tone Y, Kitagawa H, Imiya K, Oka S, Kawasaki T, Sugahara K. Characterization of recombinant human glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. FEBS Lett. 1999; 459(3):415-420.
70. Moses J, Oldberg A, Eklund E, Fransson LA. Biosynthesis of the proteoglycan decorin - identification of intermediates in galactosaminoglycan assembly. Eur. J. Biochem. 1997; 248(3):767-774.
71. Moses J, Oldberg A, Fransson LA. Initiation of galactosaminoglycan biosynthesis - separate galactosylation and dephosphorylation pathways for phosphoxylosylated decorin protein and exogenous xyloside. Eur. J. Biochem. 1999; 260(3):879-884.
72. Sugahara K, Yamashina I, Dewaard P, Vanhalbeek H, Vliegenthart JFG. Structural studies on sulfated glycopeptides from the carbohydrate-protein linkage region of chondroitin 4-sulfate proteoglycans of swarm rat chondrosarcoma - demonstration of the structure Gal(4-O-Sulfate)-β-1-3-Gal-β-1-4Xyl-β-1-O-Ser. J. Biol. Chem. 1988; 263(21):10168-10174.
73. Lauder RM, Huckerby TN, Brown GM, Bayliss MT, Nieduszynski IA. Age-related changes in the sulphation of the chondroitin sulphate linkage region from human articular cartilage aggrecan. Biochem. J. 2001; 358(2):523-528.
74. Gulberti S, Lattard V, Fondeur M, Jacquinet JC, Mullier G, Netter P, Magdalou J, Ouzzine M, Fournel-Gigleux S. Phosphorylation and sulfation of oligosaccharide substrates critically influence the activity of human β1,4-galactosyltransferase 7 (GalT-I) and β1,3-glucuronosyltransferase I (GlcAT-I) involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 2005; 280(2):1417-1425.
75. Nadanaka S, Kitagawa H, Goto F, Tamura J, Neumann KW, Ogawa T, Sugahara K. Involvement of the core protein in the first β-N-acetylgalactosamine transfer to the glycosaminoglycan- protein linkage-region tetrasaccharide and in the subsequent polymerization: the critical determining step for chondroitin sulphate biosynthesis. Biochem. J. 1999; 340(2):353-357.
76. Uyama T, Kitagawa H, Tanaka J, Tamura J, Ogawa T, Sugahara K. Molecular cloning and expression of a second chon- droitin N-acetylgalactosaminyltransferase involved in the initiation and elongation of chondroitin/dermatan sulfate. J. Biol. Chem. 2003; 278(5):3072-3078.
77. McCormick C, Duncan G, Goutsos KT, Tufaro F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. U.S.A. 2000; 97(2):668-673.
78. Aikawa J, Esko JD. Molecular cloning and expression of a third member of the heparan sulfate/heparin GlcNAc N-deacetylase/N-sulfotransferase family. J. Biol. Chem. 1999; 274(5):2690- 2695.
79. Aikawa J, Grobe K, Tsujimoto M, Esko JD. Multiple isozymes of heparan sulfate/heparin GlcNAc N-deacetylase/GlcN N-sulfotransferase - structure and activity of the fourth member, NDST4. J. Biol. Chem. 2001; 276(8):5876-5882.
80. Humphries DE, Lanciotti J, Karlinsky JB. cDNA cloning, genomic organization and chromosomal localization of human heparan glucosaminyl N-deacetylase/N-sulphotransferase-2. Biochem. J. 1998; 332(2):303-307.
81. Pikas DS, Eriksson I, Kjellen L. Overexpression of different isoforms of glucosaminyl N-deacetylase/N-sulfotransferase results in distinct heparan sulfate N-sulfation patterns. Biochemistry 2000; 39(15):4552-4558.
82. Kusche-Gullberg M, Eriksson I, Pikas DS, Kjellen L. Identification and expression in mouse of two heparan sulfate glucosaminyl N-deacetylase/N-sulfotransferase genes. J. Biol. Chem. 1998; 273(19):11902-11907.
83. Toma L, Berninsone P, Hirschberg CB. The putative heparin-specific N-acetylglucosaminyl N-deacetylase/N-sulfotransferase also occurs in non-heparin-producing cells. J. Biol. Chem. 1998; 273(35):22458-22465.
84. Pinhal MAS, Smith B, Olson S, Aikawa J, Kimata K, Esko JD. Enzyme interactions in heparan sulfate biosynthesis: uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc. Natl. Acad. Sci. U.S.A. 2001; 98(23):12984-12989.
85. Habuchi H, Tanaka M, Habuchi O, Yoshida K, Suzuki H, Ban K, Kimata K. The occurrence of three isoforms of heparan sulfate 6-O-sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J. Biol. Chem. 2000; 275(4):2859-2868.
86. Shworak NW, Liu JA, Petros LM, Zhang LJ, Kobayashi M, Copeland NG, Jenkins NA, Rosenberg RD. Multiple isoforms of heparan sulfate D-glucosaminyl 3-O-sulfotransferase - isolation, characterization, and expression of human cDNAs and identification of distinct genomic loci. J. Biol. Chem. 1999; 274(8):5170-5184.
87. Jemth P, Smeds E, Do AT, Habuchi H, Kimata K, Lindahl U, Kusche-Gullberg M. Oligosaccharide library-based assessment of heparan sulfate 6-O-sulfotransferase substrate specificity. J. Biol. Chem. 2003; 278(27):24371-24376.
88. Smeds E, Habuchi H, Do AT, Hjertson E, Grundberg H, Kimata K, Lindahl U, Kusche-Gullberg M. Substrate specificities of mouse heparan sulphate glucosaminyl 6-O-sulphotransferases. Biochem. J. 2003; 372(2)371-380.
89. Kitagawa H, Izumikawa T, Uyama T, Sugahara K. Molecular cloning of a chondroitin polymerizing factor that cooperates with chondroitin synthase for chondroitin polymerization. J. Biol. Chem. 2003; 278(26):23666-23671.
90. Kitagawa H, Uyama T, Sugahara K. Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 2001; 276(42):38721-38726.
91. Malmstrom A, Aberg L. Biosynthesis of dermatan sulfate - assay and properties of the uronosyl C-5 epimerase. Biochem. J. 1982; 201(3):489-493.
92. Kang HG, Evers MR, Xia GQ, Baenziger JU, Schachner M. Molecular cloning and characterization of chondroitin-4-O-sulfotransferase-3 - a novel member of the HNK-1 family of sulfotransferases.J. Biol. Chem. 2002; 277(38):34766-34772.
93. Evers MR, Xia GQ, Kang HG, Schachner M, Baenziger JU. Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 2001; 276(39):36344-36353.
94. Kobayashi M, Sugumaran G, Liu JA, Shworak NW, Silbert JE, Rosenberg RD. Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan chondroitin sulfate. J. Biol. Chem. 1999; 274(15):10474-10480.
95. Nadanaka S, Fujita M, Sugahara K. Demonstration of a novel sulfotransferase in fetal bovine serum, which transfers sulfate to the C6 position of the GalNAc residue in the sequence iduronic acid α1-3GalNAc β1-4iduronic acid in dermatan sulfate. FEBS Lett. 1999; 452(3):185-189.
96. Funderburgh JL. Keratan sulfate: structure, biosynthesis, and function. Glycobiology 2000; 10(10):951-958.
97. Tai GH, Nieduszynski IA, Fullwood NJ, Huckerby TN. Human corneal keratan sulfates. J. Biol. Chem. 1997; 272(45):28227- 28231.
98. Nilsson B, Nakazawa K, Hassell JR, Newsome DA, Hascall VC. Structure of oligosaccharides and the linkage region between keratan sulfate and the core protein on proteoglycans from monkey cornea. J. Biol. Chem. 1983; 258(10):6056-6063.
99. Ziegler C, Mersmann G. Influence of effectors of the complex- type-oligosaccharide biosynthesis on the formation of proteokeratan sulfate in bovine cornea. Biochim. Biophys. Acta 1984; 799(3):203-208.
100. Plaas AHK, Wongpalms S. Biosynthetic mechanisms for the addition of polylactosamine to chondrocyte fibromodulin. J. Biol. Chem. 1993; 268(35):26634-26644.
101. Oeben M, Keller R, Stuhlsatz HW, Greiling H. Constant and variable domains of different disaccharide structure in corneal keratan sulfate chains. Biochem. J. 1987; 248(1):85-93.
102. Tai GH, Huckerby TN, Nieduszynski IA. Multiple non-reducing chain termini isolated from bovine corneal keratan sulfates. J. Biol. Chem. 1996; 271(38):23535-23546.
103. Nieduszynski IA, Huckerby TN, Dickenson JM, Brown GM, Tai GH, Morris HG, Eady S. There are 2 major types of skeletal keratan sulfates. Biochem. J. 1990; 271(1):243-245.
104. Brown GM, Huckerby TN, Abram BL, Nieduszynski IA. Characterization of a non-reducing terminal fragment from bovine articular cartilage keratan sulphates containing a (2-3)-linked sialic acid and a (1-3)-linked fucose. Biochem. J. 1996; 319(1):137-141.
105. Krusius T, Finne J, Margolis RK, Margolis RU. Identification of an O-glycosidic mannose-linked sialylated tetrasaccharide and keratan sulfate oligosaccharides in the chondroitin sulfate proteoglycan of brain. J. Biol. Chem. 1986; 261(18):8237-8242.
106. Sasaki K, Kurata-Miura K, Ujita M, Angata K, Nakagawa S, Sekine S, Nishi T, Fukuda M. Expression cloning of cDNA encoding a human β-1,3-N-acetylglucosaminyltransferase that is essential for poly-N-acetyllactosamine synthesis. Proc. Natl. Acad. Sci. U.S.A. 1997; 94(26):14294-14299.
107. Christner JE, Distler JJ, Jourdian GW. Biosynthesis of keratan sulfate - purification and properties of a galactosyltransferase from bovine cornea. Arch. Biochem. Biophys. 1979; 192(2):548- 558.
108. Zhou DP, Dinter A, Gallego RG, Kamerling JP, Vliegenthart JFG, Berger EG, Hennet T. A β-1,3-N-acetylglucosaminyltransferase with poly-N-acetyllactosamine synthase activity is structurally related to β-1,3-galactosyltransferases. Proc. Natl. Acad. Sci. U.S.A. 1999; 96(2):406-411.
109. Cai CX, Gibney E, Gordon MK, Marchant JK, Birk DE, Linsenmayer TF. Characterization and developmental regulation of avian corneal β-1,4-galactosyltransferase RNA. Exp. Eye Res. 1996; 63(2):193-200.
110. Torii T, Fukuta M, Habuchi O. Sulfation of sialyl N-acetyllactosamine oligosaccharides and fetuin oligosaccharides by keratan sulfate Gal-6-sulfotransferase. Glycobiology 2000; 10(2):203-211.
111. Akama TO, Misra AK, Hindsgaul O, Fukuda MN. Enzymatic synthesis in vitro of the disulfated disaccharide unit of corneal keratan sulfate. J. Biol. Chem. 2002; 277(45):42505-42513.
112. Akama TO, Nakayama J, Nishida K, Hiraoka N, Suzuki M, McAuliffe J, Hindsgaul O, Fukuda M, Fukuda MN. Human corneal GlcNAc 6-O-sulfotransferase and mouse intestinal GlcNAc 6-O-sulfotransferase both produce keratan sulfate. J. Biol. Chem. 2001; 276(19):16271-16278.
113. Shao L, Haltiwanger RS. O-fucose modifications of epidermal growth factor-like repeats and thrombospondin type 1 repeats: unusual modifications in unusual places. Cell. Mol. Life Sci. 2003; 60(2):241-250.
114. Chiba A, Matsumura K, Yamada H, Inazu,T, Shimizu T, Kusunoki S, Kanazawa I, Kobata A, Endo T. Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve α-dystroglycan: the role of a novel O-mannosyl-type oligosaccharide in the binding of α-dystroglycan with laminin. J. Biol. Chem. 1997; 272(4):2156-2162.
115. Samuel J, Bertozzi CR. Chemical tools for the study of polysialic acid. Trends Glycosci. Glycotech. 2004; 16(91):305-318.
116. Myllyla R, Anttinen H, Risteli L, Kivirikko KI. Isolation of collagen glucosyltransferase as a homogeneous protein from chick embryos. Biochim. Biophys. Acta 1977; 480(1):113-121.
117. Hang HC, Yu C, Ten Hagen KG, Tian E, Winans KA, Tabak LA, Bertozzi, CR. Small molecule inhibitors of mucin-type O-linked glycosylation from a uridine-based library. Chem. Biol. 2004; 11(3):337-345.
118. Tian E, Ten Hagen KG, Shum L, Hang HC, Imbert Y, Young WW, Bertozzi CR, Tabak LA. An inhibitor of O -glycosylation induces apoptosis in NIH3 T3 cells and developing mouse embryonic mandibular tissues. J. Biol. Chem. 2004; 279(48):50382- 50390.
119. Hettkamp H, Bause E, Legler G. Inhibition by nojirimycin and 1-deoxynojirimycin of microsomal glucosidases from calf liver acting on the glycoprotein oligosaccharides Glc1-3Man9GlcNAc2. Biosci. Rep. 1982; 2(11):899-906.
120. Saunier B, Kilker RD, Tkacz JS, Quaroni A, Herscovics A. Inhibition of N-linked complex oligosaccharide formation by 1-deoxynojirimycin, an inhibitor of processing glucosidases. J. Biol. Chem. 1982; 257(23):4155-4161.
121. Gross V, Steube K, Tran-Thi TA, McDowell W, Schwarz RT, Decker K, Gerok W, Heinrich PC. Secretion of high-mannose-type α1-proteinase inhibitor and α1-acid glycoprotein by primary cultures of rat hepatocytes in the presence of the mannosidase I inhibitor 1-deoxymannojirimycin. Eur. J. Biochem. 1985; 150(1):41-46.
122. Tulsiani DRP, Harris TM, Touster O. Swainsonine inhibits the biosynthesis of complex glycoproteins by inhibition of Golgi mannosidase-II. J. Biol. Chem. 1982; 257(14):7936-7939.
123. Miao HQ, Fritz TA, Esko JD, Zimmermann J, Yayon A, Vlodavsky I. Heparan sulfate primed on β-D-xylosides restores binding of basic fibroblast growth factor. J. Cell. Biochem. 1995; 57(2):173-184.
Further Reading
Taylor ME, Drickamer K. Introduction to Glycobiology. 2003. Oxford University Press, New York.
Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J. Essentials of Glycobiology. 2002. Cold Spring Harbor Laboratory Press, New York.
See Also
Glycosyltransferases, Chemistry of
Golgi Trafficking, Glycoengineering