CHEMICAL BIOLOGY
Hemes in Biology
Paul R. Ortiz de Montellano, University of California at San Francisco,
doi: 10.1002/9780470048672.wecb221
Heme (iron protoporphyrin IX) is a fundamental factor in biology with many physiologic roles. Its biosynthesis and degradation are regulated tightly because heme in excess of that required for incorporation into the available apoproteins is toxic. Heme itself is the dominant form in biology, but it is modified additionally to satisfy specific biochemical requirements. Both the biosynthesis of heme and its distribution into the various compartments of the cell and organism require the intervention of heme transporters. In contrast to the passive transport afforded by proteins like albumin, the transfer of heme across membranes is mediated by energy-dependent transporters. One role of heme is to function as a regulatory molecule whose concentration controls the translational expression of multiple genes directly. As the prosthetic group of receptors that respond to oxygen, nitric oxide (NO), and other iron ligands, it is also involved in signaling. Finally, it is the prosthetic group of diverse families of hemoproteins, including the globins, cytochromes, P450 monooxygenases, NO synthases, dioxygenases, peroxidases, peroxygenases, and catalases.
Iron is an essential element for life because its oxidation-reduction properties are in the biologically accessible range, are readily modulated, and can be used to satisfy a diversity of functions. These functions include electron transfer, ligand binding and sensing, ligand transport, and catalytic functions that involve the activation of molecular oxygen or peroxides. However, iron itself is highly insoluble in the ferric state, and in the ferrous state it supports the formation of deleterious species such as the hydroxyl radical. Therefore, nature has evolved stratagems for the chelation and encapsulation of iron that allow the exploitation of its useful features but minimize the associated toxicity. The most effective and ubiquitous of these solutions is to incorporate the iron into a porphyrin, producing heme. Incorporation of the iron into heme not only allows better control of its solubility and spatial localization, but also enables fine-tuning of its redox properties for specific tasks.
The field of hemes in biology is enormous and it is possible only to touch on the salient points in this article, which focuses primarily on the biosynthesis, degradation, transport, and regulatory role of heme. The catalytic functions of heme are diverse and fascinating, but many of the relevant enzymes are summarized only briefly here.
Heme Structures and Nomenclature
Heme (heme B) is a highly conjugated cyclic tetrapyrrole in which an iron is coordinated to the four nitrogen atoms of a protoporphyrin IX framework. In heme, four methyls, two vinyls, and two propionic acid substituents are distributed asymmetrically around the porphyrin periphery (Fig. 1). The four pyrrole rings are labeled A to D, the four meso-carbons are denoted by Greek letters, and the positions of the eight substituents are numbered. Crystallographers sometimes use a different A-D order for the four pyrrole rings. The formal IUPAC nomenclature for heme (Fig. 1) is used by organic chemists but is not often used in biology. The compound is called heme when the iron is in the ferrous state and is called hemin when it is in the ferric state, but chemists do not always adhere strictly to this distinction.
Heme is the most common biologic form, but additional elaborations of the porphyrin skeleton are required for certain purposes. Among these more baroque iron porphyrins are heme A, the structure found in cytochrome c oxidase; heme C, the prosthetic group of cytochrome c in which the covalently bound cysteines are part of the protein structure; and less common structures such as the heme D in the catalase from Escherichia coli (Fig. 2). The 5-methyl group of heme is modified additionally by the formation of an ester link to an active-site carboxyl group in most members of the CYP4 family of cytochrome P450 enzymes (1), and by both the 1-methyl and the 5-methyl groups in the mammalian peroxidases, including myeloperoxidase, lactoperoxidase, eosinophil peroxidase, and thyroid peroxidase. In myeloperoxidase, a third covalent bond is formed in addition to these ester links that links a methionine to the 2-vinyl group. Other modifications are known that tailor the heme for specific biologic functions.
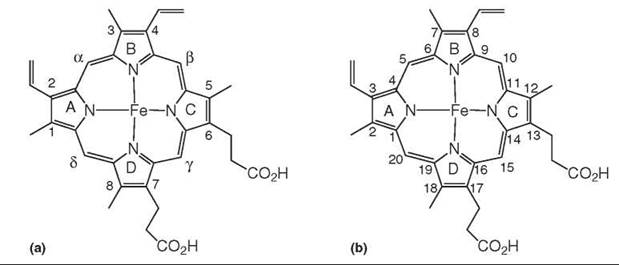
Figure 1. The structures of heme (Fe2+) and hemin (Fe3+). The substituents and pyrrole rings are numbered (A) as most commonly used in biology and (B) according to the IUPAC nomenclature convention.

Figure 2. The structures of heme A, heme C, heme D, and the modified heme found in the CYP4 family of P450 enzymes.
Heme Biosynthesis
Mammalian heme biosynthesis traverses eight enzymatic steps and requires shuttling of the first intermediate, 5-aminolevulinic acid from the mitochondrion into the cytosol, and later transferring coproporphyrinogen III back into the mitochondrion for the three final biosynthetic steps (2).
Pre-porphyrin steps
The first and rate-limiting step in heme biosynthesis is the condensation of glycine and succinyl-CoA to give 5-aminolevulinic acid by aminolevulinic acid synthase (ALAS) (Fig. 3). Two mammalian forms of this enzyme are known: ALAS1, which is ubiquitously expressed, and ALAS2, which is specifically expressed in erythroid precursors (3, 4). Both enzymes are homodimers and use pyridoxal 5-phosphate as a cofactor. Although the crystal structures of mammalian ALAS1 and ALAS2 have not been determined, the structure of a highly homologous enzyme from Rhodobacter capsulatus has been (5). The condensation reaction catalyzed by these enzymes involves the formation of an imine bond between the pyridoxal and glycine followed by deprotonation, the displacement of the CoA from succinyl-CoA, decarboxylation, and the hydrolytic release of 5-aminolevulinic acid from the pyridoxal cofactor. ALAS1 has three putative heme-binding motifs (HRMs), two in the N-terminus of its mitochondrial targeting domain and one in the N-terminus of the mature protein. Binding of heme to one or more of these HRMs inhibits mitochondrial import of the protein and thus provides end-product feedback regulation of heme biosynthesis (6). This regulation complements the direct heme-dependent downregulation of ALAS expression at the transcriptional level. Mutations in ALAS2 result in X-linked sideroblastic anemia (5).
In plants and in most bacteria, 5-aminolevulinic acid is produced by an alternative pathway involving tRNA-bound glutamate and two enzymatic steps catalyzed by glutamyl-t-RNA reductase and by glutamate-1-semialdehyde-2,1-aminomutase (7, 8).
The 5-ALA formed in mammalian mitochondria is transferred to the cytosol, where aminolevulinate dehydratase (ALAD) catalyzes the condensation of two monomers to give porphobilinogen (Fig. 3), the monopyrrole building block of the porphyrin skeleton. ALAD is a zinc-dependent tetramer of homodimers with one catalytic site per dimer (9, 10). The condensation reaction requires the formation of an enamine link, tautomerization of the enamine, closure of the five-membered ring by an aldol-like reaction, and dehydration to give the pyrrole ring. Mutations in ALAD are associated with the rare recessive autosomal ALAD porphyrias. Deficiencies in ALAD activity are also involved in hepatorenal tyrosinemia and lead poisoning, the former because the defect produces succinyl acetone that inhibits ALAD and the latter because lead displaces essential zinc ions from the protein complex.
The next stage of heme biosynthesis is the formation of the linear tetrapyrrole hydroxymethylbilane by porphibilinogen deaminase (PBGD). The crystal structure of PBGD shows that a dipyrrole cofactor is covalently within the active site of the enzyme (11). This dipyrrole is condensed with four additional porphobilinogen monomers to form a hexameric polymer. Cleavage of the tetrapyrrole unit from the dipyrrole cofactor produces hydroxymethylbilane (Fig. 3). Mutations in the PBGD gene are responsible for the disorder known as acute intermittent porphyria (12). Acute episodes of this disorder are treated by the administration of hematin (the Fe-OH complex of hemin), which leads to restoration of heme levels, the downregulation of ALAS1, and a decrease in the synthesis of toxic porphyrin precursors.

Figure 3. The synthesis of heme from glycine and succinyl-CoA. The enzymes are ALAS, 5-aminolevulinic acid (ALA) synthase; ALAD, 5-aminolevulinic acid dehydratase; PBGD, porphobilinogen deaminase; UROIIIS, uroporphyrinogen III synthase; UROD, uroporphyrinogen decarboxylase; CPO, coproporphyrinogen oxidase; PPO, protoporphyrinogen oxidase; and FECH, ferrochelatase.
Porphyrins and heme
Hydroxymethylbilane is converted by uroporphyrinogen III synthase into uroporphyrinogen III in a remarkable ring-closing reaction that inverts the orientation of pyrrole ring D of hydroxymethylbilane (Fig. 3). This ring flip exchanges the positions of the propionic and acetic acid side chains and introduces the substituent asymmetry characteristic of heme. The crystal structure has not yet provided a clear understanding of how this transformation is achieved (13). Mutations in uroporphyrinogen III synthase cause a rare disease known as congenital erythropoietic porphyria (14). This disease results in the accumulation of uroporphyrin I, the abnormal porphyrin without the pyrrole ring D flip. Uropophyrin I is formed by an abnormal, nonenzymatic closure of hydroxymethylbilane and cannot be processed further. The accumulated porphyrin acts as a photosensitizer that causes the cutaneous lesions associated with the disease.
Uroporphyrin III is the branching point in the pathway at which heme synthesis diverges from those of chlorophyll and the corrins. In heme biosynthesis, uroporphyrinogen III is next decarboxylated by uroporphyrinogen III decarboxylase to give coproporphyrinogen III (Fig. 3) (15). This enzyme promotes the decarboxylation of the four acetic acid side chains to give the methyl groups at positions 1, 3, 5, and 8 of the heme group. The decarboxylation sequence at physiologic substrate concentrations proceeds clockwise, commencing with the acetic acid side chain of ring D. A mechanism has been proposed that involves the protonation of a carbon of the pyrrole ring to be decarboxylated to give an iminium isomer that is neutralized by decarboxylation (16). Familial porphyria cutanea tarda, a disease that is also characterized by porphyrin accumulation and cutaneous photosensitivity, is caused by mutations in the uroporphyrinogen III decarboxylase gene (17).
Coproporphyrinogen oxidase decarboxylates the pyrrole ring A and B propionic acid residues of coproporphyrinogen III to produce the vinyl groups of protoporphyrinogen IX (Fig. 3). The other two propionic acid residues are retained in the final protoporphyrin IX framework. Mammals use a form of this enzyme that is oxygen-dependent and releases CO2 and H2O2, but an oxygen-independent form is found in bacteria. The crystal structures of the human and the yeast enzymes have led to both acid-base and free radical mechanistic proposals (18, 19). This decarboxylation reaction occurs in the mitochondria and thus requires the translocation of coproporphyrinogen III from the cytosol into this organelle. Hereditary coproporphyria, the disorder in humans that is caused by mutations in the coproporphyrinogen oxidase gene, is treated by the administration of hematin.
Protoporphyrinogen oxidase converts protoporphyrinogen IX to the fully desaturated porphyrin in a reaction that uses O2 as the terminal electron acceptor (Fig. 3). The crystal structure of the homodimeric enzyme shows it has one FAD per monomer, which presumably mediates the porphyrin oxidation reaction (19). Like the decarboxylation mediated by coproporphyrinogen oxidase, this reaction also occurs in the mitochondrion. Mutations in the protoporphyrinogen oxidase gene are responsible for variegate porphyria (21). Acute attacks of this disease can be effectively treated by intravenous administration of hematin.
The synthesis of heme (Fig. 1) is completed in the mitochondrion by insertion of iron into the protoporphyrin IX framework by ferrochelatase. Ferrochelatases from various organisms have been crystallized and their structures determined. The human enzyme contains one 2Fe-2S cluster in each of the two subunits of the functional dimer (22), possibly as a mechanism to link heme synthesis to iron availability. Erythropoietic protoporphyria, which is characterized by cutaneous photosensitivity, is caused by mutations in the ferrochelatase gene (23).
Additional elaboration of the heme framework
The formation of heme C requires the addition of a cysteine thiol to each of the two vinyl groups of heme B (Fig. 2). The thiol links are usually formed with a cysteine in a CXXCH sequence, but small variations of this motif are known (24). Three systems catalyze the formation of these bonds and are known imaginatively as systems I, II, and III (25). System I occurs commonly in Gram-negative bacteria and in plant mitochondria, system II in Gram-negative and Gram-positive bacteria as well as in thylakoids, and System III in fungi, invertebrates, and vertebrates. In all three systems, an addition of the cysteine to the vinyl group is mediated enzymatically, with a supporting cast of several proteins required in systems I and II but not in system III. Despite its ubiquitous nature, the catalytic or structural advantage of the heme C covalent links remains unclear (26). Interestingly, a Synechocystis hemoglobin is known in which the heme is covalently linked through a vinyl group to a histidine residue (27).
The synthesis of heme A (Fig. 2) involves the initial addition of the farnesyl moiety to the heme 2-vinyl group by heme O synthase, which generates heme O that only has this modification. In a second step, heme A synthase oxidizes the 8-methyl of heme O to an aldehyde, which generates heme A. An electron transfer mechanism (rather than double hydroxylation) has been proposed for this final biosynthetic step (28).
In contrast to the biosyntheses of heme A and heme C, which require dedicated protein catalysts, the ester groups that link methyl groups of heme B with protein carboxyl groups are generated autocatalytically. As has been clearly shown for lactoperoxidase (29) and for several CYP4 cytochrome P450 enzymes (30), the ester links are generated in the initial catalytic turnovers of the proteins in question. Investigation of the reaction details, and mimicry of the reaction in horseradish peroxidase (31), a protein that normally does not form such bonds but does upon introduction of an active site carboxylic acid residue, strongly supports a carboxyl radical/hydrogen abstraction mechanism (Fig. 4).

Figure 4. Autocatalytic formation of an ester bond between a heme methyl group and the carboxyl group of an active-site aspartate or glutamate residue. Only the ester link of the final structure is shown (see Fig. 2). The iron-nitrogen bonds are not shown for clarity.
Heme Catabolism
The only physiologic (as opposed to pathologic) mechanism for the degradation of heme in mammals is catalyzed by heme oxygenase. Analogous heme oxygenases are found in plants, bacteria, and fungi. Two heme oxygenases, known as HO1 and HO2, are present in mammals. HO1 is distributed widely, is induced by a diversity of agents, and is critical for the removal of the heme released by erythrocyte lysis and the degradation of hepatic hemoproteins. The levels of HO1 expression are regulated in a feedback manner at the transcription level by heme. HO2, although widely distributed, is concentrated in tissues such as the testes and the brain, is only induced by hormones, and has been postulated to fulfill physiologic roles beyond the simple removal of excess heme. For example, HO2 has been reported to function as an oxygen sensor that controls a potassium channel in the carotid body (32). The structures of rat and human HO1 (33, 34), and of heme oxygenases from several microorganisms (35), have been determined, but HO2 has not been crystallized. One of the significant differences between the sequences of HO1 and HO2 is the presence in HO2 of two HRMs analogous to those in 5-aminolevulinate synthase. The role of these HRMs in HO2 is unknown.
The oxidation of heme by heme oxygenase requires O2 and seven electrons. The electrons in mammals are provided by cytochrome P450 reductase, but alternative electron donors are employed in the plant and bacterial systems. However, the actual transformation is similar in all cases. The first step is hydroxylation of the meso-carbon at which the porphyrin ring will be cleaved (Fig. 5). In mammals, this position is the α-meso carbon, but alternative meso-positions can be oxidized in bacteria. The resulting α-meso-hydroxyheme is further oxidized by O2 to α-verdoheme and CO. Additional catalytic turnover produces ferric biliverdin that, after reduction of the iron, loses the iron atom and dissociates from the protein. In mammals, the biliverdin is further reduced to bilirubin by biliverdin reductase and is then excreted as a glucuronic acid conjugate. In situations where the relevant glucuronyl transferase activity is low, as in some neonates or in Criggler-Najar syndrome, bilirubin accumulates and causes jaundice or, at higher levels, causes neurological toxicity. Agents such as tin protoporphyrin IX can be used to inhibit heme oxygenase and thus to decrease the formation of biliverdin and bilirubin.
All the products of the oxidation of heme by heme oxygenase are important physiologically. Biliverdin and its reduction product bilirubin are powerful antioxidants and, at nontoxic concentrations, contribute to cellular protection. CO, the second product, also has potent biologic activities, although it is often unclear how these activities are mediated. Finally, the ferrous iron that is released causes upregulation of the iron-binding protein ferritin and can also have an antioxidant effect. These products, singly or in combination, have been shown to confer protection against oxidative injury and cellular stress. Induction of HO1, or administration of CO or bilirubin, inhibits apoptosis, whereas inhibition of HO1 stimulates apoptosis. Therefore, the inhibition of heme oxygenase may be relevant to the treatment of cancer (36). In another context, oxidized low density lipoprotein (LDL) is a key player in the development of atherosclerotic plaques, and LDL oxidation is inhibited by HO1 and its antioxidant products. The manipulation of the HO1 system is therefore of potential interest in cardiovascular disease (37). The heme oxygenases and/or their products are relevant to the amelioration of reperfusion injury, avoidance of transplant rejection, and a variety of inflammation-based disorders (38).
In bacteria, heme oxygenases play a role in iron acquisition. A greater variety is seen in the bacterial heme oxygenases than is seen in man, but the essential catalytic features are preserved and the mechanism of the oxidation is the same (35, 39). The presence of two heme oxygenases in Pseudomonas aeruginosa, one that cleaves the heme at the α-meso-carbon and the other at the 8-position, may reflect the fact that one of the two provides the tetrapyrrole for a bacterial phytochrome response regulator (40, 41). It may be desirable to generate a different regioisomer of biliverdin in the iron acquisition pathway so that it does not interfere with the regulatory system.
Under oxidative stress conditions, heme can also be degraded nonenzymatically to nonporphyrin monopyrrole and dipyrrole products, although the mechanistic details of this degradation remain obscure (42).

Figure 5. The multistep conversion of heme to α-biliverdin catalyzed by heme oxygenase. The electrons from NADPH are transferred to heme oxygenase by cytochrome P450 reductase or other electron donor proteins.
Heme Transport
Heme (hemin) at physiologic pH is relatively insoluble and is found bound to proteins or membranes except for a low concentration (<10-7 M) of so-called “labile” heme (43). Unbound heme is potentially cytotoxic and its levels are controlled tightly by feedback regulation of its biosynthesis at the level of 5-aminolevulinic acid synthase, and of its degradation by heme oxygenase, which is a heme-inducible enzyme. As heme is lipophilic, it is able to diffuse into and through cell membranes. Neverthless, the efficient trafficking of heme involves protein transporters, some of which have been identified. Transporters of heme in the circulatory system, such as albumin, can be classified simply as heme-binding proteins. However, proteins that mediate the energy-dependent transmembrane transport of heme have been identified. Subtractive suppression hybridization has led to the identification of heme-carrier protein 1 (HCP1) in the duodenum of mice (44). This protein also transports Zn protoporphyrin IX, which indicates that the iron is not essential for recognition. Interestingly, homologous proteins are found in man and other mammals. HCP1, which is upregulated in hypoxia, has 459 amino acids and is predicted to have nine transmembrane domains (44). It has a GxxSDRxGRR motif that is found in bacterial tetracycline transporters, with which it has significant (22%) similarity, but it does not have the CP motif of the HRMs in proteins such as HO2.
Heme must be exported from the mitochondrion, where the final steps of heme synthesis occur, and a system may exist to import heme into the nucleus, as diverse genes are regulated directly at the transcriptional level by heme. The transporters that mediate this trafficking of heme have not been unambiguously identified. Recent work has shown that the mitochondrial ATP-binding cassette transporter ABCB6, a homodimeric protein that is located on the outer mitochondrial membrane, is required for porphyrin uptake into the mitochondria during heme biosynthesis (45). Heme can be transported by this protein, but it transports coproporphyrinogen III more efficiently, which implicates it as the protein involved in importation of this heme precursor. A 60-kDa protein known as FLVCR, which serves as the feline leukemia virus C receptor (thus the name), has been shown to export cytoplasmic heme from developing erythroid cells and is postulated to do so to protect them from heme toxicity (46). FLVCR has the same GxxSDRxGRR motif found in HCP1. A second heme efflux protein denoted as ABCG2, also known as breast cancer resistance protein (BCRP), has been reported (47). This 70-kDa protein, a member of the ABC transporter family, has six predicted transmembrane domains and is functional as a homodimer.
Heme that is released into the blood stream by lysis of red cells, catabolism of haptoglobin-hemoglobin complexes, or other mechanisms binds to albumin (Kd = 10-8 M) and hemopexin (Kd ~ 10-12M) (48). The hemopexin-heme complex is taken up in the liver by a receptor-mediated process. A candidate for the hemopexin-heme receptor has been identified (49). Other heme-binding proteins are α-1-microglobulin and the glutathione transferases.
The iron requirements of many pathogenic microorganisms can be met by scavenging and degrading heme. Gram-negative bacteria have receptors that interact directly with hemoproteins, such as hemoglobin and hemoglobin-haptoglobin, or that interact with a hemophore excreted by the bacterium that scavenges the heme and delivers it to the cell (50). The receptors use the TonB/ExbB/ExbD complex to transport the heme through the outer membrane. The best understood of these hemophores is HasA from Serratia marcescens, for which a crystal structure is available (51). The subsequent transport of the heme is mediated by periplasmic ABC permeases that depend on heme-binding proteins. In Gram-positive bacteria, only permease-like proteins seem to be functional, but their heme uptake systems are less understood. The recent characterization of the Isd import apparatus of Staphylococcus aureus illustrates the state of the art (52). As pathogenic organisms must acquire iron from their environment for growth, inhibition of this acquisition apparatus may provide an avenue for the development of antimicrobial agents.
Heme as a Regulatory Molecule
Heme is not only a protein prosthetic group but also it controls the activities of diverse regulatory systems directly. Heme binds to and regulates histidine kinases, heme-responsive transcription factors, cyclic nucleotide phosphodiesterases, and factors such as the eIF2α heme-regulated HRI kinase. Through these interactions, heme contributes to cellular functions such as cell growth and differentiation, oxygen and nitric oxide sensing, cell respiration, and globin gene activation. Furthermore, as illustrated by its control of heme synthesis through binding to ALAS1 and inhibition of its migration into the mitochondrion, it can influence protein maturation directly.
S. cerevisiae, a facultative aerobe, responds to changing oxygen levels by altering the expression of a battery of genes. This coordinated response to oxygen levels in S. cerevisiae is mediated by the heme activator protein Hap1 (53). The activation of Hap1 increases in parallel with the concentration of heme and reaches maximum activation at micromolar heme concentrations. The binding of heme allows Hap1 to bind to regulatory motifs in nuclear DNA and, thus, allows it to promote the transcription of genes encoding proteins that are essential for respiration and the control of oxidative damage. A repressor (ROX1) of genes involved in anaerobic growth is also regulated by Hapl. A zinc cluster and a dimerization domain are involved in its binding to DNA, whereas seven HRMs are involved in its response to heme.
In mammals, Bach 1 is a heme-activated leucine zipper protein that acts as a transcriptional repressor (54). Bach1 associates with members of the Maf-related oncogene family. The resulting heterodimers bind to the Maf recognition element of target genes, including HO1, globin genes, and ALAS2. Bach1 has six HRMs that are critical for regulation of its activity by heme. Heme binding seems to act as a signal for nuclear export of the protein, so that heme regulation is partially determined by control of Bach1 nuclear localization. A concentration of ~1 μM heme almost completely inhibits the binding of the Bachl-MafK dimer to DNA.
The maturation of reticulocytes to erythrocytes requires a massive synthesis of heme to enable the assembly of hemoglobin. It is important to couple the synthesis of heme to that of the hemoglobin α-chains and β-chains, as both the unassembled apoproteins and excess heme are toxic to the cell. The HRI kinase is responsible for coordinating heme and protein synthesis in reticulocytes. HRI inhibits globin synthesis at the translation initiation level under limited heme conditions. Under these conditions, HRI is activated and phosphorylates Ser51 of the eIF2 α-subunit. The phosphorylated protein binds tightly to eIF2B and ties up this protein, preventing it from catalyzing the replacement of GDP by GTP in eIF2α required for its role in initiating protein synthesis (55). HRI has three HRMs, one of which binds heme reversibly and is thought to couple the activity of the protein to the heme concentration and another that binds heme stably and is thought to be involved in the response of HRI to gases: NO increases HRI activity, whereas CO decreases it. The molecular chaperones Hsp90 and Hsc70 play important roles in this process, as immature HRI has no activity but becomes responsive to heme deprivation when transiently complexed to the chaperones.
Neuronal differentiation has been found in a model system to be heme-dependent (56). Inhibition of heme synthesis at the level of ALAS reduces the number and the length of neurites induced by nerve growth factor (NGF), and this effect can be overcome by the addition of exogenous heme. These consequences of heme deficiency reflect the associated inactivation of the NGF-dependent Ras-ERK 1/2 signaling pathway.
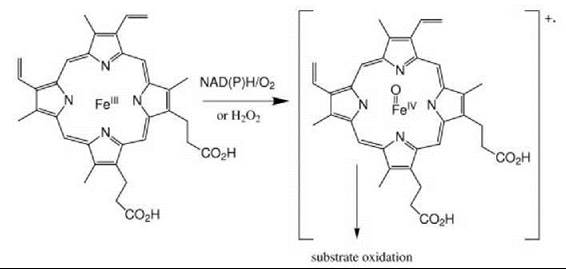
Figure 6. The most common paradigm for hemoprotein-catalyzed substrate oxidation involves heterolytic scission of the O-O bond of an iron-bound peroxo species to give an Fe(IV) = O ferryl intermediate and either a porphyrin radical or a protein radical. The peroxo intermediate is generated in the cytochromes P450 by in situ NAD(P)H-dependent reduction of O2, and in the peroxidases by reaction with H2O2.
Heme as an Enzyme Prosthetic Group
Heme is a ubiquitous prosthetic group in proteins with oxidation- reduction functions. The proteins that incorporate heme as an essential component include respiratory proteins, such as cytochrome c and cytochrome c oxidase. The oxygen carriers myoglobin and hemoglobin are members of a second class of hemoproteins that have some relationship to gas sensor hemoproteins, as represented for NO by guanylate cyclase; for O2 by Hap1, FixL, and HemAT; and for CO by CooA. The largest class of hemoproteins is made up of catalytic enzymes and includes the cytochromes P450, peroxidases, peroxygenases, catalases, NO synthases, prostaglandin synthases, and thromboxane/prostacyclin synthases.
A general scheme of the catalytic manifold of hemoproteins is outlined in Fig. 6. In this abbreviated mechanism, the reaction of the ferric heme with H2O2, or with oxygen and two electrons derived from NAD(P)H, produces a “Compound I” ferryl (FeIV = O) intermediate. This intermediate can catalyze a single two-electron oxidation (monooxygenase or peroxygenase activity) or two one-electron oxidations (peroxidase activity) (57, 58). In addition to the dominant role of the ferryl intermediate in hemoprotein catalysis, oxidations can also be mediated by intermediates that precede the ferryl species. For example, the iron peroxy anion (Fe-OO-), a precursor of the ferryl intermediate, is thought to be involved in the carbon-carbon bond cleaving reactions of cytochrome P450 enzymes and the formation of NO by NO synthases. The function of these and other catalytic hemoproteins, all of which illustrate the virtuoso catalytic power and flexibility of the heme group, are reported in more detail in other chapters of this volume.
References
1. Colas C, Ortiz de Montellano PR. Autocatalytic radical reactions in physiological prosthetic heme modification. Chem. Rev. 2003; 103:2305-2332.
2. Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim. Biophys. Acta. 2006; 1763:723-736.
3. Cotter PD, Willard HF, Gorski JL, Bishop DF. Assignment of human erythroid delta-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; autosome translocations. Genomics 1992; 13:211-212.
4. Cox TC, Bawden MJ, Abraham NG, Bottomley SS, May BK, Baker E, Chen LZ, Sutherland GR. Erythroid 5-aminolevulinate synthase is located on the X chromosome. Am. J. Hum. Genet. 1990; 46:107-111.
5. Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005; 24:3166-3177.
6. Lathrop JT, Timko MP. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science 1993; 259:522-525.
7. Hungerer C, Weiss DS, Thauer RK, Jahn D. The hemA gene encoding glutamyl-tRNA reductase from the archaeon Methanobacterium thermoautotrophicum strain Marburg. Bioorg. Med. Chem. 1996; 4:1089-1095.
8. Hennig M, Grimm B, Contestabile R, John RA, Jansonius JN. Crystal structure of glutamate-1-semialdehyde aminomutase: an alpha 2-dimeric vitamin B6-dependent enzyme with asymmetry in structure and active site reactivity. 1997; 94:4866-4871.
9. Erskine PT, Norton E, Cooper JB, Lambert R, Coker A, Lewis G, Spencer P, Sarwar M, Wood SP, Warren MJ, Shoolingin-Jordan PM. X-ray structure of 5-aminolevulinic acid dehydratase from Escherichia coli complexed with the inhibitor levulinic acid at 2.0 A resolution. Biochemistry 1999; 38:4266-4276.
10. Frankenberg N, Erskine PT, Cooper JB, Shoolingin-Jordan PM, Jahn D, Heinz DW. High resolution crystal structure of a Mg2+-dependent porphobilinogen synthase. J. Mol. Biol. 1999; 289:591-602.
11. Helliwell JR, Nieh YP, Habash J, Faulder PF, Raftery J, Cianci M, Wulff M, Hadener A. Time-resolved and static-ensemble structural chemistry of hydroxymethylbilane synthase. Faraday Discuss. 2003; 122:131-144; discussion 171-190.
12. Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 2005; 142:439-450.
13. Mathews MA, Schubert HL, Whitby FG, Alexander KJ, Schadick K, Bergonia HA, Phillips JD, Hill CP. Crystal structure of human uroporphyrinogen III synthase. EMBO J. 2001; 20:5832-5839.
14. Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Ceaudet AL, Sly WS, Valle D, Vogelstein B, Childs B, eds. 2001. McGraw-Hill, New York. pp. 2991-3062.
15. Whitby FG, Phillips JD, Kushner JP, Hill CP. Crystal structure of human uroporphyrinogen decarboxylase. EMBO J. 1998; 17:2463-2471.
16. Martins BM, Grimm B, Mock HP, Huber R, Messerschmidt A. Crystal structure and substrate binding modeling of the uroporphyrinogen-III decarboxylase from Nicotiana tabacum. Implications for the catalytic mechanism. J. Biol. Chem. 2001; 276:44108-44116.
17. Elder GH. Genetic defects in the porphyrias: types and significance. Clin. Dermatol. 1998; 16:225-233.
18. Lee DS, Flachsova E, Bodnarova M, Demeler B, Martasek P, Raman CS. Structural basis of hereditary coproporphyria. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:14232-14237.
19. Phillips JD, Whitby FG, Warby CA, Labbe P, Yang C, Pflugrath JW, Ferrara JD, Robinson H, Kushner JP, Hill CP. Crystal structure of the oxygen-dependant coproporphyrinogen oxidase (Hem13p) of Saccharomyces cerevisiae. J. Biol. Chem. 2004; 279:38960-38968.
20. Koch M, Breithaupt C, Kiefersauer R, Freigang J, Huber R, Messerschmidt A. Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. EMBO J. 2004; 23:1720-1728.
21. Whatley SD, Puy H, Morgan RR, Robreau AM, Roberts AG, Nordmann Y, Elder GH, Deybach JC. Variegate porphyria in 41. Western Europe: identification of PPOX gene mutations in 104 families, extent of allelic heterogeneity, and absence of correlation between phenotype and type of mutation. Am. J. Hum. Genet. 1999; 65:984-994.
22. Wu CK, Dailey HA, Rose JP, Burden A, Sellers VM, Wang BC. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 2001; 8:156-160.
23. Went LN, Klasen EC. Genetic aspects of erythropoietic protoporphyria. Ann. Hum. Genet. 1984; 48:105-117.
24. Stevens JM, Daltrop O, Allen JW, Ferguson SJ. C-type cytochrome formation: chemical and biological enigmas. Acc. Chem. Res. 2004; 37:999-1007.
25. Kranz R, Lill R, Goldman B, Bonnard G, Merchant S. Molecular mechanisms of cytochrome c biogenesis: three distinct systems. Mol. Microbiol. 1998; 29:383-396.
26. Barker PD, Ferguson SJ. Still a puzzle: why is haem covalently attached in c-type cytochromes? Structure 1999; 7:R281-R290.
27. Hoy JA, Kundu S, Trent JT 3rd, Ramaswamy S, Hargrove MS. The crystal structure of Synechocystis hemoglobin with a covalent heme linkage. J. Biol. Chem. 2004; 279:16535-16542.
28. Brown KR, Brown BM, Hoagland E, Mayne CL, Hegg EL. Heme A synthase does not incorporate molecular oxygen into the formyl group of heme A. Biochemistry 2004; 43:8616-8624.
29. Colas C, Kuo JM, Ortiz de Montellano PR. Asp-225 and Glu-375 in autocatalytic attachment of the prosthetic heme group of lactoperoxidase. J Biol Chem. 2002; 277:7191-7200.
30. Hoch U, Ortiz De Montellano PR. Covalently linked heme in cytochrome P4504A fatty acid hydroxylases. J. Biol. Chem. 2001; 276:11339-11346.
31. Colas C, Ortiz de Montellano PR. Horseradish peroxidase mutants that autocatalytically modify their prosthetic heme group: insights into mammalian peroxidase heme-protein covalent bonds. J. Biol. Chem. 2004; 279:24131-24140.
32. Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemeoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science 2004; 306:2093-2097.
33. Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos T. Crystal structure of human heme oxygenase-1. Nature Struct. Biol. 1999; 6:860-867.
34. Sugishima M, Omata Y, Kakuta Y, Sakamoto H, Noguchi M, Fukuyama K. Crystal structure of rat heme oxygenase-1 in complex with heme. FEBS Lett. 2000; 471:61-66.
35. Unno M, Matsui T, Ikeda-Saito M. Structure and catalytic mechanism of heme oxygenase. Nat. Prod. Rep. In press.
36. Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Kunzli B, Autschbach F, Meuer S, Buchler MW, Friess H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 2005; 11:3790-3798.
37. Morita T. Heme oxygenase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005; 25:1786-1795.
38. Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 2006; 86:583-650.
39. Frankenberg-Dinkel N. Bacterial heme oxygenases. Antioxid. Redox. Signal. 2004; 6:825-834.
40. Ratliff M, Zhu W, Deshmukh R, Wilks A, Stojiljkovic I. Homologues of neisserial heme oxygenase in gram-negative bacteria: degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J. Bacteriol. 2001; 183:6394-6403.
41. Tasler R, Moises T, Frankenberg-Dinkel N. Biochemical and spectroscopic characterization of the bacterial phytochrome of Pseudomonas aeruginosa. FEBS J. 2005; 272:1927-1936.
42. Nagababu E, Rifkind JM. Heme degradation by reactive oxygen species. Antioxid. Redox. Signal. 2004; 6:967-978.
43. Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000; 28:289-309.
44. Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT. Identification of an intestinal heme transporter. Cell 2005; 122:789-801.
45. Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG, Schuetz JD. Identification of a mammalian mitochondrial porphyrin transporter. Nature 2006; 443:586-589.
46. Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004; 118:757-766.
47. Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, Sarkadi B, Sorrentino BP, Schuetz JD. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004; 279:24218- 24225.
48. Hrkal Z, Vodrazka Z, Kalousek I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur. J. Biochem. 1974; 43:73-78.
49. Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005; 106:2572-2579.
50. Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Annu. Rev. Microbiol. 2004; 58:611-647.
51. Arnoux P, Haser R, Izadi-Pruneyre N, Lecroisey A, Czjzek M. Functional aspects of the heme bound hemophore HasA by structural analysis of various crystal forms. Proteins 2000; 41:202-210.
52. Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Passage of heme-iron across the envelope of Staphylococcus aureus. Science 2003; 299:906-909.
53. Mense SM, Zhang L. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006; 16:681-692.
54. Kitamuro T, Takahashi K, Ogawa K, Udono-Fujimori R, Takeda K, Furuyama K, Nakayama M, Sun J, Fujita H, Hida W, Hattori T, Shirato K, Igarashi K, Shibahara S. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003; 278:9125-9133.
55. Chen JJ, London IM. Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem. Sci. 1995; 20:105-108.
56. Zhu Y, Hon T, Ye W, Zhang L. Heme deficiency interferes with the Ras-mitogen-activated protein kinase signaling pathway and expression of a subset of neuronal genes. Cell Growth Differ. 2002; 13:431-439.
57. Ortiz de Montellano PR, De Voss JJ. Substrate oxidation by cytochrome P450 enzymes. In: Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed. Ortiz de Montellano Paul R, ed. 2005. Kluwer Academic/Plenum Publishers, New York.
58. Berglund GI, Carlsson GH, Smith AT, Szoke H, Henriksen A, Hajdu J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002; 417:463-468.
Further Reading
Latunde-Dada GO, Simpson RJ, McKie AT. Recent advances in mammalian haem transport. Trends Biochem. Sci. 2006; 31:182-188.
Mense SM, Zhang L. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006; 16:681-692.
Tsiftsoglou AS, Tsamadou AI, Papadopoulou LC. Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006; 111:327-345.
See Also
Hemoglobin and Myoglobin, Chemistry of
Metal Transport Through Membranes
Metalloenzymes, Chemistry of
Nitric Oxide Signaling
Oxygen Activating Enzymes, Chemistry of
Sensing and Adapting, Bacterial Mechanisms for