CHEMICAL BIOLOGY
Human Hemoglobin: Identification of a Key Intermediate
Gary K. Ackers and Jo M. Holt, Department of Biochemistry & Molecular Biophysics, Washington University School of Medicine, St. Louis, Missouri
doi: 10.1002/9780470048672.wecb222
Human hemoglobin binds four oxygen molecules with positive cooperativity in a binding cascade of eight partially ligated intermediates whose oxygen ligands are distributed in different combinations between the hemoglobin's two αβ dimers. Traditionally, it had been assumed that, given their close association in forming the hemoglobin tetramer, the two αβ dimers would always respond to oxygen binding in a synchronous and symmetric manner. Using linkage thermodynamics, the intermediate binding constants are evaluated via dimer-tetramer assembly with ligand configurations within the tetramer fixed through the use of hemesite analogs. It is observed that the free energy contribution of the asymmetrically ligated intermediate composed of one fully oxygenated αβ dimer plus one unoxygenated dimer is not the same as other doubly ligated intermediates, which contain at least one bound oxygen on both dimers. Therefore, the dimers within the tetramer respond to oxygenation differently, and cooperativity is dependent on the distribution of ligands between the two αβ dimers.
Introduction
The structural and functional properties of human hemoglobin (Hb) have been the subject of study for decades, stimulated by the intriguing characteristic of positive cooperativity. How do the four subunits that compose the Hb tetramer communicate with one another? The answer to this question has been sought primarily through the comparison of deoxy with oxy Hb. However, to understand the molecular mechanism of a chemical reaction, it is necessary to characterize the intermediate(s) of the process, and the reaction of Hb with O2 is no exception.
The binding of four O2 ligands by human Hb occurs through a series of 14 partially ligated intermediates, of which eight are unique in their combinatorial arrangement of bound O2 among the two α-subunits and two β-subunits. The well-known sigmoidal binding curve that results (see Fig. 2 for an example) is indicative of a strong positive cooperativity of oxygenation, thus, the binding constants for each Hb intermediate are changing as the O2 binding process continues.
The individual microscopic binding constants cannot, however, be measured from the binding curve: Only four average, macroscopic binding constants can be directly observed. This result is a result of several factors, foremost of which is the high cooperativity of O2 binding itself, which suppresses the concentrations of the intermediates. Thus, the binding curve is dominated by the properties of the two end-states, i.e., the fully deoxygenated tetramer and its fully oxygenated counterpart. Other factors that contribute to the low resolution of the binding curve are the lability of the bound O2 and the continuous dissociation of the tetramer to its constituent αβ dimers. Therefore, in a system that binds O2 close to equilibrium to begin with, the rearrangement of bound O2 among the heme binding sites acts only to mask the individual properties additionally (such as a microscopic binding constant) of a given intermediate.
This classic problem of disproportionation can be solved experimentally through the use of hemesite analogs that either block O2 binding or O2 dissociation in specific subunits (α1, β1, α2, β2) within the tetramer (1). The dissociation of tetramer to free dimer, and the resulting dimer rearrangement among tetramers, cannot be blocked but can be measured. The dimer→tetramer assembly free energy, ∆Gasm, can then be applied as a constraint that permits the determination of the Gibbs free energy, ∆Gij, of each intermediate binding reaction by employing thermodynamic linkage analysis.
The microscopic O2 binding constants thus determined reveal a particularly strong energetic coupling between the subunits within each αβ dimer of the tetramer (2). This intradimer cooperativity is evident particularly in the intermediate composed of one oxy dimer and one deoxy dimer, or the asymmetric doubly ligated species. Identification of this intermediate provided the first direct experimental evidence of intradimer cooperativity, which challenged the commonly held two-state model of cooperativity, in which the two dimers within the tetramer are assumed to maintain the same structural and energetic properties throughout the binding process. Rather than maintaining dimer-dimer symmetry, the αβ dimers each exhibit a unique O2 affinity and continue to modulate the O2affinity of each other.
Background
Since the determination of its crystal structure almost five decades ago, the study of cooperativity and allostery in human Hb was focused primarily on the properties of the two end-states, the deoxy and oxy tetramers. The approach to mechanistic questions of subunit-subunit coupling within the tetramer is now shifting to the characterization of the partially ligated intermediates. Although crystal structures are not yet available for the intermediates, their individual O2 binding constants are now determined for one set of solution conditions.
Structural elements of the hemoglobin tetramer
The human hemoglobin tetramer is composed of two types of polypeptide chains, designated a (with 141 amino acid residues) and p (with 146 residues). Both subunit types exhibit a high degree of a-helical content with no p structure, and each contains a noncovalently associated b-type Fe heme to which O2 binds. As a tetramer, the four subunits are organized structurally as two aP dimers held together by a polar, water-filled dimer-dimer interface (Fig. 1) (3, 4). Although the dimer-dimer interface dissociates readily under physiologic conditions to produce free aP dimers, the intradimer interface is hydrophobic and only dissociates appreciably in the presence of certain metal ions or under denaturing conditions. Therefore, the aP dimers are shared constantly and redistributed among the tetramers.
When the Hb tetramer binds O2, a large change in quaternary structure is observed in which the two aP dimers reorient relative to one another. From the deoxy or T structure, this reorientation can occur in either one of two major forms, which yields the R or the R2 structure. The R structure is observed by crystallization of oxyHb under high salt conditions, whereas the R2 structure is observed in low salt crystals. Nuclear magnetic resonance analysis has demonstrated that the R and R2 structures can coexist in solution (5). Additional crystal structure conditions have revealed that multiple oxy or R as well as deoxy or T forms are possible (6-8). All structural forms of the tetramer are ligated symmetrically (or unligated); i.e., the two dimers within the tetramer are always observed as structurally equivalent in available crystal structures.
Structural changes that take place in the αβ dimers themselves are referred to in the Hb literature as “tertiary” and include the movement of the heme Fe into the plane of the heme when oxygenation begins; subsequent movement of helices are close to the heme and to the dimer-dimer interface. A significant structural change in the intradimer interface is not observed in crystal structures, which has led to the conclusion that oxygenation-induced tertiary structural changes are not communicated between the subunits within a dimer, i.e., between α1 and β1 or between α2 and β2. Although this belief has spanned the course of several decades, more subtle structural changes in the intradimer interface have not been ruled out. Recently, Arnone et al. have pointed out that the intradimer structure has not been analyzed thoroughly in modern crystal structures of Hb (6). Very few attempts to crystallize the partially ligated intermediates of Hb have been reported, and structural information is still not available.
When O2 binding occurs, the quaternary reorientation of the two dimers forms the primary basis for the popular two-state model of Hb cooperativity. In this model, O2 binding to a heme Fe causes significant structural change only in the dimer-dimer interface. The bonds of the deoxy or T interface, held by the model to be significantly stronger than those of the oxy or R interface, maintain the tetramer in the low-affinity conformation. Oxygenation of deoxy Hb causes bonds in the T dimer-dimer interface to break, which weakens the low-affinity T state relative to the high-affinity R state. Therefore, the O2 affinity of the tetramer is controlled by the strength of the dimer-dimer interface, as modulated by the number of O2 ligands bound to the subunits. The particular configuration of the bound O2 among the four hemesites is not significant in this model. For example, in a tetramer that bears two bound O2, six possible configurations of the bound ligands exist among the four hemesites (see Fig. 1). In the two-state model, all six of the doubly ligated tetramers have the same O2 affinity because the presence of two ligands results in the same number of bonds broken in the dimer-dimer interface, regardless of the exact distribution of the ligands among the four hemesites.

Figure 1. The cascade of O2 binding to the four subunits of the human Hb tetramer. The polar dimer-dimer interface is composed of α1β2 plus α2β1 contacts. Two intradimer interfaces exist, the α1β1 and the α2β2; both are nonpolar. Each tetramer is assigned a species designation, which begins with deoxy Hb (species 01) and ending with oxy Hb (species 41). The first O2 can bind to any one of four subunits; however, because oxygenation of the a 1 subunit is indistinguishable from oxygenation of the a 2 subunit, the two isomeric tetramer species that result are designated "11a" and "11b." This labeling is likewise the case for the p subunits. Similar isomeric oxygenation microstate tetramers are also generated in the second and third binding steps. Crystal structures are from the Arnone laboratory 14, 15.
Relationship of macroscopic to microscopic binding constants
Oxygen-binding curves can be analyzed directly to yield four macroscopic binding constants: K1, K2, K3, and K4. Usually, the macroscopic constants are defined as product constants, i.e., products of the stepwise macroscopic constant Ki→i+1 (where i is the number of bound O2):
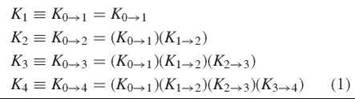
The constant Ki→i+1 is composed of microscopic constants, as each O2 binding step is composed of multiple microscopic reactions, which is illustrated by the reaction arrows in Fig. 1. Thus, 4 ways exist to bind the first O2, 12 ways to bind the second O2, 12 ways to bind the third O2, and 4 ways to bind the fourth O2. Each microscopic constant is designated by the notation ij of the species formed in the binding process (Fig. 1, Table 1). For each binding step i = i,2,3, and 4, the macroscopic constant Ki→i+1 represents the average of the microstate constants kij→(i+1)j, with accompanying statistical factors that account for the different isomeric forms of the microstate tetramers, as shown in Table 1.
Table 1. Relationship between macroscopic and microscopic O2
binding constants
Any scenario in which the tetramer is assumed to maintain symmetry between the two dimers throughout the O2 binding process (such as the historic two-state model or models with multiple T and R forms) requires that the microscopic stepwise binding constants for each binding step be equal to one another:
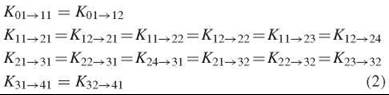
An exception to these equalities can be made to accommodate inherently different O2 affinities of α- and β-subunits; however, because differences between α- and β-subunit binding constants are not significant in normal human Hb, this variation is not additionally considered here.
Chemistry
The dimers tetramer assembly constant K asm is very sensitive to O2 binding by human Hb, which ranges over i30,000-fold among the intermediates, in comparison with the 4oo-fold change in O2 binding constant under conditions of this study (pH 7.4, 2i.5°C) (9). The equilibrium between free dimer and assembled tetramer is an integral property of Hb in solution, and has a marked impact on the O2 binding curve observed experimentally.
The concentration-dependent oxygen isotherm
The sigmoidal shape of the O2 binding isotherm, i.e., the cooperativity of O2 binding, is dependent on the concentration of the Hb solution (Fig. 2). As the solution is diluted, the relative concentration of free αβ dimers increases, and unlike the tetramer, the free dimer binds O2 noncooperatively with high affinity. Thus, the true tetramer-binding curve is observed only at the highest Hb concentrations: At lower concentrations, the experimental isotherm reports a mixture of tetramer and free dimer (1).
The thermodynamic scheme that links the ligation of the free dimer to the ligation of the assembled tetramer (Fig. 2) shows all reaction equilibriums that contribute to the concentration- dependent isotherms. Binding to the free dimer is designated by ∆Gint, which denotes the change in intrinsic (noncooperative) free energy. To solve the linkage scheme experimentally, the assembly free energy change for the deoxy tetramer, 0∆Gasm, is determined in an independent kinetic measurement using haptoglobin trapping of the free dimer. In addition, the corresponding 4∆Gasm (for the oxy tetramer) is measured independently by large-zone size-exclusion chromatography (1).
|
Overall binding step |
Microstate binding step |
Average microstate binding constants |
Macro Ki→i+1 Micro kij→(i+1)j |
|
0 → 1
1 → 2
2 → 3
3 → 4
|
01 → 11 01 → 12 11 → 21 12 → 21 11 → 22 12 → 22 11 → 23 12 → 24 21 → 31 22 → 31 24 → 31 21 → 32 22 → 32 23 → 32 31 → 41 32 → 41
|
|
|


Each binding step is composed of multiple microstate binding steps, and each microstate binding step represents an average of isomeric forms of the microstates, yielding the average microstate binding constants. The macrostate binding constant is then the average of the binding constant for each microstate.
The macroscopic thermodynamic linkage scheme
The concentration dependence of the O2 binding curve is a result of thermodynamic linkage between O2 binding and dimers→tetramer assembly. Consider the first binding step as illustrated in the linkage scheme in Fig. 2. Conservation of free energy dictates that the change in free energy during assembly followed by ligation must equal the change in free energy during ligation followed by assembly:
![]()
Therefore, the change in the tetramer assembly free energy during O2 binding is equal to the change in the O2 binding constant during tetramer assembly:
![]()
Each stepwise microscopic binding reaction follows the same formula, as thermodynamic linkage holds for all binding steps.

Figure 2. The dependence of O2 binding on Hb concentration. Binding curves are shown (solid black lines) for Hb concentrations of 0.005, 0.04, 0.10, 0.27, 1.0, 5.4, and 38 μM (from left to right). Theoretical binding curves (broken red lines) are shown for a pure tetramer solution and a pure dimer solution. The macroscopic, thermodynamic linkage scheme relates the dimer → tetramer assembly constants to the O2 binding constants for free dimer and assembled tetramer. The brackets around figurines indicate that the O2 ligand may be bound at any one of the available deoxy hemesites. Thus, the macroscopic constants are average values for multiple microscopic processes.
Forming hybrid tetramers from parent tetramers
The equilibrium between free dimer and tetramer can be exploited to provide a means of forming partially ligated hybrid tetramers by mixing any two-parent tetramers. However, it is necessary to fix the hemesite ligand to prevent disproportionation caused by ligand rearrangement among the hemesites. Hemesite analogs employed for either the deoxy heme (which replaces Fe2+) or the oxy heme (which replaces Fe2+O2) are:
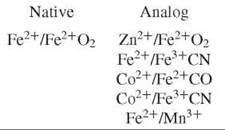
Each hemesite analog perturbs the Hb tetramer in some manner: The Fe3+ CN and Mn3+ analogs are susceptible to electron exchange over very long incubation periods (10). The Co2+ analog exerts a specific effect on α-subunit binding constants (11), and the use of Zn2+ imparts a light sensitivity to the solution (12). However, the relative relationship between each measured microstate-binding constant is found to be invariant among the analog species (9).
Using the Zn2+/Fe2+O2 analog as an example, when deoxy ZnHb (species 01) is mixed with an equimolar amount of native FeHb (species 41), a mixture is formed that contains the asymmetrically doubly ligated species 21 (Fig. 3a). Likewise, species 11 or 12 are formed by mixing species 01 with 23 or 24, respectively (see Fig. 1 for illustrations of each species). Species 22 is formed by mixing species 23 with 24. And species 31 or 32 are formed by mixing species 41 with 24 or 23, respectively. In this way, all possible combinatorial forms of the partially ligated intermediates can be formed. Only the parent tetramers 01, 23, 24, and 41 are present in pure form in solution: All other species are present in equilibrium with their respective parent tetramers.
Assembly of tetramers from free dimers occurs very rapidly with a rate constant kon of 1.1 ± 0.1 x 106 M-1s-1. This assembly is referred to as the “consensus on constant,” which is not dependent on the number of bound ligands or their configuration, the presence of hemesite analogs, or the presence of mutations. Therefore, it is in the tetramer → free dimer dissociation constant, koff, that the sensitivity of the assembly constant is manifest, because
![]()
Key Experiments and Observations
Two experimental approaches are taken to measure the assembly free energy of partially ligated Hb intermediates: an equilibrium method and a kinetic protocol. In the equilibrium method, symmetrically ligated tetramers are mixed to generate asymmetrically ligated hybrid tetramers. Then, the relative stability of the hybrid to its parents is measured, which permits the hybrid assembly free energy to be calculated from the independently measured ij∆Gasm of the parents. In the kinetic approach, the tetramers → dimer dissociation constant is measured by trapping free dimers kinetically with the plasma protein haptoglobin.
Assembly free energy of hybrid tetramers
Low-temperature isoelectric focusing
Species 21 represents a unique halfway point in oxygenation of Hb in that one of its dimers is fully ligated and the other is fully deoxygenated. The 21 hybrid is formed in vitro by mixing species 01 and 41, as in the example in Fig. 3. One of the two parent Hbs carries an electrophoretic tag to enhance separation based on charge, typically the HbS variant (β6 Glu→Val). At equilibrium, the assembly free energy of species 21 is related to the assembly free energies of each parent by

where δ21 is the free energy deviation from the average of the parent tetramer assembly free energies. The deviation free energy is measured directly from the fraction of each tetramer at equilibrium:
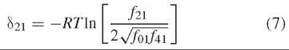
The relative fractions of hybrid and parent tetramers are measured by quenching the dissociation of tetramer to free dimers with low temperature (-25° to —35°C). Electrophoretic focusing is then conducted at the same low temperature to maintain a quenching environment.
The equilibrium population of species 21 lies far from the average of the two parents, which would be observed as a 1:2:1 binomial distribution and 48% of the mixture as hybrid. Instead, species 21 is observed at only 3% of the hybrid mixture (Fig. 3), which translates to a deviation free energy of 2.0 kcal/mol and an assembly free energy 21∆Gasm of —9.2 ± 0.2 kcal/mol (10). In contrast, the assembly free energy of species 22, which is also determined by low-temperature isoelectric focusing, was measured at —7.7 ± 0.3 kcal/mol, a value similar to the other doubly ligated species 23 and 24. The results from low-temperature isoelectric focusing for the remaining partially ligated intermediates yielded the following pattern of microscopic binding constants for each binding step: For the first binding step, ![]() for the second binding step,
for the second binding step, ![]()
![]() for the third step,
for the third step, ![]()
![]() and for the final step,
and for the final step, ![]()
![]() These results show that binding two ligands to the same dimer within a tetramer occurs with a greater positive cooperativity than binding one ligand to each dimer. This distinction between the distribution of O2 ligands within the tetramer does not agree with Equation 2, which demonstrates that one of the basic tenets of all symmetric (multistate or two-state) models of cooperativity is not verified by experiment.
These results show that binding two ligands to the same dimer within a tetramer occurs with a greater positive cooperativity than binding one ligand to each dimer. This distinction between the distribution of O2 ligands within the tetramer does not agree with Equation 2, which demonstrates that one of the basic tenets of all symmetric (multistate or two-state) models of cooperativity is not verified by experiment.

Figure 3. Hybridization of deoxy and oxy Hb to form the asymmetrically ligated Hb species 21. (a) Each parent tetramer is in equilibrium with two free dimers. The free dimers may reassemble to the original parent tetramer or may assemble with one another to form the hybrid. The oxy Hb tetramer carries the sickle Hb mutation, on the surface of each p-subunit (yellow). (b) The populations of the three-tetramer species are at equilibrium. Their electrophoretic separation is made possible by their charge difference. If the assembly free energy of the hybrid was the average of that of the two parents, the hybrid population would be 50% of the hybrid mixture, or additive, rather than the 3% observed experimentally.
Haptoglobin trapping
The tetramer → free dimer dissociation constant, koff, is measured in the presence of haptoglobin (Hp), which is a plasma protein that binds two free dimers rapidly and essentially irreversibly (13). The reaction of Hp with Hb that contains deoxy subunits can be followed by UV/visible spectroscopy, whereas the Hp reaction with fully ligated Hb, which has no appreciable change in absorbance, can be monitored by fluorescence spectroscopy. Thus, mixing Hb with a slight excess of Hp results in complete conversion to the Hp∙(dimer)2 complex by pulling the tetramer dissociation reaction to the right:
![]()
Because both kon and kHp are very rapid (essentially diffusion-controlled) processes, the overall rate-limiting step in Equation 8 is koff.
The koff for the asymmetrically ligated species 21 was measured first by forming the unligated version of the hybrid by mixing native Fe-heme deoxy Hb with Zn Hb (Fig. 4). Because both parents and hybrid (the unligated species 21 or 21u) have the same assembly free energy, 01∆Gasm, mixing the parent tetramers in a 1:1 ratio generates an equilibrium hybrid mixture that contains approximately 50% hybrid 21u. Because of the slow koff for both parents and hybrid (7.5 hours), equilibrium is attained after 3 days of incubation. In practice, the amount of hybrid present after 24 hours is sufficient for detection in the reaction with Hp.
The anaerobic hybrid mixture is mixed with an oxygenated solution of Hp in a stopped-flow instrument. The absorbance is monitored for 20 seconds, and the resulting observed rate constant is measured at 0.20 ± 0.02 s—1. This results in an assembly free energy, 21∆Gasm, of —9.1 ± 0.1 kcal/mol when combined with the consensus on constant in Equation 6 (9). This value is in excellent agreement with the results of the equilibrium low-temperature isoelectric focusing experiment.

Figure 4. Chemical strategy for the measurement of the dissociation rate constant for the hybrid tetramer species 21. The unligated version of the hybrid is formed from a mixture of Fe Hb and Zn Hb. When exposed to an oxygenated solution of Hp, the Fe hemes bind O2, which destabilizes the tetramer immediately, and it begins to dissociate into free dimers. Hp traps all free dimers, but the absorbance change is caused by the production of free dimer by dissociation of the hybrid.
Model-independent distribution of ∆Gc among the hemoglobin intermediates
The measurements described here show that binding O2 ligands to only one αβ dimer within the Hb tetramer occurs with a unique binding constant that differs significantly from that for the other doubly ligated intermediates (Table 2). This finding does not agree with the historically dominant presumption of a symmetric T/R-based model for cooperativity, which requires that the hemesites in both dimers maintain equal O2 affinity at each binding step. This asymmetric doubly ligated Hb intermediate, species 21, is considered a key intermediate in that its unique configuration of ligands reveals the presence of functional differences between the two dimers.
Symmetric models for cooperativity have been supported by the observation that only two functional states of the Hb tetramer, the low-affinity T and high-affinity R state, are required to describe O2 binding curves obtained over a range of solution conditions. However, the O2 binding curves are dominated by the properties of the two end-states, largely because of the presence of strong cooperativity, and thus cannot provide a clear distinction between most allosteric models. To begin to understand the rules for coupling between the subunits in Hb, it is necessary to measure the microscopic binding constants experimentally, as they cannot be determined from the binding curves.
An extensive kinetic analysis of ligand binding in normal Hb carried out by Goldbeck et al. has demonstrated agreement with the unique binding constant for the asymmetric doubly ligated Hb (14). Thermodynamic experiments from the Ackers laboratory that employs asymmetrically modified human Hbs have confirmed the asymmetric character of Hb cooperativity (15). This discovery generates critical energetic and structural questions, particularly with respect to the relationship of intradimer to cross-dimer cooperativity, in a classic system that was once thought to be well understood.
Table 2. The distribution of binding free energy among the individual binding steps in Hb
|
Binding steps |
|
Stepwise binding |
Stepwise microscopic |
|
|
free energy, |
binding constant, |
|
|
|
∆G(i→i+1)j kcal/mol |
k(i→i+1)j M-1 |
|
|
Oxygenation through the asymmetric doubly ligated tetramer |
|
||
|
1 01 + O2 |
→ 11 or 12 |
-5.5 ± 0.3 |
1 e + 4 |
|
2 (11 or 12) + O2 |
→ 21 |
-6.1 ± 0.4 |
3e + 4 |
|
3 21 + O2 |
→ 31 or 32 |
-6.6 ± 0.4 |
8e + 4 |
|
4 (31 or 32) + O2 |
→ 41 |
-8.9 ± 0.3 |
400 e + 4 |
|
Oxygenation through the symmetric doubly ligated tetramer |
|
||
|
1 01 + O2 |
→ 11 or 12 |
-5.5 ± 0.3 |
1 e + 4 |
|
2 (11 or 12) + O2 |
→ 22,23 or 24 |
-4.8 ± 0.5 |
1 e + 4 |
|
3 (22,23 or 24) + O2 |
→ 31 or 32 |
-7.9 ± 0.5 |
70 e + 4 |
|
4 (31 or 32) + O2 |
→ 41 |
-8.9 ± 0.3 |
400 e + 4 |
The binding cascade shown in Fig. (1) is essentially composed of two different pathways, passing through either an asymmetrically ligated species at the second binding step or a symmetrically ligated species.
References
1. Ackers GK, Holt JM, Burgie ES, Yarian CS. Analyzing intermediate state cooperativity in hemoglobin. In: Methods in Enzymology. Volume 379. Energetics of Biological Macromolecules Part D. 2004. Holt Jo M., Johnson, MJ, and Ackers GK, eds. Elsevier, San Diego, CA. pp. 3-28.
2. Ackers GK, Doyle ML, Myers D, Daugherty MA. Molecular code for cooperativity in hemoglobin. Science 1992; 255:54-63.
3. Kavanaugh JS, Rogers PH, Case DA, Arnone A. High resolution x-ray study of deoxyhemoglobin Rothschild 37b Trp to Arg: a mutation that creates an intersubunit chloride-binding site. Biochemistry 1992; 31:4111-4121.
4. Silva MM, Rogers PH, Arnone A. A third quaternary structure of human hemoglobin A at 1.7 A resolution. J. Biol. Chem. 1992; 267:17248-17256.
5. Lukin J, Kontaxis G, Simplaceanu V, Yuan Y, Bax A, Ho C. Quaternary structure of hemoglobin in solution. Proc. Nat. Acad. Sci. USA 2003; 100:517-520.
6. Kavanaugh JS, Rogers PH, Arnone A. Crystallographic evidence for a new ensemble of ligand-induced allosteric transitions in hemoglobin: the T-to-T(high) quaternary transitions. Biochemistry 2005; 44:6101-6121.
7. Mueser T, Rogers P, Arnone A. Interface sliding as illustrated by the multiple quaternary structures of liganded hemoglobin. Biochemistry 2000; 39:15353-15364.
8. Samuni U, Juszczak L, Dantsker D, Khan I, Friedman AJ, Perez-Gonzalez-de-Apodaca J, Bruno S, Hui HL, Colby JE, Karasik E, Kwiatkowski LD, Mozzarelli A, Noble R, Friedman JM. Functional and spectroscopic characterization of half-liganded iron-zinc hybrid hemoglobin: evidence for conformational plasticity within the T state. Biochemistry 2003; 42:8272-8288.
9. Holt JM, Klinger AL, Yarian CS, Keelara V, Ackers GK. Asymmetric distribution of cooperativity in the binding cascade of normal human hemoglobin. 1. Cooperative and noncooperative oxygen binding in Zn-substituted hemoglobin. Biochemistry 2005; 44:11925-11938.
10. Ackers GK, Holt JM, Huang Y, Grinkova Y, Klinger AL, Denisov I. Confirmation of a unique intra-dimer cooperativity in the human hemoglobin a1b1 half-oxygenated intermediate supports the symmetry rule model of allosteric regulation. PROTEINS: Struct. Func. Gen. 2000; (Suppl. 4):23-43.
11. Huang Y, Ackers GK. Transformation of cooperative free energies between ligation systems of hemoglobin: resolution of the carbon monoxide binding intermediates. Biochemistry 1996; 35:704-718.
12. Scholler DM, Wang M-YR, Hoffman BM. Metal-substituted hemoglobin and other hemoproteins. Meth. Enzymol. 1972; 76:487-493.
13. Nagel RL, Gibson QH. The hemoglobin-haptoglobin reaction as a probe of hemoglobin conformation. Biochem. Biophys. Res. Commun. 1972; 48:959-966.
14. Goldbeck RA, Esquerra RM, Holt JM, Ackers GK, Kliger DS. The molecular code for hemoglobin allostery revealed by linking the thermodynamics and kinetics of quaternary structural change. 1. Microstate linear free energy relations. Biochemistry 2004; 43:12048-12064.
15. Ackers GK, Dalessio PM, Lew GH, Daugherty MA, Holt JM. Single residue modification of only one dimer within the hemoglobin tetramer reveals autonomous dimer function. Proc. Natl. Acad. Sci. USA 2002; 99:9777-9782.
Further Reading
Ackers GK. Deciphering the molecular code of hemoglobin cooperativity. Adv. Prot. Chem. 1998; 51:185-253.
Ackers GK, Holt JM. Asymmetric cooperativity in a symmetric tetramer: human hemoglobin. J. Biol. Chem. 2006; 281:11441-11443.
Jayaraman V, Spiro TG. Structure of a third cooperativity state of hemoglobin: ultraviolet resonance Raman spectroscopy of cyanomethemoglobin ligation microstates. Biochemistry 1995; 34:4511-4515.
Royer WEJ, Zhu H, Gorr T, Flores J, Knapp J. Allosteric hemoglobin assembly: diversity and similarity. J. Biol. Chem. 2005; 280:27477-27480.
Wyman J Jr. Linked functions and reciprocal effects in hemoglobin: a second look. Adv. Protein Chem. 1964; 19:223-286.
Wyman J, Gill SJ. Binding and Linkage. Functional Chemistry of Biological Macromolecules. 1990. University Science Books, Mill Valley, CA.
See Also
Bioenergetics of Self-Assembly
Energetics of Protein Folding
Ligand-Operated Membrane Channels
Protein-Protein Interactions
Thermodynamics in Living Systems