CHEMICAL BIOLOGY
B12-Dependent Enzyme Reactions, Chemistry of
Wolfgang Buckel, Laboratorium fur Mikrobiologie, Fachbereich Biologie, Philipps-Universitat, Marburg, Germany
Bernard T. Golding, School of Chemistry, Newcastle University, Newcastle upon Tyne, United Kingdom
doi: 10.1002/9780470048672.wecb032
The vitamin B12 cofactor called coenzyme B12 (adenosylcobalamin) assists enzymes called mutases and eliminases in the catalysis of molecular rearrangements. The mutases comprise reactions in which a substrate equilibrates with a product by migration of an amino group (as with (3-lysine 5,6-aminomutase) or a carbon-based group (e.g., COSCoA in methylmalonyl-CoA mutase, which interconverts methylmalonyl-CoA with succinyl-CoA). The eliminases (e.g., propane-1,2-diol dehydratase) have either hydroxyl or amino as the migrating group, but they differ from the mutases by affording an intermediate that eliminates water or ammonia to give the observed product (e.g., propanal from propane-1,2-diol via propane-1,1-diol). Methylcobalamin is essentially an intermediary for synthetic reactions catalyzed by methyltransferases. These reactions depend on the "supernucleophilicity" of reduced vitamin B12 (cob(I)alamin) and in humans provide for the synthesis of the amino acid methionine.
The pursuit of the “antipernicious anemia factor” seemingly ended with the publication in Science in 1948 by Karl Folkers (1906-1997) of a paper entitled “Crystalline vitamin B12.” Just a few weeks later, Lester Smith (1904-1992), who had been guided by testing column fractions on pernicious anemia patients, independently obtained crystals of the vitamin [for a review of the early history of B12isolation and characterization, and references to the work described in this section see Lester Smith’s monograph (1)]. He identified cobalt in B12 and gave the crystals to Dorothy Hodgkin (1910-1994) that led to the structure of the isolated vitamin as cyanocobalamin (CN-Cbl: see Fig. 1 for cobalamin structures and other aspects of nomenclature of so-called corrinoids). In 1958, Horace Albert Barker (1907-2000) discovered coenzyme B12 (adenosylcobalamin, AdoCbl); the structure determination of which, again by Dorothy Hodgkin, revealed the presence of a Co-C a-bond. Finally, methylcobalamin (MeCbl) was recognized as another member of this very exclusive club of natural organometallic compounds. In this article, we review the fundamental chemistry of the B12 cofactors in selected enzymatic reactions for which they are obligatory participants.
Biological Context
Human cobalamins and dietary requirements
Quantitative assays have shown that the plasma of nonsmoking, healthy adults contains MeCbl (250pg cm-3), AdoCbl, and hydroxocobalamin (OH-Cbl) [AdoCbl + OH-Cbl (125 pg cm-3 with AdoCbl > OH-Cbl)]. The blood of smokers contains CN-Cbl (≈ 2% of total cobalamin), which is derived from hydrogen cyanide in tobacco smoke. CN-Cbl can also develop from the consumption of foods (e.g., cassava) that release cyanide ions. In erythrocytes, the chief cobalamin is AdoCbl (> 50% of total cobalamin) followed by OH-Cbl (25%), MeCbl (10-15%), and small amounts of CN-Cbl. Cobalamin-dependent enzymatic reactions in animals and microorganisms (see below) involve AdoCbl and MeCbl and the reduced form of OH-Cbl, cob(I)alamin, whereas CN-Cbl has no established role.
The total human body store of the corrinoids described is ~5 mg, and the recommended daily requirement of corrinoids is ~2 μg; this nutrient is provided by a typical “Western” diet but may not be included in a vegan diet. Whether 2 μg per day is sufficient to maintain health and protect against disease, especially of a degenerative kind, is a subject of current debate in which some believe it wise to consume larger amounts of B12 (2). Corrinoids are sequestered from food sources by a glycoprotein of mass ~45kDa called “intrinsic factor,” which is secreted in the stomach and binds B12 derivatives very tightly (for CN-Cbl, K = 1.5 x 1010 mol-1 dm3). Several other proteins bind and transport B12 into cells (3). The disease pernicious anemia has been recognized since the early nineteenth century and linked to a deficiency of what William Castle called “extrinsic factor” (i.e., vitamin B12) in 1928 (1). This disease develops because of the failure of the patient to secrete sufficient intrinsic factor.
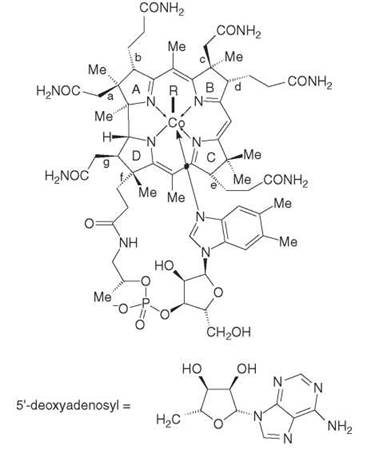
Figure 1. In the above structure, R = CN denotes cyanocobalamin (CN-Cbl), whilst R = OH is hydroxocobalamin (OH-Cbl); R = 5’deoxyadenosyl is coenzyme B12 (adenosylcobalamin, AdoCbl) and R = Me is methylcobalamin (MeCbl). By definition all cobalamins contain 5,6-dimethylbenzimidazole, which is the so-called 6th ligand to cobalt in the above structure. Substances containing the corrin ligand, i.e. the planar 14 electron p-system embracing cobalt in the above structure, are also called corrinoids.
Overview of cobalamin-dependent reactions
With the identification of vitamin B12's cofactors, their mechanisms of action could begin to be understood; today one may ask, “What do we know some 50 years after their discovery?” We know that coenzyme B12 assists a group of enzymes (Table 1, entries 1-9) in the catalysis of molecular rearrangements, which can all be described by Scheme 1. In Table 1, these enzymes are subdivided into mutases and eliminases. The mutases catalyze equilibrations and comprise reactions in which a carbon skeleton rearranges (entries 1-4, sometimes called Class I reactions; substituted carbon atom as migrating group) as well as the amino mutases (entries 5 and 6, Class III reactions; amino as migrating group) (4). The eliminases [entries 7-9, Class II reactions (4)] have either hydroxyl or amino as the migrating group, but they differ from the mutases in that the rearrangement affords an intermediate that eliminates water or ammonia to give the observed product. Coenzyme B12-dependent ribonucleotide reductase (entry 10) is included in Table 1 because its catalytic reaction exhibits some features of the diol and glycerol dehydratase reactions (i.e., elimination of OH from a 1,2-diol moiety), although the overall reaction is a redox process and not a rearrangement. How the reactions of Table 1 occur remains in part a puzzle, but this review intends to enlighten the reader. The coenzyme B12-dependent mutases and eliminases are a distinctive class of so-called “radical enzymes” (Fig. 2) (5), which are unique among enzymes because their catalytic pathways function via species with an unpaired electron (i.e., radicals). These species are normally highly reactive, but containment within a protein “straightjacket” enables their reactivity to be tamed and harnessed without destructive side reactions. The term “negative catalysis” was coined by Janos Retey in 1990 to describe this phenomenon.
An important structural difference between eliminases and mutases concerns the axial base coordinated to the cobalt, perpendicular to the plane of the corrin ring. Whereas in all the eliminases, the axial base is the dimethylbenzimidazole of the coenzyme itself (Fig. 1), the mutases use a conserved histidine residue of the enzyme for this purpose. On binding of the coenzyme to the apo-enzyme, the axial base is replaced by the histidine and moves into a distinct pocket of the protein. Methyltransferases (see below) also use a protein histidine as axial base, whose reactivity is fine tuned by protonation. Possibly the mutases and methyltransferases have a common evolutionary origin, whereas the eliminases evolved separately.
Methylcobalamin is completely different from adenosylcobalamin because it is essentially a conduit for synthetic reactions catalyzed by methyltransferases, illustrated in Scheme 2 for the case of methionine. These reactions depend on the “supernucle-ophilicity” of cob(I)alamin. In one case, this species removes a methyl group from N 5-methyltetrahydrofolate with the formation of methylcobalamin, and then transfers this group to the acceptor homocysteine, which results in the synthesis of methionine (see Scheme 2).
Examples of mutases, eliminases, and methyltransferases are discussed in detail below.
Coenzyme B12: Nature's Most Complex Cofactor
Coenzyme B12 (adenosylcobalamin, Fig. 1) has the most complex structure of all of the cofactors used by nature to aid enzymatic catalysis. Although many unusual features are observed in the structure, it is the cobalt-carbon sigma (a) bond that is vital, above all, for the mode of action of the coenzyme. The bond dissociation energy (BDE) of ~130 kJ mol-1 (6) is typical for a metal-carbon bond; although this value is much lower than for a-bonds from carbon to hydrogen, nitrogen or oxygen, or another carbon (BDE ~ 350 kJ mol-1), the coenzyme is rather stable in water provided light is excluded. The rate constant for decomposition of coenzyme B12 at 30°C in water was determined to be 1.16 x 10-8 s-1, which corresponds to a half-life of 1.9 years (4). The power of the primary organic radical 5'-deoxyadenosyl that develops from homolysis of the coenzyme’s Co-C bond is only unleashed when the coenzyme is bound to a partner enzyme in the presence of a substrate molecule. The 5'-deoxyadenosyl radical initiates the group of reactions summarized by Scheme 1 (4). In these reactions, a protein-bound radical is initially formed from a substrate molecule. The basic pathway requires at least two participating radicals, with one of these (S●) being structurally related to the substrate (SH) and the other (P●) structurally related to the product (PH). In addition, there may be an intermediate radical (I●) in some, if not all, cases. Homolysis of coenzyme B12 also releases cob(II)alamin, a paramagnetic (d7) species, which is easily detected by electron spin resonance (EPR) spectroscopy of reaction mixtures created by incubating enzyme, coenzyme, and substrate molecules for a few seconds and then freezing in liquid nitrogen.
Table 1. Classification of Coenzyme B12-dependent Radical Enzymesa


a Examples of either the natural or a typical substrate + product are given.
b “Base-off, his-on” means that the 5,6-dimethylbenzimidazole ligand (cf. Fig. 1) is detached and replaced by a histidine residue from the protein partner; “base-on” means that the 5,6-dimethylbenzimidazole ligand is still ligated to cobalt.
Coenzyme B12-Dependent Mutases
A common feature of the six known mutases is that a hydrogen atom has to be reversibly abstracted by the 5'-deoxyadenosyl radical from a nonactivated methyl group to yield a methylene radical, which is S● or P● depending on the identity of the substrate and product and is not stabilized by any adjacent group. However, the partner radical is always stabilized by a neighboring group, which is a carboxylate or CoA-ester for entries 1-5 and methyl for entry 6.
Glutamate mutase: the first coenzyme B12-dependent mutase
Glutamate mutase was discovered by Barker, who showed that the enzyme catalyzes the equilibration of (S)-glutamate with (2S,3S)-3-methylaspartate (Entry 1, Table 1; ∆G°’ = + 6.3kJ mol-1, K = 0.095) (for a review of glutamate mutase see Reference 7). This reaction is one of three distinct methods that nature uses to ferment glutamate to butyrate (5). Surprisingly, they all proceed through intermediate radicals. The coenzyme B12-dependent fermentation is the only one of the three that includes a molecular rearrangement, the point of which is to enable the elimination of ammonia, which might be entertained for glutamate but is only realistic for 3-methylaspartate (Scheme 3). The reason is because the C-3 protons of glutamate have an estimated pKa > 40 and cannot therefore be removed by an enzyme base, whereas the C-3 proton in 3-methylaspartate has an estimated pKa < 20 and so it can participate in an E2-type elimination.
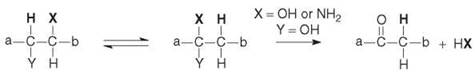
Scheme 1. Coenzyme B12-dependent enzyme-catalyzed rearrangements (for examples of a, b, X, and Y see Table (1)).

Figure 2. Possible side reactions for a radical (R-) with protein components and dioxygen; energy profile illustrating the concept of negative catalysis.
The substrate and product, (S)-glutamate and (2S ,3S)-3-methylaspartate, and their derived radicals, are held at the active site, approximately 6 A apart from the cobalt atom of cob(II)alamin by three arginine residues (8). This special architecture was termed the “arginine claw” by our collaborator Antonio Pierik. The 5'-deoxyadenosyl radical abstracts the 4-Si hydrogen atom of glutamate to give 5'-deoxyadenosine and the 4-glutamyl radical (S●), which has been postulated to fragment to a glycyl radical (I●) and acrylate (Scheme 4) (7). Now, a realignment of these species could enable their recombination to the 3-methylene-aspartate radical (P●), which reclaims a hydrogen atom from the methyl group of 5'-deoxyadenosine to afford 3-methylaspartate (cf. pathways for 2-methyleneglutarate mutase, Scheme 5). The most convincing experimental evidence for this “fragmentation-recombination” mechanism was the isolation of similar amounts of acrylate and glycine (~0.06mol of each per mol of enzyme) when the working enzyme was interrupted by addition of trifluoroacetic acid (9).
To achieve its catalytic reaction, glutamate mutase faces the problem of how to surmount the two relatively high transition-state energy barriers that lead from the 4-glutamyl radical to acrylate and the glycyl radical and then the recombination of these radicals to the 3-methylene-aspartate radical (Scheme 4). These barriers were computed as AH = + 59.9 and + 66.5 kJ mol-1 (10), respectively, and the 3-methylene-aspartate radical was found to be significantly less stable than the resonance-stabilized 4-glutamyl radical (AH = + 20.3 kJ mol-1). Likewise, with methylmalonyl-CoA mutase and 2-methyleneglutarate mutase the substrate and product radicals are interconnected via transition states of relatively high energy. In each case, the intermediate methylene radical (P● for glutamate and 2-methyleneglutarate mutase, S● for methylmalonyl-CoA mutase) is much less stable than the partner methine radical, which is stabilized by a carboxylate or CoA-ester group. It was recently suggested that stabilization of the methylene radicals by cob(II)alamin in the role of “conductor” might be required to lower the activation energy for the process of radical exchange (11). However, this proposal lacks experimental support.
2-Methyleneglutarate mutase: glutamate mutase in disguise?
The equilibration catalyzed by 2-methyleneglutarate mutase is similar to the glutamate mutase reaction but with a methylidene group (= CH2) in 2-methyleneglutarate/3-methylitaconate in place of the amino center of glutamate/3-methylaspartate. However, this feature enables a different mechanism (addition-elimination, Scheme 5, path a) to be followed, although an analogous mechanism (fragmentation-recombination, Scheme 5, path b) to that of glutamate mutase is also possible in principle. The mechanism of path a and possibly that of path b are consistent with the results of a study in which 2-methyleneglutarate was shown to catalyze the equilibration of (Z)-3-methyl(2’-2H1) itaconate with an equal quantity of its (E )-isomer, as well as with a 1:1 mixture of (E)- and (Z)-2-methylene(2’-2H1)glutarate (12). Newcomb and Miranda (13) performed model studies in which radicals that correspond to S●, P●, and U (Scheme 5) were generated, and their rates of interconversion were measured. It was concluded that these rates were too slow by a factor of ~105 relative to the known enzymatic kcat (30 s-1) for either of the mechanisms of Scheme 5 to be kinetically plausible. To explain this discrepancy, there could be a role for cob(II) alamin in the rearrangement as mentioned above and discussed below.
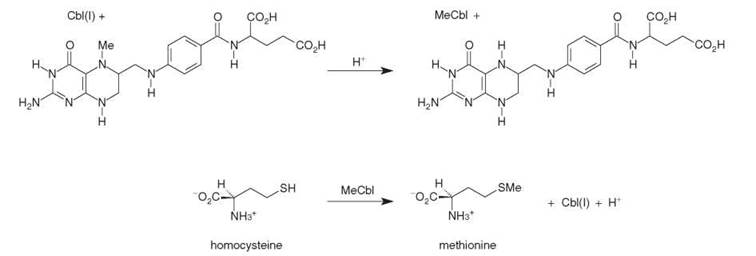
Scheme 2. Two-step synthesis of methionine from homocysteine catalyzed by methionine synthase and using methylcobalamin (MeCbl) derived from N5-methyltetrahydrofolate and cob(I)alamin (Cbl(I)).
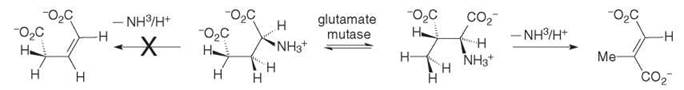
Scheme 3. Interconversion of (S)-glutamate with (25,35)-3-methylasparate catalysed by glutamate mutase that shows two options for ammonia elimination, one possible and the other not.
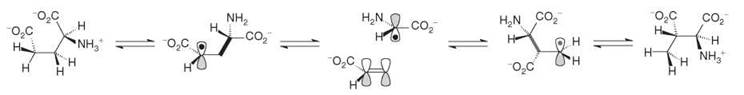
Scheme 4. Intermediate radicals in the glutamate mutase reaction.
Methylmalonyl-CoA mutase
This enzyme’s role in humans is to assist the detoxification of propionate derived from the degradation of the amino acids methionine, threonine, valine, and isoleucine. Propionyl-CoA is carboxylated to (S)-methylmalonyl-CoA, which is epimerized to the (R)-isomer. Coenzyme B12-dependent methylmalonyl-CoA mutase isomerizes the latter to succinyl-CoA (Fig. 2), which enters the Krebs cycle. Methylmalonyl-CoA mutase was the first coenzyme B12-dependent enzyme to be characterized crystallographically (by Philip Evans and Peter Leadlay). A mechanism for the catalytic reaction based on ab initio molecular orbital calculations invoked a “partial protonation” of the oxygen atom of the substrate thioester carbonyl group that facilitated formation of an oxycyclopropyl intermediate, which connects the substrate-derived and product-related radicals (14). The partial protonation was supposed to be provided by the hydrogen bonding of this carbonyl to His 244, which was inferred from the crystal structure of the protein. The ability of the substrate and product radicals to interconvert even in the absence of the enzyme was demonstrated by model studies (15).
The role of Cob(II)alamin in mutases
In mutases, the separation between cob(II)alamin and the intermediate radicals is ~ 6 A (cf. ~ 11 A in eliminases—see below), which was taken to suggest the participation of cob(II) alamin during turnover. In all known mutases, but not in the eliminases, a methylene radical may need to be stabilized, possibly by interaction of the dz2-orbital of cob(II)alamin with the disappearing p-orbital of the 5'-deoxyadenosyl radical and the emerging p-orbital of the substrate radical during the reversible transposition of the hydrogen atom. No coenzyme B12-independent alternative to the mutases is known at this time, and such enzymes were predicted (11) not to exist at all.
Unusual isotope effects and hydrogen tunneling
Unusually high isotope effects have been observed for several coenzyme B12-dependent reactions. For methylmalonyl-CoA mutase acting on (CD3)methylmalonyl-CoA, a primary deuterium isotope effect of 35.6 at 20°C was recorded (16). This effect was ascribed to quantum tunneling in the transition state for hydrogen (or deuterium) atom abstraction by the 5'-deoxyadenosyl radical. To probe the nature of the hydrogen abstraction steps with glutamate, mutase experiments were performed using labeled coenzyme, substrate, or product (17). The results were interpreted as providing evidence for hydrogen tunneling and a coupled motion of the hydrogen atoms at the adenosyl C-5' with the hydrogen atom being transferred from substrate/product.
Coenzyme B12-Dependent Eliminases
Diol and glycerol dehydratase
Soon after the discovery of glutamate mutase, Robert Abeles (1926-2000) recognized that the apparently simple conversion of glycerol to 3-hydroxypropionaldehyde catalyzed by diol dehydratase, was a coenzyme B12-dependent process (18). It had been long been known that acrolein could develop from the fermentation of glycerol. During the production of whiskey, infection of the broth may lead to the production of 3-hydroxypropionaldehyde and hence to acrolein on distillation. Isotopic labeling studies with glycerol dehydratase showed a remarkable control of the movement of the oxygen atoms in the rearrangement of 1,2-ethanediol to acetaldehyde and glycerol to 3-hydroxypropionaldehyde, which necessitates the postulate of intermediate geminal diols (Table 1, reactions 7 and 8). Substrate-derived radicals, which were diagnosed by EPR spectroscopy, were implicated as intermediates.

Scheme 5. Two pathways for 2-methyleneglutarate mutase (substrate: 2-methyleneglutarate; product: (R)-3-methylitaconate). Path a: Addition-elimination via substrate radical S+ and product radical P+ with the participation of an intermediate radical I+ (1-methylene-1,2-cyclopropanedicarboxylate, (1 S,2R)-isomer shown). Path b: Fragmentation-recombination that involves S+ and P+ and an intermediate radical P (2-acrylate radical accompanied by a 2-acrylate molecule) (AdoCH2 = 5’-deoxyadenosyl; AdoCH3 = 5’-deoxyadenosine).
Homolysis of adenosylcobalamin affords cob(II)alamin and the 5'-deoxyadenosyl radical, which abstracts a H-atom from C-1 of the diol giving 5'-deoxyadenosine and a substrate-derived radical in which the pK of the C-1 hydroxyl group has decreased from 16 to about 11. Deprotonation of this hydroxyl group affords a ketyl radical that could eliminate the adjacent hydroxyl, which yields a resonance-stabilized enoxy radical. Then, water (or hydroxide) is re-added to obtain a more reactive product-related radical that can reabstract an H-atom from the transiently formed 5'-deoxyadenosine. Finally, elimination of water from the intermediate 1,1-diol affords the aldehyde product. Alternatively, the substrate-derived radical rearranges to the product-related radical via an oxirane-like transition state in which there is “partial protonation” of the migrating hydroxyl group and “partial deprotonation” of the nonmigrating hydroxyl. The latter proposal was supported by evidence from protein crystallography of diol dehydratase and ab initio molecular orbital calculations of possible reaction pathways (19). In the eliminases, cob(II)alamin seems to act as a mere spectator that is not involved in the rearrangement. This view was supported by the EPR spectroscopy of diol dehydratase, which indicated a relatively large distance of ~ 11 A between cob(II)alamin and the substrate-derived radical intermediate(s).
Recently, a glycerol dehydratase was discovered in the anaerobic bacterium Clostridium butyricum, whose active site contains a glycyl radical formed by the action of the 5'-deoxyadenosyl radical on a specific glycine residue of the protein (20). The 5'-deoxyadenosyl radical is generated not from coenzyme B12, but by one-electron reduction of the structurally much simpler molecule S-adenosylmethionine (SAM), named “poor man’s B12” by Barker. Hence, this glycerol dehydratase performs the same reaction, probably with a similar pathway, to the coenzyme B12-dependent glycerol dehydratase.
Ribonucleotide reductase
The building blocks for the synthesis of DNA are 2'-deoxyribo-nucleotides, which are obtained from ribonucleotides by reductive elimination of their 2'-hydroxyl group. Three distinctive ribonucleotide reductases catalyze these reactions: the reductase of aerobic organisms (e.g., humans, Escherichia coli) uses dioxygen with a di-iron or an iron-manganese center for radical formation; organisms that thrive under anaerobic conditions (e.g., E. coli and most strict anaerobes) contain a SAM-derived glycyl radical in their ribonucleotide reductase; and finally, anaerobic organisms that can live in the presence of dioxygen (e.g., Lactobacillus and some algae) deploy coenzyme B12 in their reductase. With SAM and coenzyme B12, it is the adenosyl radical derived from these cofactors that is responsible for hydrogen abstraction from a conserved cysteine (Cys408) to give a thiyl radical. This species abstracts a hydrogen atom from C-3' of the ribose unit that regenerates the cysteine. Deprotonation of the 3'-hydroxyl group of the resulting C-3' radical affords a ketyl radical, which eliminates the 2'-hydroxyl group with formation of an enoxy radical, cf. the mechanism of diol dehydratase. Consecutive reductions that involve a pair of cysteine residues yield a deoxyribose unit with a C-3' radical center to which a hydrogen atom is donated by Cys408. This process regenerates the thiyl radical and affords the product (21).
The participation of a cysteine thiol in the abstraction of hydrogen from ribose C-3' coenzyme B12, which is not a feature of any other of the reactions of Table 1, leads to exchange of H between this thiol group, the 5'-methylene group of coenzyme B12 and water (22). It was also shown that ribonucleotide reductase catalyzes the conversion of adenosylcobalamin labeled with one deuterium atom at C-5' (initial R/S ratio = 3:1) to monodeuterated coenzyme with R/S ratio = 1. This result shows that the cobalt-carbon a-bond is reversibly cleaved to a 5'-deoxyadenosyl radical, which permits rotation about the C-4'/C-5' σ-bond.
Methyltransferases
Methionine synthase
The crucial steps in the pathway catalyzed by human cytosolic enzyme methionine synthase (MetH) are the transfer of a methyl group, first from N5-methyltetrahydrofolate to the cobalt of cob(I)alamin to give methylcobalamin (MeCbl), and then from cobalt to the sulfur of homocysteine to give methionine (Scheme 2). Our knowledge of MetH is largely from the incisive experiments of Liptak et al. (23), with crystallographic characterization of the enzyme by Martha Ludwig and Catherine Drennan underpinning all else. One large polypeptide (1227 amino acids, 136 kDa) comprises all functions of MetH, which has four modules. The substrates N5-methyltetrahydrofolate and homocysteine bind to a module each. The cobalamin cofactor binds to a third module with its dimethylbenzimidazole replaced by His759. The function of the fourth module is to reactivate oxidized cobalamin to MeCbl using SAM as source of the methyl group and flavodoxin as reductant (see below). Profound conformational changes bring the reacting components together in turn.
The key intermediate in the catalytic pathway is the “supernucleophile” cob(I)alamin, which attacks N5-methyltetrahydrofolate, generating tetrahydrofolate and MeCbl. Then homocysteine (probably as its thiolate) attacks MeCbl, which yields methionine and regenerates cob(I)alamin (Scheme 2). The demethylation of N5-methyltetrahydrofolate is not trivial, even for the “supernucleophilic” cob(I)alamin, and considerable efforts have been invested into understanding this reaction, dubbed “improbable” by Duilio Arigoni. The obvious mode of activation is by proton transfer to N-5 of N5-methyltetrahydrofolate, but as this is weakly basic (pKa 5.1) the nature of the proton source and mode of transfer has been difficult to pin down. Recent research from the Matthews group has shown how the reactivities of cob(I)alamin and methyl- cobalamin are modulated by the ligand trans to the lone pair of cob(I)alamin and methyl group of methylcobalamin (21).
It is interesting that E. coli contains two genes that code for methionine synthase: metH for the cobalamin-dependent enzyme and metE for a cobalamin-independent enzyme that depends on an active site Zn2+ to stabilize deprotonated homocysteine (24). This thiolate species demethylates N5-methyltetrahydrofolate, which is activated by proton transfer to N-5. MetE is less active (~ 100 x) than MetH, and so in the absence of B12 E. coli it produces much more MetE to compensate for the lack of MetH.
Cobalamin-dependent methionine synthase contains a built-in repair mechanism. If accidental oxidation of cob(I)alamin leads to inactive cob(II)alamin, then the enzyme employs SAM and reduced flavodoxin to regenerate cob(I)alamin. Although the redox equilibrium below lies mainly on the left side, any cob(I)alamin formed is trapped by SAM-dependent methylation to yield methylcobalamin.
Cob(II)alamin + flavodoxin hydroquinone = Cob(I)alamin + flavodoxin semiquinone
The anesthetic nitrous oxide (N2O) inhibits MetH by reacting with cob(I)alamin, which probably yields reactive hydroxyl radicals that damage the MetH protein (25). Model studies have shown that N2O reacts with cob(I)alamin, but not cob(II)alamin or cob(III)alamin complexes, which affords dinitrogen (N2) and hydroxocobalamin. Repeated anesthesia with N2O over a few days can be life threatening because the production of methionine is suppressed, and more seriously, turnover of folate cofactors stops because folates are trapped as N5-methyltetrahydrofolate (“methyl trap hypothesis”). This mechanism leads to inhibition of DNA and protein synthesis, and hence cell death.
Other methyltransferases
Corrinoids participate in the global C1 carbon cycle through the synthesis of methane and acetyl-CoA. Methane formation by methanogenic archaea exhibits a cobalamin-dependent step in which an overall methyl transfer occurs from N5-methyltetrahy-dromethanopterin to coenzyme M (26). This methionine synthase-like process is exergonic (∆G°’ = -30 kJ mol-1) and is catalyzed by a multienzyme complex that comprises eight different subunits. The subunit MtrA binds a cob(I)amide cofactor, which reacts with N5-methyltetrahydromethanopterin during the catalytic cycle to give a methylcobamide. This reaction undergoes a Na+-dependent demethylation by coenzyme M. The methylcobamide has a formal Co(III) center with the corrinoid in a base-off/His-on state, whereas the cob(I)amide is in the four-coordinate His-off state. The conformational change between these two states may drive the Na+-pump of a Na+-translocating, membrane-associated process.
The synthesis of acetyl-CoA by the Ljungdahl-Wood pathway of autotrophic carbon fixation in diverse bacteria and archaea is catalyzed by a Co- and Fe-containing corrinoid iron-sulfur protein (CoFeSP). This protein participates in the transfer of a methyl group from N 5-methyltetrahydrofolate to the cob(I)amide of CoFeSP to give a methylcob(III)amide, from which the methyl group is transferred to the reduced Ni-Ni-(4Fe-4S) active site cluster A of acetyl-CoA synthase (27).
The astonishing reactivity of cob(I)alamin is exploited in several other demethylation reactions, for example, the transfer of the methyl group of methanol to 2-mercaptoethanesulfonic acid (coenzyme M) catalyzed by methanol:2-mercaptoethanesulfonic acid methyltransferase. In this case, protein crystallography has shown that the methanol molecule is activated for nucleophilic attack by cob(I)alamin by coordination of its hydroxyl group to a Zn2+ ion (28). Even more remarkable is the ability of cob(I)alamin to affect the dechlorination of vinyl halides, for example, tetrachloroethene to (Z)-1,2-dichloroethene catalyzed by a dehalogenase from Sulfurospirillum multivorans (29). Given the wide range of reactions dependent on cob(I)alamin, it would seem better to designate this species as a coenzyme, rather than methylcobalamin, which is an intermediate in some cob(I)alamin reactions.
References
1. Smith LE. Vitamin B12. 1965. Methuen & Co. Ltd., London.
2. Watson WP, Munter T, Golding BT. A new role for glutathione: protection of vitamin B12 from depletion by xenobiotics. Chem. Res. Toxicol. 2004; 17:1562-1567.
3. Morkbak AL, Poulsen SS, Nexo E. Haptocorrin in humans. Clin. Chem. Lab. Med. 2007; 45:1751-1759.
4. Brown KL. Chemistry and enzymology of vitamin B12. Chem. Rev. 2005; 105:2075-2149.
5. Buckel W, Golding BT. Radical enzymes in anaerobes. Annu. Rev. Microbiol. 2006; 60:27-49.
6. Finke RG. Coenzyme B12-based chemical precedent for Co-C bond homolysis and other key elementary steps. In: Vitamin B12 and B12 Proteins. Krautler B, Arigoni D, Golding BT, eds. 1998. Wiley VCH, Weinheim, Germany.
7. Buckel W, Golding BT. Glutamate and 2-methyleneglutarate mutase: from microbial curiosities to paradigms for coenzyme B12-dependent enzymes. Chem. Soc. Rev. 1996; 26:329-337.
8. Gruber K, Reitzer R, Kratky C. Radical shuttling in a protein: ribose pseudorotation controls alkyl radical transfer in the coenzyme B12 dependent glutamate mutase. Angew. Chem. Int. Ed. 2001; 40:3377-3380.
9. Chih HW, Marsh ENG. Mechanism of glutamate mutase: identification and kinetic competence of acrylate and glycyl radical as intermediates in the rearrangement of glutamate to methylaspartate. J. Am. Chem. Soc. 2000; 122:10732-10733.
10. Wetmore SD, Smith DM, Golding BT, Radom L. Interconversion of (S)-glutamate and (2S, 3S)-3-methylaspartate: a distinctive B-12-dependent carbon-skeleton rearrangement. J. Am. Chem. Soc. 2001; 23:7963-7972.
11. Buckel W, Golding BT, Kratky C. Stabilisation of methylene radicals by cob(II)alamin in coenzyme B12 dependent mutases. Chem. Eur. J. 2006; 12:352-362.
12. Pierik AJ, Ciceri D, Broker G, Edwards CH, McFarlane W, Winter J, Buckel W, Golding BT. Rotation of the exo-methylene group of (R)-3-methylitaconate catalyzed by coenzyme B12-dependent 2-methyleneglutarate mutase from Eubacterium barkeri. J. Am. Chem. Soc. 2002; 124:14039-14048.
13. Newcomb M, Miranda N. Kinetic results implicating a polar radical reaction pathway in the rearrangement catalyzed by a-methyleneglutarate mutase. J. Am. Chem. Soc. 2003; 125:4080-4086.
14. Smith DM, Golding BT, Radom L. Understanding the mechanism of B-12-dependent methylmalonyl-CoA mutase: partial proton transfer in action. J. Am. Chem. Soc. 1999; 121:9388-9399.
15. Darbre T, Keese R, Siljegovic V, WollebGygi A. Model studies for the coenzyme-B-12-catalyzed methylmalonyl-succinyl rearrangement. The importance of hydrophobic peripheral associations. Helv. Chim. Acta. 1996; 79:2100-2113.
16. Dybala-Defratyka A, Paneth P, Banerjee R, Truhlar DG. Coupling of hydrogenic tunneling to active-site motion in the hydrogen radical transfer catalyzed by a coenzyme B12-dependent mutase. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:10774-10779.
17. Cheng MC, Marsh ENG. Evidence for coupled motion and hydrogen tunneling of the reaction catalyzed by glutamate mutase. Biochemistry 2007; 46:883-889.
18. Speranza G, Buckel W, Golding BT. Coenzyme B12-dependent enzymatic dehydration of 1,2-diols: simple reaction, complex mechanism! J. Porphyrins Phthalocyanines. 2004; 8:290-300.
19. Kamachi T, Toraya T, Yoshizawa K. Computational mutation analysis of hydrogen abstraction and radical rearrangement steps in the catalysis of coenzyme B12-dependent diol dehydratase. Chem. Eur. J. 2007; 13:7864-7873.
20. O’Brien JR, Raynaud C, Croux C, Girbal L, Soucaille P, Lanzilotta WN. Insight into the mechanism of the B12-independent glycerol dehydratase from C. butyricum; preliminary and structural characterization. Biochemistry 2004; 43:4635-4645.
21. Lawrence CC, Stubbe J. The function of adenosylcobalamin in the mechanism of ribonucleoside triphosphate reductase from Lactobacillus leichmannii. Curr. Opin. Chem. Biol. 1998; 2:650-655.
22. Chen D, Abend A, Stubbe J, Frey PA. Epimerization at carbon-5' of (5'R)-(5'-2H)adenosylcobalamin by ribonucleoside triphosphate reductase: cysteine 408-independent cleavage of the Co-C5' bond. Biochemistry 2003; 42:4578-4584.
23. Liptak MD, Fleischhacker AS, Matthews RG, Brunhold TC. Probing the role of the histidine 759 ligand in cobalamin-dependent methionine synthase. Biochemistry 2007; 46:8024-8035.
24. Taurog RE, Matthews RG. Activation of methyltetrahydrofolate by cobalamin-independent methionine synthase. Biochemistry 2006; 45:5092-5102.
25. Drummond JT, Matthews RG. Nitrous oxide degradation by cobalamin-dependent methionine synthase: characterization of the reactants and products in the inactivation reaction. Biochemistry 1994; 33:3732-3741.
26. Gottschalk G, Thauer RK. The Na+-translocating methyltrans- ferase complex from methanogenic archaea. Biochim. Biophys. Acta 2001; 1505:28-36.
27. Svetlitchnaia T, Svetlitchnyi V, Meyer O, Dobbek H. Structural insights into methyltransfer reactions of a corrinoid iron-sulfur protein involved in acetyl-CoA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:14331-14336.
28. Hagemeier C, Rriier M, Thauer RK, Warkentin E, Ermler U. Insight into the mechanism of biological methanol activation based on the crystal structure of the methanol-cobalamin methyl- transferase complex. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18017-18022.
29. Diekert G, Gugova D, Limoges B, Robert M, Saveant J-M. Electroenzymatic reactions. Investigation of a reductive dehalogenase by means of electrogenerated redox cosubstrates. J. Am. Chem. Soc. 2005; 127:13583-13588.
Further Reading
Banerjee R. Chemistry and Biochemistry of B12. 1999. Wiley, New York.
Green R, Miller JR. Vitamin B12. In: Handbook of Vitamins. Rucker RB et al., eds. 2007. Taylor & Francis, Boca Raton, FL.
Krautler B, Arigoni D, Golding BT, eds. Vitamin B12 and B12 Proteins. 1998. Wiley VCH, Weinheim, Germany.
Ragsdale SW. Metals and their scaffolds to promote difficult enzymatic reactions. Chem. Rev. 2006; 106:3317-3337.
See Also
Amino Acids
Enzyme Catalysis, Chemical Strategies for
Enzyme Catalysis, Roles of Structural Dynamics in
Enzymatic Cofactors
Vitamins