CHEMICAL BIOLOGY
High Throughput Screening (HTS) Techniques: Overview of Applications in Chemical Biology
James Inglese and Douglas S. Auld, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland
doi: 10.1002/9780470048672.wecb223
The rapid testing of chemical libraries for biological activity is the primary aim of high throughput screening (HTS). Advances in HTS have paralleled those in molecular biology, instrumentation and automation, and informatics, and the increased availability of arrayed compound libraries. Sophisticated high sensitivity assays and the associated technologies required to implement these assays in HTS have been largely developed within the pharmaceutical industry for the identification of new chemical matter for drug development. However, HTS approaches are now widely applied to the broader questions within biological research. By way of introduction, we will describe the components of HTS and provide examples of strategies used to identify novel chemtypes for specific biological targets using large chemical libraries. We then will illustrate how more narrowly defined compound collections (e.g., targeted libraries or bioactive compounds) have been profiled against related targets or cell types for the purpose of discovering or defining, compound class/gene family selectivity, off-target activity or ''hidden phenotypes'', toxic fingerprints, or any other relationship between chemical structure and bioactivity. In this way, HTS systems can expand the scope of an experimental hypothesis to address questions of chemical biology, be they at the level of an isolated enzymatic activity or that of a complex cellular phenotype.
High throughput screening (HTS) is a technologically enabled field of applied science that creates an interface between chemical libraries and biological assays for the purpose of rapidly exploring and identifying chemical bioactivity. The technologies that make HTS of large compound collections possible have been supported principally by the pharmaceutical industry because of the needs inherent to drug discovery but are increasingly being used to expand the boundaries of traditional academic disciplines such as enzymology and cell biology. “Chemical biology” that emphasizes the application of synthetic chemistry in the study of biological processes or “chemical genetics” in which compounds are used to recapitulate the effect of genetic mutations are particularly enabled by HTS as it allows the identification and profiling of wide-ranging chemotypes that modulate individual gene products or cellular phenotypes (1, 2). HTS laboratories are now an integral part of many universities, and in 2004 the U.S. National Institutes of Health (NIH) implemented a specific initiative to expand access to HTS for translational research (3-5). Whether the aim is chemical biology or drug discovery, HTS is most effective when biological assay systems are designed and screening results interpreted with the technological capabilities and limitations of the entire process in mind.
As a primer to this area, we outline the basic process of HTS and synergisms between pharmaceutical and chemical biology research endeavors and provide examples of platforms that use HTS technologies to enable chemical biology.
Bridging Chemistry and Biology with HTS
Compound discovery or profiling that uses HTS involves merging diverse types of chemical libraries and biological assays (6) (Fig. 1) and generally involves four major components that include the compound library itself, high-quality assays, process engineering platforms, and informatics procedures to track, analyze, and annotate the results. As a consequence, implementation of HTS requires expertise from multiple disciplines. In this section, we highlight some of the important features for each of these processes and how these technologies currently are being applied to the goals of chemical biology.
Chemical libraries
Multiple categories, including natural products (7) and synthetic bioactives [e.g., metabolites, carcinogens, and approved drugs (8)]; privileged scaffold-based libraries [e.g., untested analogs of synthetic drugs or natural products such as benzodiazepines (9), indoloquinolizidine (10), or diketopiperazines (11)]; biologically uncharacterized compounds of low diversity but high density [e.g., combinatorial chemistry-derived libraries (12, 13)]; and consolidated samples/collections that represent extensive structural diversity [e.g., Molecular Libraries Small Molecule Repository, see PubChem (14)] have been used to describe the general character of library collections in use today (Fig. 1a). These libraries range in size from small focused or diverse sample collections of ~1K compounds or less to very large collections that may contain a million or more compounds. The cost can range from between a few dollars per mg for specialty sets of < 1500 compounds to the well over one billion dollars spent by Pfizer to enrich its corporate library (15) (Table 1) (16-22). Modern collections used in pharmaceutical companies are constructed considering multiple factors including DMSO solubility and purity, “lead” or “drug-likeness,” and synthetic tractability (23). Common measures of “drug-likeness” are “Lipinksi’s rule of 5 (Ro5)” or variations thereof that define the physio-chemical characteristics of well-absorbed drugs (24). However, for chemical biology, the library may encompass compounds that are not Ro5 compliant but are nonetheless potentially useful research tools to address biological questions, for example, compounds of a peptidic nature or that contain reactive groups (see Fig. 2b and 2c for examples) (25-30).
The nature of the library is dependent on the aim of the experiment. For example, if an inhibitor of a novel protein kinase is desired, then a targeted library derived from the amino-quinazoline scaffold (31) (Fig. 1a, i and ii) might be considered, whereas an enzyme that belongs to a gene family with no precedent of small-molecule modulation may benefit from a more random or diverse collection (32). Also, although identifying chemical matter that allows a proprietary or patentable structure can require screening large diverse collections (> 1 million compounds), useful chemical matter for research purposes can be identified from smaller collections (21). Aiming for many dissimilar scaffolds (the central rigid part of the molecule capable of positioning key interactions) and analogs could lead rapidly to very large libraries, and, therefore, the complexity and the redundancy of the library needs to be carefully managed. The quality of the compounds should also be determined with respect to the stability, synthetic tractability, resupply, and purity of the samples. Various chemical descriptors can be used to measure library diversity; however, a universal method has not been found (21, 33). General screening compound collections aim to contain many different scaffolds with few analogs and are expanded in size by similarity searches using parameters such as the Tanimoto coefficient (34) to consider how new compounds may complement or add to the novelty of the collection. At the other extreme are combinatorial libraries that contain few scaffolds but potentially thousands of related analogs that systematically explore the activity relationships between two or more structural variables. Several thousand different combinatorial libraries have been described since 1992 (35, 36). Combinatorial libraries are synthesized using a variety of methods and can be very large in size (105), and although these libraries are not classically diverse, they provide the advantage of containing many subtle changes in structure that can be enormously important for biological activity (37). Knowledge around compound classes that target specific members of a gene family, as in the case of protein kinases or G protein-coupled receptors (GPCRs), can form the basis of targeted libraries that can increase the probability of finding useful leads (12, 38). More specialized libraries are often required to address certain target classes, such as protein-protein interactions that have been historically refractory to conventional small-molecule approaches. For example, fragment library approaches in which small scaffolds (MW 150-300) are screened at very high concentration to identify weak interactions using nuclear magnetic resonance (NMR)- or X-ray crystallography-based methods have been successful for these difficult target classes (16). Once active fragments are found, the affinity is improved by merging or growing fragments into nearby sites on the protein through the use of structure-guided techniques.
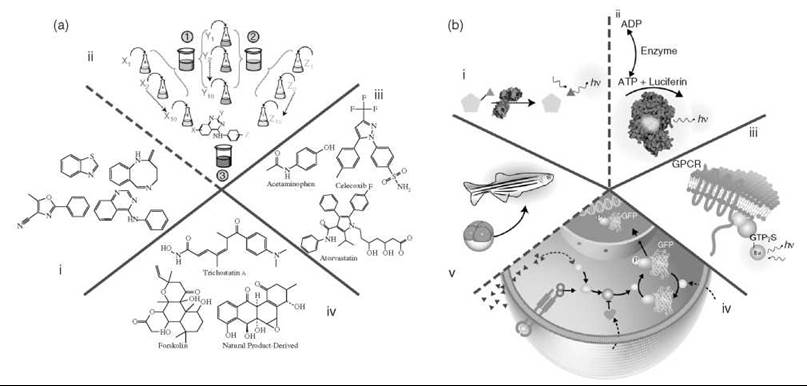
Figure 1. Chemistry and biology. a) Categories of compounds. i, ii) Representative chemical scaffolds that could be present either as part of a diverse set of compounds (i) or as a combinatorial library template, illustrated with the 4-amino-quinazoline scaffold (ii). Here a ''split and mix'' strategy is used in which the library is synthesized on solid support beads that contain the scaffold. Chemical side chains are added to the scaffold by reacting building blocks (or synthons) in separate batches and then pooling all the batches and dividing the beads equally for the next round of synthesis. In the example shown, 10 unique synthons are added at each of three positions (X, Y, and Z) to create a library of 10 x 10 x 10 = 1000 different 4-amino-quanzolines in three steps. iii) Compounds that represent known drugs. Shown are anti-inflammatory compounds (acetaminophen and celecoxib) and the HMG-CoA reductase inhibitor, Atorvastatin. (7) iv) Natural product libraries represented by trichostatin A, forskolin, or a compound from a natural-product-derived synthetic library (Boston University). b) Categories of different assay methodologies based on purified (i, ii) or partially purified (iii) targets and intact cellular systems (iv). Examples of purified molecular targets (i, ii) in which an enzyme catalyzes turnover of a profluorescent red emitting substrate (i) or a substrate/product (e.g., ATP) of the target enzymatic reaction catalyzed, for example, by an ATPase (ii) is detected by the ''reporter'' enzyme firefly luciferase via luminescent output (hv). iii) A membrane preparation used to measure the activity of a GPCR through the ligand-dependent activation of associated G proteins as measured by fluorescent GTPyS analogue binding. iv, v) Phenotypic cellular and whole-organism assays in which (iv) illustrates a prototypical cell-based assay designed to probe a signaling pathway. In this example, a conditional activation of an intracellular protein (blue dashed arrow) with a pharmacological agent (A) is required to identify potentiators of the pathway that act downstream of the extracellular receptor as measured by the nuclear translocation of a GFP-labeled sentinel protein. Such potentiators could be envisioned to act, for example, by inhibiting proteins that act to repress the pathway (black dashed arrows). v) A zebra fish assay where organogenesis can be monitored by GFP expression in a particular organ system (6).
Chemical library handling
Collection sample preparation, formatting for screening (39), short- and long-term storage, and analytical quality control (QC) (40) comprise important variables in the management of HTS libraries. Generally, libraries are arrayed with a single compound per well, but strategies that employ pooling of compounds have been described to compensate for limited screening capability (41). For the purpose of HTS, compounds are routinely dissolved in DMSO, a so-called “universal solvent” at high (typically 10 mM) concentrations, and stored in polypropylene microtiter plates. Critical to the “stability” of these collections is the control of humidity and temperature. DMSO is highly hygroscopic, and when it absorbs water, because of nonideal behavior, it becomes more structured and viscous and limits compound solubility, which can lead to compound precipitation (42). That precipitation, rather than compound degradation, is the central factor determining library integrity has been supported by studies on modern pharmaceutical screening collections (40, 43, 44). Therefore, refined compound storage units use desiccated inert chambers (e.g., argon saturated environments) and dry conditions. If dry conditions cannot be maintained, then the compound collection can be stored at room temperature (never cooled) and exchanged with a new copy at time intervals compatible with the plate lid/sealing method [e.g., every 4 to 6 months for Kalypsys lidded plates (39) or after the plate has been reheat-sealed a predetermined number of times]. Storage at -80 °C is preferred for long-term (> 6 months) archiving, as the samples will remain solid even if water has been absorbed. Evaporation of DMSO is also a concern, particularly for low-volume (< 10 pL) stock solutions, and several specially designed microtiter plate lids and plate sealing technologies have been developed to limit evaporation from compound sample wells. The complexity of a compound archive and retrieval system depends on the size, frequency of use, and types of access requirements of the chemical library. Systems range from relatively low-tech manual storage of sealed replicate copies thawed and used as needed (repeated freeze-thaw cycles of a single library plate is not advised) to highly engineered systems capable of compound dissolution, environmentally controlled storage, plate thawing, and individual compound or “cherry picking” capabilities (45).
Compound structure and purity is assessed before samples enter a screening collection by supplier QC data and/or independently by the user (46). Ongoing monitoring of compound integrity, typically by liquid chromatography-mass spectrometry (LC-MS) analysis, throughout the lifetime of the library can be accomplished on a randomly selected fraction of the collection (i.e., spot-checking) and during retest of samples entering post-HTS assays, for example. Comprehensive and continuous library QC is currently an area under evaluation (47, 48).
Table 1. Example Chemical Libraries Used in Drug Discovery and Chemical Biology
|
Library |
Category |
Size |
Comments |
Reference |
|
Sigma LOPAC |
Pharmacologically active. |
1208 |
Major target classes: GPCRs, kinases, ion channels, nuclear receptors, metabolic enzymes, cell signaling, apoptosis, cell cycle, etc. Used extensively for assay/HTS validation. |
* |
|
ChemBridge Fragment Set |
Low MW (≤ 300), and cLogP (≤ 3) designed for high aqueous solubility (~3 mM). |
5000 |
Useful for high concentration screening (e.g., NMR/SPR-based approaches). |
16 |
|
TimTech Natural compound library (NPL400) |
Purified natural products. |
480 |
Structures retrievable from website. Mainly plant derived, ~16% flavonoids. |
17 |
|
|
||||
|
Angular epoxyquinol librarya ∆2-Pyrazoline libraryb |
Diversity oriented synthesis (DOS). |
a244 b80 |
Chemical Methodology and Library Development (CMLD) initiative to develop novel libraries. |
18, 19 |
|
|
||||
|
National Toxicology Program (NTP1408) |
Toxic agents. Includes compounds tested in traditional in vivo and in vitro toxicologic assays. |
1408 |
Chemical descriptions are publicly available in PubChem. |
20 |
|
Commercial screening libraries |
Range from low scaffold diversity (e.g., combinatorial libraries) to high diversity. |
100s to > 100 K |
Used widely in academia. |
21 |
|
NIH Molecular Libraries Small |
Diverse collection: procured, QC’ed, stored and distributed to 10 network labs. |
> 250 K ^500 K |
Chemical descriptions are publicly available in PubChem. |
22 |
|
Molecule Repository (ML SMR) |
||||
|
Pfizer compound file |
Large pharma collection. Outsourced from ArQule, ChemRx, ChemBridge, and Tripos. |
> 2 x 106 |
~U.S.$ 1 billion file enrichment program to expand diversity, drug likeness, synthetic tractability, and rapid analog access. |
15 |
* http://www.sigmaaIdrich.com/Area_of_Interest/Chemistry/Drug_DiscoveryAiiIidation_Libraries/LOPAC1280.htmI.
Nature of high-quality HTS assays
The configuration and nature of the assay format is critical to any HTS experiment and must be coordinated with subsequent assays that evaluate biological relevance/mechanism of action. One must consider that even with an assay having 99% accuracy, the number of false positives can match or greatly exceed the number of true positives (typically between 0.001-0.1%) (49), which complicates interpretation of the experimental results and creates significant follow-up efforts that lead to dead ends. In addition, compound-mediated phenomena independent of the target biology is a major source of HTS artifacts. Examples include, compound aggregation (50), fluorescence (51), and inhibition or activation of assay reporter enzymes (52). Therefore, primary assay selection and design must consider the nature of potential assay liabilities (e.g., sensitivity to compound library fluorescence) and match counter or orthogonal assays (e.g., an assay that is insensitive to compound fluorescence) to stringently and accurately verify or eliminate the primary assay findings. Ideally, a counterscreen is integrated into the primary HTS assay either as multiple measurements from the same assay well or as independent assays run in close proximity. As an example of the latter, a 1536-well robotic screen used fluorescent polarization to measure the interaction between the C-terminus of the tumor suppressor protein BRCA1 and a phosphopeptide derived from an interacting protein pBACH. Here, two assays that differed only in the labeled phosphopeptide ligand, carrying either a green or red fluorescent probe, were interleaved into a single HTS (53). The experiment was designed to discriminate signal changes from fluorescent samples, likely to affect the assay pair differentially, from genuine inhibitors of the protein●phosphopeptide complex that should have an unbiased effect on the assays. For cell-based assays, dual-luciferase reporter systems, for example, where one luciferase is constitutively expressed to track cell number, have been used to segregate compounds that modulate the biology in question from nonspecific effects on the assay format or cytotoxicity (54, 55).
Assay technologies for modem drug discovery have been fueled by achievements in molecular biology and the numerous genome sequencing projects that have lead to the cloning and expression of novel proteins, highly engineered cell lines (54, 56, 57), and model organisms (58, 59) with convenient reporters such as luciferases (55), fluorescent proteins (57), or enzymes that act on fluorescent substrates such as P-lactamase (60). In current drug discovery, HTS involves a variety of assay types (Fig. 1b) based on purified molecular targets, reconstituted enzyme cascades, cell extracts (61), and cellular/organism phenotypes (62). The use of reconstituted systems or extracts is particularly attractive as multiple molecular targets/interactions can be tested without the complication of the test compounds binding to serum, having limited cell penetration, or undergoing rapid cellular efflux. Also, such systems can be used to identify the target through techniques analogous to genetic suppression (63).
Decisions surrounding specific HTS assay technologies will take into consideration factors including pharmacological relevance, HTS compatibility, follow-up strategy, and costs, among others. For example, whether a membrane receptor antagonist is sought using a SPA-based radioligand binding assay with an enriched membrane preparation or through a cell-based reporter gene assay may depend on the availability of a suitable radioligand and ability to prepare sufficient plasma membrane versus developing a stable cell-based assay responsive to receptor activation. Also, in this example, a consideration of the cost of assay implementation including handling/disposal of radioactivity versus the counter or orthogonal assays required to sort out the often multiple possible mechanisms of action from reporter gene responses are critical evaluation criteria. In a recent review (64), we provide detailed information and references on technologies used in HTS assays and the considerations in assay protocol design and reporting (65) to aid in evaluating the options for HTS assay development and communication of protocols and results.
Optimizing assays for HTS performance involves several steps including reducing the number of required reagent additions; assessing QC and availability of the reagents; determining the stability and batch variability testing of the reagents; determining the DMSO tolerance of the assay (particularly important for cell-based assays); minimizing incubation and measurement times; and choosing and optimizing detector settings. Therefore, an HTS assay can differ greatly from typical laboratory assays, and not all assays that function adequately in bench-top experiments will be adaptable to HTS. Typically, libraries are screened at one concentration in a manner in which every compound is assayed once. This “n = 1” experiment is rapidly performed on as many as 1 million samples, and, therefore, the assay must show excellent precision, but this does not guarantee nor should it be confused with biological fidelity (66). Therefore, during HTS implementation, the entire procedure is validated with test plates to assess the assay precision. The goal is to minimize variation and provide adequate signal:background (S:B) without adversely effecting the sensitivity or biological relevance of the assay. A measure of the assay performance or quality is typically made using a parameter, the Z-factor (67, 68) (Table 2) (67-71), that is a measure of both assay variation and S:B in which assays that exhibit Z-factors > 0.5 are generally considered acceptable. The background can be taken from specified control wells (Z’) or the median of the sample field (Z).
To obtain statistical significance, these parameters should only be reported from entire microtiter plates (e.g., 96-wells or higher densities) and are best measured by performing the experiment in triplicate on different days.* Also, the accuracy of the assay response in reflecting the biology under investigation can be measured when control compounds are present. IC50/EC50s can be compared with accepted values typically obtained from the literature and should include a consideration of the assay precision. For this purpose, the minimum-significant ratio can be calculated from control titrations across individual assay plates, which provides the smallest potency ratio between two compounds that can be considered real based on the precision of the assay (72).

Figure 2. Chemical probes versus drugs. Structures across the top are a) monastrol (25), an inhibitor of the kinesin Eg5 identified through forward chemical genomics, b) an activity-based chemical probe (26) used to label cellular metalloproteases, c) the natural product fumagillin (27) from Aspergillus fumigatus that acts as an angiogenesis inhibitor through inhibition of a methionine aminopeptidase, d) CKD-731, a semisynthetic analog of fumagillin currently in clinical trials as an anticancer agent, e) LG335 (i, 28), an inactive analog of the drug Targretin®(ii, Bexarotene), a pharmaceutical developed to bind RXR for the treatment of cutaneous T-cell lymphoma, paired with a designer RXR nuclear receptor to enable conditional gene expression (29), f) the approved HMG-CoA reductase inhibitor Atorvastatin (8), g) the natural product toxin Taxol used as an anticancer agent, h) structures of the opioid analgesics fentanyl—(i) (8) and 1 -methyl-4-phenyl-4-propionoxypiperidine, (MPPP, ii) and its metabolite 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP, iii) that is neurotoxic resulting in Parkinson disease-like symptoms (30)—and i) a polychlorinated dibenzodioxin (TCDD) that represents an environmental pollutant that can reach toxic levels by accumulating in fatty tissues.
Engineering processes in HTS
To achieve a “high-throughput” process requires that systems are available to test compounds rapidly by processing of 96-well or higher density plates (73). The throughput of HTS can vary from low (10,000-50,000 data points/day) to ultrahigh (uHTS) where > 100,000 data points are collected/day. Efficiency in HTS requires highly engineered systems and components, such as high-density microtiter plates, precision, low-volume liquid handlers for dispensing assay reagents (nL to pL) or concentrated compound solutions (pL to nL) (74-76), microtiter plate readers varying in sophistication from reporting changes in a well’s total absorbance to the subcellular distribution of target proteins within a cell (77, 78), and robotic systems to integrate these components (Fig. 3). Fully integrated systems that leverage miniaturized parallel assay processing (e.g., 1536-well plates) served by robotic arms having direct access to compound libraries, reagent/compound dispensers, and plate incubators and readers can achieve throughputs as fast as 10 samples/sec (79, 80). Operating within a thin margin for error, automated assays by virtue of their speed and parallelism should be validated carefully on the robotic system, and the entire process of assay implementation should be managed to minimize costly loss of reagents that can occur quickly if the screen is improperly initiated and monitored (73).

Figure 3. Dispensing and detection instrumentation. a) Example of liquid handling instrumentation. Top: Piezo-electric tip plumbing and pulse control for pL dispensing of fluids. Bottom: Plumbing and valve control of a solenoid-based for nL dispenser. b) Methods for nL dispensing of compound solutions in DMSO (beige wells) to assay plate wells (blue wells) using contact-based dispensing with pin-tools (i) that may contain slots as shown or noncontact-based dispensing as in using a focused acoustic beam (ii). For acoustic dispensing, it is necessary for the destination plate to be inverted over the source plate. c) Reader modalities (i-iii) are depicted for population averaged detection, (i) transmittance using microtiter plates made of clear plastic (e.g., polystyrene or cyclic olefin polymers), (ii) luminescence, typically using opaque white microtiter plates, and (iii) fluorescence where in the example shown epifluorescence is collected by using focusing optics for excitation (green arrow) and emission (red arrow) above the well of an opaque black microtiter plate. d) Detection output (i, ii) for single object enumeration or imaging. (i) Cell cytometry in HTS is possible using either flow or microtiter plate systems (78). Here, the population distribution of green and blue fluorescent cells in a p-lactamase reporter gene assay is analyzed. (ii) High-content screening using automated wide-field or confocal microscopes. High-resolution confocal image of G2 M cell cycle sensor (GE Healthcare) expressed in U2OS cells. Actin and microtubular staining using Texas red- and Alex488-labeled antibodies, respectively, and nuclei stained with DAPI.
HTS data analysis and informatics
Several independent streams of data are tracked and integrated throughout the HTS lifecycle. The library samples’ structures, plate locations, and array positions, concentrations, and other parameters and annotations comprise one group; a second group is the detector output readings for the assay responses, and a third group include process information, such as time stamps and trace files that track compound and assay plate histories. For smaller-scale screens (a few 1000 compounds) Excel spreadsheets and an Access database can suffice, but as the scale of the HTS increases, organizing this large amount of chemical and biological assay data requires efficient laboratory information management systems (LIMS) that cover and provide tools to facilitate the entire experimental process. Flexibility has been cited as one of the primary requirements in LIMS design as both the processes and nature of the information content will change over time (81).
HTS assay data is statistically analyzed to identify genuine outliers from systematic or random assay noise, which allows the level of activity for each compound tested in the assay to be determined. The values used in the final analysis are derived from a normalization procedure that converts the raw data to the percent activity relative to the control values (Table 2). Wells that contain controls can be placed in any number of wells on an assay plate, but a common practice is to use ≤ 10% of the plate for controls, often in columns along the left or right edges (Fig. 4a).
A threshold cut off is the simplest method to select putatively active compounds (Table 2). For example, in Fig. 4, samples to the left of (Fig. 4b) the 3 standard deviation (a) threshold or below it (Fig. 4c) are considered “active” (red values; often referred to as “hits”) and are selected for “confirmation” or prioritized with additional criteria (e.g., clogP < 4 or availability of analogs). However, several problems have been noted with this method (70). For example, the controls may be unstable or, in the case of uncharacterized biology, may be absent entirely, which makes it difficult to derive accurate normalized percent activity values. Therefore, methods such as the z-score (69) have been used where the test samples act as the controls (Table 2).
Several methods have been employed to correct for positional variations within the screening data that can be very common in HTS and include edge effects, dispensing artifacts, and changes in reagent stability during the course of the screen. Correction of systematic errors requires special software and is facilitated by incorporating blank plates (e.g., containing DMSO alone without test compounds) uniformly throughout the screen to capture a sample-independent signature of the plate signal variation (Fig. 4). One method that considers positional effects uses the b-score (for “better” score) that is analogous to the z-score except positional and plate effects are additionally taken into account and result in a marked reduction in false positives (Table 2) (70). However, such methods can fail in screening-focused collections, such as combinatorial libraries in which many analogs around a common scaffold are present, because the assumption that the majority of the tested wells are inactive may not hold when many genuine actives occur on the same plate. Therefore, for combinatorial libraries, the activity is better addressed using multiple assays to help rank or profile the actives or screening at multiple concentrations where potencies can be resolved (see below, Fig. 4e). More attempts to improve active selection from large HTS data sets include the use of structure-activity relationships (SAR) (82) to help reduce false negatives and advanced population analysis to construct predictive models of activity (83).
Table 2. Assay Diagnostic and Hit Scoring Methods Used in HTS
|
Equation |
Explanation |
Reference |
|
Assay Diagnostic Z'-factor
Scoring Methods %Inhibition=
Threshold value (T)
z-score |
Factor used to evaluate assay performance where a value > 0.5 represents an acceptable assay. Measured for control wells. δ = standard deviation of the assay signal μ = mean of the assay signal
Control-based. Normal distribution assumed. xi = raw value for the ith sample well on a plate μmin = mean of the minimum control values μmax = mean of the maximum control values
Used with or without data correction. Normal distribution assumed. Values below (inhibition assay, —nδx) or above (activation assay, +nδx) threshold are scored. x = median value of the entire sample field. n = typically between 3 and 6. δx = standard deviation of the tested compound field values
Method to score the relative potency of actives on a plate-by-plate basis without correction for row and column effects. Controls not used. Normal distribution assumed. xi = raw value for the ith sample well on a plate μx = mean of all raw values for the samples tested on a single plate. δx = standard deviation of the tested compound field values
Row (R) and column (C) effects are estimated using a median polish procedure to score the relative potency of actives on a plate-by-plate basis. yijp = value at the ith row and jth column on the pth plate μp = plate center mean Ri = row effect Cj = column effect εijp = error in measurement
Row and column effects are estimated using a robust linear model to score the relative potency of actives on a plate-by-plate basis. All parameters are defined as in the b-score above. |
67, 68 |
|
69 |
||
|
b -score |
70 |
|
|
r-score |
71 |
|
|
|
|
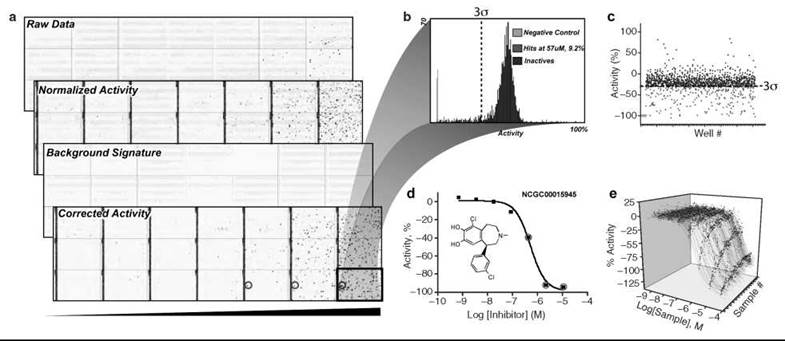
Figure 4. Data processing and Informatics. a) Heat maps depicting 1536-well plate activity for a library of 1408 compounds that was screened from low to high concentrations using qHTS (black bar indicates increasing concentration) in triplicate with each plate containing a different compound concentration. Controls are contained in the left four columns (128 wells) and used to obtain normalized activity from the raw data. Background signatures are derived from plate sample wells containing DMSO/assay buffer alone (not shown) and used to correct for systematic artifacts before scoring and selection of ''actives.'' The final corrected activity shows actives increasing with concentration (blue: inhibitory activity in well, red: activating activity in well, and white: inactive). The background signature shown (stripped pattern) indicates a systematic artifact because of liquid dispenser tip variability. This background signature was subtracted from the normalized data to yield the final corrected activity. b) Traditional HTS is typically conducted at a single concentration (e.g., 10 μM), and treatment of this data is shown. Distribution of activity at one concentration using the highest tested concentration of the qHTS data. A 3a threshold differentiates the active (red) and inactive (blue) data sets. c) Scatterplot representation of the single concentration-based data set. Example CRC data sets are shown in (d) for a single compound that showed a concentration-response relationship or where (e) multiple CRCs are obtained through the use of qHTS.
Goals of chemical biology
The goals of chemical biology have been stated in various ways. In the “gene-centric” view, the aims are borrowed from classical genetic studies in which genes were mutated to determine their role in phenotypes. In this analogy, small molecules rather than gene mutations are used to affect protein function and perturb the biological system. Terms have been coined such as “forward chemical genomics” to represent phenotypic or response assays that are used to identify compounds that generally affect a cellular or model organism phenotype (Fig. 5). However, no single technology is sufficient to identify a target for a small molecule from such phenotypic assays, and target identification remains a challenging area in chemical genomics. In “reverse chemical genomics,” the starting point is often based on an assay that uses an isolated protein, and with subsequently identified compounds, one backs away from the “gene product” to determine the effect on phenotype. In this respect, classical drug discovery resembled forward chemical genomics in which compound testing was often conducted in physiological model systems (e.g., organ bath), whereas modern drug discovery follows a reverse chemical genomics paradigm in which isolated molecular target-based assays are used for compound discovery, and these then are progressed into disease models and eventually to testing in humans.
It is now widely appreciated that the proteome is considerably larger than the sum of its genes, and defining what constitutes a “gene” has become increasingly complex (84). Mechanisms such as alternative splicing result in an average of three protein isoforms/gene leading to a larger and more complex proteome. In this “proteomic view,” chemical biology has the goal of annotating every protein with compounds capable of inhibiting, activating, or allosterically regulating their function. Ultimately, both the gene-centric and the proteomic viewpoints converge to create a pharmacological database that relates compound activity to biological effects (85). This “chemical probe directory” will list compounds useful for the interrogation of basic biological questions and initiation points to develop novel therapeutics. Furthermore, as the pharmacological database becomes populated with diverse biological assays and grows in structural scope, it will act to guide the design of higher-quality libraries that will access the darkest recess of the genome.

Figure 5. Reverse and Forward Drug Discovery/Chemical Biology. Reverse chemical genomics starts with a target-small-molecule interaction and progresses to determine the pharmacologic effect on an organism's phenotype, whereas forward chemical genomics begins with a chemically induced phenotype and aims to identify the target(s) responsible for the phenotypic effect. HTS is enabling to both processes.
Chemical probes
To understand the nature of chemical probes, we can evaluate these probes relative to the current drug discovery process. Although the goals of drug discovery have a clear end point, the development of a therapeutic, the goals of chemical biology are broader so that although all drugs are probes because of the special requirements of drugs, not all probes are intended or can be developed into drugs (Fig. 2). Chemical probes can be drawn from the embryonic stages of the drug development pipeline, for example, in the lead optimization stage, once activity has been confirmed and compounds of sufficient potency have been obtained. Early stage absorption, distribution, metabolism, elimination (ADME)-toxicity data such as plasma serum binding, cytochrome P450 inhibition/induction, and cell permeability data can be obtained around chemical probes to determine their usefulness for in vivo testing. Although cell permeability and low serum binding are important, fully optimized ADME-toxicity parameters are not a requirement for many chemical probes. For example, the natural product forskolin (Fig. 1a, iv), an activator of adenylyl cyclase, has been used extensively and primarily in the study of Gi-coupled or “inhibitory” GPCRs in many cellular model systems, with no need to consider ADME-toxicity data around such a tool compound. Furthermore, although selectivity is an important consideration for drug development and many tool compounds, a pan-inhibitor such as staurosporine that is broadly active against the kinome (86) has proven useful in the validation of assays for protein kinases. Also, such pan-inhibitors have been used to develop generic protein kinase assays (87). However, the hydroxamate-containing antifungal antibiotic trichostatin A (TSA; Fig. 1a, iv) has been used as an investigational tool to study histone-deacetylases (88) (more than 2000 publications have resulted from the use of TSA since the initial discovery in 1976) and as a proof-of-principle compound in the validation of novel cancer drug targets. Also, drugs have been used as probes to uncover new targets as in the case of the identification of cyclooxygenase-3 (COX3), a splice isoform of COX-1, through the investigation of the pharmacology associated with analgesic/antipyretic drugs (e.g., acetaminophen and coxibs, Figure 1a, iii) (89).
Academic chemical biology and pharmaceutical drug discovery synergies
Finally, it is worth noting where chemical biology can support the discovery of novel therapeutics. For example, many rare genetic and infectious diseases are not currently the focus of the pharmaceutical industry but provide fertile ground for public-private partnerships that use chemical biology as the vector to initiate and coordinate programs around these disease areas. The unprecedented open access to screening data enabled by the National Center for Biotechnology Information that manages PubChem (14), where results are found from a diversity of bioassays tested against a growing compound library, will provide new avenues and opportunities for drug discovery and development. Such initiatives have now made a large amount of chemical information available on the worldwide web (90). The open access model also brings together laboratories that possess the proper expertise both in the know-how of HTS and the required knowledge of the biology under investigation that is critical to advance compounds in an efficient manner. Recent broad access to HTS and chemical screening data should act to alleviate bottlenecks in our knowledge around new target classes and their potential modulation by small molecules and possibly lower the investment for development by industry.
Example HTS Platforms that Support Research in Chemical Biology
HTS can serve the needs of chemical biology by functioning in the traditional mode of “mining” large chemical libraries with individual assays to identify specific chemical matter for development (91, 92) or “profiling” a specific small ensemble of compounds with bioassay panels (80, 93) or combining both to populate a pharmacological database (94). In this section, we will touch on specific examples from the literature on the profiling of annotated chemical libraries and show the power automation can bring to increase the scope of these experimental designs. Lastly, we discuss several areas of chemical biology that have been beneficiaries of HTS-derived leads, which, with tighter integration to HTS, will enhance the value of new chemical probes.
Profiling assays for understanding mechanism of action
Defining the spectrum of activity for a collection of compounds can be achieved by profiling bioassay panels focused, for example, on gene families (86) or distributed across diverse signaling pathways (95). Bioactivity profiling can be most efficiently performed when the assays share a common format such as a luciferase-reporter system to streamline assay implementation, results quantification, and interpretation. Also, with format homogeneity, nonspecific effects across the profile can be readily identified. As an example, we describe experiments using the cell-based protein complementation assay (PCA) technology (96, 97). In this system, cells are engineered to express two interacting proteins where each protein is fused to complementary fragments of a split yellow fluorescent protein (EYFP). Interaction of the two protein fragments yields a reconstituted EYFP whose signal is at least 10 times brighter than either fragment alone. This system has been used to place cellular “sentinels” along points in signaling pathways where modulation of the sentinel complex reports on a compound’s effect in that pathway. In the work of MacDonald and colleagues (95), a collection of 107 drugs from six therapeutic classes was screened with 49 PCA assays and provided 127 different measurements per drug, designed to monitor a range of biological pathways. Drugs with similar mechanisms of action revealed similar cellular response profiles, for example, PPARy agoinsts induced PPARγ●SRC-1 complexes, and differences in profile signatures often attributable to chemical structure. Additionally, the hierarchal clustering of compound activity exposed a supercluster of drugs that did not share any common therapeutic target or mechanism of action but showed a “hidden phenotype” of antiproliferative activity. Therefore, pharmacological profiling enabled with assays such as PCA can be used at an early stage of chemical characterization or optimization to identify such hidden phenotypes, understand off-target activity, or decipher the mechanism of action.
Pharmacologically defined libraries and their uses
Drugs and bioactives from compound collections, such as the Library of Pharmacology Active Compounds (LOPAC, Sigma-Aldrich; Table 1), or chemical probe collections available from vendors, such as Tocris, represent libraries that are annotated with biological activity. These types of libraries often show more activity across a broader range of HTS assays compared with nonbiologically biased libraries (e.g., based on synthons and scaffolds selected for synthetic tractability alone) and therefore are useful in identifying controls for assay development and validation. Annotated information can be used to generate a hypothesis around the mechanism of action for a given response phenotype (98). However, the reliability of the annotation, often derived from multiple databases or fragmented literature sources, can be misleading and should be “triangulated” or corroborated with structurally distinct chemical classes. A group at Amphora, Inc. has approached this problem by employing microfluidic technologies to construct an annotated database in-house by measuring IC50s for 88 protein kinases against a set of 130K compounds (99). Collecting and improving the annotation of large compound collections against a wide range of biological targets is an active area of research in cheminformatics (100), but ultimately, the activity profile of library molecules will be confirmed experimentally owing to the advances in HTS such as those described below.
Automation of compound profiling
Once an assay platform configurable with highly engineered HTS robotic systems is developed, large-scale compound profiling becomes possible. An example of such an experiment was described by scientists at the Genomics Institute of the Novartis Research Foundation where automated cell culturing through dispensing in either 384-well or 1536-well plates was used to test 1400 small-molecule kinase inhibitors to generate concentration-response curves (CRCs) against 35 activated tyrosine-kinase-dependent cellular assays (80). Similar to the PCA system described above, the results of these screens were used to clusters the kinase inhibitors based on the phenotypes observed in the cell-based assays.
At the NIH Chemical Genomics Center (NCGC), we have leveraged the advances in HTS technology to determine concentration-response relationships of large chemical libraries routinely across diverse biological systems, a process called quantitative HTS or qHTS (94). To date, > 5 million CRCs have been generated in > 100 unique assays within 3 years of operation [see PubChem (14)] using qHTS at the NCGC. The relatively short timeframe under which such large-scale experiments can be conducted has enabled the needs of scientists with diverse interests and backgrounds, for example, scientists searching for antiparasitic agents (79, 101), comparing experimental with in silico screening results (102), and testing hierarchical clustering algorithms for cheminformatics (103). The large data sets achievable from methodologies like qHTS will be indispensible to progress in chemical biology and often will serve as the only means by which a foothold at the interface of chemistry and biology can be made.
Chemoproteomic methods: profiling gene families and downstream uses for chemical probes
For investigating the function of enzyme gene families within cells, activity-based probes (ABPs) are particularly useful (104). Requirements for an ABP include sufficient affinity (< 100 nM), pan-selectivity for the enzyme family, an expressed active enzyme to modify covalently, and a reporter tag such as a fluorophore for detection. In a recent exploration of metalloproteases, ABPs were synthesized around two alkyne-tagged hydroxamate-benzophenone libraries, and metalloproteases that bound the probes could be covalently labeled through the benzopheonone group (Fig. 2b) (26). The alkyne was placed distal to the metal-binding hydroxamate moiety and readily coupled via click-chemistry with an azido-containing tag (e.g., rho- damine or biotin). The two libraries identified metalloproteases from nearly all branches of this enzyme superfamily, and the authors used the library to detect differences in metalloprotease activity from invasive and noninvasive melanoma cells.
Another method that enables large-scale profiling is the development of generic platforms for entire gene families. Researchers at Ambit have developed a competitive-binding assay that involves the expression of human kinases as fusions to T7 bacteriophage and a set of adenosine triphosphate (ATP) competitive pan-inhibitors tagged with biotin that enable selectivity profiles of kinase inhibitors to be determined on hundreds of protein kinases (more than 350 now available) (86). This format shows exquisite sensitivity with the ability to measure binding affinities as low as 1 pM.
Researchers at Serenex (now part of Pfizer) have turned the compound discovery process on its head using affinity chromatography to “proteome mine” and asking what proteins bind to the compounds rather than searching the library against a single protein (105). In this system, a chemical scaffold such as purine is tethered to a solid support, and a cell, tissue, or organ extract is passed over the surface so that the purine-binding proteome is specifically captured by the affinity resin. Then, library members are added to displace (specifically elute) the targets competitively at single or multiple concentrations, and the proteins that bind to these targets are identified by chromatographic separation and MS analysis. Some considerations in these affinity-based panels are that the affinity of the interaction must be high (Kd < 100 nM), the targets must have relatively good abundance (> 100 copies/cell with 108 cells typically harvested), and the identification of specific binding requires optimization of the washing protocols.
Ongoing HTS advancement includes systems that more closely replicate physiological context in the assay format. For example, techniques in which cells are grown under 3-D culture conditions better mimic the biological and pharmacological consequence of the response after compound treatment. An excellent review using 3-D culture of a multicellular tumor spheroid model was recently published (106). The use of 3-D cultures in these tumor models recapitulates the morphological, functional, and mass transport characteristics of the tumor tissue in vivo (107). Such 3-D cultures are also being used in high throughput toxicity screening and in the analysis of their cytochrome P450-generated metabolites (108). Simple metazoan model organisms (e.g., Caenorhabditis elegans and zebrafish) offer the opportunity to identify substances that modulate complex physiological systems through HTS (58, 59). As these organisms are suitable to both reverse and forward genetics, screens based on target-directed or phenotypic assays are possible. Furthermore, rapid organogenesis (hours to a few days) in these metazoans allows, for example, developmental neurotoxicity and teratoge- nesis to be studied on the timescales compatible with HTS (6).
Biophysical and analytical measurements adapted to HTS systems have broadened the sophistication of assays formats, as witnessed by the rapid evolution of electrophysiological assay technology to measure ion channel function (109). The high sensitivity and multiparametric output inherent in flow cytometry (110) has also been applied to HTS and provides a platform for assay multiplexing that uses either beads or intact cells (111). For example, in one study, a three laser flow cytometry system was used to enumerate over 15 different parameters from phosphorylation events within multiple cell types including primary cells to define both pathway and cell type-specific compounds (112). Such technologies enable the simultaneous correlated measurements of signal transduction networks that provide physiological relevant compound mechanism and selectivity data. Other methods are being developed, based on principles of microcalorimetry, that will enable the mass screening of compounds against targets with unknown function or ligands (113). These advances are but a few of the biological and technological advances that are impacting the future direction and uses of HTS in chemical biology.
Summary and Future Perspective
HTS developed rapidly in the pharmaceutical sector after the molecular biology revolution changed the drug discovery paradigm from a forward to a reverse genetics model (Fig. 5). Technological breakthroughs that allow sensitive in vitro assay designs (64) combined with the explosion in the availability and quality of chemical libraries (Table 1) accelerated the evolution and need for efficient HTS (114). Broad commercialization of HTS platforms and components coupled with an emigration of many “discovery” scientists from industry to academia in recent years has created a unique environment for HTS technology and expertise to enable academic chemical biology. Within a setting less restrained by market pressures, scientists will continue to find novel uses for the experimental power of HTS. Breaching the > 90% of the human genome not currently interdicted by small molecules, particularly the “dark matter” of the genome whose functions remain undefined, is a key challenge for HTS-assisted chemical biology. The current drug pharmacopeia targets only ~330 proteins although as many as 8000 may be involved with disease (115). Adding to this situation, polymorphic variations between individuals will demand increased intricacy in chemical genomics research that will fuel the growing field of pharmacogenomics and ultimately usher in the era of personalized medicine. Redirecting the fate of pluripotent stem cells, the holy grail of regenerative medicine, will likewise find an ally in HTS (116). Understanding the disposition and effects of small molecules on biological systems through research in chemical biology should provide more optimal prediction of in vivo efficacy (64) and toxicity (117). The new HTS approaches are poised to have a transformative impact on biology and medicine, to remove many historical limitations to experimental designs, and to bring the promise of the genomic revolution closer to realization. Finally, the advent of synthetic biology, which spans from designer receptors (118, 29) (e.g., Receptor Activated Solely by a Synthetic Ligand (RASSL) and Receptor Exclusively Activated by Designer Drugs (DREADDs)) to genome transplantation (119) to future man-made life forms, will require novel ligands and designer drugs to complement and control (or keep in check) this new biological frontier.
Acknowledgments
This work was supported by the Molecular Libraries Initiative of the NIH Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, NIH. We thank Daryl Leja for the illustrations used in this work.
References
1. Schreiber SL, Nicolau KC. Crossing the boundaries. Chem. Biol. 1994; 1:1.
2. Spring DR. Chemical genetics to chemical genomics: small molecules offer big insights. Chem. Soc. Rev. 2005; 34:472-482.
3. Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science 2004; 306:1138-1139.
4. Lazo JS. Roadmap or roadkill: a pharmacologist’s analysis of the NIH Molecular Libraries Initiative. Mol. Interv. 2006; 6:240-243.
5. Gordon EJ. Small-molecule screening: it takes a village. ACS Chem. Biol. 2007; 2:9-16.
6. Rubinstein AL. Zebrafish assays for drug toxicity screening. Exp. Opin. Drug Metab. Toxicol. 2006; 2:231-240.
7. Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002. J. Nat. Prod. 2003; 66:1022-1037.
8. Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 10th ed. 2001. McGraw-Hill Professional, New York. p. 1825.
9. Bunin BA, Plunkett MJ, Ellman JA. The combinatorial synthesis and chemical and biological evaluation of a 1,4-benzodiazepine library. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:4708-4712.
10. Noren-Muller A, Reis-Correa I Jr, Prinz H, Rosenbaum C, Saxena K, Schwalbe HJ, Vestweber D, Cagna G, Schunk S, Schwarz O, Schiewe H, Waldmann H. Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:10606-10611.
11. Dandapani S, Lan P, Beeler AB, Beischel S, Abbas A, Roth BL, Porco JA Jr, Panek JS. Convergent synthesis of complex diketopiperazines derived from pipecolic acid scaffolds and parallel screening against GPCR targets. J. Org. Chem. 2006; 71:8934-8945.
12. Guo T, Hobbs DW. Privileged structure-based combinatorial libraries targeting G protein-coupled receptors. Assay Drug Dev. Technol. 2003; 1:579-592.
13. Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. Application of combinatorial chemistry science on modern drug discovery. J. Comb. Chem. 2008.
14. PubChem. http://pubchem.ncbi.nlm.nih.gov/.
15. Clark DE. Outsourcing lead optimization: constant change is here to stay. Drug Discov. Today 2007; 12:62-70.
16. Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007; 450:1001-1009.
17. Fullbeck M, Michalsky E, Dunkel M, Preissner R. Natural products: sources and databases. Nat. Prod. Rep. 2006; 23:347-356.
18. Lei X, Zaarur N, Sherman MY, Porco JA Jr. Stereocontrolled synthesis of a complex library via elaboration of angular epoxyquinol scaffolds. J. Org. Chem. 2005; 70:6474-6483.
19. Manyem S, Sibi MP, Lushington GH, Neuenswander B, Schoenen F, Aube J. Solution-phase parallel synthesis of a library of delta(2)-pyrazolines. J. Comb. Chem. 2007; 9:20-28.
20. Xia M, Huang R, Witt KL, Southall N, Fostel J, Cho MH, Jadhav A, Smith CS, Inglese J, Portier CJ, Tice RR, Austin CP. Compound cytotoxicity profiling using quantitative high throughput screening. Environ. Health Perspect. 2008; 116:284-291.
21. Krier M, Bret G, Rognan D. Assessing the scaffold diversity of screening libraries. J. Chem. Inf. Model 2006: 46:512-524.
22. MacNeil JS. Diving deep into the chemical genome. Genome Technology Magazine 2005, January/February, 26-35.
23. Jacoby E, Schuffenhauer A, Popov M, Azzaoui K, Havill B, Schopfer U, Engeloch C, Stanek J, Acklin P, Rigollier P, Stoll F, et al. Key aspects of the Novartis compound collection enhancement project for the compilation of a comprehensive chemogenomics drug discovery screening collection. Curr. Top. Med. Chem. 2005; 5:397-411.
24. Lipinski CA, Lonbardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discover and development settings. Adv. Drug Del. Rev. 1997; 23:3-25.
25. Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999; 20:971-974.
26. Sieber SA, Cravatt BF. Analytical platforms for activity-based protein profiling-exploiting the versatility of chemistry for functional proteomics. Chem. Communicat. 2006; 2311-2319.
27. Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H, Folkman J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature 1990; 348:555-557.
28. Doyle DF, Braasch DA, Jackson LK, Weiss HE, Boehm MF, Mangelsdorf DJ, Corey DR. Engineering orthogonal ligand-receptor pairs from “near drugs”. J. Am. Chem. Soc. 2001; 123:11367-11371.
29. Schwimmer LJ, Rohatgi P, Azizi B, Seley KL, Doyle DF. Creation and discovery of ligand-receptor pairs for transcriptional control with small molecules. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:14707-14712.
30. Langston JW, Ballard PA Jr. Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 1983; 309:310.
31. Bridges AJ. Chemical inhibitors of protein kinases. Chem. Rev. 2001; 101:2541-2572.
32. Gudkov AV. Cancer drug discovery: the wisdom of imprecision. Nat. Med. 2004; 10:1298-1299.
33. Martin EJ, Blaney JM, Siani MA, Spellmeyer DC, Wong AK, Moos WH. Measuring diversity: experimental design of combinatorial libraries for drug discovery. J. Med. Chem. 1995; 38:1431-1436.
34. Tanimoto TT. IBM Internal Report 1957, November 17.
35. Dolle RE, Le Bourdonnec B, Morales GA, Moriarty KJ, Salvino JM. Comprehensive survey of combinatorial library synthesis: 2005. J. Comb. Chem. 2006; 8:597-635.
36. Wess G. How to escape the bottleneck of medicinal chemistry. Drug Discov. Today 2002; 7:533-535.
37. Sanchez-Martin RM, Mittoo S, Bradley M. The impact of combinatorial methodologies on medicinal chemistry. Curr. Top. Med. Chem. 2004; 4:653-669.
38. Auld DS, Diller D, Ho KK. Targeting signal transduction with large combinatorial collections. Drug Discov. Today 2002; 7:1206-1213.
39. Yasgar A, Shinn P, Jadhav A, Auld DS, Michael S, Zheng W, Austin CP, Inglese J, Simeonov A. Compound management for quantitative high-throughput screening. J. Assoc. Lab. Automat. 2008; 13:79-89.
40. Isbell JJ, Zhou Y, Guintu C, Rynd M, Jiang S, Petrov D, Micklash K, Mainquist J, Ek J, Chang J, et al. Purifying the masses: integrating prepurification quality control, high-throughput LC/MS purification, and compound plating to feed high-throughput screening. J. Comb. Chem. 2005; 7:210-217.
41. Ferrand S, Schmid A, Engeloch C, Glickman JF. Statistical evaluation of a self-deconvoluting matrix strategy for high-throughput screening of the CXCR3 receptor. Assay Drug Dev. Technol. 2005; 3:413-424.
42. Oldenburg K, Pooler D, Scudder K, Lipinski C, Kelly M. High throughput sonication: evaluation for compound solubilization. Comb. Chem. High Throughput Screen. 2005; 8:499-512.
43. Cheng X, Hochlowski J, Tang H, Hepp D, Beckner C, Kantor S, Schmitt R. Studies on repository compound stability in DMSO under various conditions. J. Biomolec. Screen. 2003; 8:292-304.
44. Kozikowski BA, Burt TM, Tirey DA, Williams LE, Kuzmak BR, Stanton DT, Morand KL, Nelson SL. The effect of room-temperature storage on the stability of compounds in DMSO. J. Biomolec. Screen. 2003; 8:205-209.
45. Archer JR. History, evolution, and trends in compound management for high throughput screening. Assay Drug Dev. Technol. 2004; 2:675-681.
46. Isabell J. Changing requirements of purification as drug discovery programs evolve from hit discovery. J. Comb. Chem. 2008:104.
47. Chaudhry P, Schoenen F, Neuenswander B, Lushington GH, Aube J. One-step synthesis of oxazoline and dihydrooxazine libraries. J. Comb. Chem. 2007; 9:473-476.
48. Lewis KC, Simpkins JD. An interview with Kenneth C. Lewis, Ph.D., CEO, and Joseph D. Simpkins, Vice President, Software, OpAns (Optimized Analytical Solutions), Durham, NC. Interview by Vicki Glaser. Assay Drug Dev Technol 2007; 5:695-702.
49. Glickman JF, Wu X, Mercuri R, Illy C, Bowen BR, He Y, Sills M. A comparison of ALPHAScreen, TR-FRET, and TRF as assay methods for FXR nuclear receptors. J. Biomolec. Screen. 2002; 7:3-10.
50. Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007; 50:2385-2390.
51. Simeonov A, Jadhav A, Thomas CJ, Wang Y, Huang R, Southall NT, Shinn P, Smith J, Austin CP, Auld DS, Inglese J. Fluorescence spectroscopic profiling of compound libraries. J. Med. Chem. 2008; 51:2363-2371.
52. Auld DS, Southall NT, Jadhav A, Johnson RL, Diller DJ, Simeonov A, Austin CP, Inglese J. Characterization of chemical libraries for luciferase inhibitory activity. J. Med. Chem. 2008; 51:2372-2386.
53. Simeonov A, Yasgar A, Jadhav A, Lokesh GL, Klumpp C, Michael S, Austin CP, Natarajan A, Inglese J. Dual-fluorophore quantitative high-throughput screen for inhibitors of BRCT- phosphoprotein interaction. Anal. Biochem. 2008; 375:60-70.
54. Davis RE, Zhang YQ, Southall N, Staudt LM, Austin CP, Inglese J, Auld DS. A cell-based assay for IkappaBalpha stabilization using a two-color dual luciferase-based sensor. Assay Drug Dev. Technol. 2007; 5:85-104.
55. Fan F, Wood KV. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007; 5:127-136.
56. Dinger MC, Beck-Sickinger AG. Reporter gene assay systems for the investigation of G protein-coupled receptors. In Molecular Biology in Medicinal Chemistry. Dingermann T, Steinhiber D, Folkers G, eds. 2004. Wiley-VCH Verlag GmbH & Co., Weinheim, Germany. pp. 73-94.
57. Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science 2006; 312:217-224.
58. Tran TC, Sneed B, Haider J, Blavo D, White A, Aiyejorun T, Baranowski TC, Rubinstein AL, Doan TN, Dingledine R, Sandberg EM. Automated, quantitative screening assay for antiangiogenic compounds using transgenic zebrafish. Cancer Res. 2007; 67:11386-11392.
59. Burns AR, Kwok TC, Howard A, Houston E, Johanson K, Chan A, Cutler S.R, McCourt P, Roy PJ. High-throughput screening of small molecules for bioactivity and target identification in Caenorhabditis elegans. Nat. Protoc. 2006; 1:1906-1914.
60. Zlokarnik G. Fusions to beta-lactamase as a reporter for gene expression in live mammalian cells. Methods Enzymol. 2000; 326:221-244.
61. Verma R, Peters NR, D’Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 2004; 306:117-120.
62. Naylor LH. Reporter gene technology: the future looks bright. Biochem. Pharmacol. 1999; 58:749-757.
63. Peterson JR, Lebensohn AM, Pelish HE, Kirschner MW. Biochemical suppression of small-molecule inhibitors: a strategy to identify inhibitor targets and signaling pathway components. Chem. Biol. 2006; 13:443-452.
64. Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld D. S. High-throughput screening assays for the identification of chemical probes. Nat. Chem. Biol. 2007; 3:466-479.
65. Inglese J, Shamu CE, Guy RK. Reporting data from high- throughput screening of small-molecule libraries. Nat. Chem. Biol. 2007; 3:438. Screen. 2003; 8:381-392.
67. Iversen PW, Eastwood BJ, Sittampalam GS, Cox KL. A comparison of assay performance measures in screening assays: signal window, Z’ factor, and assay variability ratio. J. Biomol. Screen. 2006; 11:247-252.
68. Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999; 4:67-73.
69. Fox J, Long JS. Modern Methods of Data Analysis. 1990. Sage, Newbury Park, NJ.
70. Brideau C, Gunter B, Pikounis B, Liaw A. Improved statistical methods for hit selection in high-throughput screening. J. Biomol. Screen. 2003; 8:634-647.
71. Wu Z, Liu D, Sui Y. Quantitative assessment of hit detection and confirmation in single and duplicate high-throughput screenings J. Biomol. Screen. 2008; 13:159-167.
72. Eastwood BJ, Farmen MW, Iversen PW, Craft TJ, Smallwood JK, Garbison KE, Delapp NW, Smith GF. The minimum significant ratio: a statistical parameter to characterize the reproducibility of potency estimates from concentration-response assays and estimation by replicate-experiment studies. J. Biomol. Screen. 2006; 11:253-261.
73. Cohen S, Trinka RF. Fully automated screening systems. Methods Mol. Biol. 2002; 190:213-228.
74. Niles WD, Coassin pj. piezo- and solenoid Valve-Based Liquid Dispensing for Miniaturized Assays. Assay Drug Devel. Technol. 2005; 3:189-202.
75. Mere L, Bennett T, Coassin P, England P, Hamman B, Rink T, Zimmerman S, Negulescu P. Miniaturized FRET assays and microfluidics: key components for ultra-high-throughput screening. Drug Discov. Today 1999; 4:363-369.
76. Harris D, Mutz M, Sonntag M, Stearns R, Shieh J, Pickett S, Ellson R, Olechno J. Low nanoliter acoustic transfer of aqueous fluids with high precision and accuracy of volume transfer and positional placement. JALA 2008; 13:97-102
77. Beggs M, Blok H, Diels A. The high throughput screening infrastructure: the right tools for the task. J. Biomol. Screen. 1999; 4:143-149.
78. Bowen WP, Wylie PG. Application of laser-scanning fluorescence microplate cytometry in high content screening. Assay Drug Dev. Technol. 2006; 4:209-221.
79. Simeonov A, Jadhav A, Sayed AA, Wang Y, Nelson ME, Thomas CJ, Inglese J, Williams DL, Austin CP. Quantitative high-throughput screen identifies inhibitors of the Schistosoma mansoni redox cascade. PLoS Negl. Trop. Dis. 2008; 2:e127.
80. Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:3153-3158.
81. Rasmussen L, Maddox CB, Harten B, White EL. A successful LIMS implementation: case study at southern research institute. JALA 2007; 12:384-390
82. Engels MF, Wouters L, Verbeeck R, Vanhoof G. Outlier mining in high throughput screening experiments. J. Biomol. Screen. 2002; 7:341-351.
83. Fogel P, Collette P, Dupront A, Garyantes T, Guedin D. The confirmation rate of primary hits: a predictive model. J. Biomol. Screen. 2002; 7:175-190.
84. Pearson H. Genetics: what is a gene?. Nature 2006; 441:398-401.
85. Lazo JS, Brady LS, Dingledine R. Building a pharmacological lexicon: small molecule discovery in academia. Mol. Pharmacol. 2007; 72:1-7.
86. Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008; 26:127-132.
87. Zaman GJ, van der Lee MM, Kok JJ, Nelissen RL, Loomans EE. Enzyme fragment complementation binding assay for p38alpha mitogen-activated protein kinase to study the binding kinetics of enzyme inhibitors. Assay Drug Dev. Technol. 2006; 4:411-420.
88. Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. Journal of Biological Chemistry 1990; 265:17174-17179.
89. Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:13926-13931.
90. Richard AM, Gold LS, Nicklaus MC. Chemical structure indexing of toxicity data on the internet: moving toward a flat world. Curr. Opin. Drug Discov. Devel. 2006; 9:314-325.
91. Pereira DA, Williams JA. Origin and evolution of high throughput screening. Br. J. Pharmacol. 2007; 152:53-61.
92. Rognan D. Chemogenomic approaches to rational drug design. Br. J. Pharmacol. 2007; 152:38-52.
93. McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:19936-19941.
94. Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:11473-11478.
95. MacDonald ML, Lamerdin J, Owens S, Keon BH, Bilter GK, Shang Z, Huang Z, Yu H, Dias J, Minami T, Michnick SW, West- wick JK. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat. Chem. Biol. 2006; 2:329-337.
96. Yu H, West M, Keon BH, Bilter GK, Owens S, Lamerdin J, Westwick JK. Measuring drug action in the cellular context using protein-fragment complementation assays. Assay Drug Dev. Technol. 2003, 1, 811-822.
97. Michnick SW. Protein fragment complementation strategies for biochemical network mapping. Curr. Opin. Biotechnol. 2003; 14:610-617.
98. Root DE, Flaherty SP, Kelley BP, Stockwell BR. Biological mechanism profiling using an annotated compound library. Chem. Biol. 2003; 10:881-892.
99. Janzen WP, Hodge CN. A chemogenomic approach to discovering target-selective drugs. Chem. Biol. Drug Des. 2006; 67:85-86.
100. Zhou Y, Zhou B, Chen K, Yan SF, King FJ, Jiang S, Winzeler EA. Large-scale annotation of small-molecule libraries using public databases. J. Chem. Inf. Model 2007; 47:1386-1394.
101. Sayed AA, Williams DL. Biochemical characterization of 2-Cys peroxiredoxins from Schistosoma mansoni. J. Biol. Chem. 2004; 279:26159-26166.
102. Babaoglu K, Simeonov A, Irwin JJ, Nelson M E, Feng B, Thomas CJ, Cancian L, Costi MP, Maltby DA, Jadhav A, Inglese J, Austin CP, Shoichet BK. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-Lactamase. J. Med. Chem. 2008; 51:2502-2511.
103. Schuffenhauer A, Ertl P, Roggo S, Wetzel S, Koch MA, Waldmann H. The scaffold tree-visualization of the scaffold universe by hierarchical scaffold classification. J. Chem. Inf. Model 2007; 47:47-58.
104. Nomanbhoy TK, Rosenblum J, Aban A, Burbaum JJ. Inhibitor focusing: direct selection of drug targets from pro- teomes using activity-based probes. Assay Drug Dev. Technol. 2003; 1:137-146.
105. Hall SE. Chemoproteomics-driven drug discovery: addressing high attrition rates. Drug Discov. Today 2006; 11:495-502.
106. Kunz-Schughart LA, Freyer JP, Hofstaedter F, Ebner R. The use of 3-D cultures for high-throughput screening: the multicellular spheroid model. J. Biomol. Screen. 2004; 9:273-285.
107. Reininger-Mack A, Thielecke H, Robitzki AA. 3D-biohybrid systems: applications in drug screening. Trends Biotechnol. 2002; 20:56-61.
108. Lee MY, Kumar RA, Sukumaran SM, Hogg MG, Clark DS, Dordick JS. Three-dimensional cellular microarray for high- throughput toxicology assays. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:59-63.
109. Zheng W, Spencer RH, Kiss L. High throughput assay technologies for ion channel drug discovery. Assay Drug Dev. Technol. 2004; 2:543-552.
110. Robinson JP. Flow cytometry. In Encyclopedia of Biomaterials and Biomedical Engineering. Bowlin GL, Wnek G, eds. 2004. Informa Healthcare, New York. pp. 630-640.
111. Edwards BS, Oprea T, Prossnitz ER, Sklar LA. Flow cytometry for high-throughput, high-content screening. Curr. Opin. Chem. Biol. 2004; 8:392-398.
112. Krutzik PO, Crane JM, Clutter MR, Nolan GP. High-content single-cell drug screening with phosphospecific flow cytometry. Nat. Chem. Biol. 2008; 4:132-142.
113. Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:15835-15840.
114. Inglese J. Expanding the HTS paradigm. Drug Discov. Today 2002; 7:S105-106.
115. Landry Y, Gies JP. Drugs and their molecular targets: an updated overview. Fundam. Clin. Pharmacol. 2008; 22:1-18.
116. Chen S, Takanashi S, Zhang Q, Xiong W, Zhu S, Peters EC, Ding S, Schultz PG. Reversine increases the plasticity of lineage-committed mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:10482-10487.
117. Collins FS, Gray GM, Bucher JR. Toxicology. Transforming environmental health protection. Science 2008; 319:906-907.
118. Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:5163-5168.
119. Lartigue C, Glass JI, Alperovich N, Pieper R, Parmar PP, Hutchison CA 3rd, Smith HO, Venter JC. Genome transplantation in bacteria: changing one species to another. Science 2007; 317:632-638.
_______________________
* Additional reading on the subject of assay validation and specific protocols can be found online at the NCGC web site: Assay Guidance Manual (http://www.ncgc.nih.gov/guidance/index.html).