CHEMICAL BIOLOGY
Integrin Signaling, Bidirectional-Signaling of
Xiaoyang Wu and Jun-Lin Guan, Cornell University, Ithaca, New York
doi: 10.1002/9780470048672.wecb253
Integrins are a large family of heterodimeric receptors that mediate the adhesive behavior of cells. Most integrins bind to extracellular matrix (ECM) molecules, and they transmit signals that are critical in growth, development, tissue homeostasis, and host defense. A central feature of these receptors is their ability to transduce bidirectional signals into and out of the cell. In this article, we will give an overview of our current understanding of the structure and cellular signaling functions of integrins.
An amazing feature of life is that cells, the tiny units of an organism, can develop such elaborate molecular systems to sense all kinds of environmental information and translate it into various cellular responses to survive. The extracellular matrix (ECM) is composed of a complex mixture of polysaccharides and large fibrous proteins such as fibronectin, collagen, and laminin. ECM serves as a key environmental cue for cells in multicellular organisms, because many fundamental cellular processes, including proliferation, survival, migration, and differentiation, are regulated by the cells’ adherence to the ECM and the composition of the ECM (1, 2). In the 1980s, integrins were recognized as the major metazoan receptors for the ECM (3). Ever since, intensive efforts have been made to unravel its complex functions as an important signal transducer. Importantly, the underlying structure-activity relationships have also started to be elucidated by resolving the high resolution three-dimensional (3-D) structure of integrins. It is now widely appreciated that integrins serve as a pivotal module to mediate a bidirectional signal transduction, namely outside-in and inside-out signaling pathways (4). Upon engagement of ECM ligands, integrins make transmembrane connections to cytoskeleton and regulate many intracellular responses through the outside-in signaling. Conversely, intracellular signaling pathways can modulate integrin-mediated cell adhesion to ECM via the inside-out signaling. In this article, we summarize recent progress in this area by discussing the bidirectional signaling of integrins.
Integrin Family (An Overview)
A functional integrin is a heterodimeric protein complex consisting of an α and a β subunit. So far, the mammalian integrin family consists of 8 β subunits and 18 α subunits, which are known to assemble into 24 distinct integrins (Fig. 1). The different combination of subunits accounts for heterodimer specificity, with certain integrins showing preference for particular ECM molecules (4-6). Each subunit has a large extracellular domain, a single transmembrane (TM) segment, and a short cytoplasmic tail (with the exception of β4). The N-terminal portion of the a and p subunits associate to form the headpiece, which contains the ligand-binding site, whereas the C -terminal segments traverse the plasma membrane and mediate interaction of the integrin with the cytoskeleton and other signaling molecules. Hence, the exterior and interior of a cell are physically linked by integrins, which allows the bidirectional transmission of mechanical and biochemical signals across the plasma membrane, and leads to a cooperative regulation of cellular functions, including adhesion, migration, growth, survival, and differentiation.
Most integrins bind to ECM components. The ECM is a complex network of polysaccharides and proteins with high molecular weight, such as laminins, collagens, vitronectin, and fibronectin (1, 2). ECM proteins are secreted and organized by the cells. They, in turn, provide structural support for cells. In addition, the ECM functions as a physical barrier to or as a selective filter for soluble molecules (e.g., glomerular basement membrane). It has also been demonstrated that the ECM sequesters growth factors and plays a critical role in the differentiation and growth of various cell types. Integrins containing the α4, α5, α8, αIIb, or αV subunits recognize the RGD (Arg-Gly-Asp) motif of ECM components, namely fibronectin and vitronectin (4-6). Although laminins and collagens also contain the same RGD sequences, these are normally cryptic and inaccessible to the integrin receptors. Instead, these ECM proteins are recognized by integrins containing the α3, α6, and α7 subunits (laminin-binding receptors) or the α1, α2, α10, and an subunits (collagen-binding receptors).
In addition to serving as ECM receptors, some integrins (e.g., integrins of hematopoietic cells) can bind to counter-receptors on other cells, such as intercellular adhesion molecules (ICAMs) and the vascular cell adhesion molecule-i (VCAM-i) (7). Integrins of hematopoietic cells also recognize plasma proteins that are deposited at sites of injury (e.g., fibrinogen or von Willebrand factor) and complement factors. Other ligands of integrins include inhibitors of platelet aggregation that are secreted by leeches and ticks and the disintegrins present in snake venoms. Interestingly, several bacterial pathogens such as Borrelia burgdorferi (Lyme disease), Bordetella pertussis (whooping cough), and Yersinia spp. as well as viruses such as adenovirus, coxsackievirus, echovirus, foot-and-mouth disease virus, HIV (Tat protein), hantavirus, and papillomavirus use integrins as their receptors (8). Interactions between integrins and ADAMs (A disintegrin and metalloprotease) have also been documented (9).

Figure 1. Different combinations of 18 α and 8 β subunits in mammals give rise to 24 functional heterodimeric receptors.
Integrin Regulation of Outside-In Signaling
Integrins recognize positional cues encoded by the ECM and convert them into biochemical signals that control a wide spectrum of cellular behaviors. ECM binding to integrins leads to integrin clustering and recruitment of actin filaments and signaling of molecules to the cytoplasmic domain of integrins (4). These specialized, cell adhesion organelles and signaling centers are named focal complexes (nascent adhesion structures) or focal adhesions (FAs; fully formed, mature adhesion structures). It has been documented that more than 20 different important signaling proteins are recruited to the ECM/integrin-binding site. Because of the length limit of this article, we will only focus on the role of the MAPK (mitogen-activated protein kinase) pathway, FAK, ILK, and Rho family small G proteins in mediating the outside-in signaling of integrin (summarized in Fig. 2).

Figure 2. Integrin receptors relay signals from ECM to regulate a wide spectrum of cellular processes, including migration, cell cycle, survival, and cell differentiation. (Please see text for details of the outside-in signaling of integrins.)
MAPK Pathway
Activation of the MAPK pathway provides a common route leading to transcriptional regulation of genes that are crucial for cell-cycle progression and differentiation (10). In a classic model, activation of Ras via growth factor receptors (GFRs) leads to sequential stimulation of the protein kinases Raf, MEK, and, finally, the MAP kinases Erk1 and Erk2. It is now well established that, in addition to growth factors, many normal cells require adhesion to the ECM to proliferate (ii). Without cell adhesion to ECM, soluble mitogens alone cause a transient and relatively modest activation of Erk. On the other hand, integrin-mediated adhesion also causes a weak activation of Erk in cells deprived of growth factors. Transient activation of Erk is not sufficient to promote transcription of Cyclin D, but it induces transcription of the CDK inhibitor p21 (12). Combined activation of both integrins and GFRs is thus required to activate Ras-MAPK signaling beyond the threshold necessary for transcription of cyclin D (13). Interestingly, an in vivo study revealed that integrin αvβ3 antagonists reduced long-term Erk activation in endothelial cells, which supports the biologic relevance of the in vitro results (14).
Several signaling proteins involved in this pathway have been found in focal adhesion complexes. Based on current understanding, integrin signaling activates ERK through two major mechanisms: the SFK (Src Family Kinase)/FAK pathway, which is activated by most, perhaps all, integrins (will be discussed in details later); and the SFK/Shc pathway (15), which is activated by a subset of integrins through the transmembrane segment of their α subunits. Both pathways are required for efficient joint integrin/GFR signaling to ERK, as supported by the effect of mutations that inhibit either one of the two pathways (16-18). The a subunit-dependent SFK/Shc signaling is necessary and sufficient for activation of ERK in several cell types (18, 19). Shc is an adaptor protein containing SH2 and a phosphotyrosine binding (PTB) domain, which links tyrosine-phosphorylated signal transducers to Ras (15). Upon binding to activated receptors, Shc is phosphorylated on tyrosine, which serves as a binding site to the Grb2/mSOS complex. This process leads to the juxtaposition of the GTP exchange factor domain of mSOS to Ras, and it subsequently activates Ras. In addition, recent results indicate that Shc is also a potent PI3K/Akt activator (20). In hematopoietic cells, phosphorylated Shc recruits gab2 through Grb2. Gab2 in turn recruits the regulatory subunit of PI3K, which leads to PI3K activation.
In addition to the Shc-mediated response, some integrins may directly cooperate with GFRs. For example, the aVp3 integrin receptor has been found to uniquely associate with insulin receptor substrate 1 (IRS-1), which is a cytoplasmic signal transducer of the insulin and insulin-like growth factor receptors (IGFRs) (21). IRS-1 is tyrosine phosphorylated by the activated IGFR and subsequently binds to a variety of other signaling molecules. In cells that have adhered to ECM, such as vitronectin through the αVβ3 integrin, a subset of IRS-1 binds to the integrin. This interaction substantially enhances the growth-stimulating function of insulin and IGF. In another example, a 190-Kd protein that is tyrosine phosphorylated as a result of PDGF receptor activation also binds to αVβ3 receptor, which suggests that there could be cooperation between this integrin and the PDGF signaling pathway (22). Previous studies have also shown that engagement of β1 or αV integrin can induce tyrosine phosphorylation of EGFR (epidermal growth factor receptor) and partially activate EGFR signaling, which leads to subsequent activation of ERK1 (23, 24).
Integrin-mediated adhesion not only activates ERK but also JNK (25-27). JNK is another member of the MAPK family, which is activated by stress stimuli, such as UV-radiation, hyperosmolar conditions, and inflammatory cytokines (28). After activation, JNK translocates into the nucleus and phosphorylates the transcription factor c-Jun, thereby activating c-Jun-dependent transcription (29). c-Jun activation plays an indispensable role in initiation of DNA synthesis (30). Thus, cells derived from mice lacking JNK1 and JNK2 or c-Jun display defective proliferation (31, 32). Integrins can activate signaling to JNK in the absence of a significant contribution from GFRs. α6β4 can activate JNK through Shc and a downstream pathway involving Ras, PI3-K, and Rac (25), whereas β1 and αv integrins signal to JNK through the FAK/Cas/Rac pathway (27, 33). In addition, the β2 integrins are associated with JAB1, a coactivator of c-Jun. Upon their association, a subset of JAB1 moves to the nucleus and transactivates c-Jun-dependent transcription (34). The existence of multiple mechanisms of integrin-dependent regulation of c-Jun further highlights the importance of this pathway in integrin signaling.
FAK
Early studies on integrin-mediated cell adhesion and signaling demonstrated that cell adhesion to the ECM was accompanied by integrin aggregation, and that this clustering could trigger increased tyrosine phosphorylation of various intracellular proteins (35). As integrins lack intrinsic tyrosine kinase activity, proteins in the integrin/ECM-binding site (focal adhesions) were dissected carefully to identify potential tyrosine kinases that could mediate this signaling event. Interestingly, a predominant protein in focal adhesions, which was shown to undergo rapid tyrosine phosphorylation after integrin ligation and clustering, is a 120-kDa nonreceptor tyrosine kinase known as FAK (25, 36, 37). FAK lacks SH2 and SH3 domains, but contains an N-terminal FERM domain, a central kinase domain, and a C-terminal focal adhesion targeting (FAT) domain (38, 39). Direct association of FAK and integrin cytoplasmic tails has been demonstrated (40). However, the role of this direct interaction remains unclear. The integrin-binding site in FAK was mapped to the region N-terminal to the central kinase domain; however, this region is not required for localization of FAK to focal adhesions. Instead, the C -terminal FAT domain is both required and sufficient for FAK localization to focal adhesions. It has been shown that talin binds directly to the C -terminal domain of FAK (41) and also interacts with vinculin and thereby paxillin (42). Thus, the recruitment of FAK to activated integrin clusters could be indirect.
A tempting model has been proposed that, upon recruitment to focal adhesion complexes, FAK undergoes conformational change and interacts through its amino terminal domain with the integrin cytoplasmic tail (43-45). The amino terminal domain of FAK could fold back onto the kinase domain and play a negative autoregulatory role. Thus, this conformational change may be a prerequisite for FAK’s catalytic activity. The phosphorylation of FAK then initiates a cascade of phosphorylation events and new protein-protein interactions. Phosphotyrosine 397 serves as a binding site for Src family kinases via the SH-2 domain (46-48). Conversely, it has also been shown that tyrosine 397 is phosphorylated at a significantly lower level in cells lacking SFKs (49), suggesting that the SFK can phosphorylate FAK at Tyr 397 and then bind to it. Hence, SFKs could function both upstream and downstream of FAK. In addition, Src can phosphorylate FAK at other tyrosine residues, such as tyrosine 925, which creates a binding site for the Grb2-mSOS complex, resulting in activation of MAPK (50, 51).
The two major substrates of SFKs at focal adhesions are p130CAS and paxillin. CAS associates with FAK through the SH3 domain of CAS and the proline-rich motif at FAK C terminus (52). CAS contains a C-terminal proline-rich segment, through which it interacts with the SH3 domain of Src and likely other SFKs, and a large substrate domain, which contains several phosphotyrosine sites mediating the interaction with adaptor proteins, such as Crk and Nck (53). Crk associates with DOCK180 in many cells (54). DOCK180 is the mammalian counterpart of C. elegans gene, ced-5, which is involved in the regulation of phagocytosis and cell migration in nematode (55). Overexpression of DOCK180, along with CAS and Crk, can stimulate membrane ruffling, which is a hallmark of Rac activation. Crk also binds to the GTP exchange factor (GEF) C3G, which activates the Ras-related small G protein Rap1 (56, 57). Rap1 in turn reinforces integrin adhesion to the ECM by an inside-out activation mechanism (58-60). Additionally, because B-Raf is a target of Rap-1 and, like other Raf isoforms, promotes signaling to Erk, Crk/C3G signaling at focal adhesions results in activation of Erk in cells that express B-Raf (16). Paxillin is phosphorylated by SFKs at Tyr 31 and 118 and can recruit Crk as CAS (61). Furthermore, paxillin can associate with Csk, which suppresses SFKs (62), and PTP-PEST (63), which can dephosphorylate CAS (64). Thus, paxillin could potentially participate in a negative feedback loop. Paxillin also associates with p120Ras GAP (GTPase-activating protein) and prevents it from binding to and suppressing p190RhoGAP (65). Therefore, paxillin may inhibit Rho activity during the initial phase of cell adhesion. Furthermore, paxillin is associated with a complex containing the adaptor PKL, the GEF Pix/Cool, and the Cdc42/Rac target-effector PAK, and it may play a role in linking Cdc42 and Rac to PAK and its downstream targets, such as LIMK and MLCK (66, 67). Lastly, paxillin can recruit the Abl tyrosine kinase from the nucleus to focal adhesions upon integrin ligation (68). At focal adhesions, Abl phosphorylates Ena/VASP proteins, thus regulating actin cytoskeleton (69).
Phosphotyrosine 397 of FAK associate with the SH2 domain of p85 subunit of PI3-K (70). This direct interaction could activate PI3-K and thus promote cell survival. The same residue on FAK also mediated association with other proteins containing SH2 domain, including p120 Ras GAP, PLCγ, and GRB7 (71-73). In addition, FAK contains two proline-rich motifs at the C-terminal portion, which mediates interaction with various SH3-domain-containing proteins, such as CAS, Graf, ASAP1, and Endophilin A2 (74-77). N-terminal region of FAK has also been shown to associate with signaling proteins, such as N-WASP (78). Thus, FAK can relay the integrin signaling to affect a wide variety of pathways, which in turn regulate various cellular processes, including growth, survival, differentiation, and migration. In addition to the study on in vitro cell culture systems, the analysis of FAK knockout mice has provided important insight into the biological function of FAK in vivo. Constitutive as well as tissue-specific knockout of FAK have implicated it in many physiological and pathological processes, ranging from embryonic development, angiogenesis, and cortical basement membrane assembly or remodeling to tumorigenesis in skin, and heart hypertrophy (79-84).
ILK
ILK was identified by yeast two-hybrid screen for proteins that could bind to the cytoplasmic tail of integrin (85). The N -terminal domain of ILK contains three ankyrin repeats, which mediate protein interactions, and a putative fourth ankyrin repeat that lacks some conserved residues. The C-terminal portion shares significant sequence homology to Ser/Thr protein kinases. A pleckstrin homology (PH) domain is situated between these two domains. It is now widely accepted that ILK functions as the central component to organize a heterotrimeric protein complex named IPP (ILK, PINCH, and Parvin) complex (86, 87). ILK binds PINCH through the N-terminal ankyrin-repeat domain and parvins through the kinase domain. ILK may also link the IPP complex to the cytoplasmic tails of integrins. The IPP complex functions both as an adaptor between integrins and the actin cytoskeleton and as a hub that regulates various signaling pathways.
It remains controversial whether ILK is a real protein kinase. The kinase domain of ILK shows significant homology to Ser/Thr protein kinases, except residues within the catalytic loop and the conserved DXG motif (88). Thus, ILK lacks a conventional catalytic base and Mg2+-chelating residues. However, recombinant, purified ILK has been shown to be able to phosphorylate several substrates in vitro (89-91), including GSK3β and AKT, which regulate many different signaling pathways. It is well established that AKT activation requires phosphorylation of Thr at position 308 by phosphatidylinositol 3-kinase (PI3K)-dependent kinase-1 (PDK1) and Ser at position 473 by PDK2 [which is also known as hydrophobic-motif kinase (HMK)]. Recent studies suggest that ILK could function as an HMK, and this notion is further supported by immuno- precipitation assays showing that ILK directly binds to AKT (90). However, it is not clear whether ILK possesses sufficient activity to function as a physiologically relevant kinase in vivo.
The function of the IPP complex as a signaling platform could be achieved mainly through its interaction with other proteins. Many IPP functions are consistent with a role for the IPP complex at focal adhesions. For example, the regulation of podocyte adhesion and spreading (92), platelet aggregation (93, 94), neuronal spreading and outgrowth (95, 96), and leukocyte recruitment (97) all support the notion that the IPP complex regulates actin-cytoskeleton dynamics and integrin activation in focal adhesions. As mentioned, ILK binds directly to the cytoplasmic tails of integrins (85), and it is connected to the actin cytoskeleton through its interaction with parvins (98). Interactions with the cytoskeleton can also be mediated by the adaptor protein paxillin, which binds to ILK through a paxillin-binding site (PBS) within the kinase domain of ILK. Paxillin binds to F-actin via interactions with α-parvin and the actin-binding adaptor molecule vinculin (61, 98, 99). In addition, ILK binds to UNC-112, the Caenorhabditis elegans orthologue of vertebrate MIG2/kindlin-2, which binds to migfilin (100). Migfilin in turn binds to filamin—an adaptor protein that interacts with several molecules, including F-actin and integrins.
The presence of the different PINCH or parvin isoforms provides the signaling specificity of the IPP complexes. For example, PINCH2 can compete with PINCH1 for ILK binding, but it failed to relay integrin signals to cell spreading and migration (101). Functional differences between PINCH1 and PINCH2 might develop at least partially from differential interaction of the Ras-suppressor protein RSU1. RSU1 is a negative regulator of JNK activation (102-105). It associates with the LIM5 domain of PINCH1, but not PINCH2. In addition, PINCH1, but not PINCH2, associates with Thymosin-P4 through the LIM domains -4 and -5 of PINCH1 (106). This interaction can increase ILK activity and positively influences migration and survival of cardiac cells. PINCH1 also binds to NCK2, an adaptor protein in vitro through a LIM4-SH3-domain (Src-homology-3 domain) interaction (107, 108). However, the physiological relevance of this interaction is not clear. α-Parvin can bind to F-actin directly. It also interacts with paxillin, which could further bridge it with actin cytoskeleton. HIC5, a paxillin-related protein, also associates with α-parvin (98). Interestingly, HIC5 shuttles to the nucleus, where it modulates the expression of several genes (109, 110). Furthermore, α-parvin specifically binds to TESK1, a Ser/Thr kinase that phosphorylates cofilin (111). β-Parvin can bind to the actin-cross-linking protein α-actinin and the guanine nucleotide-exchange factor α-PIX (112, 113). It therefore provides a connection between the IPP complex and the Rho family GTPases, Rac1, and Cdc42. α-PIX, in turn, binds to PAK1 (114), a Rac1/Cdc42 effector that regulates cytoskeletal dynamics. Furthermore, α-PIX can associate with the protease subunit calpain-4 (115). Calpain-4 has been shown to cleave talin, which serves as the rate-limiting step in the disassembly of focal adhesions (116).
Cytoskeleton and Rho Family Small GTPase
The initial study from Alan Hall’s group described that addition of serum or LPA stimulates the formation of stress fibers and focal adhesion in serum-starved Swiss 3 T3 cells, which retain only a few stress fibers and are devoid of focal adhesions (117-120). Later, it was found that the induction of cortical actin assemblies and formation of focal adhesions can be ascribed to the action of different Rho family proteins. Rho family of proteins are Ras-related small GTPase consisting of Rho A, B, C, D, and E; Racs 1, 2, and E; and Cdc42, Rho G, and TC10 (121). It is now well established that Rho is involved in the organization of focal adhesions and stress fibers; Rac is responsible for the membrane ruffling and extension of lamellipodia; and Cdc42 controls the formation of filipodia (121).
In addition to the growth factor receptors, Rho family members can also be activated by integrins. During adhesion and spreading on an ECM, cells develop filopodia and lamellipodia, which are regulated by Cdc42 and Rac, respectively. Integrin-mediated adhesion can activate Cdc42 and Rac (122). Like other small GTPase, Rho family protein is active when GTP-bound and inactive when GDP-bound. Activation of Rho proteins is catalyzed by GEFs and inactivation is induced by GAPs that stimulate the intrinsic GTPase activity of the Rho proteins. Vav1, a hematopoietic cell-specific GEF, is activated upon integrin engagement (123). However, the closely related GEF Vav2, which is widely distributed, is activated downstream from growth factor receptors but not from integrins (124, 125). Nevertheless, a dominant-negative mutant of Vav2 blocked lamellipodium formation and spreading on fibronectin, suggesting that Vav2 plays a role in Rac activation after integrin engagement (126). Furthermore, integrins may activate Rac through other pathways. P130cas and paxillin associate with FAK, and both of them have been linked to Rac activation. As mentioned above, tyrosine phosphorylation of p130cas promotes the formation of a functional complex consisting of Crk, DOCK180, and ELMO (127). DOCK180 can function as a Rac GEF, even though it lacks the Dbl-homology/pleckstrin-homology tandem domains characteristic of conventional Rho family GEFs (127, 128). Paxillin associates with another complex consisting of PKL/GIT and Pak-interacting exchange factor (PIX), the latter being a conventional Rac GEF (61). Interestingly, recent work demonstrated that, in addition to their effect on GTP loading, integrins independently control the translocation of active Rac to the plasma membrane (129). This step is required for Rac binding to its downstream effectors. Integrins can increase membrane affinity for Rac, leading to RhoGDI dissociation and effector coupling locally, in the vicinity of integrins. Furthermore, integrin-regulated Rac binding sites are within cholesterol-enriched membrane microdomains or lipid rafts. Integrins control Rac signaling by preventing the internalization of Rac-binding sites in lipid rafts.
Regulation of Rho A activity by integrins is more complicated. It has been shown that integrin engagement leads to a transient depression in RhoA activity, followed by activation (130). The depression could play an essential role in lamellipodial extension during cell migration (131). The mechanism for this depression is not completely understood, but it may involves Src, FAK, paxillin, and p190RhoGAP (64, 65, 132, 133). However, different integrins may initiate different response of Rho A activity. Engagement of α6β4 resulted in stimulation of RhoA activity, in contrast to the depression induced by clustering of β1 integrins (134). Overexpression of β3 integrin in CHO cells resulted in a pronounced increase in Rho-GTP levels when the cells were plated on fibronectin or fibrinogen, whereas β1 integrin overexpression had no effect (135). In contrast, re-expression of β1 subunits in β1-deficient cells stimulated RhoA activity, whereas β3 had no effect (136). This controversy could be due to the difference of cell types used in these two studies.
Rho family proteins serve as important mediators for integrin outside-in signaling. Integrin clustering and focal complex formation requires the activity of Rho family proteins (137). It has been shown that integrins can stimulate the production of Phosphatidyl Inositol biphosphate (PIP2), and recent studies have provided evidence that this regulation is mediated by Rho family proteins (138, 139), possibly through its interaction with a type I isoform of phosphatidyl inositol 4-phosphate 5-kinase (PIP4-5K) (140). The increase in PIP2 synthesis by Rho A could be relevant to focal adhesion assembly because the actin-binding activity of several cytoskeletal proteins, such as profilin and gelsolin, is modulated by PIP2 (138) and PIP2 is enriched in focal adhesion plaques. As a result, Gilmore et al. reported that the association of PIP2 with vinculin induces a conformational change of vinculin, allowing it to associate with talin, which in turn binds actin (141). Consistent with this notion, injection of anti-PIP2 antibodies into fibroblasts inhibited Rho-mediated stress fiber and focal adhesion formation. Furthermore, dominant-negative mutant of Rho suppresses the activation of Erk in response to ligation of integrins, suggesting that a Rho-mediated signaling pathway is necessary for the activation of ERK after integrin-mediated cell adhesion (142). In addition to PIP4-5K, RhoA associates with and activates Rho kinase (143, 144). Rho kinase plays an important role in the assembly of focal adhesions. It phosphorylates the myosin-binding subunit of myosin light chain (MLC) phosphatase, thereby suppressing the enzymatic activity of the phosphatase (145). It has also been shown that Rho kinase can phosphorylate MLC at the same site phosphorylated by MLC kinase (146). The resulting increase in MLC phosphorylation could induce a conformational change in myosin, which would increase its binding to actin filaments, promote actomyosin contractility, and induce the formation of focal complexes and stress fibers (147).
Rho family proteins play a central role in remodeling of actin cytoskeleton. First, Rac/Cdc42 can stimulate new actin filament formation via activation of Arp2/3 complex, which initiates the formation of new actin filaments on the sides of existing filaments to form a branching actin filament network (148). Rho family protein can also activate Diaphanous-related formins (DRFs) to promote polymerization of actin monomers (149-151). Interestingly, Butler et al. have recently found that purifed aVp3 integrin complex can dramatically accelerate the rate of actin assembly, which is dependent on the sequestration of DRFs (152). In addition, Rac can stimulate actin polymerization by promoting the uncapping of cortical actin filaments (138). In resting state, actin filaments are capped at their barbed ends with capping proteins to prevent spontaneous actin polymerization. Similar to Rho, Rac acts via a phosphatidylinositide 4-phosphate 5-kinase (PIP 5-kinase) to induce the formation of PIP2 in platelets. PIP2 then displaces capping proteins from the barbed ends of actin filaments (153, 154). PAK (p21-activated kinase) is an important target of Rac/Cdc42 (155). Like Rho kinase, PAK can affect the phosphorylation of both myosin II heavy chain (156) and MLC (157, 158). As well as phosphorylating myosin, PAK can stimulate the activity of LIM-kinase (159). LIM-kinase phosphorylates, and inactivates cofilin, a protein that depolymerizes F-actin (160). There is strong evidence that cofilin is involved in lamellipodium extension and cell migration (161, 162), through promoting release of actin monomers that can then be reincorporated into growing actin filaments at the plasma membrane and/or by severing actin filaments and thereby providing more barbed ends for actin polymerization (163, 164).
Regulation of Integrin Through Inside-Out Signaling, a Structural View
As the signals that are transduced by integrins from the outside of the cell to the inside were being elaborated, parallel investigations on the regulation of integrin affinity revealed that information also flowed in the opposite direction—from integrin cytoplasmic tails to the extracellular ligand-binding domain, usually named the “inside-out” signaling of integrin. This process was actually described even before the molecular definition of integrins. Bennett and Vilaire found that the binding of platelets to fibrinogen is subject to regulation by agonists (165). It was then shown that this regulation was not dependent on the recruitment of receptors to the surface, rather to an increase in the binding activity of the receptor, and that leukocyte adhesion receptors were subjected to a similar regulation of ligand-binding affinity (166, 167). After cloning of integrins, it became clear that most leukocyte and platelet integrins, including β1-containing integrins, exist in a resting state until activated, and that a variety of agonists could regulate this process (4, 6). The physiological relevance of this regulation is obvious. It prevents the spontaneous adhesion of platelets and leukocytes within the circulation or to the blood vessel wall, but allows the rapid response upon injury.
Earlier studies using biophysical approches and antibodies that specifically recognize the activated state of integrins demonstrated that their activation involves alterations in integrin conformation (168-172) and that the cytoplasmic tails of integrins regulate ligand-binding activity (173). Regulation of inside-out signaling is multifaceted and has been well characterized in hematopoietic cells. For example, in lymphocytes, several signaling pathways have been implicated in regulation of integrin inside-out signaling, including 1) the pathway involving phosphatidylinositol 3-kinase, Rho, RAP1 (a Ras-related small GTPase), RAPL (regulator of adhesion and cell polarization enriched in lymphoid tissues), and DOCK2 (dedicator of cytokinesis 2), which together mediate chemokine-induced modulation of LFA1 (lymphocyte function-associated antigen 1) activity; 2) the pathway that involve TEC-family kinases, VAV1, ADAP (adhesion- and degranulation-promoting adaptor protein), RAP1, and RAPL, which are responsible for T-cell-receptor-triggered modulation of LFA1 avidity (174). Recent data has also focused attention on talin as an indispensable mediator of inside-out signaling (175). Changes in ligand-binding affinity are largely due to changes in the conformation of the extracellular domain, which are initiated by interactions at the integrin’s cytoplasmic face. Understanding the details of integrin activation is greatly facilitated by recent studies resolving the 3-D structure of integrins. Talin binds to most integrin β cytoplasmic tails through a structurally conserved phosphotyrosine-binding domain (PTB)-like interaction (176). Overexpression of talin fragments containing this PTB-like domain activates integrins, whereas knocking down talin expression with small interfering RNAs or sequestration of talin blocks β1 and β3 integrin activation in different cells (175, 177). Furthermore, mutations in conserved residues in talin or the integrin p tail that disrupt their interaction prevent activation (175). Thus, the binding of the talin PTB-like domain to the integrin p tail serves as a common step in integrin activation.
The conserved membrane-proximal portions of integrin α and β cytoplasmic tails control integrin activation. Mutation or truncation of this region results in constitutive integrin activation (178). Furthermore, replacement of the regions by heterodimeric coiled-coil peptides or a covalent cross-linking of the tails inactivates integrins, whereas breaking the coiled-coil or the covalent bond activates them (179). It has been proposed that the membrane-proximal regions of α and β cytoplasmic tails associate through a salt bridge between a conserved Arg in the α tail and an Asp in the β tail, thereby keeping the integrin heterodimer in an inactive state (178). The talin head domain can disrupt the salt bridge formed between αIIb-Arg995 and β3-Asp723 (180) (Figure 3). Therefore, talin binding may activate an integrin by disrupting a “clasp” between the α and β cytoplasmic tails, leading to tail separation and integrin activation. However, this clasp has not been consistently observed (179) in structural studies of isolated integrin cytoplasmic domains, and the interaction between tails was of low affinity. Therefore, although a and P tail interactions may occur via a salt bridge, the strength of this interaction appears to be modest.
In addition to the “clasp” model, a “piston” model has been proposed. In this model, it is the displacement of the membrane-proximal domain from the membrane that leads to integrin activation by shortening of the TM domain. Integrin cytoplasmic domains contain remarkably conserved sequences consisting of a Trp-Lys (Arg) doublet that is predicted to terminate the TM sequence. This doublet is followed by the membrane-proximal region, a hydrophobic stretch of four or five residues terminated by strongly polar residues, which are hotspots for mutations that activate integrins (181, 182). It has been shown that fragments of talin that activate integrins perturb NMR resonances in the membrane-proximal region, whereas a subdomain of talin that binds to the tail but fails to activate αIIbβ3 perturbs the NMR resonances to a much lesser extent. In addition, the αIIβ cytoplasmic tail, which blocks talin interaction with the membrane-proximal region of the β3 tail, prevents αIIbβ3 activation (183). Glycosylation mapping have indicated that the membrane-proximal domains of the α and β subunits can reside within the membrane and that certain activating mutations in this region can displace them from the membrane, thereby shortening the TM domains (184,185). Various targeted and random activating mutants of integrin αIIbβ3 would be predicted to shorten the TM domain in a similar manner (173, 181, 182, 186). However, studies with isolated αIIβ and β3 peptides and liposomes suggest that an upward movement of membrane-proximal helices upon talin binding could contribute to integrin activation (187). Resolution of this controversy will require further structural analysis of integrin.
Propagation of the inside-out activation signal across the plasma membrane is mediated by the TM helices. The TM helices of the inactive, but not active, integrin show a periodic disulfide cross-linking pattern (188), suggesting that the TM helices specifically interact in the inactive state and that this interaction is disrupted upon activation. Many membrane-embedded activating mutations identified by random and site-directed mutagenesis studies further support this notion (181, 189, 191). However, the molecular details of these interactions are not fully understood. Furthermore, CFP and YFP fluorophores fused to the C -termini of α and β subunits showed decreased fluorescence resonance energy transfer (FRET) upon integrin activation by a variety of stimuli (191), implying cytoplasmic domain separation during integrin activation. So the cytoplasmic domain separation, and therefore TM separation, might be the critical step in activation. However, another possibility cannot be ruled out—that mutations of the TM domain might lead to altered orientation of the TM domains instead of actual separation; such an alteration of orientation is also consistent with the reported FRET results (191).
A number of key questions concerning the integrin activation remain unresolved. Although we know that the membrane-proximal region and TM region of integrins controls the activation, other mutagenesis results indicate that the C-terminal membrane distal region of the α or β cytoplasmic tails is also important in regulating integrin activation via a mechanism that is yet unknown. Thus, the whole picture for the inside-out activation may be substantially more complicated. In addition, there may exist other proteins besides talin that bind to the cytoplasmic tails and that regulate the conformational change required for integrin activation. The current “clasp” or “piston” model explains how the alterations of the cytoplasmic tail and TM region of integrins relieve the structural constraint and allow the unbending of the extracellular domain to attain the high affinity ligand-binding state (192). However, a thorough molecular understanding of integrin inside-out signaling awaits further structural analysis of the intact receptor in inactive and active states.
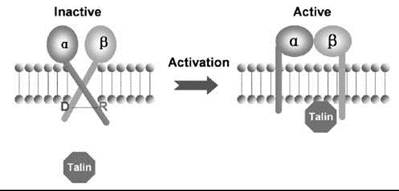
Figure 3. Talin binds to the cytoplasmic tail of the integrin β subunit and activates the ligand-binding affinity of integrin. The potential mechanisms could involve the disruption of ''clasp'' (the salt bridge between two polar residues, Asp and Arg, at the membrane proximal region), or shortening the transmembrane region of integrins (the ''piston'' model).
Concluding Remarks
Recent years have witnessed exciting progress in understanding integrin signaling, owing to structural analyses and characterization of proteins that interact with integrin. The role of integrins in vivo has also been revealed by the functional studies of gene-targeted mice (193). Interestingly, despite the fact that the binding specificities of many of the integrins overlap, the loss of almost any integrin subunit leads to defects with varying severity in knockout mice, providing the strongest evidence for biological relevance of integrin signaling. However, many molecular details of integrin signaling remain elusive. Answers are likely to come from using advanced techniques to study the structure of integrins in their native membrane environments, from determining how integrin-binding proteins receive and relay signals to imaging dynamic integrin interactions at the molecular level. Importantly, studies over the past two decades in this field have led to the development of clinically useful or promising integrin antagonists combating diseases such as cancer, cardiovascular defect, and inflammatory diseases (194). Thus, a thorough understanding of integrin signaling should lead to even greater opportunity for novel therapies.
References
1. Hay ED. Extracellula. matrix. J. Cell. Biol. 1981; 91:205s-223s.
2. Juliano RL, Haskill S. Signa. transduction from the extracellular matrix. J. Cell. Biol. 1993; 120:577-585.
3. Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science 1995; 268:233-239.
4. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002; 110:673-687.
5. Hynes RO. Integrins: a family of cell surface receptors. Cell 1987; 48:549-554.
6. Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992; 69:11-25.
7. Dejana E, Lauri D. Biochemical and functional characteristics of integrins: a new family of adhesive receptors present in hematopoietic cells. Haematologica 1990; 75:1-6.
8. Kerr JR. Cel. adhesion molecules in the pathogenesis of and host defence against microbial infection. Mol. Pathol. 1999; 52:220-230.
9. Bridges LC, Bowditch RD. ADAM-Integri. interactions: potential integrin regulated ectodomain shedding activity. Curr. Pharm. Des. 2005; 11:837-847.
10. Qi M, Elion EA. MAP kinase pathways. J. Cell. Sci. 2005; 118:3569-3572.
11. Lee JW, Juliano R. Mitogenic signal transduction by integrin- and growth factor receptor-mediated pathways. Mol. Cells 2004; 17:188-202.
12. Bottazzi ME, Zhu X, Bohmer RM, Assoian RK. Regulation of p21(cip1) expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell. Biol. 1999; 146:1255-1264.
13. Roovers K, Davey G, Zhu X, Bottazzi ME, Assoian RK. Alpha5beta1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol. Biol. Cell. 1999; 10:3197-3204.
14. Eliceiri BP, Klemke R, Stromblad S, Cheresh DA. Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell. Biol. 1998; 140:1255- 1263.
15. Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene 2001; 20:6322-6330.
16. Barberis L, Wary KK, Fiucci G, Liu F, Brancaccio M, Altruda F, Tarone G, Giancotti FG. Distinct roles of the adaptor protein Shc and focal adhesion kinase in integrin signaling to ERK. J. Biol. Chem. 2000; 275:36532-36540.
17. Hirsch E, Barberis L, Brancaccio M, Azzolino O, Xu D, Kyriakis JM, Silengo L, Giancotti FG, Tarone G, Fassler R, Altruda F. Defective Rac-mediated proliferation and survival after targeted mutation of the beta1 integrin cytodomain. J. Cell. Biol. 2002; 157:481-492.
18. Pozzi A, Wary KK, Giancotti FG, Gardner HA. Integrin alpha1beta1 mediates a unique collagen-dependent proliferation pathway in vivo. J. Cell. Biol. 1998; 142:587-594.
19. Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 1996; 87:733-743.
20. Gu H, Maeda H, Moon JJ, Lord JD, Yoakim M, Nelson BH, Neel BG. New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. Mol. Cell. Biol. 2000; 20:7109-7120.
21. Vuori K, Ruoslahti E. Association of insulin receptor substrate-1 with integrins. Science 1994; 266:1576-1578.
22. Schneller M, Vuori K, Ruoslahti E. Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J. 1997; 16:5600- 5607.
23. Moro L, Venturino M, Bozzo C, Silengo L, Altruda F, Beguinot L, Tarone G, Defilippi P. Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998; 17:6622-6632.
24. Bill HM, Knudsen B, Moores SL, Muthswamy SK, Rao VR, Brugge JS, Miranti CK. Epidermal growth factor receptor-dependent regulation of integrin-mediated signaling and cell cycle entry in epithelial cells. Mol. Cell. Biol. 2004; 24:8586-8599.
25. Mainiero F, Murgia C, Wary KK, Curatola AM, Pepe A, Blumemberg M, Westwick JK, Der CJ, Giancotti FG. The coupling of alpha6beta4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO J. 1997; 16:2365-2375.
26. MacKenna DA, Dolfi F, Vuori K, Ruoslahti E. Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Invest. 1998; 101:301-310.
27. Oktay M, Wary KK, Dans M, Birge RB, Giancotti FG. Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J. Cell. Biol. 1999; 145:1461-1469.
28. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002; 298:1911-1912.
29. Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr. Opin. Cell. Biol. 1997; 9:240-246.
30. Riabowol K, Schiff J, Gilman MZ. Transcription factor AP-1 activity is required for initiation of DNA synthesis and is lost during cellular aging. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:157-161.
31. Johnson RS., van Lingen B, Papaioannou VE, Spiegelman BM. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 1993; 7:1309-1317.
32. Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000; 288:870-874.
33. Dolfi F, Garcia-Guzman M, Ojaniemi M, Nakamura H, Matsuda M, Vuori K. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:15394-15399.
34. Bianchi E, Denti S, Granata A, Bossi G, Geginat J, Villa A, Rogge L, Pardi R. Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature 2000; 404:617-621.
35. Guan JL, Trevithick JE, Hynes RO. Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell. Regul. 1991; 2:951-964.
36. Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:5192-5196.
37. Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc. Natl. Acad Sci. U.S.A. 1992; 89:8487-8491.
38. Guan JL. Focal adhesion kinase in integrin signaling. Matrix Biol. 1997;16:195-200.
39. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell. Biol. 2005; 6:56-68.
40. Schaller MD, Otey CA, Hildebrand JD, Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J. Cell. Biol. 1995; 130:1181-1187.
41. Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. Interaction of focal adhesion kinase with cytoskeletal protein talin. J. Biol. Chem. 1995; 270:16995-16999.
42. Brown MC, Perrotta JA, Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J. Cell. Biol. 1996; 135:1109-1123.
43. Cooper LA, Shen TL, Guan JL. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol. Cell. Biol. 2003; 23:8030-8041.
44. Cohen LA, Guan JL. Mechanisms of focal adhesion kinase regulation. Curr. Cancer Drug Targets 2005; 5:629-643.
45. Cohen LA, Guan JL. Residues within the first subdomain of the FERM-like domain in focal adhesion kinase are important in its regulation. J. Biol. Chem. 2005; 280:8197-8207.
46. Parsons JT, Parsons SJ. Src family protein tyrosine kinases: cooperating with growth factor and adhesion signaling pathways. Curr. Opin. Cell. Biol. 1997; 9:187-192.
47. Schlaepfer DD, Hunter T. Integrin signalling and tyrosine phosphorylation: just the FAKs Trends Cell. Biol. 1998; 8:151-157.
48. Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci. 1999; 4:D102-D113.
49. Cary LA, Klinghoffer RA, Sachsenmaier C, Cooper JA. SRC catalytic but not scaffolding function is needed for integrin-regulated tyrosine phosphorylation, cell migration, and cell spreading. Mol. Cell. Biol. 2002; 22:2427-2440.
50. Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin- mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994; 372:786-791.
51. Schlaepfer DD, Broome MA, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130cas, and Nck adaptor proteins. Mol. Cell. Biol. 1997; 17:1702-1713.
52. Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J. Cell. Biol. 1998; 140:211-221.
53. O’Neill GM, Fashena SJ, Golemis EA. Integrin signalling: a new Cas(t) of characters enters the stage. Trends Cell. Biol. 2000; 10:111-119.
54. Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol. Cell. Biol. 1996; 16:1770-1776.
55. Wu YC, Horvitz HRC. Elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 1998; 392: 501-504.
56. Gotoh T, Hattori S, Nakamura S, Kitayama H, Noda M, Takai Y, Kaubuchi K, Matsui H, Hatase O, Takahashi H, Kurata T, Matsuda M. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol. Cell. Biol. 1995; 15:6746-6753.
57. Tanaka S, Morishita T, Hashimoto Y, Hattori S, Nakamura S, Shibuya M, Matuoka K, Takenawa K, Kurata T, Nagashima K, Matsuda M. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:3443-3447.
58. Ohba Y, Ikuta K, Ogura A, Matsuda J, Mochizuki N, Nagashima K, Kurokawa K, Mayer BJ, Maki K, Miyazaki J-I, Matsuda M. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 2001; 20:3333-3341.
59. Shimizu Y. Putting the rap on integrin activation. Immunol. Today 2000; 21:597.
60. Tsukamoto N, Hattori M, Yang H, Bos JL, Minato N. Rap1 GTPase-activating protein SPA-1 negatively regulates cell adhesion. J. Biol. Chem. 1999; 274:18463-18469.
61. Turner CE. Paxillin interactions. J. Cell. Sci. 2000; 113:4139- 4140.
62. Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Analysis of the binding of the Src homology 2 domain of Csk to tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-Src. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:3984-3988.
63. Shen Y, Schneider G, Cloutier JF, Veillette A, Schaller MD. Direct association of protein-tyrosine phosphatase PTP-PEST with paxillin. J. Biol. Chem. 1998; 273:6474-6781.
64. Angers-Loustau A, Cote JF, Charest A, Dowbenko D, Spencer S, Lasky LA, Tremblay ML. Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J. Cell. Biol. 1999; 144:1019-1031.
65. Tsubouchi A, Sakakura J, Yagi R, Mazaki Y, Schaefer E, Yano H, Sabe H. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell. Biol. 2002; 159:673-683.
66. Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J. Cell. Biol. 1999; 145:851-863.
67. West KA, Zhang H, Brown MC, Nikolopoulos SN, Riedy MC, Horwitz AF, Turner CE. The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J. Cell. Biol. 2001; 154:161-176.
68. Lewis JM, Schwartz MA. Integrins regulate the association and phosphorylation of paxillin by c-Abl. J. Biol. Chem. 1998; 273:14225-14230.
69. Reinhard M, Jarchau T, Walter U. Actin-based motility: stop and go with Ena/VASP proteins. Trends Biochem. Sci. 2001; 26:243-249.
70. Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phos- phatidylinositol 3-kinase. J. Biol. Chem. 1996; 271:26329-26334.
71. Hecker TP, Ding Q, Rege TA, Hanks SK, Gladson CL. Overexpression of FAK promotes Ras activity through the formation of a FAK/p120RasGAP complex in malignant astrocytoma cells. Oncogene 2004; 23:3962-3971.
72. Zhang X, Chattopadhyay A, Ji QS, Owen JD, Ruest PJ, Carpenter G, Hanks SK. Focal adhesion kinase promotes phospholipase C-gamma1 activity. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:9021-9026.
73. Han DC, Guan JL. Association of focal adhesion kinase with Grb7 and its role in cell migration. J. Biol. Chem. 1999; 274:24425-24430.
74. Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:10678-10682.
75. Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain- containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol. Cell. Biol. 1996; 16:3169- 3178.
76. Liu Y, Loijens JC, Martin KH, Karginov AV, Parsons JT. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol. Biol. Cell. 2002; 13:2147-2156.
77. Wu X, Gan B, Yoo Y, Guan JL. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev. Cell 2005; 9:185-196.
78. Wu X, Suetsugu S, Cooper LA, Takenawa T, Guan JL. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J. Biol. Chem. 2004; 279:9565-9576.
79. Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 1995; 377:539-544.
80. Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J. Cell. Biol. 2005; 169:941-952.
81. Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J. Cell. Biol. 2006; 172: 151-162.
82. Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 2003; 40:501-514.
83. McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, Hodivala-Dilke K, Metzger D, Chambon P, Grant SG, Frame MC. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004; 18:2998-3003.
84. Peng X, Kraus MS, Wei H, Shen T-L, Pariaut R, Alcaraz A, Ji G, Cheng L, Yang Q, Kotlikoff MI, Chen J, Chien K, Gu H, Guan J-L. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J. Clin. Invest. 2006; 116:217-227.
85. Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 1996; 379:91-96.
86. Wu C. PINCH, N(i)ck and the ILK: network wiring at cell-matrix adhesions. Trends Cell. Biol. 2005; 15:460-466.
87. Wu C. The PINCH-ILK-parvin complexes: assembly, functions and regulation. Biochim. Biophys. Acta 2004; 1692:55-62.
88. Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 1988; 241:42-52.
89. Deng JT, Van Lierop JE, Sutherland C, Walsh MP. Ca2+- independent smooth muscle contraction. a novel function for integrin-linked kinase. J. Biol. Chem. 2001; 276:16365-16373.
90. Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J. Biol. Chem. 2001; 276:27462-27469.
91. Persad S, Troussard AA, McPhee TR, Mulholland DJ, Dedhar S. Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J. Cell. Biol. 2001; 153:1161-1174.
92. Yang Y, Guo L, Blattner SM, Mundel P, Kretzler M, Wu C. Formation and phosphorylation of the PINCH-1-integrin linked kinase-alpha-parvin complex are important for regulation of renal glomerular podocyte adhesion, architecture, and survival. J. Am. Soc. Nephrol. 2005; 16:1966-1976.
93. Yamaji S, Suzuki A, Kanamori H, Mishima W, Takabayashi M, Fujimaki K, Tomita N, Fujisawa S, Ohno S, Ishigatsubo Y. Possible role of ILK-affixin complex in integrin-cytoskeleton linkage during platelet aggregation. Biochem. Biophys. Res. Commun. 2002; 297:1324-1331.
94. Pasquet JM, Noury M, Nurden AT. Evidence that the platelet integrin alphaIIb beta3 is regulated by the integrin-linked kinase, ILK, in a PI3-kinase dependent pathway. Thromb. Haemost. 2002; 88:115-122.
95. Chun SJ, Rasband MN, Sidman RL, Habib AA, Vartanian T. Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J. Cell. Biol. 2003; 163:397-408.
96. Mills J, Digicaylioglu M, Legg AT, Clint E, Young CE, Young SS, Alasdair M, Barr AM, Fletcher L, O’Connor TP, Dedhar S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J. Neurosci. 2003; 23:1638-1648.
97. Friedrich EB, Sinha S, Li L, Dedhar S, Force T, Rosenweig A, Gerszten RE. Role of integrin-linked kinase in leukocyte recruitment. J. Biol. Chem. 2002; 277:16371-16375.
98. Nikolopoulos SN, Turner CE. Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J. Cell. Biol. 2000; 151:1435-1448.
99. Turner CE, Glenney JR Jr, Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. J. Cell. Biol. 1990; 111:1059-1068.
100. Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 2003; 113:37-47.
101. Zhang Y, Chen K, Guo L, Wu C. Characterization of PINCH-2, a new focal adhesion protein that regulates the PINCH-1-ILK interaction, cell spreading, and migration. J. Biol. Chem. 2002; 277:38328-38338.
102. Tsuda T, Marinetti MR, Masuelli L, Cutler ML. The Ras suppressor RSU-1 localizes to 10p13 and its expression in the U251 glioblastoma cell line correlates with a decrease in growth rate and tumorigenic potential. Oncogene 1995; 11:397-403.
103. Vasaturo F, Dougherty GW, Cutler ML. Ectopic expression of Rsu-1 results in elevation of p21CIP and inhibits anchorage- independent growth of MCF7 breast cancer cells. Breast Cancer Res. Treat. 2000; 61:69-78.
104. Kadrmas JL, Smith MA, Clark KA, Pronovost SM, Muster N, Yates JR, Beckerle MC. The integrin effector PINCH regulates JNK activity and epithelial migration in concert with Ras suppressor 1. J. Cell. Biol. 2004; 167:1019-1024.
105. Dougherty GW, Chopp T, Qi SM, Cutler ML. The Ras suppressor Rsu-1 binds to the LIM 5 domain of the adaptor protein PINCH1 and participates in adhesion-related functions. Exp. Cell. Res. 2005; 306:168-179.
106. Bock-Marquette I, Saxena A, White MD, Dimaio JM, Srivastava D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature 2004; 432:466-472.
107. Velyvis A, Vaynberg J, Yang Y, Vinogradova O, Zhang Y, Wu C, Qin J. Structural and functional insights into PINCH LIM4 domain-mediated integrin signaling. Nat. Struct. Biol. 2003; 10:558-564.
108. Vaynberg J, Fukuda T, Chen K, Vinogradova O, Velyvis A, Tu Y, Ng L, Wu C, Qin J. Structure of an ultraweak protein-protein complex and its crucial role in regulation of cell morphology and motility. Mol. Cell. 2005; 17:513-523.
109. Kim-Kaneyama J, Shibanuma M, Nose K. Transcriptional activation of the c-fos gene by a LIM protein, Hic-5. Biochem. Biophys. Res. Commun. 2002; 299:360-365.
110. Shibanuma M, Kim-Kaneyama JR, Sato S, Nose K. A LIM protein, Hic-5, functions as a potential coactivator for Sp1. J. Cell. Biochem. 2004; 91:633-645.
111. LaLonde DP, Brown MC, Bouverat BP, Turner CE. Actopaxin interacts with TESK1 to regulate cell spreading on fibronectin. J. Biol. Chem. 2005; 280:21680-21688.
112. Yamaji S, Suzuki A, Kanamori H, Mishima W, Yoshimi R, Takasaki H, Takabayashi M, Fujimaki K, Fujisawa S, Ohno S, Ishigatsubo Y. Affixin interacts with alpha-actinin and mediates integrin signaling for reorganization of F-actin induced by initial cell-substrate interaction. J. Cell. Biol. 2004; 165:539-551.
113. Rosenberger G, Jantke I, Gal A, Kutsche K. Interaction of alphaPIX (ARHGEF6) with beta-parvin (PARVB) suggests an involvement of alphaPIX in integrin-mediated signaling. Hum. Mol. Genet. 2003; 12:155-167.
114. Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell. 1998; 1:183-192.
115. Rosenberger G, Gal A, Kutsche K. AlphaPIX associates with calpain 4, the small subunit of calpain, and has a dual role in integrin-mediated cell spreading. J. Biol. Chem. 2005; 280:6879- 6889.
116. Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell. Biol. 2004; 6:977-983.
117. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992; 70:401-410.
118. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response 137. to growth factors. Cell 1992; 70:389-399.
119. Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995; 81:53-62.
120. Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem. Soc. Trans. 1995; 23:456-459.
121. Hall A. Rho GTPases and the actin cytoskeleton. Science 1998; 279:509-514.
122. Price LS, Leng J, Schwartz MA, Bokoch GM. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell. 1998; 9:1863-1871.
123. Miranti CK, Leng L, Maschberger P, Brugge JS, Shattil SJ. Identification of a novel integrin signaling pathway involving the kinase Syk and the guanine nucleotide exchange factor Vav1. Curr. Biol. 1998; 8:1289-1299.
124. Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Mol. Cell. Biol. 2000; 20:7160-7169.
125. Moores SL, Selfors LM, Fredericks J, Breit T, Fujikawa K, Alt FW, Brugge JS, Swat W. Vav family proteins couple to diverse cell surface receptors. Mol. Cell. Biol. 2000; 20:6364-6373.
126. Marignani PA, Carpenter CL. Vav2 is required for cell spreading. J. Cell. Biol. 2001; 154:177-186.
127. Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell. Biol. 2002; 4:574-582.
128. Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell. Sci. 2002; 115:4901-4913.
129. Grande-Garcia A, Echarri A, Del Pozo MA. Integrin regulation of membrane domain trafficking and Rac targeting. Biochem. Soc. Trans. 2005; 33:609-613.
130. Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999; 18:578-585.
131. Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol. Biol. Cell. 2001; 12:2711-2720.
132. Ren XD, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell. Sci. 2000; 113:3673- 3678.
133. Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr. Biol. 2000; 10:719-722.
134. O’Connor KL, Nguyen BK, Mercurio AM. RhoA function in lamellae formation and migration is regulated by the alpha6beta4 integrin and cAMP metabolism. J. Cell. Biol. 2000; 148:253-258.
135. Miao H, Li S, Hu YL, Yuan S, Zhao Y, Chen BP, Puzon-McLaughlin W, Tarui T, Shyy JY, Takada Y, Usami S, Chien S. Differential regulation of Rho GTPases by beta1 and beta3 integrins: the role of an extracellular domain of integrin in intracellular signaling. J. Cell. Sci. 2002; 115:2199-2206.
136. Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J. Cell. Biol. 2002; 159:1071-1086.
137. Hotchin NA, Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J. Cell. Biol. 1995; 131:1857-1865.
138. Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell 1995; 82:643-653.
139. Chong LD, Traynor-Kaplan A, Bokoch GM, Schwartz MA. The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell 1994; 79:507-513.
140. Ren XD, Bokoch GM, Traynor-Kaplan A, Jenkins GH, Anderson RA, Schwartz MA. Physical association of the small GTPase Rho with a 68-kDa phosphatidylinositol 4-phosphate 5-kinase in Swiss 3T3 cells. Mol. Biol. Cell. 1996; 7:435-442.
141. Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature 1996;3 81:531-535.
142. Renshaw MW, Toksoz D, Schwartz MA. Involvement of the small GTPase rho in integrin-mediated activation of mitogen- activated protein kinase. J. Biol. Chem. 1996; 271:21691-21694.
143. Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, Kaibuchi K. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science 1997; 275:1308- 1311.
144. Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997; 11:2295-2322.
145. Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996; 273:245-248.
146. Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996; 271:20246-20249.
147. Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell. Biol. 1996; 133:1403-1415.
148. Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000; 29:545-576.
149. Schirenbeck A, Bretschneider T, Arasada R, Schleicher M, Faix J. The Diaphanous-related formin dDia2 is required for the formation and maintenance of filopodia. Nat. Cell. Biol. 2005; 7:619-625.
150. Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell. Biol. 2006; 16:522-529.
151. Kovar DR. Molecular details of formin-mediated actin assembly. Curr. Opin. Cell. Biol. 2006; 18:11-17.
152. Butler B, Gao C, Mersich AT, Blystone SD. Purified integrin adhesion complexes exhibit actin-polymerization activity. Curr. Biol. 2006; 16:242-251.
153. Carpenter CL, Tolias KF, Van Vugt A, Hartwig J. Lipid kinases are novel effectors of the GTPase Rac1. Adv. Enzyme Regul. 1999; 39:299-312.
154. Tolias KF, Hartwig JH, Ishihara H, Shibasaki Y, Cantley LC, Carpenter CL. Type Ialpha phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr. Biol. 2000; 10:153-156.
155. Zhao ZS, Manser E. PAK and other Rho-associated kinases— effectors with surprisingly diverse mechanisms of regulation. Biochem. J. 2005; 386:201-214.
156. van Leeuwen FN, van Delft S, Kain HE, van der Kammen RA, Collard JG. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat. Cell. Biol. 1999; 1:242-248.
157. Kiosses WB, Daniels RH, Otey C, Bokoch GM, Schwartz MA. A role for p21-activated kinase in endothelial cell migration. J. Cell. Biol. 1999; 147:831-844.
158. Daniels RH, Bokoch GM. p21-activated protein kinase: a crucial component of morphological signaling Trends Biochem. Sci. 1999; 24:350-355.
159. Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell. Biol. 1999; 1:253-259.
160. Stanyon CA, Bernard O. LIM-kinase1. Int. J. Biochem. Cell. Biol. 1999; 31:389-394.
161. Chen J, Godt D, Gunsalus K, Kiss I, Goldberg M, Laski FA. Cofilin/ADF is required for cell motility during Drosophila ovary development and oogenesis. Nat. Cell. Biol. 2001; 3:204-209.
162. Aizawa H, Sutoh K, Yahara I. Overexpression of cofilin stimulates bundling of actin filaments, membrane ruffling, and cell movement in Dictyostelium. J. Cell. Biol. 1996; 132:335-344.
163. Zebda N, Bernard O, Bailly M, Welti S, Lawrence DS, Condeelis JS. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J. Cell. Biol. 2000; 151:1119-1128.
164. Chan AY, Bailly M, Zebda N, Segall JE, Condeelis JS. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J. Cell. Biol. 2000; 148:531-542.
165. Bennett JS, Vilaire G. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J. Clin. Invest. 1979; 64:1393-1401.
166. Wright SD, Silverstein SC. Tumor-promoting phorbol esters stimulate C3b and C3b’ receptor-mediated phagocytosis in cultured human monocytes. J. Exp. Med. 1982; 156:1149-1164.
167. Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 1989;341:619- 624.
168. Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J. Biol. Chem. 1985; 260:11107-11114.
169. Altieri DC, Edgington TS. The saturable high affinity association of factor X to ADP-stimulated monocytes defines a novel function of the Mac-1 receptor. J. Biol. Chem. 1988; 263:7007-7015.
170. Keizer GD, Visser W, Vliem M, Figdor CG. A monoclonal antibody (NKI-L16) directed against a unique epitope on the alpha-chain of human leukocyte function-associated antigen 1 induces homotypic cell-cell interactions. J. Immunol. 1988; 140:1393-1400.
171. Parise LV, Helgerson SL, Steiner B, Nannizzi L, Phillips DR. Synthetic peptides derived from fibrinogen and fibronectin change the conformation of purified platelet glycoprotein IIb-IIIa. J. Biol. Chem. 1987; 262:12597-12602.
172. Sims PJ, Ginsberg MH, Plow EF, Shattil SJ. Effect of platelet activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J. Biol. Chem. 1991; 266:7345-7352.
173. O’Toole TE, Mandelman D, Forsyth J, Shattil SJ, Plow EF, Ginsberg MH. Modulation of the affinity of integrin alpha IIb beta 3 (GPIIb-IIIa) by the cytoplasmic domain of alpha IIb. Science 1991; 254:845-847.
174. Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 2005; 5:546-559.
175. Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: a final common step in integrin activation. Science 2003; 302:103-106.
176. Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, Ginsberg MH. The phosphotyrosine binding-like domain of talin activates integrins. J. Biol. Chem. 2002; 277:21749-21758.
177. Calderwood DA, Tai V, Dis Paolo G, De Camilli P, Ginsberg MH. Competition for talin results in trans-dominant inhibition of integrin activation. J. Biol. Chem. 2004; 279:28889-28895.
178. Liddington RC, Ginsberg MH. Integrin activation takes shape. J. Cell. Biol. 2002; 158:833-839.
179. Campbell ID, Ginsberg MH. The talin-tail interaction places integrin activation on FERM ground. Trends Biochem. Sci. 2004; 29:429-435.
180. Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas T, Plow E, Qin J. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell 2002; 110:587-597.
181. Partridge AW, Liu S, Kim S, Bowie JU, Ginsberg MH. Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J. Biol. Chem. 2005; 280:7294-7300.
182. Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J. Biol. Chem. 1996; 271:6571-6574.
183. Ulmer TS, Calderwood DA, Ginsberg MH, Campbell ID. Domain-specific interactions of talin with the membrane-proximal region of the integrin beta3 subunit. Biochemistry 2003; 42:8307-8312.
184. Stefansson A, Armulik A, Nilsson I, von Heijne G, Johansson S. Determination of N- and C-terminal borders of the transmembrane domain of integrin subunits. J. Biol. Chem. 2004; 279:21200-21205.
185. Armulik A, Nilsson I, von Heijne G, Johansson S. Determination of the border between the transmembrane and cytoplasmic domains of human integrin subunits. J. Biol. Chem. 1999; 274:37030-37034.
186. O’Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell. Biol. 1994; 124:1047-1059.
187. Vinogradova O, Vaynberg J, Kong X, Haas TA, Plow EF, Qin J. Membrane-mediated structural transitions at the cytoplasmic face during integrin activation. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:4094-4099.
188. Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS. Biol. 2004; 2:e153.
189. Luo BH, Carman CV, Takagi J, Springer TA. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:3679-3684.
190. Li W, Metcalf DG, Gorelik R, Li R, Mitra N, Nanda V, Law PB, Lear JD, Degrado WF, Bennett JS. A push-pull mechanism for regulating integrin function. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:1424-1429.
191. Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 2003; 301:1720-1725.
192. Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002; 110:599-611.
193. Bouvard D, Brakebusch C, Gustafsson E, Aszodi A, Bengtsson T, Berna A, Fassler R. Functional consequences of integrin gene mutations in mice. Circ. Res. 2001; 89:211-223.
194. Hamm CW. Anti-integrin therapy. Annu. Rev. Med. 2003; 54:425-435.