CHEMICAL BIOLOGY
Intramembrane Proteolysis
Michael S. Wolfe, Center for Neurologic Diseases, Harvard Medical School and Brigham and Women's Hospital, Boston, Massachusetts
doi: 10.1002/9780470048672.wecb258
Evolutionarily conserved membrane-embedded enzymes somehow use water to hydrolyze peptide bonds that reside within the lipid bilayer. These proteases are stitched into the membrane and bear little or no sequence similarity to other known proteases with one exception: All known intramembrane proteases apparently have converged on catalytic mechanisms that resemble those of proteases that reside in the aqueous milieu. The identities of these residues suggest that the intramembrane proteases use catalytic functionalities similar to those found in their soluble counterparts. However, in contrast to classical proteases, residues essential for catalysis lie within predicted hydrophobic transmembrane domains. Current research is focused on elucidating the range of biologic roles of these proteases, identifying their substrates, and understanding their structure and mechanism of action. Such knowledge is important because the intramembrane proteases discovered thus far play critical regulatory roles in biology and contribute to human disease.
Proteases catalyze the hydrolysis of the amide bonds that link amino acids together in peptides and proteins, and this process requires the concerted effort of key residues within the active site of the enzyme. These hydrolytic enzymes are classified into four general types based on their catalytic residues and mechanism of action: 1) serine/threonine proteases, 2) cysteine proteases, 3) aspartyl proteases, and 4) metalloproteases. Each of these four main protease categories contains hundreds of known examples and has representatives in all forms of life (1). Until recently, all the identified proteases had been water-soluble enzymes: Either the entire enzyme normally is found in an aqueous environment or a membrane anchor holds down an otherwise aqueous protease. In recent years, however, new proteases have been discovered that apparently are embedded within the hydrophobic environment of the lipid bilayer and yet somehow carry out hydrolysis on the transmembrane region of their substrates in the generally water-excluding environment of the membrane. Another unusual feature of this process are the substrates, which typically are folded into an a-helix, a conformation that makes the backbone amide bonds inaccessible to nucleophilic attack because of steric hindrance by the amino acid side chains. These intramembrane-cleaving proteases (I-CLiPs) (2) therefore must create an environment for water and the hydrophilic residues needed for catalysis and must bend or unwind their substrates to make the amide bonds susceptible to hydrolysis. Supporting these mechanistic notions is the observation that these newly discovered I-CLiPs apparently are variations on familiar themes in protease biochemistry: Despite the novelty of being membrane-embedded and cleaving transmembrane domains, the residues essential for catalysis by these I-CLiPs are virtually the same as those found in aqueous proteases.
The S2P Family of Metalloproteases
The first discovery of an I-CLiP arose from studies on the regulation of sterol and fatty acid metabolism. Sterol regulatory element binding proteins (SREBPs) are transcription factors that promote the expression of genes involved in the synthesis of cholesterol and fatty acids (3). Coordinated gene expression is controlled through negative feedback inhibition by cholesterol to ensure that lipids and sterols are produced only when needed. SREBPs are synthesized as a precursor protein that contains three distinct domains: a domain exposed to the cytosol that binds DNA and activates transcription, two transmembrane regions, and a regulatory domain involved in the feedback control by cholesterol (Fig. 1). When cholesterol levels are high, the SREBP precursor is kept in the endoplasmic reticulum (ER) by a multipass membrane protein called SCAP (SREBP cleavage-activating protein) (4) in a complex with a small membrane protein called Insig (5). Reduced cholesterol levels result in the dissociation of Insig from SCAP, which allows SCAP to shepherd SREBP to the Golgi apparatus. Proteolysis of SREBP in the Golgi results in the release of the transcription factor and its translocation to the nucleus.
Proteolytic release of SREBPs occurs in two steps (Fig. 1). First, the luminal loop between the two transmembrane regions is cleaved by the membrane-tethered Site-1 protease (S1P) (6). The release of the transcription factor requires subsequent cleavage by the Site-2 protease (S2P), which performs a hydrolysis of an amide bond predicted to lie three residues within the transmembrane domain (7). The requirement for a prior proteolytic event is a common theme with I-CLiPs. Complementation cloning identified S2P as a multipass membrane protein that contains a conserved HEXXH sequence characteristic of zinc metalloproteases (8). The two histidines and the glutamate are required for S2P activity, consistent with known metallo-protease biochemistry in which the two histidines coordinate with zinc and the zinc in turn activates the glutamate for interaction with the catalytic water. Additional analysis led to the discovery of a conserved aspartate located ~300 residues from the HEXXH sequence that is likewise critical for S2P activity and thought to be a third residue involved in zinc coordination (9). The involvement of zinc in S2P activity has not been demonstrated, and a cell-free assay for S2P activity has not been reported yet; therefore, S2P has not been shown directly to act as a protease (however, see below about bacterial S2Ps). Nevertheless, extensive genetic analysis has not uncovered any other proteins required for S2P cleavage of SREBP. Similar to SREBP, sequential processing by S1P and S2P of the otherwise membrane-associated transcription factor ATF6 are essential steps in the ER stress response (10).
More support for the proteolytic function of S2P comes from the discovery of a family of related proteins in bacteria (11). These prokaryotic proteins play an essential role in the proteolysis of otherwise membrane-bound transcription factors needed for sporulation. These factors control gene expression in the mother cell after engulfment of the forespore. The cleavage of pro-ok and the release of the transcription factor requires the multipass membrane protein SpoIVFB in Bacillus subtilis, and this protein likewise contains the HEXXH motif and a second conserved region with an aspartate, both of which are essential for proteolysis. Another bacterial S2P family member, YaeL (also called RseP) in Escherichia coli, similarly requires HEXXH and a conserved aspartate to play a role in coordinating cell growth and cell division, through intramembrane proteolysis of RseA, a factor critical for responding to extracytoplasmic stress (12). Interestingly, the membrane orientations of the substrates SREBP and ok are opposite to each other, which correlates with that of their respective enzymes, S2P and SpoIVFB, which similarly are thought to have opposite orientations (11). This opposition implies that the catalytic region must align with the peptide substrate with proper relative directionality. Although SpoIVFB and YaeL are both S2P-like enzymes that cleave transmembrane proteins during sporulation, the regulation of this key intramembrane proteolytic event for these two I-CLiPs is quite different. For cleavage of RseA by YaeL/RseP, the regulation is similar to that in SREBP cleavage by S2P: Intramembrane proteolysis requires a prior cleavage event outside the membrane by another protease called DegS (13). In contrast, SpoIVFB apparently does not require a prior proteolysis and regulation occurs more directly at the level of SpoIVFB. Two membrane proteins, BofA and SpoIVFA, serve to inhibit SpoIVFB activity, and this inhibition is released by the proteolysis of SpoIVFA by other proteases (14, 15) (Fig. 2).
As mentioned above, the a-helical conformation of the transmembrane substrate renders the amide bonds inaccessible to attack by a catalytic residue or water, which requires some bending or unwinding of the helix before proteolysis can occur. The SREBP substrate contains a conserved asparagine-proline (NP) sequence within its transmembrane region that is critical for proteolytic processing by S2P (16). These two residues have the lowest propensity to form a-helices, which suggests that the NP-containing SREBP transmembrane region may be metastable. After S1P cleavage and dissociation of the other transmembrane region, the NP sequence may facilitate unwinding of the residues immediately upstream, including the leucine-cysteine bond that gets cleaved. Unwinding may result in a protrusion of this bond to the membrane surface and access by the active site residues of S2P.
So far, one member of the S2P family has been purified to homogeneity with preservation of proteolytic activity (17). The purification of this protease, the E. coli YaeL/RseP, should allow a more rigorous and direct determination of substrate sequence requirements. Moreover, the ability to purify an I-CLiP and reconstitute activity (in appropriate detergent and usually in the presence of added lipids) is an important step toward studying these unusual membrane-embedded proteases in terms of structural biology. Indeed, one such protease has been seduced to crystallize, which provided the first detailed structures of an I-CLiP (see the section below on the Rhomboid serine proteases).

Figure 1. S2P contains conserved HEXXH and LDG motifs found in metalloproteases. SREBP first is cleaved by S1P in the luminal loop. The regulatory domain (Reg) interacts with the cholesterol-sensing SCAP to ensure that S1P proteolysis only occurs when cholesterol levels are low. Subsequent intramembrane proteolysis releases this transcription factor for the expression of genes essential to cholesterol and fatty acid synthesis.

Figure 2. Bacillus subtilis S2P-like protease SpoIVFB and sporulation. When the mother cell engulfs the forespore, a signaling pathway that involves the transcription factor σG is initiated in the forespore that triggers the synthesis of the IVB serine protease. This protease degrades SpoIVFA, which along with BofA serves to inhibit SpoIVFB. With the inhibition of the S2P-like protease released, SpoIVFB cleaves pro-σK, which allows this transcription factor to signal in the mother cell for more factors needed for spore maturation.
γ-Secretases: Presenilin-Containing Aspartyl Protease Complexes
A key step in the pathogenesis of Alzheimer’s disease is APP proteolysis that results in the formation of the amyloid-β peptide (Aβ), the principle protein component of the characteristic cerebral plaques of the disease (18). The N-terminus of Aβ is produced from the amyloid β-protein precursor (APP) by the action of β-secretase, which leads to membrane shedding of the large luminal/extracellular APP domain (Fig. 3a). The 99-residue remnant (C99) then is cleaved in the middle of its transmembrane region by y-secretase, which releases AP and is cleaved again near the inner leaflet at the 8 site to release the APP intracellular domain (AICD). As described below, chemical probes played important roles in the characterization, identification, purification, and mechanistic understanding of the I-CLiP that now is known as the γ-secretase complex.

Figure 3. Presenilin, the γ-secretase complex, and the proteolysis of APP to Aβ. (a) Presenilin is processed into two pieces, an N-terminal fragment (NTF, dark portion) and a C-terminal fragment (CTF, light portion) that remain associated. Each fragment donates one aspartate that is essential for γ-secretase activity. APP is cleaved first in the extracellular domain by β-secretase, and the remnant is cleaved twice within the membrane by γ-secretase to produce the Aβ peptide of Alzheimer's disease (secreted) and the intracellular domain (AICD, freed into the cytosol). (b) Presenilin interacts with three other membrane proteins, nicastrin, Aph-1, and Pen-2, to form active γ-secretase.
Two contemporaneous observations provided critical clues for the identification of the elusive γ-secretase, a subject of intense interest as a potential therapeutic target. First, the knockout of presenilin genes eliminated the γ-secretase cleavage of APP (19). Second, the types of compounds that could inhibit γ-secretase contained moieties typically found in aspartyl protease inhibitors (20). These findings led to the identification of two conserved transmembrane aspartates in the multipass presenilin that are critical for γ-secretase cleavage of APP (Fig. 3a), which suggests that presenilins might be the responsible aspartyl proteases (21). Presenilin is cut into two pieces, an N-terminal fragment (NTF) and a C-terminal fragment (CTF), the formation of which is gated by limiting cellular factor(s) (22). NTF and CTF remain physically associated in a high-molecular weight complex and are metabolically stable (23-25). These and other results suggested that the NTF-CTF heterodimer is the biologically active form (26). Intriguingly, the NTF and CTF each contribute one of the critical and conserved aspartates, which suggests that the y-secretase active site might be at the interface between these two presenilin fragments. In strong support of this hypothesis, transition-state analog inhibitors of γ-secretase, compounds designed to interact with the active site of the protease (Fig. 4), were found to bind directly to presenilin NTF and CTF (27, 28). However, presenilins apparently are part of a larger multiprotein complex that constitutes γ-secretase (see below).

Figure 4. Chemical tools for the study of γ-secretase. Transition-state analog inhibitors include hydroxyl-containing moieties that interact with the catalytic aspartates of aspartyl proteases. Helical peptides mimic the APP transmembrane domain and interact with the substrate docking site on the protease. These potent inhibitors were converted into affinity labeling reagents that contain a chemically reactive bromoacetyl or photoreactive benzophenone for covalent attachment to the protein target and a biotin moiety to allow isolation and detection of the labeled protein. Both types of chemical probes interacted with the two presenilin subunits but at distinct locations, which suggests that both the active site and the docking site of y-secretase lie at the interface between these subunits.
At the same time that presenilins were discovered as susceptibility loci for Alzheimer’s disease, they also were shown to be required for Notch signaling (29), a pathway essential for cell differentiation during development and beyond (30). After Notch is synthesized in the ER, the receptor is cleaved in its extracellular domain during its passage through the secretory pathway and the two pieces so generated remain associated (31). When interacting with a cognate ligand, Notch becomes susceptible to a second extracellular proteolysis near the membrane (S2 in Fig. 5) (32, 33). The membrane-associated remnant is then cleaved at the S3 site by γ-secretase (34), which releases the Notch intracellular domain (NICD). NICD translocates to the nucleus and activates transcription after associating with the nuclear partner CSL (CBP/RBPjk, Su(H), Lag-1) (35). The knockin of a Notch-1 transmembrane mutation that greatly reduces presenilin-mediated proteolysis at S3 leads to a lethal phenotype in mice similar to that seen in Notch-1 knockout mice, which indicates that efficient γ-secretase cleavage is essential for Notch signaling during development (36).

Figure 5. Proteolytic processing and signaling of the Notch receptor. In the ER, Notch is cleaved at SI by a furin-like protease to produce a stable heterodimeric receptor that is trafficked to the cell surface. Interaction with ligands such as the proteins Delta and Jagged triggers a shedding of the ectodomain by membrane-tethered metalloprotease-mediated cleavage at S2. The remnant then is cleaved at least twice, at the S3 and S4 sites, to release the Notch counterpart of Aβ (Nβ) and the intracellular domain (NICD). The latter translocates to the nucleus where it interacts with transcription factors to influence gene expression relevant to cell differentiation.
The highly conserved role of γ-secretase in Notch signaling and its importance in development made possible genetic screens in worms that identified two Notch modifiers, a single-pass membrane protein APH-2 (nicastrin) and a multipass protein APH-1 (37-39). Nicastrin independently was isolated biochemically as a presenilin-associated protein and later found to be essential for γ-secretase processing of both APP and Notch (40). A saturation screen in C. elegans for presenilin modifiers netted all these proteins and added Pen-2. All four proteins (presenilin, nicastrin, Aph-1, and Pen-2) associate with one another (41, 42) and with an immobilized y-secretase inhibitor (42, 43). Moreover, their coexpression increased γ-secretase activity in both Drosophila and mammalian cells (41, 42) and reconstituted activity in yeast (44). Because yeast have no such protease activity and contain no apparent orthologs of these metazoan proteins, these findings strongly suggest that this quartet of proteins is necessary and sufficient for γ-secretase activity. γ-Secretase is so far unique among intramembrane proteases in being composed of several different proteins; all others (see below) apparently work alone as single proteins. Coexpression, RNA interference, and the identification of assembly intermediates suggest the order in which these four subunits come together (41, 45, 46), and partial dissociation of the protease complex with detergent offers a model for how these subunits interact (Fig. 3b) (47). Nicastrin and Aph-1 together can stabilize full-length presenilin, and the final addition of Pen-2 apparently triggers presenilin endoproteolysis and γ-secretase activity (41). Pen-2 also is required to stabilize the presenilin subunits (48). The specific biochemical functions of these presenilin cofactors have been mostly enigmatic; however, nicastrin recently has been discovered to play a role in substrate recognition (see below).
Since the discovery that Notch is cleaved by γ-secretase, a plethora of other substrates have been identified, including Erb-B4, E-and N-cadherins, CD44, the low-density lipoprotein receptor, Nectin-1, and the Notch ligands Delta and Jagged (49). Knowledge of the cellular functions of these proteolytic events vary, but in the case of N-cadherin, the produced intracellular domain associates with the transcriptional activator CBP (CREB binding protein) and promotes its migration to the cytosol and its degradation by the proteasome (50). Although cellular function can be ascribed in some cases, the ability of y-secretase to cleave so many different substrates and its apparently poor sequence specificity raises the question of whether a major role of this enzyme is to serve as a general degrading protease for membrane-bound protein remnants. Indeed, γ-secretase seems to be unique among intramembrane proteases in its ability to process so many different substrates. The broad substrate recognition by γ-secretase likely is related to the fact that, unlike the other intramembrane proteases, the enzyme does not require helix-breaking residues near the cleavage sites within the substrates.
Among the more intriguing questions about the entire emerging family of I-CLiPs is how they handle substrates and cleave within their TMDs. Because it presumably contains water and uses hydrophilic residues, the membrane-embedded active site should be sequestered from the hydrophobic environment of the surrounding lipid tails. Thus, the active site might be envisioned to be part of a pore or channel that could allow the entry of water (2). However, the substrate passes through the membrane and cannot enter such a pore or channel directly; docking on the outer surface of the protease, with lateral gating to bring the substrate into the internal active site, might be required (2). Initial evidence for such a mechanism came from the isolation of the γ-secretase complex with an immobilized transition-state analog inhibitor (43). Detergent-solubilized membranes from human HeLa cells were passed through this affinity matrix, which resulted in the copurification of γ-secretase complex members and an endogenous membrane-bound APP stub found in HeLa cells. This stub results from the alternative processing of APP by α-secretases, and like the stub produced by β-secretase it also is a γ-secretase substrate. Thus, an endogenous substrate copurified with the γ-secretase complex while the protease active site was blocked by the immobilized transition-state analog inhibitor, which suggests the existence of a separate substrate binding site. Substrate bound to this special type of exosite, dubbed the “docking site,” could copurify without being subject to proteolysis.
Designed peptides based on the transmembrane domain of APP and constrained in a helical conformation potently can inhibit γ-secretase, apparently by interacting with this docking site (51). Conversion of these helical peptide inhibitors into affinity labeling reagents (Fig. 4) led to the localization of the substrate docking site to the presenilin NTF/CTF interface (52). Transition-state analog inhibitors also bind directly to the NTF/CTF interface but at a site distinct from that of the helical peptide inhibitors (Fig. 6). These findings suggest a pathway for γ-secretase substrate from docking site to active site: When binding to the outer surface of presenilin at the NTF/CTF interface, the substrate can pass, either in whole or in part, between these two presenilin subunits to access the internal active site (Fig. 6). Interestingly, the extension of a 10-residue helical peptide inhibitor by just 3 additional residues resulted in a potent inhibitor (53) apparently capable of binding both docking site and active site (52), which suggests that these two substrate binding sites are relatively close.

Figure 6. Model for how inhibitors and substrates interact with presenilin. Helical peptides are docking site inhibitors (DSIs) and interact on the outside of the presenilin molecule at the NTF/CTF heterodimeric interface. Transition-state analog inhibitors (TSAs) interact on the inside of the presenilin molecule where the active site resides. The active site, which contains water and two aspartates, is thought to be sequestered away from the hydrophobic environment of the lipid bilayer. These findings have implications for how substrate interacts with the enzyme. The transmembrane domain of the substrate (S) interacts with the docking site and passes either in whole or in part into the active site for proteolysis.
Up until recently, all the action seemed to be taking place on presenilin. However, an elegant study has demonstrated that nicastrin also plays a critical role in substrate recognition (54). The ectodomain of nicastrin bears sequence resemblance to aminopeptidases, although certain catalytic residues are not conserved. Nevertheless, nicastrin recognizes the N-terminus of γ-secretase substrates derived from APP and Notch, and mutation of the aminopeptidase domain prevents this interaction. One conserved glutamate is especially important probably because this residue forms an ion pair with the amino terminus of the substrate. The sequence of the substrate N-terminus apparently is not critical for the interaction, but a free amino group is. Indeed, the simple formylation of the substrate N-terminus is enough to prevent effective substrate interaction and proteolytic processing. Thus, nicastrin can be thought of as a kind of gatekeeper for the y-secretase complex: Type I membrane proteins that have not shed their ectodomains cannot interact properly with nicastrin and do not gain access to the active site.
Although γ-secretase has in many ways been an attractive target for Alzheimer therapeutics (see inhibitor in clinical trials, Fig. 7), interference with Notch processing and signaling may lead to toxicities that preclude the clinical use of inhibitors of this protease. Long-term treatment with γ-secretase inhibitors causes severe gastrointestinal toxicity and interferes with the maturation of B-and T-lympocytes in mice, effects that are indeed because of the inhibition of Notch processing and signaling (55, 56). However, compounds that can modulate the enzyme to alter or block Aβ production with little or no effect on Notch would bypass this potential roadblock to therapeutics. Recent studies suggest that the protease complex contains allosteric binding sites that can alter substrate selectivity and the sites of substrate proteolysis. Certain nonsteroidal anti-inflammatory drugs (NSAIDs; e.g., ibuprofen, indomethacin, and sulindac sulfide) can reduce the production of the highly aggregation-prone AP42 peptide and increase a 38-residue form of AP, a pharmacologic property independent of inhibition of cyclooxygenase (57). The alteration of the proteolytic cleavage site is observed with isolated or purified y-secretase (58-60), which indicates that the compounds can interact directly with the protease complex to exert these effects. Enzyme kinetic studies and displacement experiments suggest that the selective NSAIDs can be noncompetitive with respective to APP substrate (60) and to a transition-state analog inhibitor, which suggests an interaction with a site distinct from the active site (61). The site of cleavage within the Notch transmembrane domain is affected similarly, but this subtle change does not inhibit the release of the intracellular domain and thus does not affect Notch signaling (62). For this reason, these agents may be safer as Alzheimer therapeutics than inhibitors that block the active site or the docking site. Indeed, one compound, R-flurbiprofen (Fig. 7), has advanced recently to Phase III clinical trials. Surprisingly, the site of proteolytic cleavage by the presenilin homologue SPP also can be modulated by the same NSAIDs that affect y-secretase. Because SPP apparently does not require other protein cofactors, these findings suggest that presenilin is the site of NSAID binding within the γ-secretase complex and that SPP and presenilin share a conserved drug binding site for the allosteric modulation of substrate cleavage sites (63).
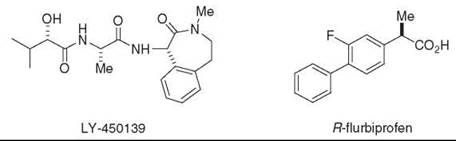
Figure 7. γ-Secretase inhibitor (LY-450,139) and NSAID-like modulator (R-Flurbiprofen) in clinical trials for Alzheimer's disease.
Another type of allosteric modulator is compounds that resemble kinase inhibitors and interact with a nucleotide binding site on the γ-secretase complex. The discovery that ATP can increase AP production in membrane preparations prompted the testing of a variety of compounds that interact with ATP binding sites on other proteins (64). In this focused screen, the Abl kinase inhibitor Gleevec emerged as a selective inhibitor of AP production in cells without affecting the proteolysis of Notch. In light of these findings, ATP and other nucleotides were tested for effects on purified y-secretase preparations and found to increase selectively the proteolytic processing of a purified recombinant APP-based substrate without affecting the proteolysis of a Notch counterpart (65). Furthermore, certain compounds known to interact with ATP binding sites were found to inhibit selectively the APP processing vis-a-vis Notch in purified protease preparations. These and other results suggest that the γ-secretase complex contains a nucleotide binding site and that this site allows the allosteric regulation of y-secretase processing of APP with respect to Notch. Whether this regulation is physiologically important is unclear, but the pharmacologic relevance is profound and may lead to new therapeutic candidates for Alzheimer's disease.
The purification of the γ-secretase complex (59) has allowed the first glimpse into its structure. Electron microscopy and single particle analysis reveals that the complex has a globular structure that at low resolution (10-15 A) appears rather amorphous (66). [Another structure, elucidated in a similar manner but of poorer resolution (~45 A), also has been reported (67).] Nevertheless, two important features can be gleaned. The first is a rather large interior cavity of ~20 A diameter that is presumably where the active site resides, a characteristic reminiscent of the proteasome. The second is the presence of two small openings that may be the site of entry for water. Other structural features have been revealed by cysteine mutagenesis with the cross-linking of chemical probes (68, 69). The generation of a cysteine-less version of presenilin (that retains the ability to assemble with other complex members, to undergo endo-proteolysis to NTF and CTF, and to process APP) allowed incorporation of single cysteine resides at various sites near the key aspartates. Disulfide formation with thiol-containing reagents then provided information about the relative accessibility of these sites from the aqueous milieu, which allowed the construction of a model in which water can funnel down to where the aspartates reside. More detailed information likely will require a crystal structure of presenilin or a presenilin homologue (see below).
SPP Aspartyl Proteases
The concept of presenilin as the catalytic component for y-secretase was strengthened considerably when signal peptide peptidase (SPP) was found to be a similar intramembrane aspartyl protease. SPP clears remnant signal peptides from the membrane after their production by signal peptidase (Fig. 8). However, this process apparently also plays a role in immune surveillance, in which signal peptides from the major histocompatibility complex (MHC) type I are cleaved by SPP and the peptide products are presented onto the cell surface as an indication to natural killer cells whether MHC synthesis is proceeding normally (70). In addition, SPP is exploited by the hepatitis C virus for the maturation of its core protein, which suggests that this protease may be a suitable target for antiviral therapy (71). SPP was identified by affinity labeling with a peptidomimetic inhibitor, and the protein sequence displayed intriguing parallels with presenilin (Fig. 8) (72). SPP contains two conserved aspartates, each predicted to lie in the middle of a transmembrane domain, and the aspartate-containing sequences resemble those found in presenilins. The predicted topology of SPP also resembles that of presenilins, placing the key aspartates in the same relative position to each other in the membrane. As with S2P compared with its bacterial relatives, the orientation of the aspartate-containing transmembrane domains of SPP apparently is opposite that of presenilins, again in correlation with the orientation of SPP substrates, which is opposite that of y-secretase substrates. Interestingly, before the identification of SPP, a computational search for presenilin-like proteins netted an entire family of so-called presenilin homologs (PSHs) (73); however, it is not yet clear if all these proteins have catalytic activity. Two homologs, SPP-like proteases SPPL2a and SPPL2b, recently have been found to cleave tumor necrosis factor a (74), although the biologic role of this proteolysis is unknown.
SPP seems to be less complicated than y-secretase. The expression of human SPP in yeast reconstituted the protease activity, which suggests that the protein has activity on its own and does not require other mammalian protein cofactors (72). Moreover, unlike presenilins, SPP is not processed into two pieces. Thus, SPP may be a more tractable enzyme for understanding this type of intramembrane aspartyl protease and may shed light on γ-secretase structure and function. Indeed, the catalytic sites of the two proteases seem remarkably similar; their activities are inhibited by some of the same active site-directed peptidomimetics (75, 76) and helical peptides (77), and activity can be modulated by the same NSAIDs that affect γ-secretase (77). SPP forms a homodimer very rapidly in cells, and this dimer is stable enough to allow isolation and analysis (78). Moreover, this dimer can be labeled specifically by a transition-state analog inhibitor, which suggests that the dimer is catalytically active. The functional importance of this dimer, however, is unclear; dimerization may not be necessary for proteolytic activity. In terms of substrate recognition, however, SPP does display an important difference with γ-secretase: the apparent requirement for helix-breaking residues that should facilitate the ability of the enzyme to access the site of hydrolysis (79).

Figure 8. Comparison of signal peptide peptidase (SPP) with presenilin and the γ-secretase complex. Signal peptides are removed from membrane proteins via signal peptidase (SP), and these peptides are released from the membrane by SPP-mediated intramembrane proteolysis. SPP, like presenilin, contains two aspartates that are essential for protease activity, but the conserved aspartate-containing motifs are in the opposite orientation compared with their presenilin counterparts. Consistent with the flipped orientation of SPP vis-a-vis presenilin, the substrates of these two proteases also run in the opposite direction. Unlike presenilin, SPP apparently does not require other protein cofactors or cleavage into two subunits for proteolytic activity.
Rhomboid Serine Proteases
The study of a conserved growth factor signaling pathway also led to intramembrane proteolysis. Epidermal growth factor (EGF) receptor ligands are synthesized as single-pass membrane proteins, but signaling requires the proteolytic release and secretion of the ligand for interaction with its cognate receptor. In vertebrates, this release and secretion is accomplished by membrane-tethered metalloproteases. Genetic analysis in Drosophila, however, identified two essential players, dubbed Star and Rhomboid-1, in the proteolysis of an EGF ortholog Spitz. No other components apparently are required. Full-length Spitz remains in the ER until it is ushered by Star to the Golgi apparatus where it encounters Rhomboid-1 (80). Rhomboid-mediated proteolysis in the Golgi then is followed by secretion for intercellular communication. But how does Rhomboid allow cleavage of Spitz?
Mutational analysis of conserved nonglycine residues revealed a tantalizing requirement for a serine, a histidine, and an asparagine, which together might serve as a catalytic triad typically found in serine proteases (81) [although subsequent studies supported Ser-His dyad (82)] (Fig. 9). These three residues were predicted to reside about the same depth within the membrane and thus have the potential to interact with each other. Consistent with this idea, the cleavage site of Spitz was estimated to be at an equivalent depth in the transmembrane region, and Spitz cleavage was sensitive only to serine protease inhibitors. Moreover, a careful analysis of concentration dependence revealed that expression of catalytic amounts of Rhomboid-1 still allowed Spitz proteolysis. Taken together, Rhomboid-1 apparently is a novel intramembrane serine protease.

Figure 9. Rhomboids contain a conserved serine and histidine, which comprise a putative catalytic dyad of a serine protease. Rhomboid-1 cleaves within the transmembrane region of the Drosophila EGF-like growth factor Spitz.
What determines Rhomboid substrate specificity, and how is this proteolytic event regulated? Most of the Spitz transmembrane region could be swapped with that of a nonsubstrate protein without affecting cleavage by Rhomboid; however, the N-terminal quarter of the transmembrane region was critical for substrate recognition (83). Indeed, incorporation of this substrate motif into Delta allowed this Notch ligand to be processed by Rhomboid. Additional examination of the substrate motif led to the tentative identification of a critical glycine-alanine, which suggests that, as with S2P and SPP, Rhomboid seems to require helix-destabilizing residues within the transmembrane domain of its substrates. Rhomboid activity is distinguished from that of the other I-CLiPs because Rhomboid does not require prior substrate cleavage by another protease. Rhomboid regulation apparently occurs mainly by the translocation of the substrate from the ER to the Golgi (mediated by Star) and the spatial control of Rhomboid transcription.
Like S2P, Rhomboid genes have been conserved throughout evolution. Surprisingly, despite overall low homology with eukaryotic Rhomboids, several bacterial Rhomboids could cleave Drosophila Rhomboid substrates and mutation of the putative catalytic triad residues abolished protease activity, which illustrates the evolutionary conservation of the serine protease function of Rhomboid (84). The natural substrates for the bacterial Rhomboids are unknown. As for substrates of eukaryotic Rhomboid-1 homologues, two mitochondrial membrane proteins have been identified as substrates for the yeast Rhomboid RBD1 (85-87). The RBDl-mediated release of one of these substrates is essential for the remodeling of the mitochondrial membrane, and the human ortholog of RBD1, PARL, could restore the substrate proteolysis and proper growth rates and mitochondrial morphology in a yeast RBD1 mutant (86), which suggests that the role of these Rhomboids in mitochrondrial function has been conserved evolutionarily. Indeed, a recent study identified a mitochondrial protein OPA1 as a likely substrate for PARL, the cleavage of this substrate being critical to cristae remodeling and cytochrome c release during apoptosis (88). In Toxoplasma, TgROM5, one of five nonmitochondrial Rhomboids in these parasites, cleaves a cell surface adhesion protein as a key step in cell invasion; similar findings in the related Plasmodium falciparum, the malarial parasite, recently have been reported (89), which suggests that Rhomboids are potential targets for treating infections by these deadly pathogens.
Most recently, the first crystal structures of an I-CLiP have been reported by three different research groups, all on the E. coli Rhomboid GlpG (90-92). These structures show remarkable similarities and important differences that provide insight into how this class of membrane-embedded protease carries out hydrolysis in the lipid bilayer. The structures all reveal that the key serine and histidine implicated as the catalytic dyad indeed are coordinated with each other and lie at a depth within the membrane consistent with where Rhomboids cleave their transmembrane substrates (Fig. 10). A cavity is open to the periplasmic space with the catalytic dyad at the bottom of this opening, and this cavity contains multiple water molecules. How substrate enters this cavity is not entirely clear, but the position of the transmembrane domain 5 varies in the different structures and the movement of this domain can provide a space through which substrate may reach the catalytic dyad. Indeed, one reported structure contains a bound lipid in this space (92) with the phosphate group residing near the Ser-His dyad and a key Asn residue that may contribute to the oxyanion hole that stabilizes intermediates and transition-states during serine protease catalysis. These exciting structural findings validate the molecular and biochemical studies on Rhomboids and suggest that such approaches have been providing true mechanistic insight into the workings of other I-CLiPs. Moreover, these structures offer details that inspire specific hypotheses about how Rhomboids handle substrates to hydrolyze transmembrane domains.

Figure 10. Structure of E. coli Rhomboid GlpG. The serine in transmembrane domain 4 and the histidine in transmembrane domain 6 are coordinated in a manner consistent with known serine proteases and at a depth within the membrane consistent with the site of proteolysis of Rhomboid substrates.
Perspective
I-CLiPs are membrane-embedded enzymes that hydrolyze transmembrane substrates, and the residues essential to catalysis reside within the boundaries of the lipid bilayer. These proteases seem to recapitulate the mechanisms of soluble proteases, and the first crystal structures of an E. coli Rhomboid support this notion, at least for the serine I-CLiPs. All I-CLiPs would be predicted to contain an initial substrate docking site, but to date evidence for such a docking site has been provided only for y-secretase. The I-CLiPs discovered so far each play critical roles in biology and are regulated closely, but the means of control vary. They all are involved in cell signaling but do so in a variety of ways. Membrane topology seems to dictate the types of substrates that can be cleaved, but this concept remains speculative. Most I-CLiPs seem to require helix-breaking residues near the cleavage sites of their substrates, although y-secretase may be a notable exception.
Critical remaining issues include the identity of substrates for the I-CLiP family members whose roles are unknown. For instance, although an entire family of PSHs and Rhomboids have been discovered, natural substrates are known only for a handful of these proteins. The conservation of putative catalytic residues implies a conservation of the proteolytic function, but the search for substrates is far from trivial. A computational approach for sequence motifs that apparently are required for substrate proteolysis by Rhomboids led to the identification of adhesion proteins in Toxoplasma as potential substrates (83). Most typically, genetic and cell biologic studies suggest a connection between protease and putative substrate, with follow-up molecular and biochemical studies for validation. Another key issue is understanding the specific mechanisms of these proteases (e.g., elucidating conformational changes that take place in both enzyme and substrate during proteolysis, determining if these changes require the input of energy, and identifying enzyme residues that directly interact with substrate). Structural biology clearly is the emerging frontier in the study of I-CLiPs, with Rhomboid providing the first fruits of such endeavors. A detailed structural understanding should provide clearer appreciation for how these remarkable enzymes work. The development of small molecule tools ultimately should dovetail with these structural studies and allow cocrystal structures that offer more insight into their mechanism and that pave the way for structure-based design in cases where the target has high therapeutic relevance.
Acknowledgment
I would like to acknowledge a postdoctoral fellow in my lab, Dr. Raquel Lieberman, for rendering Fig. 10.
References
1. Handbook of Proteolytic Enzymes 1998. Academic Press, New York.
2. Wolfe MS, De Los Angeles J, Miller DD, Xia W, Selkoe DJ. Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer’s disease. Biochemistry 1999; 38:11223-11230.
3. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997; 89:331-340.
4. Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:11235-11240.
5. Rawson RB. The SREBP pathway-insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 2003; 4:631-640.
6. Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL, Brown MS. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol. Cell 1998; 2:505-514.
7. Duncan EA, Dave UP, Sakai J, Goldstein JL, Brown MS. Second-site cleavage in sterol regulatory element-binding protein occurs at transmembrane junction as determined by cysteine panning. J Biol Chem 1998; 273:1780 1-17809.
8. Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1997; 1:47-57.
9. Zelenski NG, Rawson RB, Brown MS, Goldstein JL. Membrane topology of S2P, a protein required for intramembranous cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999; 274:21973-21980.
10. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000; 6:1355-1364.
11. Rudner DZ, Fawcett P, Losick R. A family of membrane- embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:14765-14770.
12. Kanehara K, Akiyama Y, Ito K. Characterization of the yaeL gene product and its S2P-protease motifs in Escherichia coli. Gene 2001; 281:71-79.
13. Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev. 2002; 16:2156-2168.
14. Zhou R, Kroos L. Serine proteases from two cell types target different components of a complex that governs regulated intramembrane proteolysis of pro-sigmaK during Bacillus subtilis development. Mol. Microbiol. 2005; 58:835-846.
15. Campo N, Rudner DZ. A branched pathway governing the activation of a developmental transcription factor by regulated intramembrane proteolysis. Mol. Cell 2006; 23:25-35.
16. Ye J, Dave UP, Grishin NV, Goldstein JL, Brown MS. Asparagineproline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:5123-5128.
17. Akiyama Y, Kanehara K, Ito K. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J. 2004; 23:4434-4442.
18. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297:353-356.
19. De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998; 391:387-390.
20. Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Donkor IO, Selkoe DJ. Peptidomimetic probes and molecular modeling suggest Alzheimer’s γ-secretases are intramembranecleaving aspartyl proteases. Biochemistry 1999; 38:4720-4727.
21. Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999; 398:513-517.
22. Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J. Biol. Chem. 1997; 272:28415-28422.
23. Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, et al. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin. J. Biol. Chem. 1998; 273:16470-16475.
24. Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C. The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J. Biol. Chem. 1998; 273:3205-3211.
25. Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH. Subcellular distribution and turnover of presenilins in transfected cells. J. Biol. Chem. 1998; 273:12436-12442.
26. Laudon H, Mathews PM, Karlstrom H, Bergman A, Farmery MR, Nixon RA, Winblad B, Gandy SE, Lendahl U, Lundkvist J, et al. Co-expressed presenilin 1 NTF and CTF form functional gamma-secretase complexes in cells devoid of full-length protein. J. Neurochem. 2004; 89:44-53.
27. Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000; 405:689-694.
28. Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai J-Y, Rahmati T, Xia W, Selkoe DJ, Wolfe MS. Transition-state analogue inhibitors of y-secretase bind directly to presenilin-1. Nature Cell Biol. 2000; 2:428-434.
29. Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 1995; 377:351-354.
30. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science 1999; 284:770-776.
31. Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israel A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. U.S.A. 1998;95: 8108-8112.
32. Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. A novel proteolytic cleavage involved in notch signaling: the role of the disintegrin-metalloprotease TACE. Mol. Cell 2000; 5:207-216.
33. Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A Ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of notch1. Mol. Cell 2000; 5:197-206.
34. De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. A presenilin-1-dependent y-secretase-like protease mediates release of Notch intracellular domain. Nature 1999; 398:518-522.
35. Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998; 393:382-386.
36. Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, Kopan R. Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature 2000; 405:966-970.
37. Goutte C, Hepler W, Mickey KM, Priess JR. aph-2 encodes a novel extracellular protein required for GLP-1-mediated signaling. Development 2000; 127:2481-2492.
38. Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 2002; 3:85-97.
39. Goutte C, Tsunozaki M, Hale VA, Priess JR. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:775-779.
40. Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 2000; 407:48-54.
41. Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003;422:438-441.
42. Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:6382-6387.
43. Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS. Activity-dependent isolation of the presenilin/γ-secretase complex reveals nicastrin and a γ substrate. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:2720-2725.
44. Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 2003 5:486-488.
45. LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ. Assembly of the gamma-Secretase Complex Involves Early Formation of an Intermediate Subcomplex of Aph-1 and Nicastrin. J. Biol. Chem. 2003; 278:37213-37222.
46. Hu Y, Fortini ME. Different cofactor activities in gamma-secretase assembly: evidence for a nicastrin-Aph-1 subcomplex. J. Cell. Biol. 2003; 161:685-690.
47. Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, Wolfe MS. Detergent-dependent dissociation of active y-secretase reveals an interaction between Pen-2 and PS1-NTF and offers a model for subunit organization within the complex. Biochemistry. In Press.
48. Prokop S, Shirotani K, Edbauer D, Haass C, Steiner H. Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the gamma-secretase complex. J. Biol. Chem. 2004; 279:23255-23261.
49. De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003; 38:9-12.
50. Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PSl/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 2003; 114:635-645.
51. Das C, Berezovska O, Diehl TS, Genet C, Buldyrev I, Tsai JY, Hyman BT, Wolfe MS. Designed helical peptides inhibit an intramembrane protease. J. Am. Chem. Soc. 2003; 125:11794-11795.
52. Kornilova AY, Bihel F, Das C, Wolfe MS. The initial substratebinding site of gamma-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:3230-3235.
53. Bihel F, Das C, Bowman MJ, Wolfe MS. Discovery of a subnanomolar helical D-tridecapeptide inhibitor of y-secretase. J. Med. Chem. 2004; 47:3931-3933.
54. Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE 3rd, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005; 122:435-447.
55. Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, Gao H, Higgins MA, May PC, Ryan TP. Adipsin: a biomarker of gastrointestinal toxicity mediated by a functional gamma secretase inhibitor. J. Biol. Chem. 2003; 29:29.
56. Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee HJ, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004; 279:12876-12882.
57. Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 2001; 414:212-216.
58. Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J. Biol. Chem. 2003; 278:31831-31837.
59. Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, Van Dorsselaer A, Wang R, Selkoe DJ, Wolfe MS. Purification and Characterization of the Human gamma-Secretase Complex. Biochemistry 2004; 43:9774-9789.
60. Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, Natsugari H, Fukuyama T, Tomita T, Iwatsubo T. Sulindac sulfide is a noncompetitive gamma-secretase inhibitor that preferentially reduces Abeta 42 generation. J. Biol. Chem. 2003; 278:18664-18670.
61. Beher D, Clarke EE, Wrigley JD, Martin AC, Nadin A, Churcher I, Shearman MS. Selected non-steroidal anti-inflammatory drugs and their derivatives target gamma-secretase at a novel site. Evidence for an allosteric mechanism. J. Biol. Chem. 2004; 279:43419-43426.
62. Okochi M, Fukumori A, Jiang J, Itoh N, Kimura R, Steiner H, Haass C, Tagami S, Takeda M. Secretion of the Notch-1 Abeta-like peptide during Notch signaling. J. Biol. Chem. 2006; 281:7890- 7898.
63. Sato T, Nyborg AC, Iwata N, Diehl TS, Saido TC, Golde TE, Wolfe MS. Signal peptide peptidase: biochemical properties and modulation by nonsteroidal anti-inflammatory drugs. Biochemistry. In Press.
64. Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:12444-12449.
65. Fraering PC, Ye W, Lavoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. gamma -Secretase substrate selectivity can be modulated directly via interaction with a nucleotide binding site. J. Biol. Chem. 2005; 280:41987-41996.
66. Lazarov VK, Fraering PC, Ye W, Wolfe MS, Selkoe DJ, Li H. Electron microscopic structure of purified, active gamma-secretase reveals an aqueous intramembrane chamber and two pores. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:6889-6894.
67. Ogura T, Mio K, Hayashi I, Miyashita H, Fukuda R, Kopan R, Kodama T, Hamakubo T, Iwatsubo T, Tomita T, et al. Three-dimensional structure of the gamma-secretase complex. Biochem. Biophys. Res. Commun. 2006; 343:525-534.
68. Tolia A, Chavez-Gutierrez L, De Strooper B. Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the gamma -secretase complex. J. Biol. Chem. 2006; 281:27633-27642.
69. Sato C, Morohashi Y, Tomita T, Iwatsubo T. Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J. Neurosci. 2006; 26:12081-12088.
70. Lemberg MK, Bland FA, Weihofen A, Braud VM, Martoglio B. Intramembrane proteolysis of signal peptides: an essential step in the generation of HLA-E epitopes. J. Immunol. 2001; 167: 6441-6446.
71. McLauchlan J, Lemberg MK, Hope G, Martoglio B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002; 21:3980-3988.
72. Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 2002; 296:2215-2218.
73. Ponting CP, Hutton M, Nyborg A, Baker M, Jansen K, Golde TE. Identification of a novel family of presenilin homologues. Hum. Mol. Genet. 2002; 11:1037-1044.
74. Fluhrer R, Grammer G, Israel L, Condron MM, Haffner C, Friedmann E, Bohland C, Imhof A, Martoglio B, Teplow DB, et al. A gamma-secretase-like intramembrane cleavage of TNFalpha by the GxGD aspartyl protease SPPL2b. Nat. Cell Biol. 2006; 8:894-896.
75. Kornilova AY, Das C, Wolfe MS. Differential effects of inhibitors on the gamma-secretase complex. mechanistic implications. J. Biol. Chem. 2003; 278:16470-16473.
76. Weihofen A, Lemberg MK, Friedmann E, Rueeger H, Schmitz A, Paganetti P, Rovelli G, Martoglio B. Targeting presenilin-type aspartic protease signal peptide peptidase with gamma -secretase inhibitors. J. Biol. Chem. 2003; 278:16528-16533.
77. Sato T, Nyborg AC, Iwata N, Diehl TS, Saido TC, Golde TE, Wolfe MS. Signal peptide peptidase: biochemical properties and modulation by nonsteroidal antiinflammatory drugs. Biochemistry 2006; 45:8649-8656.
78. Nyborg AC, Kornilova AY, Jansen K, Ladd TB, Wolfe MS, Golde TE. Signal peptide peptidase forms a homodimer that is labeled by an active site directed y-secretase inhibitor. J. Biol. Chem. 2004; 5:5.
79. Lemberg MK, Martoglio B. Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol. Cell 2002; 10:735-744.
80. Lee JR, Urban S, Garvey CF, Freeman M. Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell 2001; 107:161-171.
81. Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell 2001; 107:173-182.
82. Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 2005; 24:464-472.
83. Urban S, Freeman M. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol. Cell 2003; 11:1425-1434.
84. Urban S, Schlieper D, Freeman M. Conservation of intramembrane proteolytic activity and substrate specificity in prokaryotic and eukaryotic rhomboids. Curr. Biol. 2002; 12:1507-1512.
85. Esser K, Tursun B, Ingenhoven M, Michaelis G, Pratje E. A novel two-step mechanism for removal of a mitochondrial signal sequence involves the mAAA complex and the putative rhomboid protease Pcp1. J. Mol. Biol. 2002; 323:835-843.
86. McQuibban GA, Saurya S, Freeman M. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 2003; 423:537-541.
87. Herlan M, Vogel F, Bornhovd C, Neupert W, Reichert AS. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J. Biol. Chem. 2003; 278:27781-27788.
88. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006; 126:177-189.
89. Baker RP, Wijetilaka R, Urban S. Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog. 2006; 2:e113.
90. Wang Y, Zhang Y, Ha, Y. Crystal structure of a rhomboid family intramembrane protease. Nature 2006; 444:179-180.
91. Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat. Struct. Mol. Biol. 2006; 13:1084-1091.
92. Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc. Natl. Acad. Sci. U.S.A. In Press.
See Also
Peptidomimetics
PeptidomimeticsProtease Pathways, Small Molecules to Elucidate
Proteolysis and Disease, Chemical Biology of
Signal Transduction Across Membranes