CHEMICAL BIOLOGY
Bacterial DNA polymerases, Chemistry of
Richard T. Pomerantz and Mike O'Donnell, Rockefeller University, New York, New York
doi: 10.1002/9780470048672.wecb033
All DNA polymerases share a common two-metal ion catalyzed chemistry of nucleotide incorporation. Structure analysis, however, suggests that DNA polymerases share one of two different ancestors, which converged to employ the same mechanism. Escherichia coli, the prototypical bacterium, encodes five different DNA polymerases. The chromosomal replicase functions closely with clamps, clamp-loaders, and other proteins. Oxidative damage to DNA during normal cell growth requires interplay among the several distinct DNA polymerases, which enable the replicase to circumvent these obstacles and complete chromosomal replication. Additional processes involving DNA polymerases are brought into action during heightened levels of DNA damage. We review here DNA polymerase structure, catalytic mechanism, and several pathways in which various bacterial DNA polymerases act.
Introduction
E. coli DNA polymerase (Pol) I is so named because it was the first DNA polymerase to be isolated and characterized (1). Pol I remains the most intensively studied DNA polymerase, and its general structure and properties generalize to other DNA polymerases. Sequence comparisons of DNA polymerases derived from many organisms indicate the presence of at least five different classes of DNA polymerase (Table 1) (2). For simplicity, we will focus in this review on the DNA polymerases of Escherichia coli. E. coli contains five different DNA polymerases that assort into four polymerase families (Table 1). Pol I is a member of the A-family; it is ubiquitous among bacteria and plays a major role in DNA repair. Pol II is a B-family DNA polymerase that seems to be involved in repair. Interestingly, eukaryotic and archael chromosomal replicases are all members of the B-family. The C-family of DNA polymerase is specific to bacteria and functions as the chromosomal replicase. E. coli contains one C-family polymerase, which is referred to as Pol III. Some bacteria contain two C-family DNA polymerases, which are referred to as Pol C (the replicase) and DnaE polymerase (after the gene encoding it) (3). E. coli contains two Y-family DNA polymerases, Pol IV and Pol V. Y-family DNA polymerases have relatively low fidelity and thus differ from typical DNA polymerases, which usually have very high fidelity in DNA synthesis (2, 4). For example, DNA polymerases I, II, and III make mistakes only once every 104-106 nucleotide additions, and fidelity is assisted in these enzymes by the presence of a 3'-5' exonuclease, which is referred to as a proofreader that removes most of the mistakes made by the polymerase (i.e., misincorporated nucleotides) (2, 4). In contrast, Y-family polymerases have intrinsically high error rates (10-1-10-3) and are the only E. coli DNA polymerases that lack 3'-5' exonuclease activity (5, 6). These unique properties allow Pol IV and Pol V to pass over DNA lesions and thus enable chromosome duplication in the face of DNA damage, but they do so at the expense of creating mutations (5, 7). Pols II, IV, and V are induced by DNA damage and are proposed to function collectively and to enable replication over DNA lesions (4, 8-10)
In this review we summarize the general architecture of DNA polymerases and the chemistry of the DNA polymerase and 3'-5' exonuclease activities (see also DNA replication). The ultimate function of DNA polymerases is the duplication of genetic material, and therefore, we also describe how Pol III functions at a replication fork. Lastly, we present a brief overview of the different repair reactions in which the remaining DNA polymerases act.
Table 1. DNA polymerase families
|
Family |
Examples |
|
A family |
Bacterial Pol I, phage T5, T7 |
|
B Family |
Bacterial Pol II, Eukaryotic Pol α, Pol δ, Pol ε |
|
C Family |
Bacterial Replicases, Pol III, Pol C, DnaE |
|
Y Family |
E. coli Pol IV, Pol V |
|
X Family |
Eukaryotic Pol β, Nucleotidyl Transferases |
Chemistry of DNA synthesis and 3'-5' exonucleolytic proofreading
The substrates for DNA synthesis are a 3' primed site and deoxyribonucleoside triphosphate (dNTP) (see Fig. 1). DNA polymerase catalyzes a phosphoryl transfer reaction that adds a dNMP moiety to a 3' terminus of an existing DNA strand, releasing pyrophosphate (Fig. 1a). The reaction is catalyzed exclusively by two metal ions (e.g., Mg++) (11, 12). Metal ion A extracts a proton from the DNA primer 3' terminal hydroxyl group to produce a oxyanion nucleophile, which attacks the α-phosphate of the incoming dNTP that is base-paired to the template. Metals A and B stabilize the resulting penta-coordinate transition state, and metal B also stabilizes the pyrophosphate leaving group.
The two metal ions are held in place by three conserved aspartic acid residues. The fact that catalysis is mediated exclusively by metal ions with no direct participation of amino acid side chains suggests that nucleotide polymerization may have originated before the evolution of proteins. Specifically, the two metals may have been chelated by RNA in the primordial “RNA World” that is thought to have operated before the evolution of proteins.
The 3'-5' proofreading exonuclease is called into action when an incorrect dNMP is added to the 3' terminus. The 3'-5' exonuclease is located in a separate domain with a distinct active site. The chemical reaction of the exonuclease proceeds by hydrolysis (see Fig. 1b), but it is remarkably similar to the polymerase reaction. Specifically, two metal ions catalyze the reaction; metal ion A activates water to form a hydroxyanion nucleophile that attacks the phosphodiester bond of the 3' terminal mismatched nucleotide. Metal B stabilizes the developing charge on the dNMP leaving group.

Figure 1. Chemical mechanism of DNA polymerase and 3'-5' exonuclease. (a) DNA polymerase reaction. The enzyme chelates two metal ions using three aspartic acid residues (only two are shown). Metal ion A abstracts the 3' hydroxyl proton of the primer terminus to generate a nucleophile that attacks the a-phosphate of an incoming dNTP substrate. The phosphoryl transfer results in production of a pyrophosphate leaving group, which is stabilized by metal ion B. (b) The 3'-5' exonuclease proofreading activity is located in a site that is distinct from the polymerase site; yet it uses two-metal-ion chemistry similar to DNA synthesis. The reaction type is hydrolysis in which metal ion A activates water to form the hydroxy anion nucleophile. Nucleophile attack on the phosphate of the mismatched nucleotide releases it as dNMP (dGMP in the case shown).
Structure of DNA polymerases
The crystal structures of multiple representatives of each class of DNA polymerase have been solved and compared (12). In all cases, the overall shape is that of a right hand, and it contains a minimum of three subdomains, which are referred to as the palm, fingers, and thumb. These subdomains are indicated for the structure of Pol I shown in Fig. 2a. Although the chain folding patterns of the fingers and thumb are different among the different polymerase families, the core architecture of the palm domain contains the catalytic site for DNA polymerization and is structurally conserved among the A, B, and Y families. The chain topology of the palm, and the relative location of the three Asp residues that hold the catalytic metal ions in polymerases of the A, B, and Y families, are shown in Fig. 2b. This conservation of structure in the palm suggests that the members of the A, B, and Y family share a common evolutionary ancestor.
Interestingly, C-family DNA polymerases (bacterial replicases) have a different chain topology in the palm, and the location of the Asp residues are also unique (13, 14). The palm architecture is shared by the X-family of DNA polymerases that include eukaryotic Pol P and certain nucleotidal transferases (Table 1). Presumably X-family polymerases share a common ancestor with DNA polymerases of the C-family.
In all DNA polymerases, the DNA lies on the palm of the polymerase and also interacts with the thumb (see Fig. 2c). The finger domain binds the incoming dNTP for incorporation into the DNA chain. The A-T and G-C base pairs have very similar geometry, and during binding of a dNTP, the fingers domain closes over the palm to match the dNTP to the template strand forming a tight enclosure into which only the geometry of a correct base pair can fit (15). Incorrect base pairs do not have the correct geometry to fit into this tight enclosure and are usually released instead of incorporated. In the rare instance of incorrect nucleotide incorporation, the primed site usually enters the 3'-5' exonuclease site for excision of the incorrect terminal nucleotide (see Fig. 2c).
The 3'-5' exonuclease intrinsic to DNA polymerase I, and most DNA polymerases, contributes to high fidelity by the kinetic scheme shown in Fig. 3. Most often, the DNA polymerase selects and incorporates the correct dNTP (pathway 1 in Fig. 3). Correct incorporation events are rapid. Incorporation of an incorrect dNTP (pathway 2 in Fig. 3) is slow, several orders of magnitude slower than incorporation of a correct dNTP (16, 17). Misincorporation is slow because the incorrect dNTP does not form a normal base pair with the template strand, and thus, the geometry of the incorrect base pair does not induce the correct fit in the enzyme active site needed to bring the two active site metal atoms into the proper juxtaposition for the chemical step. Furthermore, if the DNA polymerase incorporates the wrong nucleotide, the resulting mismatched 3' terminus presents yet another slow kinetic barrier to the DNA polymerase. The polymerase is very slow to extend the mismatched product by another nucleotide, because the correct geometry of the active site metals depends on the DNA substrate being correctly base paired. Therefore, the slow kinetics of two steps, misincorporation and extension of a mispaired 3' terminus, result in very few mistakes relative to the rapid incorporation of correct dNTP substrates. The kinetic pauses associated with incorporation of an incorrect dNTP give time for a mismatched primed template to melt and reposition into the separate active site of the 3'-5' exonuclease (Fig. 2c and Fig. 3). Once in the 3'-5' exonuclease active site, the proofreading 3'-5' exonuclease rapidly excises the incorrect 3' terminal nucleotide and thereby restores the substrate to one that is fully base paired (dashed arrow in the scheme of Fig. 3). Thus, the slow steps involved in misincorporation, combined with the rapid proofreading exonuclease removal of mismatched nucleotides, provide the DNA polymerase with a second try at chain extension. Most retrials have a positive outcome, which results in the incorporation of a correct nucleotide base.

Figure 2. Structure of DNA polymerases. (a) The structure of Pol I represents the general right-hand shape of all DNA polymerases (PDB ID 1QSY). Subdomains and the 3'-5' exonuclease domain are indicated. (b) The chain topology in the palm domain of DNA polymerases indicates their evolutionary heritage. Left: The A, B, and Y family have four antiparallel sheets supported by two a helices. The three aspartic residues that chelate the metal ions are indicated by dots. Right: The C-family and X-family DNA polymerases contain a set of parallel strands on which the acidic residues that chelate metal are oriented differently from the A-, B-, and Y-family DNA polymerases. (c) During misincorporation of an incorrect nucleotide, the DNA must leave the polymerase active site (left) and enter the 3'-5' exonuclease active site (right). This action requires the melting of three to four base pairs.
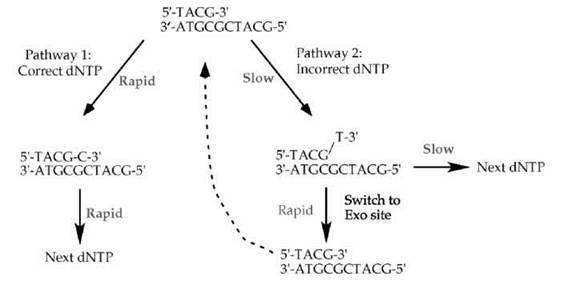
Figure 3. Kinetic steps during DNA synthesis favor incorporation of correct dNTPs. Most often the DNA polymerase selects the correct dNTP that forms a correct Watson-Crick base pair with the template strand (pathway 1, left). The chemistry of correct dNTP incorporation is rapid, and it allows the polymerase to proceed rapidly to incorporate subsequent dNTPs. The chemistry of incorporating an incorrect dNTP is slow (pathway 2, right), and subsequent elongation of the mispaired 3' terminus is also slow. These two kinetic barriers provide time for the primed template to switch into the proofreading 3'-5' exonuclease active site, where removal of the mispaired 3' terminus is rapid. The excised and fully base paired primed site then switches back to the DNA polymerase active site (dashed arrow).
DNA polymerase III holoenzyme
High-fidelity chromosomal replication in E. coli is executed by a multicomponent complex referred to as DNA polymerase III holoenzyme (see Fig. 4a) (18-21). Pol III holoenzyme consists of three main subcomponents: Pol III core, β-clamp, and γ-complex clamp-loader. Pol III core is the replicative DNA polymerase that consists of three subunits (α, ε, θ): a exhibits DNA polymerase activity, e performs 3'-5' exonuclease activity necessary for proofreading, and the function of θ is currently unclear.
Pol III core requires direct association with the β-clamp to perform processive DNA synthesis. β tethers the polymerase to its respective template by binding the α subunit of Pol III core while encircling DNA immediately behind the polymerase (see Fig. 4a). Coupling of Pol III core to β results in an increase in both the catalytic rate (~1kb s-1) and processivity (>50 kb).
The structure of the β-clamp resembles a symmetrical ring with sufficient space to accommodate double-stranded DNA (see Fig. 5a) (22). β is composed of a homodimer with each protomer consisting of three globular subdomains. The structure of the eukaryotic clamp, proliferating cell nuclear antigen (PCNA), is similar to β (23, 24). However, PCNA is composed of a homotrimer with each protomer consisting of two subdomains. The head-to-tail protomer organization of β results in two structurally distinct “faces”; the C-terminal face is involved in intermolecular interactions (i.e., binding Pol III core α subunit) (see Fig. 5a and 5b). β also binds Pol I, II, IV, and V and is therefore thought to play a role in polymerase trafficking during translesion synthesis (which is discussed in more detail below). Lastly, recent studies indicate that β interacts with DNA repair proteins MutS, MutL, and DNA ligase (25). The function of β in DNA repair is currently unclear.
The γ-complex clamp-loader is a heteropentameric ATPase responsible for assembling β onto DNA at primed sites (26-28). The clamp loading process is illustrated in Fig. 5b. The γ-complex binds and opens the ring-shaped β-clamp in the presence of ATP. The ATP-bound γ-complex selectively binds to a primed site that stimulates ATP hydrolysis and results in release of the clamp-loader and closure of β around DNA (20).
γ-complex is a member of the AAA+ (ATPases Associated with various cellular Activities) family of proteins that are involved in molecular remodeling activities (29). The “minimal” γ-complex includes only the subunits required for β-clamp assembly (γ3, δ, δ'). Importantly, the subunit composition of γ-complex exists in various states within the cell (reviewed in References 18 and 20) (which is discussed in more detail below). γ is the only subunit to exhibit ATPase activity and is therefore referred to as the “motor.” δ is responsible for opening the P-clamp and is referred to as the “wrench.” δ' is proposed to act as a “stator” (a part against which other parts of a machine move) because it is thought to be rigid relative to the other subunits.
The five clamp loading subunits (γ3, δ, δ') of the γ-complex are arranged in a spiral (see Fig. 5c) (26). A similar spiral-like subunit organization is found within the pentameric eukaryotic clamp-loader, RFC (30). The three ATP binding sites of γ-complex lie at the subunit interfaces and contain SRC (serine-arginine-cysteine) motifs common among all clamp-loaders, although mechanistic differences are proposed to exist in how ATP is coupled to the clamp loading process in bacterial and eukaryotic clamp loaders (31).
Pol III function at the replication fork
The proteins currently thought to act at the replication fork include two Pol III core molecules, two β-clamps, γ-complex, DnaB helicase, primase, and single-stranded DNA binding protein (SSB) (see Fig. 4a) (20, 32). Each Pol III core is dedicated to either the leading or the lagging strand and is tethered to its respective template via binding to the β-clamp. The unwound portion of the lagging strand is thought to form a “trombone” loop that enables codirectional movement of the lagging strand polymerase and the replisome (33).
The clamp loader acting at the replication fork is thought to be composed of subunits γ, Τ2, δ, δ', χ, ψ (32, 34, 35). In this configuration, two Y subunits are substituted by two τ subunits. γ (47kDa) and τ (71kDa) share identical N-terminal clamp loading sequences as they are encoded by the same gene, dnaX. However, γ is truncated because of an efficient translational frameshift that produces approximately equal amounts of γ and τ. The χ subunit connects to SSB and is held in the structure by ψ (36). The χ-to-SSB connection enhances processivity and enables efficient transfer of RNA primers from primase to the clamp loader (37). Each τ subunit binds to a single Pol III core thereby linking the leading and lagging strand polymerases together (see Fig. 4a) (14, 21, 38, 39).
DnaB is the replicative helicase that unwinds the parental duplex DNA ahead of the replication fork (see Fig. 4a) (40). DnaB encircles the lagging strand as a homohexamer and uses the energy of ATP hydrolysis to unwind DNA with 5'-3' polarity (see also DNA Helicases, Chemistry and Mechanisms of). DnaB is connected to Pol III holoenzyme via its interaction with the τ subunit of the clamp loader (41). This interaction greatly stimulates DnaB activity at the replication fork (42). DnaB also binds to and stimulates primase, which is a specialized RNA polymerase that synthesizes RNA primers approximately 12 nucleotides in length to initiate DNA synthesis (1, 43).
The mechanism of Pol III action at the replication fork is illustrated in Fig. 4. The lagging strand is synthesized as discontinuous sections of 1-3 kb called Okazaki fragments. An Okazaki fragment is initiated by primase action (see Fig. 4a). The clamp loader uses the energy of ATP hydrolysis to assemble P onto the newly synthesized RNA primer (see Fig. 4b). The Pol III core binds P and processively synthesizes the DNA portion of the Okazaki fragment, which creates a DNA loop (see Fig. 4c). The Okazaki fragment is completed when Pol III encounters the 5' end of the previous Okazaki fragment and dissociates from the DNA, which disassembles the loop. Pol I is required for Okazaki fragment maturation and is discussed below.
The leading strand polymerase performs DNA synthesis in a continuous fashion and therefore requires one initial priming event. However, DNA damage or other impediments along the leading strand may block progression of the replication fork and lead to additional priming events enabled by replication restart mechanisms (reviewed in Reference 44). Lagging strand blocks, such as damaged bases, are inconsequential to the progression of the replisome because the lagging strand polymerase readily cycles among numerous primed sites (45-47).
The repair of arrested replication forks involves two major pathways: recombinational repair and translesion synthesis. Recombinational repair uses RecA and a host of other recombination factors that facilitate strand exchange and non-mutagenic replication across damaged DNA (reviewed in References 48-50). Translesion synthesis employs low-fidelity DNA polymerases that often perform mutagenic replication opposite a damaged template (reviewed in References 5-7).

Figure 4. Pol III function at the replication fork. Leading and lagging strand synthesis are performed in a continuous and discontinuous fashion, respectively. The lagging strand is synthesized in contiguous sections called Okazaki fragments. (a) An Okazaki fragment is initiated as primase synthesizes a short RNA primer along the lagging strand. (b) The γ-complex clamp loader uses the energy of ATP hydrolysis to assemble p at the newly primed site. (c) The lagging strand polymerase cycles to a newly synthesized primed site after completing the previous Okazaki fragment. (d) The lagging strand polymerase synthesizes the DNA portion of the next Okazaki fragment. (Adapted with permission from Reference 20.)

Figure 5. Structure and function of β-clamp and γ-complex. (a) The structure of p resembles a ring with sufficient space to accommodate duplex DNA. β is a homodimer in which each protomer contains three globular subdomains. The protomers are arranged in a head-to-tail fashion resulting in two structurally distinct ''faces.'' The C-terminal face is involved in intermolecular interactions. Protomers are indicated in light and dark shades (PDB ID 2POL). (b) γ-complex uses the energy of ATP hydrolysis to assemble β onto DNA at primed sites. γ-complex binds to and opens β in the presence of ATP. The clamp loader selectively binds to primer-template junctions, which stimulates ATP hydrolysis and results in dissociation of γ-complex and closure of β around the DNA. DNA polymerase binds the C-terminal face of p and initiates DNA synthesis at the primed site. (c) Subunit organization of y-complex clamp loader. The γ-complex is composed of five subunits (γ3, δ', δ) that are arranged in a spiral-like fashion. γ-complex contains three ATP sites that lie at the subunit interfaces and include an ''arginine finger'' within a conserved SRC motif. The ATP site organization of the γ-complex is proposed to confer cooperativity between subunits. (Adapted with permission from Reference 20.)
Translesion synthesis by low-fidelity DNA polymerases
Chromosomal DNA is often damaged by exposure to exogenous (i.e., UV irradiation, chemical agents, etc.) and endogenous (i.e., oxidative damage) insults (see also DNA Damage, Chemical Biology of Diseases Related to (51). Spontaneous mutagenesis of nucleotide bases by cytosine deamination to yield uracil also occurs with high frequency in the cell. DNA repair mechanisms have evolved to excise and repair various types of DNA damage (discussed below) (see also DNA Damage Repair, Chemistry of). However, the replication fork sometimes encounters DNA damage that has evaded the repair machinery. Pol III and other high-fidelity DNA polymerases cannot replicate damaged DNA. Thus, the replication fork becomes arrested at the site of DNA damage. Low-fidelity lesion bypass DNA polymerases can extend DNA across various types of damaged DNA, but this often results in a mutation. Thus, switching between high-fidelity and low-fidelity DNA polymerases is thought to facilitate replication through sites of DNA damage (4, 8-10, 52).
Pol I and Pol III exhibit high-fidelity DNA synthesis and make mistakes on average of only 1 in 105-107 nucleotides (5, 16, 17). Y-family Pol IV and Pol V misincorporate nucleotides 10 to 1000 times more frequently and are error-prone lesion bypass polymerases that are associated with the mutagenic response of cells to DNA damage (5, 6, 16, 52). Structures of Y-family polymerases suggest that replication of past aberrant nucleotides is likely caused by a highly solvent exposed DNA binding cleft (53, 54). Thus, bulky lesions and nucleotide mismatches are more permissible among this class of enzyme.
Replication fork arrest by DNA damage leads to RecA mediated induction of the SOS response pathway. The SOS response involves upregulation of over 40 genes that facilitate increased viability during stress (i.e., chemical agents, UV light, and nutrient deficiency) (reviewed in References 5 and 6). The DNA polymerases that are upregulated during the SOS response include Pol II, Pol IV, and Pol V. Pol II is the first DNA polymerase to be induced (~1 minute). Interestingly, Pol II has been shown to perform high-fidelity DNA synthesis (error rate ~10-5 to 10-6 per base pair) on undamaged templates, but despite this, Pol II seems to be involved in error-prone DNA synthesis in vivo (4, 55). Pol II is somewhat of an enigmatic enzyme because it has been implicated in translesion synthesis, adaptive mutation, and recombinational repair (which is discussed below) (4-6, 56). Additional study is required to understand the relevant functions of Pol II in the cell.
Pol V is upregulated from <15 to ~200 copies per cell approximately 45 minutes after SOS induction. Pol V is a heterotrimer with a subunit composition of UmuD'2C (see Fig. 6a). UmuC contains the catalytic domain of Pol V. UmuD' is the product of RecA-mediated proteolytic cleavage of UmuD. Importantly, Pol V function requires RecA, and the mechanism by which RecA stimulates Pol V activity has been under investigation for several years (57). Recent data indicate that RecA nucleoprotein filaments act in trans to stimulate Pol V (see Fig. 6a) (57, 58). Interestingly, RecA filaments in cis (immediately 5' to Pol V on the DNA template) do not stimulate Pol V (59). Pol V and SSB may cooperate to displace RecA from DNA (57, 60). Alternatively, recent data indicate that RecA may be removed by UvrD helicase, as UvrD is also induced during the SOS response, but whether UvrD plays a direct role in translesion synthesis is currently an open question (61, 62).
Pol IV and Pol V use the β-clamp (63-66) and are capable of synthesizing DNA opposite various types of lesions (i.e., abasic site, thymine dimer, and benzo(a)pyrene) (see Table 2) (4, 8, 9, 67). Pol IV is expressed constitutively at ~250 copies per cell, suggesting a role during normal growth, and consistent with the finding that Pol IV is required for adaptive mutation under nonlethal conditions (6). Pol IV is increased 10-fold after SOS induction where it is presumably used to bypass lesions. In contrast, Pol V is undetectable during normal growth conditions and probably only plays a role during DNA damage.
All five E. coli DNA polymerases function with β, and therefore, it is perhaps not surprising that recent biochemical studies demonstrate that P plays a central role in different polymerases switching on DNA primed sites during translesion synthesis (7, 58, 68). A model for Pol IV/Pol III switching during translesion synthesis based on structural and biochemical studies is illustrated in Fig. 6b. The dimeric β-clamp can bind both Pol IV and Pol III simultaneously, and it is proposed to hold Pol IV away from the DNA template while Pol III actively synthesizes DNA (68, 69). However, during a Pol III encounter with a lesion, Pol IV is allowed to gain access to the template in place of Pol III. After the lesion is bypassed by Pol IV, the high-fidelity Pol III regains control of the primed site. These events limit the action of the low-fidelity Pol IV to the region in which it is required to bypass a lesion.

Figure 6. Mechanisms of translesion synthesis. (a) Activation mechanism of Pol V during translesion synthesis. Pol V is a heterotrimer composed of subunits UmuC, D'2: UmuC is the catalytic domain, and UmuD' is the product of RecA mediated proteolysis. Translesion synthesis by Pol V is activated by the presence of a RecA filament in trans. (b) Model of DNA polymerase switching during translesion synthesis. Pol III* and Pol IV each bind to a p protomer at a conserved hydrophobic protein binding pocket (QL[S/D]LF). 1. Pol III* is arrested at the site of DNA damage, whereas Pol IV is held in an inactive state away from the DNA. 2. Pol IV gains hold of the primer terminus from Pol III* at the stall site; Pol III* is now held away from the DNA. 3. Pol IV extends the DNA past the lesion. 4. Pol III* regains hold of the primer terminus from Pol IV.
DNA polymerase activity during repair
Oxidative damage that leads to replication fork arrest is common during bacterial growth and probably occurs at least once during each cell division even under the most favorable conditions (48, 51). Most lesions are not dealt with by mutagenic polymerases, but instead they are repaired by recombination, which is an error-free process. Recombinational repair can take many different paths and employs several different proteins, but ultimately, it involves high-fidelity DNA polymerases such as Pol I, Pol II, and/or Pol III (6, 49, 56). For additional information, we refer the reader to more thorough reviews of the various recombination repair pathways (44, 48-50, 70).
Even high-fidelity DNA polymerases sometimes incorporate an incorrect nucleotide. All cells contain a specialized mismatch repair system that catches these errors and corrects them. Mismatch repair involves excision of DNA past the mismatch, and eventually, it allows Pol III to try again (reviewed in References 71 and 72). The process of mismatch repair requires proteins MutS and MutL, among others. Mutations in the human homologues of these proteins lead to a predisposition to development of tumors (73).
Nucleotide excision repair (NER) is responsible for removing a wide array of DNA lesions that predominately occur as a result of exposure to oxygen or UV light (reviewed in Reference 74) (see also DNA Damage, Chemical Biology of Diseases Related to)). Unlike mismatch repair, NER employs Pol I to replace damaged DNA. NER also requires the action of Uvr (ultraviolet radiation) proteins that were originally discovered because of their ability to sustain cell growth after exposure to UV irradiation. Like MutS and MutL, mutations in the human homologues of the Uvr proteins predisposes individuals to cancer.
Base excision repair (BER), in which a single damaged base is excised and replaced, also uses Pol I to replace the modified nucleotide. BER is involved predominately in the repair of apurinic and apyrimidinic nucleotides [abasic (AP) sites) that frequently develop from spontaneous hydrolysis (reviewed in Reference 75). Importantly, BER is also responsible for removing uracil from DNA, which is the product of cytidine deamination. Bases modified by oxidation and alkylation are also repaired; however, these modifications occur less frequently.
A recent study suggests that Pol IV and Pol V may also be involved in BER because of their intrinsic lyase activity (76). Shen et al. demonstrate relatively weak 5'-phosphodiester bond cleavage of an abasic site (AP/5'-dRP activity) by Pol IV and Pol V. Interestingly, several DNA polymerases from the X, A, and Y families exhibit lyase activity. Human Pol P, for example, exhibits strong AP/5'-dRP activity and is thought to be involved in the excision and incorporation of nucleotides during BER (77, 78). Furthermore, eukaryotic pol i, which is a Y-family homolog of Pol IV and Pol V, has also been shown to exhibit AP/5'-dRP activity (79). Additional studies are required to determine whether Pol IV and Pol V are involved in BER.
Lastly, Pol I is responsible for repairing DNA during Okazaki fragment maturation. Pol I replaces the RNA portion of Okazaki fragments using its 5'-3' exonuclease activity and synthesizes DNA in its place. The remaining nick between adjacent Okazaki fragments is repaired by DNA ligase.
Table 2. Properties of translesion DNA polymerases
|
Lesion |
DNA polymerase |
Mutation |
|
Abasic site (2, 80) |
Pol IV |
-1 frameshift |
|
|
Pol V |
Preferential incorporation of dATP and dGTP |
|
Benzo(a)pyrene (4, 81) |
Pol IV, Pol V |
-1 frameshift |
|
N-2-acetylaminofluorene (4) |
Pol IV, Pol II |
-2 frameshift |
|
|
Pol V |
-1,-2 frameshift |
|
8-oxo-guanine (9) |
Pol IV, Pol V |
-2 frameshift |
|
6-4 Thymine-thymine photoproduct (2) |
Pol V |
T → C |
Adapted with permission from Reference 5.
Future perspectives
The work of many laboratories has provided significant insight into the detailed workings of Pol III function at the replication fork. The way in which the replisome interacts with other DNA processes (i.e., DNA compaction, transcription, and recombination, repair), however, is poorly understood. For example, how does Pol III holoenzyme replicate through high-affinity protein-nucleic acid complexes such as RNA polymerase or repressor proteins? What is the fate of the replication components after an encounter with a blocking lesion? Does the replisome directly recruit or interact with recombination factors that repair broken forks? p has been shown to bind mismatch repair proteins MutS and MutL. What is the significance of these interactions?
The presence of five DNA polymerases in E. coli suggests that access to primed sites might be regulated. Indeed, recent biochemical data indicate that p plays a major role in switching between high-fidelity and low-fidelity DNA polymerases during translesion synthesis. High-fidelity DNA polymerases Pol I and Pol II also bind to p and are expressed at relatively high levels relative to Pol III. Do high-fidelity DNA polymerases compete for primed sites during normal growth? Furthermore, we still do not understand the major role of Pol II in the cell.
Lastly, translesion synthesis by Pol V is stimulated by RecA filaments in trans. The presence of RecA filaments in cis (on the DNA template), however, inhibits the activity of Pol V. Additional work is required to elucidate the exact mechanism and organization of RecA filaments in the cell responsible for Pol V activation. Future studies will likely reveal the answers to many of the questions addressed above.
Acknowledgment
The authors are grateful to their source of funding, NI-HGM38839
References
1. Kornberg A, Baker TA. DNA Replication. DNA Replication New York 1992; 931.
2. Tang M, Pham P, Shen X, Taylor JS, O’Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature 2000; 404:1014-1018.
3. Bruck I, O’Donnell M. General features of chromosomal replicases from bacteria. Res. Adv. Biological Chem. 2001; 1:9-19.
4. Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. Embo. J. 2000; 19:6259-6265.
5. Tippin B, Pham P, Goodman MF. Error-prone replication for better or worse. Trends Microbiol. 2004; 12:288-295.
6. Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002; 71:17-50.
7. Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007; 15:70-77.
8. Jarosz DF, Godoy VG, Walker GC. Proficient and accurate bypass of persistent DNA lesions by DinB DNA polymerases. Cell Cycle 2007; 6:817-822.
9. Wagner J, Etienne H, Janel-Bintz R, Fuchs RP. Genetics of mutagenesis in E. coli: various combinations of translesion polymerases (Pol II, IV and V) deal with lesion/sequence context diversity. DNA Repair (Amst) 2002; 1:159-167.
10. Friedberg EC, Lehmann AR, Fuchs RP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 2005; 18:499-505.
11. Beese LS, Steitz TA. Structural basis for the 3'-5' exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. Embo. J. 1991; 10:25-33.
12. Steitz TA. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999; 274:17395-17398.
13. Bailey S, Wing RA, Steitz TA. The structure of T. aquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell 2006; 126:893-904.
14. Lamers MH, Georgescu RE, Lee SG, O’Donnell M, Kuriyan J. Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell 2006; 126:881-892.
15. Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature 1998; 391:251-258.
16. Kunkel TA. DNA replication fidelity. J. Biol. Chem. 2004; 279:16895-16898.
17. Kunkel TA, Bebenek K. DNA replication fidelity. Annu. Rev. Biochem. 2000; 69:497-529.
18. Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 2005; 74:283-315.
19. McHenry CS. DNA polymerase III holoenzyme. Components, structure, and mechanism of a true replicative complex. J. Biol. Chem. 1991; 266:19127-19130.
20. Pomerantz RT, O’Donnell M. Replisome mechanics: insights into a twin DNA polymerase machine. Trends Microbiol. 2007; 15:156-164.
21. McHenry CS, Johanson KO. DNA polymerase III holoenzyme of Escherichia coli: an asymmetric dimeric replicative complex containing distinguishable leading and lagging strand polymerases. Adv. Exp. Med. Biol. 1984; 179:315-319.
22. Kong XP, Onrust R, O’Donnell M, Kuriyan J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell 1992; 69:425-437.
23. Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994; 79:1233-1243.
24. Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996; 87:297-306.
25. Lopez de Saro FJ, Marinus MG, Modrich P, O’Donnell M. The beta sliding clamp binds to multiple sites within MutL and MutS. J. Biol. Chem. 2006; 281:14340-14349.
26. Jeruzalmi D, O’Donnell M, Kuriyan J. Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell 2001; 106:429-441.
27. Bowman GD, Goedken ER, Kazmirski SL, O’Donnell M, Kuriyan J. DNA polymerase clamp loaders and DNA recognition. FEBS Lett. 2005; 579:863-867.
28. Pritchard AE, Dallmann HG, Glover BP, McHenry CS. A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of deltadelta' with DnaX(4) forms DnaX(3)deltadelta'. Embo. J. 2000; 19:6536-6545.
29. Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999; 9:27-43.
30. Bowman GD, O’Donnell M, Kuriyan J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 2004; 429:724-730.
31. Neuwald AF. Hypothesis: bacterial clamp loader ATPase activation through DNA-dependent repositioning of the catalytic base and of a trans-acting catalytic threonine. Nucleic Acids Res. 2006; 34:5280-5290.
32. McHenry CS. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol. Microbiol. 2003; 49:1157-1165.
33. Sinha NK, Morris CF, Alberts BM. Efficient in vitro replication of double-stranded DNA templates by a purified T4 bacteriophage replication system. J. Biol. Chem. 1980; 255:4290-4293.
34. O’Donnell M, Jeruzalmi D, Kuriyan J. Clamp loader structure predicts the architecture of DNA polymerase III holoenzyme and RFC. Curr. Biol. 2001; 11:935-946.
35. Glover BP, McHenry CS. The DnaX-binding subunits delta' and psi are bound to gamma and not tau in the DNA polymerase III holoenzyme. J. Biol. Chem. 2000; 275:3017-3020.
36. Glover BP, McHenry CS. The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem. 1998; 273:23476-23484.
37. Yuzhakov A, Kelman Z, O’Donnell M. Trading places on DNA-a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 1999; 96:153-163.
38. Kim S, Dallmann HG, McHenry CS, Marians KJ. tau couples the leading- and lagging-strand polymerases at the Escherichia coli DNA replication fork. J. Biol. Chem. 1996; 271:21406-21412.
39. McHenry CS. Purification and characterization of DNA polymerase III'. Identification of tau as a subunit of the DNA polymerase III holoenzyme. J. Biol. Chem. 1982; 257:2657-2663.
40. LeBowitz JH, McMacken R. The Escherichia coli dnaB replication protein is a DNA helicase. J. Biol. Chem. 1986; 261:4738-4748.
41. Gao D, McHenry CS. tau binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of tau, binds the replication fork, helicase, DnaB. J. Biol. Chem. 2001; 276:4441-4446.
42. Kim S, Dallmann HG, McHenry CS, Marians KJ. Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell 1996; 84:643-650.
43. Tougu K, Marians KJ. The interaction between helicase and primase sets the replication fork clock. J. Biol. Chem. 1996; 271:21398-21405.
44. Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 2006; 7:932-943.
45. McInerney P, O’Donnell M. Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J. Biol. Chem. 2004; 279:21543-21551.
46. Pages V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 2003; 300:1300-1303.
47. Higuchi K, Katayama T, Iwai S, Hidaka M, Horiuchi T, Maki H. Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells 2003; 8:437-449.
48. Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 2002; 71:71-100.
49. Cox MM. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu. Rev. Genet. 2001; 35:53-82.
50. Kreuzer KN. Interplay between DNA replication and recombination in prokaryotes. Annu. Rev. Microbiol. 2005; 59:43-67.
51. Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature 2000; 404:37-41.
52. Kuban W, Jonczyk P, Gawel D, Malanowska K, Schaaper RM, Fijalkowska IJ. Role of Escherichia coli DNA polymerase IV in in vivo replication fidelity. J. Bacteriol. 2004; 186:4802-4807.
53. Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell 2001; 107:91-102.
54. Zhou BL, Pata JD, Steitz TA. Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol. Cell 2001; 8:427-437.
55. Cai H, Yu H, McEntee K, Kunkel TA, Goodman MF. Purification and properties of wild-type and exonuclease-deficient DNA polymerase II from Escherichia coli. J. Biol. Chem. 1995; 270:15327-15335.
56. Rangarajan S, Woodgate R, Goodman MF. A phenotype for enigmatic DNA polymerase II: a pivotal role for pol II in replication restart in UV-irradiated Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:9224-9229.
57. Schlacher K, Goodman MF. Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat. Rev. Mol. Cell Biol. 2007; 8:587-594.
58. Schlacher K, Cox MM, Woodgate R, Goodman MF. RecA acts in trans to allow replication of damaged DNA by DNA polymerase V. Nature 2006; 442:883-887.
59. Schlacher K, Leslie K, Wyman C, Woodgate R, Cox MM, Goodman MF. DNA polymerase V and RecA protein, a minimal mu- tasome. Mol. Cell 2005; 17:561-572.
60. Pham P, Bertram JG, O’Donnell M, Woodgate R, Goodman MF. A model for SOS-lesion-targeted mutations in Escherichia coli. Nature 2001; 409:366-370.
61. Centore RC, Sandler SJ. UvrD limits the number and intensities of RecA-green fluorescent protein structures in Escherichia coli K-12. J. Bacteriol. 2007; 189:2915-2920.
62. Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, Fabre F, Petit MA. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. Embo. J. 2005; 24:180-189.
63. Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. 2001; 98:11627-11632.
64. Wijffels G, Dalrymple BP, Prosselkov P, Kongsuwan K, Epa VC, Lilley PE, Jergic S, Buchardt J, Brown SE, Alewood PF, Jennings PA, Dixon NE. Inhibition of protein interactions with the beta 2 sliding clamp of Escherichia coli DNA polymerase III by peptides from beta 2-binding proteins. Biochemistry 2004; 43:5661-5671.
65. Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RP. The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 2000; 1:484-488.
66. Maor-Shoshani A, Livneh Z. Analysis of the stimulation of DNA polymerase V of Escherichia coli by processivity proteins. Biochemistry 2002; 41:14438-14446.
67. Fuchs RP, Koffel-Schwartz N, Pelet S, Janel-Bintz R, Napolitano R, Becherel OJ, Broschard TH, Burnouf DY, Wagner J. DNA polymerases II and V mediate respectively mutagenic (-2 frameshift) and error-free bypass of a single N-2- acetylaminofluorene adduct. Biochem. Soc. Trans. 2001; 29:191-195.
68. Indiani C, McInerney P, Georgescu R, Goodman MF, O’Donnell M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell 2005; 19:805-815.
69. Bunting KA, Roe SM, Pearl LH. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. Embo. J. 2003; 22:5883-5892.
70. Courcelle J. Recs preventing wrecks. Mutat. Res. 2005; 577:217-227.
71. Kunkel TA, Erie DA. DNA mismatch repair. Annu. Rev. Biochem. 2005; 74:681-710.
72. Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 2003; 57:579-608.
73. Modrich P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006; 281:30305-30309.
74. Van Houten B. Nucleotide excision repair in Escherichia coli. Microbiol. Rev. 1990; 54:18-51.
75. Seeberg E, Eide L, Bjoras M. The base excision repair pathway. Trends Biochem. Sci. 1995; 20:391-397.
76. Shen X, Woodgate R, Goodman MF. Lyase activities intrinsic to Escherichia coli polymerases IV and V. DNA Repair (Amst) 2005; 4:1368-1373.
77. Podlutsky AJ, Dianova, II, Wilson SH, Bohr VA, Dianov GL. DNA synthesis and dRPase activities of polymerase beta are both essential for single-nucleotide patch base excision repair in mammalian cell extracts. Biochemistry 2001; 40:809-813.
78. Prasad R, Beard WA, Strauss PR, Wilson SH. Human DNA polymerase beta deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J. Biol. Chem. 1998; 273:15263-15270.
79. Prasad R, Bebenek K, Hou E, Shock DD, Beard WA, Woodgate R, Kunkel TA, Wilson SH. Localization of the deoxyribose phosphate lyase active site in human DNA polymerase iota by controlled proteolysis. J. Biol. Chem. 2003; 278:29649-29654.