CHEMICAL BIOLOGY
Chemistry of Protein Kinases and Dephosphorylases
Scott M. Ulrich, Chemistry Department, Ithaca College, Ithaca, New York
doi: 10.1002/9780470048672.wecb269
Protein kinases and protein phosphatases catalyze phosphorylation and dephosphorylation of cellular proteins. Posttranslational protein phosphorylation is a major mechanism of signal transduction in eukaryotes; it serves as a reversible switch that alters the activity of target proteins as a means to link stimuli to the appropriate cellular response. In this article,
I will review the structures and mechanisms of protein kinases and phosphatases as well as describe chemical tools developed to study protein phosphorylation.
All cells must respond appropriately to external stimuli, including nutrient availability, hormones, and growth factors. A main focus of biologic research is aimed at elucidating the molecular mechanisms of how signals are relayed within cells. Protein phosphorylation is a central mechanism of signal transduction cascades in eukaryotes and is involved in nearly all regulated cellular processes. Two families of enzymes control the phosphorylation state of cellular proteins: protein kinases catalyze phosphorylation of either serine/threonine or tyrosine residues of target proteins using ATP as the phosphodonor, whereas protein phosphatases catalyze removal of the phosphate group from phosphoproteins (1). Thus, phosphorylation of cellular proteins is a reversible switch that controls the enzymatic activity, cellular localization, conformation, or macromolecular binding capacity of the target protein, ultimately culminating in cellular responses to a variety of stimuli. The families of protein kinases and phosphatases are large: The human genome contains ~600 protein kinases and ~ 150 protein phosphatases. Similarly, the extent of protein phosphorylation is immense; it is estimated that one third of all human proteins are modified by phosphorylation (2, 3). Thus, understanding protein phosphorylation is a major challenge for biologic research and also has implications for developing therapies to treat the many human diseases that originate from misregulated signal transduction. In this article, I will discuss the chemistry and enzymology of the phosphorylation/dephosphorylation reactions catalyzed by protein kinases and phosphatases as well as describe some chemical tools that have been developed to study features of protein phosphorylation that otherwise resist analysis.
Enzymology of Protein Kinases
The phosphotransfer reaction mechanism of protein kinases
Protein kinases catalyze transfer of the γ-phosphate from ATP to hydroxyl side chains of target proteins. This phosphotransfer reaction lies somewhere on a continuum between two limiting mechanistic possibilities (4). The associative mechanism is analogous to the Sn2 reaction, where the hydroxyl group of the phosphoacceptor protein attacks the γ-phosphate of ATP, followed by the formation of a -3 charged trigonal bipyramidal transition state, which is resolved then into ADP and phosphoprotein (Fig. 1a). This trigonal bipyramidal transition state has two positions around the phosphorous center. Apical positions are long, weak bonds 180° apart from each other where groups enter or depart, and equatorial positions are shorter, stronger bonds 120° apart from each other, forming a plane that is normal to the apical positions. Associative phosphotransfer reactions can be “in-line,” where the incoming nucleophile approaches from the backside of the scissile bond (as in a classic Sn2 reaction), or “adjacent,” where the nucleophile approaches the backside of a nonscissile bond. The second limiting mechanistic possibility is dissociative phosphotransfer, where the γ-phosphate ionizes (as in an Sn1 reaction) to form a -1 charged metaphosphate intermediate that subsequently is captured by the hydroxyl nucleophile (Fig. 1b).
Phosphotransferases are ubiquitous in biology and have varied mechanisms. To determine whether the phosphotransfer reaction catalyzed by protein kinases is primarily associative or dissociative, a crystal structure of cAMP-dependent protein kinase (PKA) was solved with ADP, a phosphoacceptor peptide, and AlF3, which mimics the structure of the y-phosphate being transferred (5). The structure shows that the oxygen-aluminum bond lengths are significantly shorter than their van der Waals radii (2.3 A vs. ~3.5 A), which supports a mechanism that is primarily associative. The geometry of the structure also clearly shows that the nucleophilic hydroxyl group of the phosphoacceptor peptide and the ADP are both apical about AlF3, which strongly supports an in-line phosphotransfer mechanism (Fig. 1c).

Figure 1. (a) The limiting case of purely associative phosphotransfer involves attack of the hydroxyl on the phosphoanhydride followed by release of ADP. (b) The alternative limiting case of purely dissociative phosphotransfer possible in kinase reactions involving ionization of the phosphoanhydride bond followed by capture of the metaphosphate intermediate by the hydroxyl nucleophile. (c) Structure of a transition state mimic of protein kinase A. The phosphoacceptor peptide is yellow with the nucleophilic serine in blue; the AlF3 mimic of the γ-phosphate is gray. The structure shows that the β-phosphate-aluminum-serine internuclear geometry is in-line, and the internuclear distance suggests the mechanism is closer to the associative end of the mechanistic spectrum.
Catalysis by protein kinases
How protein kinases facilitate phosphotransfer is well understood from structural studies. The overall structure of the catalytic domain of protein kinases is bilobal, consisting of a small, mostly β-sheet subdomain at the N terminus connected by a short linker to a larger, mostly α-helical C terminus. The N-terminal domain binds Mg: ATP, whereas the C terminus binds the phosphoacceptor protein substrate (Fig. 2a). The detailed architecture and catalytic residues of the active site are very highly conserved among the family.
Much structural biology analysis has been performed on protein kinase A (PKA), and its catalytic residues are conserved across the family (6, 7). In PKA, lysine 72 and glutamate 91 orient the γ-phosphate toward the protein substrate (Fig. 2b). Aspartate 166 acts as a catalytic base to accept the proton from the hydroxyl nucleophile, and Lys 168 acts as an electrostatic catalyst to stabilize the γ-phosphate during the reaction. Asparagine 171 positions a magnesium ion that coordinates the α/β phosphates (Fig. 2b).
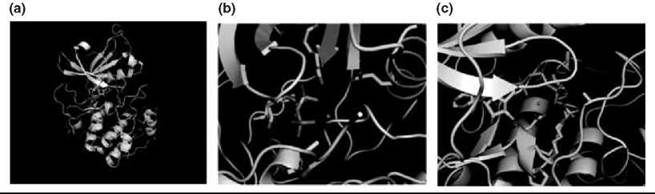
Figure 2. Snapshots of the overall structure and catalytic machinery of protein kinase A. (a) The overall fold of the catalytic domain is formed by two subdomains, a beta sheet N-terminus (gray) and a C-terminal helical domain (green). ATP binds a cleft between the two lobes, and the phosphoacceptor substrate binds the C-terminal lobe. (b) N-terminal residues Lys 72 and Glu 91 orient the phosphates toward the phosphoacceptor peptide (pink/yellow) in concert with one of two magnesium ions. (c) C-terminal residue Lys 168 acts as an electrostatic catalyst to stabilize the γ-phosphate during the reaction while asparagine 171 and aspartate 184 position the phosphates within the active site.
Enzymology of Protein Phosphatases
Phosphatases hydrolyze phosphate groups from phosphoproteins, thereby reversing the action of protein kinases. Two classes of protein phosphatases exist with distinct structures, substrate specificities, and mechanisms: protein serine/threonine phosphatases and protein tyrosine phosphatases. A few dualspecificity phosphatases also are known that are similar in structure to protein tyrosine phosphatases.
Protein tyrosine phosphatases
Protein tyrosine phosphatases (PTPs) cleave phosphate groups from phosphotyrosine residues. As is the case with protein kinases, these enzymes are well characterized structurally.
Protein tyrosine phosphatases specifically recognize phospho- tyrosine because of the presence of a P-loop binding motif, with the general sequence HCXXXXXR(S/T) (8). The P-loop arginine and several backbone amide nitrogens are the primary contacts to the phosphate oxygens through hydrogen bonds. The active site pocket is also deep, which disallows proteins phosphorylated on serine to access the P-loop. Hydrophobic side chains also pack the aryl moiety of phosphotyrosine to provide additional selectivity versus phosphoserine.
The mechanism of the PTP hydrolysis reaction has two steps. First, phosphate is transferred from tyrosine to the cysteine residue of the P-loop, which generates a phosphoenzyme intermediate with concomitant release of tyrosine. This process is followed by hydrolysis of the phosphoenzyme to free enzyme and inorganic phosphate. Two active site residues are of primary importance during the catalytic cycle: the nucleophilic cysteine of the P-loop and an aspartate on a nearby flexible loop, which serves as a general acid/base catalyst (Fig. 3). After attack of the cysteine on phosphotyrosine, tyrosine can be expelled as the protonated phenol after proton donation by the catalytic aspartic acid, which forms the phosphoenzyme intermediate and free tyrosine. The aspartate anion then deprotonates the hydrolytic water molecule that attacks phosphocysteine, which liberates inorganic phosphate (Fig. 4) (9).
The cysteine has a lowered pKa value, which amplifies its nucleophilicity. Measurement of the pH profile of the rate of alkylation of this cysteine with iodoacetate has determined its pKa to be as low as <5 (normally 8.3), which makes the residue ionized at physiologic pH (10). The aspartate also has an anomalous pKa value and is believed to be protonated at the beginning of the reaction coordinate, as evidenced by cocrystal structures with vanadate bound, where it is hydrogen-bond distance from VO4- (11).
Resolution of the phosphoenzyme intermediate into free enzyme and inorganic phosphate is accomplished by orientation and deprotonation of water by the aspartate residue and attack at the phosphothioester. Mutagenesis of the aspartate residue to alanine (as well as the Arg in the P-loop) shows its importance in this second step, as it leads to accumulation of the phosphoenzyme intermediate (12).
As was the case with other phosphotransfer reactions, the hydrolysis reaction catalyzed by PTPs lies along a mechanistic continuum between the limiting cases of associative and dissociative mechanisms. In the case of Yersina PTP, a crystal structure was solved with nitrate, a mimic of the transition state similar to the use of AlF3 with protein kinase A. In this case, analysis of the structure is proposed to be more consistent with a -1 charged metaphosphate, dissociative mechanism (11).
Interestingly, regulation of PTPs has been observed to be mediated via reversible oxidation of the catalytic cysteine to the corresponding cysteic acid, which is incompetent to carry out the reaction. Hydrogen peroxide, generated during some signaling pathways, has been shown through careful enzymology to be capable of inactivating several PTPs and is reversible by treatment with exogenous thiols, which explains the role of peroxide use as a rapid diffusable signaling molecule (13).

Figure 3. Structure of protein serine phosphatase PP2 C with bound phosphate showing the binuclear metal site (purple spheres) and the nucleophilic water (red sphere). Also shown are residues that ligate the transition metals as well as two key asparagines that coordinate the phosphate group.

Figure 4. Protein kinases with the ''gatekeeper'' residue mutated to glycine that makes them recognize N6-substituted ATP analogs also programs them to be uniquely sensitive to designed inhibitors, allowing rapid generation of potent selective kinase inhibitors of any protein kinase.
Protein serine/Threonine phosphatases
Protein phosphatases that are specific for phosphoserine/ phosphothreonine have a distinct reaction mechanism from tyrosine phosphatases. Protein serine phosphatases are transition metal-dependent, and the reaction mechanism does not involve a phosphoenzyme intermediate as in the case of PTPs. Crystal structures of multiple protein serine phosphatases have revealed how the enzymes catalyze hydrolysis of phosphoserine (14).
Serine phosphatases are metalloenzymes with two transition metals (Fe2+ and Zn2+/Mn2+ in PP2B and PP1, or 2 Mn2+ in PP2 C) coordinated by Asp, Asn, and His residues to form a binuclear metal center in the active site. This metal center is bridged by a well-ordered water molecule. In the proposed hydrolysis mechanism, the metal-bound water acts as the nucleophile to attack the phosphorous center of phosphoserine/threonine. The metal centers contribute by Lewis acid catalysis to lower the pKa of the bound water and to enhance its nucleophilicity.
The metals in some cases (PP1 and PP2B) also coordinate the phosphate of pSer/pThr, which would serve to enhance the electrophilicity of the phosphate. A second metal-bound water has been proposed to perform general acid base catalysis to shuttle protons to the departing serine hydroxyl. The phosphate of the substrate is positioned by hydrogen bonding to Arg33 residue as well as metal-bound waters (Fig. 5).

Figure 5. (a) Proposed catalytic mechanism of protein tyrosine phosphatases. (b) Structure of PTP1B (gray) with a phosphopeptide substrate (gold) showing the relative positions of catalytic residues to the phosphotyrosine moiety.
Chemical Tools to Study Protein Phosphorylation
Chemical tools have played a central role in elucidating the function of posttranslational protein phosphorylation. Kinase and phosphatase inhibitors have played a major role in the study of protein phosphorylation because they allow rapid, reversible inactivation of the target in meaningful contexts (cells and organisms) to study its biologic function. Chemical tools have also helped identify the direct substrates of individual kinases and phosphatases, and conversely, they have helped identify which kinase is responsible for phosphorylating a particular phosphoprotein. Here I will focus on chemical tools developed for protein kinases; several reviews discuss chemical tools for protein phosphatases (15, 16).
Engineering protein kinases to label direct protein substrates
A main question regarding protein phosphorylation is what the direct targets of any given protein kinase are, within a signal transduction event. If all phosphotransfers could be mapped, the sequence of events that lead to a host of important biologic functions would be known, which not only would be important for a full understanding of signal transduction but also would enable the rational design of drugs that target signaling systems with predictable outcomes. This information is very difficult to obtain because many kinases work in concert during a signaling event and phosphorylate many different proteins.
A chemical method was devised to address directly the issue of determining the direct substrates of any given protein kinase (17). The idea is centered on generating ATP analogs (A*TPs) that can function as phosphodonors in a kinase reaction only with a protein kinase that has been engineered properly to accept the modified nucleotide. Thus, the addition of radiolabeled A*TP to a cell lysate that contains one such A*TP-sensitive protein kinase would result in selective radiolabeling of its direct substrates (Fig. 6a).
Several requirements of an unnatural A*TP analog and engineered kinase exist to have substrate labeling capabilities. First, the A*TP must not be a substrate for any endogenous kinases. Second, a mutation in the ATP binding site of a kinase must be discovered such that A*TP is now used in the phosphotransfer reaction with good efficiency. Finally, the mutation that expands the nucleotide specificity of the kinase should be functionally silent, so the mutant kinase performs its roles normally, with the single exception of accepting A*TP for substrate labeling purposes.
Toward this goal, several radiolabeled N6-substituted A*TP analogs were synthesized and tested for their ability to label kinase substrates in a cell lysate rich in active kinases. Several bulky N6substituents rendered the ATPs “orthogonal” to the set of all wild-type kinases, such as N6-cyclopentyl ATP and N6-benzyl ATP. Examination of available ATP: kinase cocrystal structures then allowed design of mutations that would allow these substrates to bind productively to the active site. One residue in the linker region between the small N-terminal and larger C-terminal subdomains (M120 in protein kinase A) directly contacts N6 of ATP and is a conserved large hydrophobic residue. This hydrophobic residue serves as a “gatekeeper” to a larger hydrophobic pocket behind it (Fig. 6b). Point mutation, in which this residue is changed to glycine, allows the bulky N 6 benzyl/cyclopentyl substituent to access the hydrophobic pocket. This action rendered the mutant kinases efficient catalysts with the N6-substituted A*TPs and only modestly changed their catalytic efficiency with ATP, thereby making the mutation functionally silent. Most impressively, the mutation that sensitizes kinase toward ATPs is portable to nearly all kinases in the superfamily and in multiple organisms. Numerous groups have used this system to identify kinase substrates from many different signaling pathways in several organisms.

Figure 6. Scheme of the method to identify direct protein kinase substrates using the unnatural nucleotide N6-(benzyl) ATP and kinases engineered to accept them by mutation of the gatekeeper residue (T338G in c-Src). Introduction of either the engineered kinase or y-32P labeled ATP analog alone results in no substrate labeling, but in combination, the direct substrates of the engineered kinase are radiolabeled. (b) The cocrystal structure of wt c-Src and ADP shows direct contact between the Thr338 residue (red) and the N6 position of ATP, precluding catalytic activity with N6-substituted ATP analogs. (c) The cocrystal structure Thr338Gly c-Src and N6-(benzyl) ATP shows the this mutant exposes a hydrophobic pocket that allows recognition of the unnatural nucleotide.
Specific kinase/phosphatase inhibitors as chemical tools
Perhaps the largest contribution that chemistry has made to the study of kinase-mediated signaling is the development of chemical inhibitors of protein kinases and phosphatases. Kinase inhibitors are under intense investigation as drugs after the spectacular success of the Abl tyrosine kinase inhibitor Gleevac as a treatment for chronic mylogenous leukemia. Because of this interest in drug development, many inhibitors of kinases are known and used as biochemical tools to inactivate rapidly kinases of interest. However, chemical inhibitors are useful to study the biology of kinases and phosphatases only if the compounds are selective for one member of the larger family. If compounds are monospecific, their biologic effects can be assigned unambiguously to the inhibition of a single enzyme. For protein kinases, however, truly selective inhibitors have been difficult to discover, and because the family is so large, it has been very difficult to confirm what the selectivity of commonly used inhibitors actually is. A recent comprehensive survey of the actual specificity of commonly used kinase inhibitors showed that many compounds are not as selective as previously thought and that many have more potent targets than the kinase they are commonly used against (18). This article highlights the need for strategies that preserve the advantages of using small molecule inhibitors to probe kinase function, such as speed, ease, and tunability, with stringent verification of target specificity. The difficulty in generating specific kinase inhibitors stems from the fact that most inhibitors target the kinase ATP binding site. This site is conserved highly among the family, which makes development of specific compounds difficult.
Toward this goal, a chemical-genetic strategy for developing specific kinase inhibitors was developed (19). The “gatekeeper” residue that when mutated to alanine or glycine sensitizes protein kinases toward unnatural A*TPs also was exploited to generate kinases that are sensitive to inhibitors with similar bulky substituents oriented toward the gatekeeper residue. In this way, cells carrying the mutant kinase would be uniquely sensitive to inhibitors that require binding the hydrophobic pocket adjacent to the gatekeeper. The relatively nonselective inhibitor scaffold PP1 was synthesized with bulky substituents and ring expansions that were designed to contact the gatekeeper residue (Fig. 3). It has been shown that virtually any kinase can be “programmed” to be sensitive to these bulky inhibitors by mutating a single amino acid. The selectivity of these compounds toward “analog-sensitive” kinase alleles is remarkable (~1000 fold versus wild type kinases), as is their affinity, with IC50values in the low nanomolar range. Furthermore, because inhibition requires a specific genetic background, for example, mutation of the gatekeeper residue, off-target effects can be controlled for by comparison with drug-treated cells with an intact gatekeeper.
Another recent general strategy to generate specific kinase inhibitors involves generating compounds that probe the phosphoacceptor peptide binding groove between the small and large lobes of the catalytic domain (20). Often kinases are selective for specific peptide substrates, so exploitation of this selectivity can yield specific kinase inhibitors. Inhibitors were generated by linking the ATP moiety to kinase-specific peptide substrates to yield “bifunctional” inhibitors that capture interactions in the ATP and peptide substrate binding sites (Fig. 7). In one such study, targeted against the insulin receptor tyrosine kinase domain, γ-thio ATP was linked to an optimized IRK peptide substrate, which creates a nucleotide-peptide conjugate that has a submicromolar Ki (370 nM). The bifunctional inhibitor also shows selectivity toward IRK, as the Ki of the conjugate for the tyrosine kinase Csk was 100-fold higher. This strategy is suitable for any kinase whose peptide substrate specificity is known and has been applied for many diverse kinases.
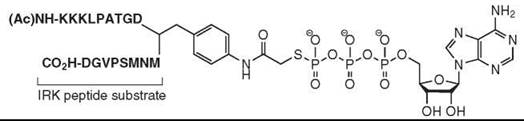
Figure 7. Structure of ATP-peptide substrate linked "bifunctional" kinase inhibitors are a general strategy to generate specific inhibitors of any protein kinase whose peptide substrate specificity is known.
Chemically cross-linking kinases and their substrates
Another chemical biology tool generated to study protein kinase signaling is a method to identify a kinase responsible for phosphorylating a particular phosphoprotein. Two methods have been devised by designing ATP molecules that form covalent bonds to both the kinase and substrate, which allows identification of the unknown partner by mass spectrometry.
In the first case, ATP analogs that have two azido groups attached at the purine and gamma-phosphate were synthesized (21). Such a molecule can bind the ATP binding site of a given kinase and allows its physiologic substrate to bind the bisazidoATP:kinase complex, which brings both kinase and target in close proximity to the azido groups. UV irradiation then uses each azide to form reactive nitrenes, which form a covalent ATP bridge between the kinase and substrate (Fig. 8a).
An alternate strategy to identify the upstream kinase responsible for phosphorylating a particular phosphoserine protein also was reported (22). In this scheme, a bis aryl aldehyde ATP analog was designed to form an imine with the lysine (Lys 72 in PKA) essential for orienting the a/p phosphates of ATP. The substrate of interest, with its target serine mutated to cysteine, provides a nucleophile to attack the imine, which then cyclizes with a second aldehyde to form a stable isoindole adduct that links kinase and substrate (Fig. 8b).

Figure 8. Two strategies to cross-link kinases with their substrates, involving either photo-cross-linking groups (a) or a mechanism-based annulation reaction (b).
References
1. Hunter T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell 1995; 80:225-236.
2. Cohen P. The regulation of protein function by multisite phosphorylation-a 25 year update. Trends Biochem. Sci. 2000; 25:596-601.
3. Hunter T. Signaling-2000 and beyond. Cell 2000;00:113-127.
4. Aqvist J, Kolmodin K, Florian J, Warshel A. Mechanistic alternatives in phosphate monoester hydrolysis: what conclusions can be drawn from available experimental data? Chem. Biol. 1999; 6:R71-80.
5. Madhusudan, Akamine P, Xuong NH, Taylor SS. Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat. Struct. Biol. 2002; 9:273-277.
6. Madhusudan, Trafny EA, Xuong NH, Adams JA, Ten Eyck LF, Taylor SS, Sowadski JM. cAMP-dependent protein kinase: crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 1994; 3:176-187.
7. Kim C, Vigil D, Anand G, Taylor SS. Structure and dynamics of PKA signaling proteins. Eur. J. Cell Biol. 2006; 85:651-654.
8. Zhang ZY, Wang Y, Wu L, Fauman EB, Stuckey JA, Schubert HL, Saper MA, Dixon JE. The Cys(X)5Arg catalytic motif in phosphoester hydrolysis. Biochemistry 1994; 33:15266-15270.
9. Kolmodin K, Aqvist J. The catalytic mechanism of protein tyrosine phosphatases revisited. FEBS Lett. 2001; 498:208-213.
10. Zhang ZY, Dixon JE. Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 1993; 32:9340-9345.
11. Fauman EB, Yuvaniyama C, Schubert HL, Stuckey JA, Saper MA The X-ray crystal structures of Yersinia tyrosine phosphatase with bound tungstate and nitrate. Mechanistic implications. J. Biol. Chem. 1996; 271:18780-18788.
12. Pannifer AD, Flint AJ, Tonks NK, Barford D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J. Biol. Chem. 1998; 273:10454-10462.
13. Denu JM, Tanner KG. Redox regulation of protein tyrosine phosphatases by hydrogen peroxide: detecting sulfenic acid intermediates and examining reversible inactivation. Methods Enzymol. 2002; 348:297-305.
14. Jackson MD, Denu JM. Molecular reactions of protein phosphatases-insights from structure and chemistry. Chem. Rev. 2001; 101:2313-2340.
15. Kumar S, Liang F, Lawrence DS, Zhang ZY. Small molecule approach to studying protein tyrosine phosphatase. Methods 2005; 35:9-21.
16. Zhang ZY. Chemical and mechanistic approaches to the study of protein tyrosine phosphatases. Acc. Chem. Res. 2003; 36:385-392.
17. Shah K, Liu Y, Deirmengian C, Shokat KM. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:3565-3570.
18. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000; 351:95-105.
19. Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000; 407:395-401.
20. Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, Cole PA. Mechanism-based design of a protein kinase inhibitor. Nat. Struct. Biol. 2001; 8:37-41.
21. Parang K, Kohn JA, Saldanha SA, Cole PA. Development of photo-crosslinking reagents for protein kinase-substrate interactions. FEBS Lett. 2002; 520:156-160.
22. Maly DJ, Allen JA, Shokat KM. A mechanism-based cross-linker for the identification of kinase-substrate pairs. J. Am. Chem. Soc. 2004; 126:9160-9161.
Further Reading
Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 1998; 27:133-164.
Cleland WW, Hengge AC. Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 2006; 106:3252-3278.
Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan M, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chem. Rev. 2001; 101:2243-2270.
Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat. Methods 2005; 2:17-25.
Walsh CT. Posttranslational Modification of Proteins: Expanding Nature’s Inventory. 2006. Roberts and Company, Greenwood Village, CO.
See Also
Cellular Communication by Signal Transduction
Mitogen Activated Protein Kinases (MAPK): ERKs and JNKs
Selective Inhibitors of Kinases
Small Molecules to Elucidate Kinase Pathways