CHEMICAL BIOLOGY
Essential Fatty Acids, Physiology and Clinical Significance of
Undurti N. Das, UND Life Sciences, Shaker Heights, Ohio
doi: 10.1002/9780470048672.wecb282
Essential fatty acids (EFAs)—linoleic acid (LA) and α-linolenic acid (ALA)—are essential for the brain growth and development of humans. EFAs are readily available in the diet, and hence their deficiency is not common. But, to provide their full benefit, EFAs have to be metabolized to their long-chain metabolites. EFAs form precursors to various prostaglandins (PGs), thromboxanes (TXs), leukotrienes (LTs), lipoxins (LXs), resolvins, neuroprotectins, isoprostanes, and hydroxy- and hydroperoxyeicosa-tetraenoates. Certain PGs, TXs, and LTs have pro-inflammatory actions, whereas LXs, resolvins, and neuroprotectins are anti-inflammatory in nature and are critical for wound healing, the resolution of inflammation, and the repair of tissues. EFAs and some of their long-chain metabolites inhibit the activities of angiotensin-converting and HMG-CoA reductase enzymes and cholesteryl ester transfer protein (CETP), enhance acetylcholine levels in the brain, increase the synthesis of endothelial nitric oxide, augment diuresis, enhance insulin action, and could regulate telomerase activity. Thus, EFAs and their metabolites may function as an endogenous ''polypill.'' In addition, EFAs and their long-chain metabolites react with nitric oxide (NO) to yield respective nitroalkene derivatives that exert cell-signaling actions via ligation and activation of peroxisome proliferator-activated receptors (PPARs). Thus, EFAs and their derivatives have varied biologic actions that may have relevance to their involvement in several physiologic processes and clinical conditions.
Essential fatty acids (EFAs) are essential for the survival of humans and other mammals; they cannot be synthesized in the body and, hence, have to be obtained in our diet and, thus, are essential (1-4). EFAs are an important constituent of cell membranes and confer on membranes properties of fluidity; thus, they determine and influence the behavior of membrane-bound enzymes and receptors. Two types of naturally occurring EFAs exist in the body: the ω-6 series derived from linoleic acid (LA, 18:2) and the ω-3 series derived from α-linolenic acid (ALA, 18:3). Both the ω-6 and the ω-3 series are metabolized by the same set of enzymes to their respective long-chain metabolites. Although some functions of EFAs require their conversion to eicosanoids and other products, in most instances the fatty acids themselves are active. The longer-chain metabolites of LA and ALA regulate membrane function and are of major importance in the brain, retina, liver, kidney, adrenal glands, and gonads.
Metabolism of Essential Fatty Acids
EFAs also are polyunsaturated fatty acids (PUFAs) because they contain two or more double bonds. PUFAs are fatty acids, some of which have at least two carbon-to-carbon double bonds in a hydrophobic hydrocarbon chain. At least four independent families of PUFAs exist, depending on the parent fatty acid from which they are synthesized. They include:
The “ω-3” series, which is derived from α-linolenic acid (ALA, 18:3, ω-3).
The “ω-6” series, which is derived from cis-linoleic acid (LA, 18:2, ω-6).
The “ω-9” series, which is derived from oleic acid (OA, 18:1, ω-9).
The “ω-7” series, which is derived from palmitoleic acid (PA, 16:1, ω-7).
LA is converted to γ-linolenic acid (GLA, 18:3, n-6) by the action of the enzyme ∆6 desaturase (d-6-d), and GLA is elongated to form dihomo-GLA (DGLA, 20:3, n-6), the precursor of the 1 series of prostaglandins (PGs). DGLA also can be converted to arachidonic acid (AA, 20:4, n-6) by the action of the enzyme ∆5 desaturase (d-5-d). AA forms the precursor of the 2 series of prostaglandins, thromboxanes, and the 4 series of leukotrienes. ALA is converted to eicosapentaenoic acid (EPA, 20:5, n-3) by d-6-d and d-5-d. EPA forms the precursor of the 3 series of prostaglandins and the 5 series of leukotrienes. LA, GLA, DGLA, AA, ALA, EPA and docosahexaenoic acid (DHA, 22:6, n-3) are all PUFAs, but only LA and ALA are EFAs (see Fig. 1 for the metabolism of EFAs). AA and EPA give rise to their respective hydroxy acids, which, in turn, are converted to their respective leukotrienes (LTs). In addition, AA, EPA, and DHA form the precursor to anti-inflammatory compounds such as lipoxins and resolvins. PGs, LTs, lipoxins (LXs), and resolvins are highly active, modulate inflammation, and are involved in several pathophysiologic processes, such as atherosclerosis, bronchial asthma, inflammatory bowel disease, and other inflammatory conditions (5-8). In general, for the sake of brevity, the term “EFAs” is used to refer to all unsaturated fatty acids: LA, GLA, DGLA, AA, ALA, EPA, and DHA; and the term polyunsaturated fatty acids (PUFAs) refers to GLA, DGLA, AA, EPA, and DHA. Although the terms EFAs and PUFAs are used interchangeably for the sake of convenience, it should be understood that all EFAs are PUFAs but all PUFAs are not EFAs. Many functions of EFAs also are brought about by PUFAs, and EFA-deficiency states can be corrected largely by PUFAs; that led to the suggestion that PUFAs are “functional EFAs.” Hence, in general, many authors use the terms EFAs and PUFAs interchangeably. This convention is followed in the current discussion also.
EFAs/PUFAs play a significant role in the pathobiology of clinical conditions such as collagen vascular diseases, hypertension, diabetes mellitus, metabolic syndrome X, psoriasis, eczema, atopic dermatitis, coronary heart disease, atherosclerosis, and cancer (1-8). This role is in addition to the role of PGs and LTs in these conditions. For instance, in ulcerative colitis, the inflammatory events are initiated and perpetuated by PGs and LTs produced from AA, whereas when significant amounts of EPA and DHA are given, the inflammatory process is abrogated to a large extent. This beneficial action of EPA/DHA when supplemented from external sources has been attributed to the displacement AA from the cell membrane phospholipid pool and formation of less pro-inflammatory PGs and LTs from them, and anti-inflammatory molecules LXs and resolvins and hence the favorable response. If the molecular mechanism(s) by which various stimuli can induce preferentially the release of AA, EPA, and/or DHA and convert them to their respective pro- and anti-inflammatory products, then it is possible to develop methods to treat various inflammatory conditions based on this knowledge. Such an understanding and knowledge is expected to lead to strategies that will help to enhance preferentially the formation of less pro-inflammatory molecules from EPA/DHA and to stimulate the synthesis and release of anti-inflammatory molecules such as LXs and resolvins that could lead to suppression of inflammation and wound healing. LXs and resolvins resolve inflammation by suppressing leukocyte infiltration and clearing cellular debris from the site of inflammation. In view of this action, it is important to know the molecular regulation of their formation and release in various cells and tissues and various diseases that is expected ultimately to develop better methods of managing various inflammatory conditions. Thus, PUFAs form precursors to both pro- and anti-inflammatory molecules, and the balance between these mutually antagonistic compounds could determine the final outcome of the disease process.
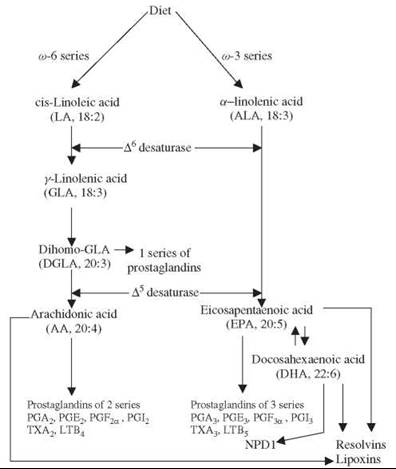
Figure 1. Scheme shows the metabolism of essential fatty acids.
Dietary Sources of EFAS
The EFAs LA and ALA are present in human diet in abundant amounts, and, hence, EFA deficiency is uncommon. In certain specific conditions, such as total parentaral nutrition (TPN) and severe malabsorption, occasionally EFA deficiency could be seen. The current TPN solutions contain adequate amounts of EFAs. The manifestations of an EFA deficiency include dry and scaly skin, hepatospleenomegaly, immunodeficiency, inappropriate water loss through the skin, dehydration, scalp dermatitis, alopecia, and depigmentation of hair (9, 10). EFAs are distributed widely in normal human diet. The main dietary sources of EFAs are as follows.
Human breast milk is rich in all types of PUFAs (11); this explains why breast-fed children are healthier compared with bottle-fed children. LA and ALA are present in significant amounts in dairy products, in organ meats such as liver, and in many vegetable oils such as sunflower, safflower, corn, and soy. GLA is present in evening primrose oil at concentrations of 7-14% of total fatty acids, in borage seed oil at 20-27%, and in black currant seed oil at 15-20%. GLA also is found in some fungal sources (1-4, 12). DGLA is found in liver, testes, adrenals, and kidneys. AA is present in meat, egg yolks, some seaweeds, and some shrimps. The average daily intake of AA is estimated to be in the region of 100-200 mg/day, more than enough to account for the total daily production of various PGs. EPA and DHA are present mainly in marine fish. Cow milk contains very small amounts of GLA, DGLA, and AA.
EFAs/PUFAs are unstable because of the presence of two or more double bonds in their structure; therefore, substantial loss occurs during food processing and the hydrogenation of oils. When exposed to high temperatures and during hydrogenation, EFAs/PUFAs are denatured and converted into trans fats that are harmful to the body (1-4, 13-15). Human diet was rich in ω-3 fatty acids in the early humans. But, with the progress in industrialization and the development of fast foods, the content of ω-3 fatty acids in the human diet dwindled, whereas that of to-6 fatty acids increased. Thus, the ratio of ω-3 to ω-6 fatty acids in the diet of early humans was >1, whereas this ratio now is believed to be about <1 and around 10:1 to 20-25:1 (16). It is recommended that the ratio between ω-3 to ω-6 fatty acids in the diet should be about 1 or >1 and preferably 2-3:1. It is believed that the fall in the intake of ω-3 fatty acids EPA and DHA in the last 50 years is responsible for the increasing incidence of modern diseases such as atherosclerosis, CHD, hypertension, metabolic syndrome X, obesity, collagen vascular diseases, and, possibly, cancer. This belief is supported by the observation that increasing the dietary α-linolenate/linoleate balance affected the ω-3/ω-6 ratio of brain phospholipid acyl chains; also, it produced changes in general behavior and changes in sensitivities to drugs known to affect behavior (17), influenced the LTs formation in polymorphonuclear leukocytes from AA and EPA and the release of histamine from mast cells that could alter the severity of allergic and inflammatory responses (18), resulted in an increased mean survival time of SHR-SP (spontaneously hypertensive-stroke prone) rats by lowering blood pressure and platelet aggregability (19), produced significant changes in Na+-K+-ATPase activity (20), and altered collagen-induced platelet aggregation and serotonin release in experimental animals (21). These results indicate that increased intake of ω-3 fatty acids is of significant benefit in various diseases.
Factors Influencing the Metabolism of EFAS
Dietary LA and ALA are metabolized by the same set of ∆6 and ∆5 desaturases and elongases to their respective metabolites (see Fig. 2), and, hence, these two fatty acids compete with one another for the same set of enzymes. ∆6 and ∆5 desaturases prefer ω-3 to ω-6. Oleic acid (OA, ω-9) that is not an EFA also is metabolized by the same ∆6 and ∆5 desaturases. But in view of the preference of these enzymes to LA and ALA under normal physiologic conditions, the metabolites of ω-9 are formed only in trivial amounts. A presence of significant amounts of 20:3 ω-9, a metabolite of OA, in the cells and plasma indicates an EFA deficiency. This fact is used to detect the presence of EFA deficiency in patients, in experimental animals, and in in vitro studies.
Several factors exist that could influence the activities of desaturases and elongases (1-4, 12, 22, 23). Saturated fats, cholesterol, trans-fatty acids, alcohol, adrenaline, and glucocorticoids inhibit ∆6and ∆5 desaturases. Pyridoxine, zinc, and magnesium are necessary cofactors for normal ∆6 desaturase activity. Insulin activates ∆6 desaturase, whereas diabetics have reduced ∆6 desaturase activity. The activity of ∆6 desaturase falls with age. Oncogenic viruses and radiation inhibit ∆6 desaturase. Total fasting, protein deficiency, and glucose-rich diets reduce the activity of ∆6 desaturase. A fat-free diet and a partial caloric restriction enhances ∆6 desaturase. Activities of ∆6 and ∆5 desaturases are decreased in diabetes mellitus, hypertension, hyperlipidemia, and metabolic syndrome X. Trans fats, saturated fatty acids, and cholesterol interfere with EFA metabolism and promote inflammation, atherosclerosis, and coronary heart disease (14, 15, 23, 24). Thus, trans fats, saturated fats, and cholesterol have pro-inflammatory actions, whereas EFAs and PUFAs possess anti-inflammatory properties. This reason explains why trans fats, saturated fats, and cholesterol are pro-atherogenic, whereas EFAs/PUFAs, especially to-3 fatty acids, are anti-atherogenic. It is likely that trans fats, saturated fats, and cholesterol interfere with the formation of AA, EPA, and DHA from dietary LA and ALA. This interference, in turn, could lead to the decreased formation of LXs, resolvins, PGI2 (prostacyclin), PGI3, and other beneficial eicosanoids that prevent platelet aggregation, leukocyte chemotaxis, and activation, that decrease the formation of pro-inflammatory cytokines, and that produce vasodilatation, events that are necessary in the prevention or arrest of atheroslcerosis. In contrast, trans fats, saturated fats, and cholesterol may activate leukocytes directly, induce the generation of free radicals, and enhance the production and release of pro-inflammatory cytokines that facilitate atherosclerosis (14, 15, 25-29). The direct action of trans fats, saturated fats, and cholesterol on leukocytes and macrophages, which activates them to produce harmful free radicals and pro-inflammatory cytokines such as IL-6, TNF-α, IL-1, IL-2, and MIF (macrophage migration inhibitory factor), could be in addition to their ability to suppress the metabolism of EFAs to their respective long-chain metabolites. It is not known yet whether trans fats, saturated fats, and cholesterol have the ability to inhibit the formation of LXs, resolvins, PGI2, and PGI3. It is likely that trans fats, saturated fats, and cholesterol are more likely to inhibit the formation of LXs, resolvins, PGI2, and PGI3. Based on these studies, it is proposed that EFAs, especially EPA and DHA, are cytoprotective to endothelial cells, whereas trans fats, saturated fats, and cholesterol produce endothelial dysfunction. It is known that AA, EPA, and DHA augment nitric oxide generation from endothelial cells (1-4, 6, 8, 30) and, thus, help in the prevention of endothelial dysfunction. In contrast, trans fats, saturated fats, and cholesterol produce endothelial dysfunction and, thus, inhibit eNO production. Furthermore, NO quenches superoxide anion; thus, it prevents the cytotoxic action of superoxide anion and protects endothelial cells from free radical-induced damage. This finding implies that for endothelial cells to be healthy, they need adequate amounts of EFAs, especially AA, EPA, and DHA so that they can generate physiologic amounts of eNO not only to prevent pathologic platelet aggregation and atherosclerosis but also to protect themselves from the cytotoxic actions of free radicals. Furthermore, NO reacts with PUFAs to yield their respective nitroalkene derivatives that can be detected in plasma. These nitroalkene derivatives, termed nitrolipids, produce vascular relaxation, inhibit neutrophil degranulation and superoxide formation, inhibit platelet activation, and show anti-atherosclerotic properties (3, 4, 31-34). Thus, a close interaction seems to exist between EFAs and their products and trans fats, saturated fats, and cholesterol with regard to the ability of endothelial cells to produce PGI2, PGI3, NO, and other anti-atheroslcerotic and beneficial molecules.
Actions of EFAS/PUFAS Relevant to Various Clinical Conditions
Cell membrane fluidity and modification of receptor properties
Cell membrane fluidity is determined by its lipid composition. The cell membrane is rendered more rigid if increased amounts of saturated fatty acids and cholesterol are incorporated into the membrane phospholipids. In contrast, increased incorporation of unsaturated fatty acids into the membrane will make it more fluid. This increase in membrane fluidity has been shown to increase the number of receptors and their affinity to their respective hormones or growth factors. For instance, an increase in the rigidity of the cell membrane reduces the number of insulin receptors and their affinity to insulin that, in turn, could cause insulin resistance. Alternatively, an increase in cell membrane fluidity because of an increase in the unsaturated fatty acid content in the membrane phospholipids increases the number of insulin receptors and their affinity to insulin and, thus, decreases insulin resistance (35-42).
This action of EFAs/PUFAs has relevance to the growth and development of the brain and to their influence on cognitive function. The growth and development of the brain during the perinatal period and adolescence is dependent on the availability of ω-3 and ω-6 fatty acids (43-45). Hence, decreased availability of ω-3 and ω-6 fatty acids during this critical period of growth may impair brain growth and the development of appropriate synaptic connections that, in turn, may lead to the development of neuropsychologic conditions such as dementia, depression, schizophrenia, Alzheimer’s disease, and neurodegenerative diseases (Huntington’s disease, Parkinson’s disease, and spinocerebellar degeneration). The alterations in the neuronal cell membrane fluidity also might influence the cognitive function. This suggestion is supported by the recent observation that EFAs/PUFAs activate syntaxin, a plasma membrane protein that has an important role in the growth of neurites.
For proper neuronal development and increase in cell membrane surface area, the growth of neurite processes from the cell body is critical (46). Nerve growth cones are highly enriched with AA-releasing phospholipases, which have been implicated in neurite outgrowth (47, 48). Cell membrane expansion occurs through the fusion of transport organelles with plasma membrane, and syntaxin 3, a plasma membrane protein that plays a significant role in the growth of neurites, has been shown to be a direct target for AA, DHA, and other PUFAs (49). AA, DHA, and other PUFAs but not saturated and monounsaturated fatty acids activate syntaxin 3. Of all the fatty acids tested, AA and DHA were found to be the most potent compared with LA and ALA. Even syntaxinl that is involved specifically in fast calcium-triggered exocytosis of neurotransmitters is sensitive to AA (50), which implies that AA is involved both in exocytosis of neurotransmitters and in neurite outgrowth. It is interesting that SNAP25 (synaptosomal-associated protein of 25 kDa), a syntaxin partner implicated in neurite outgrowth, interacted with syntaxin 3 only in the presence of AA that allowed the formation of the binary syntaxin 3-SNAP 25 complex. AA stimulated syntaxin 3 to form the ternary SNARE complex (soluble N-ethylmaleimide-sensitive factor attachment protein receptor), which is needed for the fusion of plasmalemmal precursor vesicles into the cell surface membrane that leads to membrane fusion. These results clearly demonstrated that AA and DHA change the α-helical syntaxin structure to expose the SNARE motif for immediate SNAP 25 engagement and, thus, facilitate neurite outgrowth. Hence, the availability of optimal amounts of EFAs/PUFAs during the perinatal and postnatal periods, periods of brain growth, is important for proper development of neurons, synapse formation, and cognitive function.
Second Messenger Actions of EFAS and their Metabolites
Several hormones and growth factors activate phospholipase A2 (PLA2) that, in turn, induces the release of DGLA, AA, EPA, and DHA from the cell membrane lipid pool. These fatty acids are used for the formation of various eicosanoids and bring about their actions. The inhibition of PLA2 interferes with the action of several growth factors and cytokines. For example, the tumoricidal action of TNF-α is dependent on its ability to induce PLA2, and inhibitors of PLA2 completely blocked its (TNF-a) anticancer action (51-54). Activation of PLA2 leads to the release of various PUFAs including AA.
PUFAs enhance the activity of protein kinase C (PKC), a well-known second messenger, activate macrophages and polymorphonuclear leukocytes (PMNs), and increase free radical generation (1-4, 55, 56). The effects of various types of PUFAs on the activation of macrophages and PMNs and their ability to generate free radicals depends on the type of EFAs/PUFAs used, on the degree of unsaturation, concentration(s) of the fatty acid, and on the conditions under which the experiments are performed. Thus, the effects of various PUFAs on macrophages and PMNs are complex and have to be interpreted with caution taking into consideration all of the variables involved.
Anti-Infective Properties of EFAS
Previously, I proposed that EFAs/PUFAs such as GLA, DGLA, AA, EPA, and DHA might behave as endogenous antibacterial, antifungal, antiviral, and immunostimulating agents (57, 58). LA and ALA have bacteriostatic effect on both gram-positive and gram-negative bacteria (58, 59). Staphylococcus aureus are killed rapidly by ALA, and a variety of bacteria were found to be sensitive to the growth inhibitory actions of LA and ALA in vitro (60, 61). Hydrolyzed linseed oil, which contains 52% ALA, and pure ALA were found to be capable of killing methicillin-resistant S. aureus (62). Animal herpes, influenza, Sendai, and Sindbis viruses could be inactivated by LA and AA within minutes of contact (63). Furthermore, both prostaglandin E1 (PGE1) and PGA, derived from DGLA, AA, and EPA, have the ability to inhibit viral replication and behave as antiviral compounds (64, 65). It was reported that the remission of mycosis fungoides, a rare skin disease of viral etiology, is possible with the oral administration of LA as safflower oil (which contains 76% LA) in dogs that correlated with an increase in the plasma levels of LA and AA (66).
Patients with Plasmodium falciparum showed suppressed lympho-proliferative responses to malaria antigens. This suppressed lymphocyte response has been attributed to the increased production of various prostaglandins (PGs) by monocytes/macrophages. This finding is supported by the observation that enhanced lymphocyte responses to several malaria antigens were enhanced particularly by indomethacin, a cyclo-oxygenase inhibitor, which suggests that malaria-specific T-cells are especially sensitive to the effects of PGs (67). In a related study, it was noted that during intra-erythrocytic development of the parasite, the phospholipid composition of the IEPM (infected erythrocyte plasma membrane) contained more phosphatidylcholine (38.7% versus 31.7%) and phosphatidylinositol (2.1% versus 0.8%) and less sphingomyelin (14.6% versus 28.0%) than normal uninfected erythrocytes. The percentage of PUFAs in normal erythrocyte phospholipids (39.4%) was much higher than in phospholipids from purified parasites (23.3%) or IEPM (24.0%). Large increases in palmitic acid (from 21.88% to 31.21%) and in oleic acid (from 14.64% to 24.60%) and significant decreases in AA (from 17.36% to 7.85%) and in DHA (from 4.34% to 1.8%) occurred because of the infection. The fatty acid profiles of individual phospholipid classes from IEPM resembled the fatty acid profiles of parasite phospholipids rather than those of uninfected erythrocytes, which suggests that these alterations are because of the parasite-directed metabolism of erythrocyte lipids (68). These results are interesting because it has been shown that iloprost, a synthetic prostacyclin (PGI2) analog, not only prevented the development of cerebral malaria in mice but also suppressed malaria antigen-induced tumor necrosis factor (TNF) production (69). PGI2 and PGI3 are derived from AA and EPA, respectively. Furthermore, AA, LA, EPA, and DHA inhibited the growth of P. falciparum in vitro, whereas oleic or docosenoic acids were ineffective. Parasite killing was increased significantly when oxidized forms of LA, AA, EPA, and DHA were used. Antioxidants greatly reduced the fatty acid-induced killing. Mice infected with P. berghei and treated for 4 days with DHA showed marked reduction in parasitemia (70). In this context, it is interesting to note that the cytotoxic effect of the fatty acids was very rapid: The full inhibition of nucleic acids and protein syntheses was observed in less than 30 minutes without showing any effects on RBCs such as the hemolysis of infected cells up to 500 μg/ml; also no effect on the lipid peroxidation, ATP levels, transport through the parasite-induced permeability pathways, or on the phagocytosis of the infected cells was noted (71). Furthermore, mice fed vitamin E-deficient diets that contain ω-3 fatty acids survive infection with lethal P. yoelii. Experiments performed in nu/nu mice (which lack alpha-beta T-cell-receptor-positive T cells and do not produce antimalarial antibody) and nu/+ mice revealed that animals fed casein-based diets that contain 4% menhaden oil with vitamin E supplementation for 4 weeks before infection with lethal P. yoelii developed fulminating parasitemias and quickly died, whereas both nu/nu and nu/+ mice fed diets deficient in vitamin E controlled their parasitemias for the first 18 days of infection. Thereafter, the nu/nu mice became anemic and died, whereas the nu/+ mice produced antimalarial antibodies and survived. In an extension of this study, when scid/scid.bg/bg mice (which lack B cells and alpha-beta and gamma-delta T cells and have reduced NK-cell activity) were fed the experimental diet for 6 weeks and then infected with the less virulent 17XNL strain of P. yoelii, the animals fed vitamin E-containing diets quickly died, whereas those fed the vitamin E-deficient diet survived without developing detectable parasitemias. These results suggest that under pro-oxidant dietary conditions, mice could control and even survive malaria in the absence of malaria-primed T cells and the antimalarial antibody (72), which implies that fatty acids need to be oxidized to produce their antimalarial action and eliminate malarial parasite. Our studies showed that in patients with P. falciparum malaria, the levels of lipid peroxides (a marker of free radical generation), nitric oxide (a potent free radical with immunomodulatory actions), and concentrations of LA and ALA are low, whereas those of EPA are high. Because the ability of the fatty acids to kill P. falciparum is dependent on their capacity to stimulate free radical generation in neutrophils and macrophages and EPA is more potent than LA in killing the parasite, these results imply that decreased levels of lipid peroxides and nitric oxide may contribute to the persistence of P. falciparum infection (73). In addition, it was reported that mice infected with P. berghei ANKA (which developed cerebral malaria) showed increased phospholipase A2mRNA expression in the spleen and cyclooxygenase 1 (COX1) and COX2 expression in the brain and had higher serum LTB4 levels than the control mice; also it was reported that aspirin-treated infected mice had higher serum LTB4 levels than untreated infected mice, which suggests that PGs are protective whereas LTs are detrimental in cerebral malaria (74). These results, coupled with the observations that elevated PGE2 in healthy malaria-exposed children may protect against malaria (75), that some PGs actually may reverse chloroquine-resistance of the malarial parasite (76), and that increased production of TXs may be involved in the pathogenesis of some complications associated with malaria suggest that EFAs/PUFAs and their eicosanoid metabolites play a significant role in the protection and the pathogenesis of malaria.
These observations suggest that EFAs/PUFAs and their products have antibacterial, antifungal, antiviral, and antiparasitic actions. Both lymphocytes and macrophages contain significant amounts of PUFAs and are capable of releasing them on appropriate stimulation. In addition, PUFAs stimulate NADPH-dependent superoxide production by macrophages, neutrophils, and lymphocytes that are capable of killing the invading microorganisms. In view of these evidences, it is reasonable to believe that an increased intake of LA, ALA, EPA, and DHA protects against and/or reduces the risk of various infections.
Recent studies showed that AA, EPA, and DHA could give rise to anti-inflammatory compounds such as lipoxins (LXs) and resolvins that are essential to limit and resolve inflammation (3, 4). These studies imply that a deficiency of LXs and resolvins could lead to the perpetuation of inflammation and tissue damage. In the light of these facts, it will be interesting to study whether a subclinical deficiency of PUFAs, decreased formation of LXs and resolvins, occurs in subjects who develop the various types of infections and their complications. Because, PUFAs can inactivate enveloped viruses including influenza, it is probably worthwhile to study the effect of various fatty acids on the bird flu virus, specifically, whether increased intake of these fatty acids could reduce the risk of flu.
It is interesting to note that an analog of myristic acid (14:0) showed selective toxicity to African Trypanosomes (77), PUFAs also can kill Helicobacter pylori (78), and the actions of some antibiotics could be potentiated by PUFAs (79), which lends support to the concept that PUFAs may function as endogenous anti-infective-like molecules (80, 81).
Actions that Could Qualify Pufas to be an Endogenous ''Polypill''
Coronary heart disease (CHD), stroke, peripheral vascular disease, and underlying atherosclerosis are the common cardiovascular diseases (CVD) that are responsible for considerable morbidity and mortality both in the developed and developing countries. Several studies revealed that smoking cessation, P-blockers, antiplatelet agents in the form of low-dose aspirin and other drugs, inhibitors of angiotensin-converting enzyme (ACE), and lipid-lowering agents such as statins that inhibit the activity of HMG-CoA reductase, each reduce the risk of vascular events to a moderate but important degree (82-90). In addition, observational studies suggested lower rates of fractures with statins, higher rates of obstructive airways disease at lower cholesterol concentrations, lower rates of cataracts, and lower rates of dementia with MRC/BHF-HPS study (90). The results of the MRC/BHF-HPS study led to the suggestion that about two-thirds to three-quarters of future vascular events could be prevented (see Table 1). This suggestion indicates that a combination of aspirin, P-blockers, lipid lowering (by 1-5 mmol) by the use of statins, and ACE inhibitors could reduce cardiovascular diseases by over 70-80% (91). This concept led to the suggestion that a combination pill (popularly called a “polypill”) consisting of atorvastatin 10 mg or simvastatin 40 mg; three blood pressure lowering drugs such as a thiazide, a β-blocker, and an ACE inhibitor, each at half the standard dose; folic acid 0.8 mg; and aspirin 75 mg could reduce CHD events by 88% (95% confidence interval 84% to 91%) and stroke by 80% (71% to 87%) (see Table (1) for a possible formulation of one such polypill). It was suggested that one third of the people taking such a combination pill from age 55 years of age would benefit, gaining on an average about 11 years of life free from an CHD event or stroke (92).
Table 1. Possible cumulative impact of four secondary prevention treatments in the prevention of cardiovascular diseases*
|
Drug therapy |
Relative risk reduction |
Two-year event ratio |
|
None Aspirin B-blockers Lipid lowering (by 1-5 mmol) ACE inhibitors |
— 25% 25% 30% 25% |
8% 6% 4.5% 3% 2.3% |
*Cumulative relative reduction if all four drugs are used is about 75% (see Reference 91). Events that were included in this analysis are cardiovascular death and myocardial infarction or strokes.
PUFAs suppress HMG-CoA reductase and ACE activities, inhibit platelet aggregation, enhance parasympathetic activity and, thus, enhance heart rate variability (and thus, have actions similar to that of P-blockers), and possess diuretic properties either by themselves and/or by increasing the formation of PGAs and PGEs that have been shown to increase renal blood flow and augment diuresis. These actions of PUFAs are similar to that of the polypill that is a combination of aspirin, P-blockers, statins, and ACE inhibitors, which is expected to reduce cardiovascular diseases by over 70-80% (92). This suggests that PUFAs could function as an “endogenous polypill”; the evidence for this is detailed below.
Pufas Inhibit ACE and HMG-COA Reductase Activities and Augment Endothelial Nitric Oxide Generation
PUFAs inhibited leukocyte ACE activity (93) and enhanced endothelial nitric oxide (eNO) generation (94). This finding implies that when tissue concentrations of PUFAs are low, the activity of ACE will be high, which results in increased formation of angiotensin-II and a simultaneous decrease in eNO. It was reported that transgenic rats overexpressing human renin and angiotensinogen genes (dTGR) that develop hypertension, inflammation, and renal failure showed specific renal P450-dependent AA metabolism changes that led to decreased formation of epoxy-eicosatrienoic acids (5,6-, 8,9-, 11,12- and 14,15-EETs) and hydroxyeicosa-tetraenoic acids (19- and 20-HETEs). Both EETs and HETEs inhibit IL-6 and TNF-a-induced activation of NF-KB and prevent vascular inflammation (95), which suggests that AA and other PUFAs not only regulate ACE activity and Ang-II levels in the tissues but also possess anti-inflammatory properties by generating anti-inflammatory metabolites.
In the presence of aspirin, AA, EPA, and DHA are converted to form epi-lipoxins, lipoxins, and resolvins that, in turn, enhance the formation of eNO (3, 4, 96). Lipoxins possess potent anti-inflammatory actions (reviewed in References 3 and 4). In addition, NO not only blocks the interaction between leukocytes and the vascular endothelium during inflammation but also stimulates the formation of PGI2, a potent vasodilator and platelet anti-aggregator, from AA (97, 98).
PUFAs are potent inhibitors of the HMG-CoA reductase enzyme and similar to statins are useful in the treatment of hyperlipidemias (99-102). Statins enhance plasma AA levels and decrease the ratio of EPA to AA significantly (100). This finding suggests that PUFAs mediate many actions of statins (103) and that this could be one mechanism by which they lower cholesterol levels. Statins and PUFAs have many overlap actions such as the inhibition of IL-6 and TNF-a production and NF-KB activation plus the ability to enhance eNO production; thus, both possess anti-inflammatory actions and both are useful in atherosclerosis, coronary heart disease, osteoporosis, stroke, Alzheimer’s disease, and inflammatory conditions such as lupus and cancer (3, 4, 94, 104-121). These similar and overlap actions strongly indicate that the molecular mechanisms of actions of statins and PUFAs are similar, if not identical. Furthermore, when a combination of statins and PUFAs are given together, a synergistic beneficial effect was seen in patients with combined hyperlipemia (122).
Both PUFAs and statins by inhibiting the HMG-CoA reductase enzyme reduce the formation of isoprenoid that contains compounds that are formed from mevalonate (because the HMG-CoA reductase enzyme reduces the formation of mevalonate). These isoprenoid precursors are necessary for the posttranslational lipid modification (prenylation) and, hence, the function of Ras and other small GTPases. Small GTPases, the prenylated products of the mevalonate pathway, have negative control on the expression of BMPs (bone morphogenetic proteins). BMPs are essential for neuronal growth, proliferation, and differentiation (123) and also for bone growth (105, 114). Thus, PUFAs modulate brain growth and development and neuronal differentiation. This finding explains why PUFAs are useful in the prevention and treatment of dementia and Alzheimer’s disease (115-120). Similar to PUFAs, statins also enhance the concentrations of BMPs in brain and bone and thus could be of benefit in the treatment of Alzheimer’s disease and osteoporosis (105, 109, 113). Yet another action of PUFAs and statins that contributes to their beneficial actions is their ability to enhance eNO (30, 124), a pleiotropic molecule that has many biologic actions including its ability to function as a neurotransmitter (125) and prevent osteoporosis (126, 127). But unlike statins that cannot be given during pregnancy, PUFAs can be consumed confidently during pregnancy, lactation, and infancy. In fact, PUFAs are recommended during pregnancy, lactation, and infancy to improve brain growth and development (1-4, 43, 44).
Pufas and Cholesteryl Ester Transfer Protein (CETP) Activity
Several studies suggested that HDL-cholesterol (high-density lipoprotein- cholesterol, HDL-C) is an independent risk factor for CHD. Higher plasma HDL-C is associated with a decreased incidence of CHD (128). This finding led to the suggestion that therapeutic strategies that raise HDL-C could be of benefit in preventing CHD with the hope that raising HDL-C will increase the movement of cholesterol from the periphery back to the liver (the so-called reverse cholesterol transport or RCT pathway) and protection from CHD will follow.
CETP is a hydrophobic plasma glycoprotein, mainly synthesized in the liver, that possesses the unique ability to facilitate the transfer of cholesteryl ester (CE). CETP circulates in the blood, bound predominantly to HDL. CETP mediates the transfer of cholesteryl esters from HDL to VLDL and LDL in exchange for triglycerides. CETP also promotes the transformation of HDL2 to HDL3, an action that could promote reverse cholesterol transport. CETP inhibition produces an increase in HDL by markedly delaying the catabolism of apoA-I and A-II (129), an action that increases reverse cholesterol transport. These actions of CETP suggest that CETP inhibition could prevent atherosclerosis (130-132).
In a study performed in healthy, normolipidemic men, it was observed that a lipid-lowering diet rich in monounsaturated fatty acid (oleic acid) decreased CETP concentrations to a significant degree (133). In HepG2 cells, it was noted that 0.5 mM of AA, EPA, and DHA reduced the levels of CETP mRNA by more than 50% of the control levels with a corresponding significant decrease in the CETP mass (134). This finding is supported by the observation that in type 2 diabetic subjects, CETP activity was correlated significantly with the HDL-C to apoA1 ratio and to the LDL-C to HDL-C ratio. In addition, a significant negative correlation was found between plasma CETP activity and monounsaturated fatty acid content of plasma phospholipids or free PUFAs, especially with W-3 fatty acids, which suggests that PUFAs suppress CETP activity (135).
Torcetrapib, a small molecule inhibitor of CETP, is very effective at raising HDL-C and apolipoprotein A-I and decreasing levels of LDL-C and apolipoprotein-B-100; it also showed favorable effects on increasing the size of HDL and LDL particles. Elevated CETP levels have been shown to be associated with an increased risk of future CHD in apparently healthy subjects (136). The results performed in animals wherein it was observed that the inhibition of CETP increases HDL-C and, thus, decreases the atherosclerosis process itself, led to the initiation of clinical trials. In patients with familial hypercholesterolemia, torcetrapib with atorvastatin as compared with atorvastatin alone did not result in the reduction of the progression of atheroslcerosis as measured by carotid arterial-wall thickness, despite a significant increase in HDL-C levels and a decrease in levels of LDL-C and triglycerides. In fact, it was observed that the administration of torcetrapib with atorvastatin was associated with the progression of atherosclerosis (137). In another study that involved the use of torcetrapib alone or in combination with atorvastatin, an increase in blood pressure with no significant decrease in the progression of coronary atherosclerosis was noted (138).
These results with torcetrapib and atorvastatin suggest that the simultaneous inhibition of CETP and the HMG-CoA reductase enzyme leads to an elevation of plasma HDL-C and a decrease in LDL-C, triglycerides, and cholesterol but that it does not arrest the progression of atherosclerosis. In contrast, PUFAs, especially o-EPA and DHA, not only inhibit CETP and the HMG-CoA reductase enzyme and lower plasma triglycerides, cholesterol, and LDL-C with little or no change in HDL-C but also are effective in arresting atherosclerosis and preventing CHD (139-151). In contrast to the results with torcetrapib and atorvastatin, Yokoyama et al. (152) reported that a combination of ethyl EPA and 10 mg of pravstatin or 5 mg of simvastatin prevented major coronary events and especially nonfatal coronary events in Japanese hypercholesterolemic patients with a mean period of follow up of 4.6 years. It is interesting to note that the benefits were in addition to statin treatment and that fish oil was found to be safe and well tolerated. These results once again confirm that EPA and DHA are of benefit in the prevention and treatment of cardiovascular diseases. Thus, PUFAs seem to be superior to CETP and statins in the prevention of CVD despite the fact that they do not necessarily increase plasma HDL-C levels.
Effects on Platelets and Other Hemostatic Indices
Both EPA and DHA, when given orally, are incorporated rapidly into platelets and compete with AA for the 2-acyl position of membrane phospholipid and as a substrate for the cyclo-oxygenase (CO) and lipoxygenase (LO) enzymes. As a result, when stimulated, such platelets produce less amounts of TXA2 and more of TXA3; that is less potent in inducing platelet aggregation and thrombosis (153). Increased intake of fish oil rich in EPA and DHA produces a lower platelet count, less platelet aggregation, a longer bleeding time, higher urinary PGI2 metabolites, and lower concentrations of thromboxane metabolites compared with those who were on a Western diet (154, 155); the increased intake of fish oil rich in EPA and DHA produced effects that are similar to those of low-dose aspirin and qualify to term ω-3 EPA and DHA as an “endogenous aspirin.” In general, although EPA and DHA do not seem to have a very significant effect on blood lipids, on fibrinolysis, and on the activity of plasminogen activity inhibitor-type-1 (PAI-1), they are effective still in preventing overall mortality from CAD/CVD.
Pufas and Renal Function
One component suggested to be included in the “polypill” is a thiazide, a diuretic and an anti-hypertensive drug. If PUFAs are considered to function as an endogenous “polypill,” then it is imperative that they (PUFAs) should show beneficial actions on renal function. Several studies revealed that ω-3 EPA and DHA and ω-6 GLA, DGLA, and AA have significant actions on kidney function. For instance, when healthy volunteers were given EPA (3.9 gm) and DHA (2.4 gm) per day for 6 weeks, they showed significant increase in renal plasma flow and glomerular filtration rate, decrease in renal vascular resistance, and increase in the excretion of PGE3 with no change in blood pressure and heart rate (156). Also, diets rich in evening primrose oil (a rich source of GLA and LA) and safflower oil decreased proteinuria, glomerular sclerosis, and tubular abnormalities in diabetic rats. These beneficial actions were associated with an increased ratio of renal cortical production of 6-keto-PGF3α (a metabolite of PGI2) to TXB2 with no significant changes in plasma lipid composition. In contrast, fish oil feeding decreased plasma lipids and lowered the 6-keto-PGF3α/TXB2 ratio without any effect on renal disease in diabetic rats (157). Subsequently, Singer et al (158) showed that spontaneously hypertensive rats had significantly lower systolic blood pressure when fed fish oil (EPA and DHA), evening primrose oil (a rich source of GLA), and fish oil plus evening primrose oil, which suggests that a combination of GLA, EPA, and DHA is necessary to produce optimal beneficial actions with regard to renal indices and blood pressure. Vaskonen et al. (159) noted that fish oil completely prevented a rise in blood pressure induced by a high-salt diet in stroke-prone spontaneously hypertensive rats. This beneficial effect on blood pressure was accompanied by a decrease in TXB2 formation by 75% and an increase in plasma and renal ω-3 fatty acid content. Furthermore, EPA/DHA has been shown to suppress mesangial cell proliferation, arrest progression of IgA nephropathy, and protect against cyclosporine-induced renal damage (160-162). These results suggest that when optimal amounts of GLA and EPA/DHA are given, a significant reduction occurs in blood pressure and in the preservation of renal function in diabetic and hypertensive rats. Studies performed with a 5/6 renal ablation rat model (wherein one kidney and 2/3 of the remaining portion of the other kidney were removed) that developed hypertension, albuminuria, and a decline in glomerular filtration rate revealed that renal ablation rats had significantly less glomerulosclerosis and dyslipidemia when supplemented with fish oil and flax seed oil (rich in ALA) compared with the control group at 10 and 20 weeks post surgery (163, 164). Thus, PUFAs may show actions similar to those observed with conventional, synthetic diuretics. These beneficial actions of PUFAs can be attributed to the formation of beneficial PGA, PGE3, PGI2, PGI3, and recently identified resolvins and protectins and a decrease in the production of TXA2 and LTs (165). In this context, it is interesting to note that diuretic furosemide enhances endothelial synthesis and the release of bradykinin and related kinins that, in turn, stimulates endothelial PGI2 formation via B2 kinin receptor activation (166); also, COX-2 derived PGs interact with the renin-angiotensin system to regulate renal function (167).
Pufas and the Parasympathetic Nervous System
Autonomic function can be assessed by the measurement of heart rate variability (HRV) and the evaluation of baroreflex sensitivity (BRS). HRV reflects the physiologic levels of tonic autonomic regulation, whereas BRS indicates the capacity of reflex autonomic regulation. Both low HRV and low BRS are associated with increased cardiovascular risk. Vagal stimulation by a release of acetylcholine (ACh) and adrenergic stimulation mediated by norepinephrine and epinephrine regulate the autonomic function and thus the variations in HRV and BRS. Several studies revealed that ω-3 fatty acids reduce the risk of sudden death by preventing life-threatening cardiac arrhythmias and by significantly increasing HRV (168). Furthermore, a direct positive correlation was noted between the content of DHA in cell membranes and the HRV index, which suggests an anti-arrhythmic effect of the ω-3 fatty acids (169). Because increased parasympathetic tone is responsible for an increase in the ventricular fibrillation threshold and protects against ventricular arrhythmias, it is likely that EPA/DHA supplementation enhances the parasympathetic tone. This suggestion is supported b the observation that EPA/DHA supplementation increases hippocampal ACh levels, the principal neurotransmitter of parasympathetic nerves (170). Hence, it is likely that EPA/DHA supplementation increases the brain ACh levels that lead to an increase in the parasympathetic tone and so to an increase in HRV and protection from ventricular arrhythmias.
Vagus nerve stimulation also inhibits TNF synthesis in the liver, and ACh, the principal vagal neurotransmitter, significantly attenuated the release of the proinflammatory cytokines TNF-α, IL-16, IL-1β, and IL-18 but not the anti-inflammatory cytokine IL-10 by stimulated macrophages in vitro and in vivo (171-174). Because EPA/DHA enhances brain ACh levels (170) and evidence suggests that even AA augments ACh release (175), it is possible that PUFAs enhance parasympathetic tone and, thus, increase HRV and prevent ventricular arrhythmias. These results imply that an inverse correlation could exist between plasma TNF levels and the parasympathetic tone: The higher the TNF levels are the lower the parasympathetic tone is and vice versa. Also, the higher the parasympathetic tone, the higher the brain ACh levels, and so the higher the plasma, cardiac, and brain EPA/DHA/AA levels are. Because normally a balance is maintained between the parasympathetic and sympathetic tones, it is reasonable to suggest that whenever the parasympathetic tone (vagal tone) is enhanced sympathetic tone is reduced (akin to the blocking of β-receptors as it occurs in instances of the use of P-blockers). Thus, indirectly PUFAs may function like β-blockers.
Thus, PUFAs, especially an optimal combination of EPA, DHA and possibly, GLA, DGLA, and AA, show all the qualities of the suggested “polypill,” viz they show aspirin-like action, inhibit the activities of HMG-CoA and ACE enzymes, possess diuretic and antihypertensive actions, and indirectly show β-blocker-like action.
Pufas Possess Anti-Inflammatory Actions
AA, EPA, DHA, GLA, DGLA, LXs and resolvins suppress IL-1, IL-2, IL-6, and TNF-α production by T cells (110-112, 149, 176-180). This claim suggests that EFAs/PUFAs and their metabolites function as endogenous anti-inflammatory molecules and regulate immune response and thus are likely to be of benefit in obesity, insulin resistance, atherosclerosis, metabolic syndrome X, type 2 diabetes mellitus, CHD, depression, and Alzheimer’s disease that are considered as diseases of low-grade systemic inflammation (1-8, 24, 120). Some beneficial actions of PUFAs in various inflammatory conditions are because of the formation of anti-inflammatory compounds such as lipoxins, resolvins, and neuroprotectin D1.
Role of Efas/Pufas in Some Clinical Conditions
It is evident from the preceding discussion that EFAs/PUFAs and their metabolites are useful in many clinical conditions as outlined below.
Inflammatory conditions
PUFAs and their products have the ability to modulate inflammation. The amount and type of PUFA(s) released and their products formed in response to inflammatory stimuli depend on the cell membrane phospholipid fatty acid content and the activity of the CO and LO enzymes. Because EFAs are obtained from diet, it suggests that dietary content of EFAs is one factor that modulates the degree of inflammation. Increased dietary intake of GLA, DGLA, and EPA/DHA substantially decreases inflammatory response. This beneficial action can be ascribed to the decreased formation of pro-inflammatory eicosanoids and cytokines and to an increase in the production of beneficial molecules such as PGEi, PGI2, PGI3, HPETEs, eNO, LXs, resolvins, and NPD1. When the cell membrane lipid pool is rich in GLA/DGLA/EPA/DHA and contains appropriate amounts of AA, there could occur specific activation of sPLA2 and cPLA2 (soluble and cytosolic phospholipase A2, respectively) in response to an injury/inflammatory stimuli that leads to the formation of increased amounts of LXs, PGD2 and 15deoxy∆12-14PGJ2, eNO, GSNO, PGE1, PGI2, PGI3, and HPETEs that dampen the inflammatory process and enhance the resolution of inflammation. It was demonstrated that exogenous PUFAs preferentially activate type IIA sPLA2-mediated AA release from IL-1 stimulated cells and, this activation, in turn, led to the formation of anti-inflammatory LXs, PGD2 and 15deoxy∆12-14PGJ2, which results in the prevention and the resolution of inflammation (181, 182). Several studies showed that oral or parenteral supplementation of GLA/EPA/DHA is of benefit to patients with insulin resistance, metabolic syndrome X, CHD, rheumatoid arthritis, lupus, psoriasis, sepsis, inflammatory
Atherosclerosis
Healthy endothelial cells release adequate amounts of NO, PGI2, and PGE1 to prevent the aggregation of platelets so that atherosclerosis is prevented. An increased production of pro-inflammatory cytokines and free radicals occurs because of sheer stress, hyperglycemia, clinical or subclinical infections, low-grade systemic inflammation as seen in type 2 diabetes mellitus, hypertension, hyperlipidemia, and metabolic syndrome X. EPA/ DHA/AA/DGLA inhibit free radical generation, suppress IL-6 and TNF-α synthesis and secretion, enhance eNO synthesis, and, thus, prevent oxidant stress (reviewed in References 149, 150 and 183). Endothelial cells that line atherosclerosis-free blood vessel walls have abundant concentrations of the essential fatty acid linoleate, whereas fatty streaks (an early stage of atherosclerosis) are deficient in EFAs (183-186). An EFA deficiency promotes respiratory uncoupling (187, 188) and atherosclerosis (3, 183, 189). Bernal-Mizrachi et al. (184) showed that oxidative stress increases ROS generation and decreases NO formation. These evidences suggest that endothelial cell deficiency of PUFAs increases the production of pro-inflammatory cytokines and free radicals that results in the development of insulin resistance, a decrease in plasma and cellular HDL concentrations, and a decrease in the formation of eNO, PGEi, PGI2, PGI3, LXs, resolvins, and NPD1 that ultimately may promote atherosclerosis. Providing adequate amounts of various PUFAs can restore normalcy.
Metabolic Syndrome X
Metabolic syndrome X is a low-grade systemic inflammatory condition in which plasma levels of C-reactive protein (CRP), TNF-α, and IL-6 are elevated. A negative correlation exists between plasma TNF-α and HDL cholesterol, glycosylated hemoglobin, and serum insulin concentrations. EPA, DHA, and AA, inhibit TNF-α and IL-6 production (111, 112), enhance eNO generation (30), inhibit HMG-CoA reductase (103) and ACE enzyme activities (93), function as endogenous ligands for PPARs (reviewed in References 3 and 4), modulate leptin gene expression (190, 191), enhance the production of adiponectin (192), and decrease insulin resistance (193). This finding may explain why PUFAs are useful to protect against CHD, prevent the progression of atheroslcerosis, and decrease blood pressure.
Brain Growth and Development and Cognitive Functions
Several studies showed that AA, EPA, and DHA are essential not only for brain growth and development but also to modulate the synthesis, release, and action of various neuropeptides. Because the brain is rich in AA, EPA, and DHA, one important function of these fatty acids in the brain could be to ensure the presence of an adequate number of insulin receptors; the insulin receptor number depends on the amount of PUFAs incorporated in the cell membrane phospholipids (35-42). Thus a defect in the metabolism of PUFAs or not having adequate amounts of PUFAs incorporated into the neuronal cell membranes during fetal development and infancy may cause a defect in the expression or function of insulin receptors in the brain. This defect may lead to the development of type 2 diabetes as seen in NIRKO mice (194). Furthermore, systemic injections of either glucose or insulin in ad libitum fed rats resulted in an increase in extracellular acetylcholine in the amygdala (195). Acetylcholine modulates dopamine release that, in turn, regulates appetite (196). As already discussed above, ACh inhibits the production of pro-inflammatory cytokines in the brain and, thus, protects the neurons from the cytotoxic actions of TNF-α.
For proper neuronal development and increase in cell membrane surface area, growth of neurite processes from the cell body is critical (197). Nerve growth cones are highly enriched with AA-releasing phospholipases, which have been implicated in neurite outgrowth (198, 199). Cell membrane expansion occurs through the fusion of transport organelles with plasma membrane (200), and syntaxin 3, a plasma membrane protein that has an important role in the growth of neurites, is a direct target for AA, DHA, and other PUFAs (201). It was reported that AA, DHA, and other PUFAs activate syntaxin 3. Even syntaxin1 that is specifically involved in fast calcium-triggered exocytosis of neurotransmitters is sensitive to AA (201). These results imply that AA, EPA, and DHA are involved both in the exocytosis of neurotransmitters and in neurite outgrowth. SNAP25 (synaptosomal-associated protein of 25kDa), a syntaxin partner implicated in neurite outgrowth, interacted with syntaxin 3 only in the presence of AA that allowed the formation of the binary syntaxin 3-SNAP 25 complex (soluble N-ethylmaleimide-sensitive factor attachment protein receptor), which is needed for the fusion of plasmalemmal precursor vesicles into the cell surface membrane that leads to membrane fusion (202). These results clearly demonstrated that AA and DHA change the α-helical syntaxin structure to expose a SNARE motif for immediate SNAP 25 engagement and, thus, facilitate neurite outgrowth.
Puskas and his colleagues (203-205) noted that during brain growth and development, feeding with ω-3 DHA/ALA diets altered the expression of genes involved in synaptic plasticity, cytoskeleton, signal transduction, ion channel formation, energy metabolism, and regulatory proteins. These results imply that perinatal supplementation of PUFAs may play a critical role in the pathobiology of several adult diseases including metabolic syndrome X and Alzheimer’s disease (3, 4, 43, 44, 206-208).
Alzheimer's Disease
Fish and fish oil components, EPA, and DHA are of benefit in Alzheimer’s disease (209-211). A reduction in dietary DHA in an Alzheimer’s mouse model showed a loss of postsynaptic proteins associated with increased oxidation and showed increased caspase-cleaved actin, which was localized in dendrites; however, DHA-restricted mice when given DHA were protected against dendritic pathology and behavioral deficits and showed increased antiapoptotic BAD phosphorylation, which implies that DHA could be useful in preventing Alzheimer’s disease in which synaptic loss is critical (212, 213). DHA attenuated amyloid-β secretion accompanied by the formation of neuroprotectin D1 (NPD1), a DHA-derived 10,17S-docosatriene (214). In Alzheimer’s hippocampal cornu ammonis region, DHA and NPD1 were reduced, including the expression of enzymes involved in NPD1 synthesis, cytosolic phospholipase A2 and 15-lipoxygenase. NPD1 repressed amyloid-β-induced activation of pro-inflammatory genes and upregulated the antiapoptotic genes encoding Bcl-2, Bcl-xl, and Bfl-1 (A1), which indicates its (NPD1) anti-inflammatory nature. Soluble amyloid precursor protein-α stimulated NPD1 synthesis from DHA (214).
Presenilin, a major component of γ-secretase, generates amyloid-β. Overexpression of phospholipase D1 decreases the catalytic activity of γ-secretase (215) and releases PUFAs as evidenced by the increased formation of prostaglandin E2 (216). This finding suggests that PUFAs could regulate the activity of γ-secretase. PUFAs (especially AA and DHA) enhance acetylcholine release in the brain and, thus, bring about some of their beneficial effects in Alzheimer’s. Furthermore, EPA and DHA have the ability to enhance NO generation (30), suppress production of pro-inflammatory cytokines (110-113), and enhance brain acetylcholine levels (170), a neurotransmitter whose levels are decreased in Alzheimer’s disease (217). Thus, PUFAs modulate neural function, including neurotransmission, membrane fluidity, ion channel, enzyme regulation, and gene expression, as well as prevent inflammation; thus, they bring about their beneficial actions in Alzheimer’s disease.
Fetal-Alcohol Syndrome
Ethanol exposure during brain development induces neurodevelopmental defects referred to as fetal-alcohol syndrome (FAS) that is characterized by hyperactivity, learning and memory deficits, mental retardation, psychosis, depression, and schizophrenia. Ethanol-induced neurotoxicity, oxidative stress, induction of apoptosis, excitotoxicity, interference with the action of growth factors, and EFA metabolism all could be responsible for the development of FAS. Neurons are susceptible to ethanol-induced apoptotic cell death during synaptogenesis during a brain growth spurt, which occurs during the third trimester of pregnancy and in the perinatal period.
Recent studies showed that nicotinamide enhanced the neuronal survival following free radical exposure and oxidative stress (218, 219). Nicotinamide is a cofactor in the metabolism of EFAs, which could explain the beneficial action of nicotinamide in FAS (220).
Depression
Depression is more likely to occur in individuals whose intake of PUFAs, especially of n-3 fatty acids, is lower (221). Pro-inflammatory cytokines might cause depressive illness (222). A significant decrease of ω-3 fatty acids in plasma and/or in the membranes of red blood cells in subjects with depression has been reported (223-225). Because ω-3 fatty acids suppress the production of IL-1β, IL-2, IL-6, and TNF-α, this finding suggests that these fatty acids could play a role in depression (222). In addition, antidepressants act like inhibitors of cyclo-oxygenase (222). Double-blind placebo-controlled and other studies (226-228) revealed that an addition of the ω-3 fatty acids EPA and DHA was associated with a longer period of remission among depressed patients. Thus, epidemiological, experimental, and clinical data favor the idea that PUFAs play a role in the pathogenesis and/or the treatment of depression.
Schizophrenia and Huntington's Disease
Patients with schizophrenia have increased concentrations of pro-inflammatory cytokines both in the systemic circulation and cerebrospinal fluid and showed decreased EPA and DHA in the plasma phospholipid. Clinical trials showed that supplementation of ethyl EPA is of significant benefit to these patients (228).
Huntington’s disease is an inherited neurodegenerative disorder because of a mutation in exon 1 of the Huntingtin gene that encodes a stretch of polyglutamine (poly Q) residues close to the N-terminus of the Huntingtin protein. Aggregated poly Q residues are toxic to the neuronal cells. Transgenic R6/1 mice that develop motor abnormalities of Huntington’s disease showed increased survival rates and decreased neurologic deficits when supplemented with ethyl EPA (229), which suggests that unsaturated fatty acids may prevent or arrest poly Q aggregation. These results suggest that PUFAs are useful in various neurological diseases. Understanding the molecular mechanisms of action of EPA/DHA as to why DHA is useful in Alzheimer’s disease whereas ethyl EPA is of benefit in Huntington’s disease and schizophrenia may throw more light on the pathobiology of these diseases.
Conclusions
It is evident from the preceding discussion that EFAs and their metabolites such as GLA, AA, EPA, DHA, eicosanoids, LXs, resolvins, NPD1, and nitrolipids have many actions and participate in several disease processes (Figure (2)). In this context, it is important to note that certain PUFAs, such as GLA, have both antimutagenic and anticancer actions that have been discussed in detail elsewhere (230). Mutagens and carcinogens block ∆6 and ∆5 desaturases in normal cells much before their conversion into malignant cells. Pretreatment or simultaneous treatment with GLA completely prevented DNA damage induced by mutagens and carcinogens, which implies that GLA and other PUFAs could function as endogenous antimutagenic and anticancer molecules. GLA has selective tumoricidal action and is effective against human malignant glioma and other cancers (5, 231).
NO reacts with PUFAs to yield their respective nitroalkene derivatives that can be detected in plasma. These nitroalkene derivatives of various EFAs induce vascular relaxation, inhibit neutrophil degranulation and superoxide formation, inhibit platelet activation, and have endogenous PPAR-γ ligand activity and decay in the blood to release NO. This finding suggests that EFAs/PUFAs not only form precursors to various eicosanoids, resolvins, LXs, and NPD1 but also react with various other molecules to form novel compounds that have significant biologic activity.
The major question is how these simple fatty acids can have so many biologic—and at times diametrically opposite—actions. One reason could be their ability to give rise to many metabolites that have specific biologic actions. Furthermore, these fatty acids when incorporated into the cell membrane alter its properties including fluidity that, in turn, modulates the number and affinity of various receptors to their respective growth factors, hormones, peptides, and proteins. Yet another action is their ability to form complexes with other biologically active molecules as is seen with NO to form various nitroalkene derivatives. Formation of such complexes between EFAs and other biologically active molecules could impart specific and distinctive properties to these newly formed entities that in turn may show varied biologic actions. Deciphering the formation of such complexes is not only interesting but also challenging because such complexes may form the basis of understanding certain less well understood physiologic and pathologic processes.
Although structurally EFAs are simple, they form precursors to a variety of compounds with many biologic actions. We are yet to understand the molecular triggers that facilitate the formation of specific biologically active molecules in various cells and tissues. Such an understanding may lead to the development of methods to enhance selectively the formation of the desired lipid(s) to obtain a specific function or action. In view of their varied actions, EFAs/PUFAs and their products could form the basis for the development of many drugs.
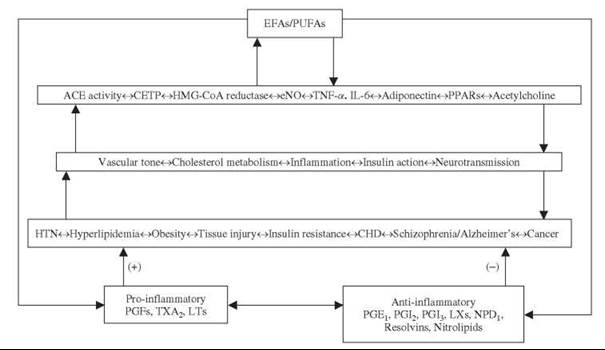
Figure 2. Scheme shows the interaction(s) between EFAs/PUFAs and ACE, HMG-CoA reductase enzymes, NO, cytokines, PPARs, and acetylcholine. The scheme is by no means exhaustive and is meant only to reflect the positive and negative interaction(s) among various endogenous molecules and their role in several diseases. (+) Indicates an increase in tissue injury, insulin resistance, and disease process. -Indicates a decrease in tissue injury, insulin resistance, and disease process. For details, see text.
References
1. Das UN, Horrobin DF, Begin ME, Huang YS, Cunnane SC, Manku MS, Nassar BA. Clinical significance of essential fatty acids. Nutrition 1988; 4:337-342.
2. Das UN. Essential fatty acids: biology and their clinical implications. Asian Pacific J. Pharmacol. 1991; 6:317-330.
3. Das UN. Essential fatty acids- a review. Curr. Pharmac. Biotech. 2006; 7:467-482.
4. Das UN. Essential fatty acids: Biochemistry, physiology, and pathology. Biotechnol. J. 2006; 1:420-439.
5. Das UN. Tumoricidal action of cis-unsaturated fatty acids and its relationship to free radicals and lipid peroxidation. Cancer Lett. 1991; 56:235-243.
6. Das UN. Long-chain polyunsaturated fatty acids interact with nitric oxide, superoxide anion, and transforming growth factor-β to prevent human essential hypertension. Eur. J. Clin. Nutr. 2004; 58:195-203.
7. Das UN. Can perinatal supplementation of long-chain polyunsaturated fatty acids prevent diabetes mellitus? Eur. J. Clin. Nutrition 2003; 57:218-226.
8. Das UN. A Perinatal Strategy for Preventing Adult Diseases: The Role of Long-Chain Polyunsaturated Fatty Acids. 2002. Kluwer Academic Publishers, Boston, MA.
9. Friedman Z, Shochat SJ, Maisels MJ, Marks KH, Lamberth EL Jr. Correction of essential fatty acid deficiency in newborn infants by cutaneous application of sunflower-seed oil. Pediatrics 1976; 58:650-654.
10. Skolnik P, Eaglstein WH, Ziboh VA. Human essential fatty acid deficiency: treatment by topical application of linoleic acid. Arch. Dermatol. 1977; 113:939-941.
11. Gibson RA, Kneebone GM. Fatty acid composition of human colostrums and mature breast milk. Am. J. Clin. Nutr. 1981; 34:252-257.
12. Horrobin DF. The regulation of prostaglandin biosynthesis by the manipulation of essential fatty acid metabolism. Rev. Pure Appl. Pharmacol. Sci. 1983; 4:339-383.
13. Cantwell MM, Flynn MA, Cronin D, O’ Neill JP, Gibney MJ. Contribution of foods to trans unsaturated fatty acid intake in a group of Irish adults. J. Hum. Nutr. Diet. 2005; 18:377-385.
14. Lopez-Garcia E, Schultze MB, Meigs JB, Manson JE, Rifai N, Stampfer MJ, Willett WC, Hu FB. Consumption of trans fatty acids is related to plasma biomarkers of inflammation and endothelial dysfunction. J. Nutr. 2005; 135:562-566.
15. Mozaffarian D, Pischon T, Hankinson SE, Rifai N, Joshipura K, Willett WC, Rimm EB. Dietary intake of trans fatty acids and systemic inflammation in women. Am. J. Clin. Nutr. 2004; 79:606-612.
16. Simopoulos AP. Omega-3 fatty acids in health and disease and in growth and development. Am. J. Clin. Nutr. 1991; 54:438-463.
17. Nakashima Y, Yuasa S, Hukamizu Y, Okuyama H, Ohhara T, Kameyama T, Nabeshima T. Effect of a high linoleate and a high alpha-linolenate diet on general behavior and drug sensitivity in mice. J. Lipid. Res. 1993; 34:239-247.
18. Hashimoto A, Katagiri M, Torii S, Dainaka J, Ichikawa A, Okuyama H. Effect of the dietary alpha-linolenate/linoleate balance on leukotriene production and histamine release in rats. Prostaglandins 1988; 36:3-16.
19. Shimokawa T, Moriuchi A, Hori T, Saito M, Naito Y, Kabasawa H, Nagae Y, Matsubara M, Okuyama H. Effect of dietary alpha-linolenate/linoleate balance on mean survival time, incidence of stroke and blood pressure of spontaneously hypertensive rats. Life Sci. 1988; 43:2067-2075.
20. Tsutsumi T, Yamauchi E, Suzuki E, Watanabe S, Kobayashi T, Okuyama H. Effect of a high alpha-linolenate and high linoleate diet on membrane-associated enzyme activities in rat brain-modulation of Na+ , K+ - ATPase activity at suboptimal concentrations of ATP. Biol. Pharm. Bull. 1995; 18:664-670.
21. Watanabe S, Suzuki E, Kojima N, Kojima R, Suzuki Y, Okuyama H. Effect of dietary alpha-linolenate/linoleate balance on collagen-induced platelet aggregation and serotonin release in rats. Chem. Pharm. Bull. 1989;37:1572-1575.
22. Brenner RR. Nutritional and hormonal factors influencing desaturation of essential fatty acids. Prog Lipid Res 1982; 20:41-48.
23. Cook HW. The influence of trans acids on desaturation and elongation of fatty acids. Lipids 1981; 16:920-926.
24. Das UN. A defect in the activity of A6 and A5 desaturases may be a factor predisposing to the development of insulin resistance syndrome. Prostaglandins Leukotrienes Essen. Fatty Acids 2005; 72:343-350.
25. Han SN, Leka LS, Lichtenstein AH, Ausman LM, Meydani SN. Effect of a therapeutic lifestyle change diet on immune functions of moderately hypercholesterolemic humans. J. Lipid Res. 2003; 44:2304-2310.
26. Han SN, Leka LS, Lichtenstein AH, Ausman LM, Schaefer EJ, Meydani SN. Effect of hydrogenated and saturated, relative to polyunsaturated, fat on immune and inflammatory responses of adults with moderate hypercholesterolemia. J. Lipid Res. 2002; 43:445-452.
27. Naruszewicz M, Daniewski M, Nowicka G, Kozlowska-Wojciechowska M. Trans-unsaturated fatty acids and acrylamide in food as potential atherosclerosis progression factors. Based on own studies. Acta Microbiol. Pol. 2003; 52:75-81.
28. Mozaffarian D. Trans fatty acids—effects on systemic inflammation and endothelial function. Atheroscler. 2006; 7:29-32.
29. Mozaffarian D, Pischon T, Hankinson SE, Rifai N, Joshipura K, Willett WC, Rimm EB. Dietary intake of trans fatty acids and systemic inflammation in women. Am J Clin Nutr 2004; 79:606-612.
30. Okuda Y, Kawashima K, Sawada T, Tsurumaru K, Asano M, Suzuki S, Soma M, Nakajima T, Yamashita K. Eicosapentaenoic acid enhances nitric oxide production by cultured human endothelial cells. Biochem. Biophys. Res. Commun. 1997; 232:487-491.
31. Baker PRS, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Swooney S, Long MH, Iles KE, Baker LMS, Branchaud BP, Chen Y, Freeman BA. Fatty acid transduction of nitric oxide signaling: Multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 2005; 280:42464-42475.
32. Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, O’Donnell VB. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils. Circ. Res. 2002; 91:375-381.
33. Lima ES, Bonim MG, Augusto O, Barbeiro HV, Souza HP, Abdalla DS. P. Nitrated lipids decompose to nitric oxide and lipid radicals and cause vasorelaxation. Free Rad. Biol. Med. 2005; 39:532-539.
34. Wright MM, Schopfer FJ, Baker PRS, Vidyasagar V, Powell P, Chumley P, Iles KE, Freeman BA, Agarwal A. Fatty acid transduction of nitric oxide signaling: Nitrolinoleic acid potently activates endothelial heme oxygenase 1 expression. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:4299-4304.
35. Das UN. Insulin resistance and hyperinsulinemia: Are they secondary to an alteration in the metabolism of essential fatty acids? Med. Sci. Res. 1994; 22:243-245.
36. Field CJ, Ryan EA, Thomson AB, Clandinin MT. Diet fat composition alters membrane phospholipid composition, insulin binding, and glucose metabolism in adipocytes from control and diabetic animals. J. Biol. Chem. 1990; 265:11143-11150.
37. Liu S, Baracos VE, Quinney HA, Clandinin MT. Dietary omega-3 and polyunsaturated fatty acids modify fatty acyl composition and insulin binding in skeletal-muscle sarcolemma. Biochem. J. 1994; 299(Pt 3):831-837.
38. Ginsberg BH, Chatterjee P, Yorek MA. Insulin sensitivity is increased in Friend erythroleukemia cells enriched in polyunsaturated fatty acid. Receptor 1991; 1:155-166.
39. Yorek M, Leeney E, Dunlap J, Ginsberg B. Effect of fatty acid composition on insulin and IGF-I binding in retinoblastoma cells. Invest. Ophthalmol. Vis. Sci. 1989; 30:2087-2092.
40. Bruneau C, Staedel-Flaig C, Cremel G, Leray C, Beck JP, Hubert P. Influence of lipid environment on insulin binding in cultured hepatoma cells. Biochim. Biophys. Acta 1987; 928:287-296.
41. Bruneau C, Hubert P, Waksman A, Beck JP, Staedel-Flaig C. Modifications of cellular lipids induce insulin resistance in cultured hepatoma cells. Biochim. Biophys. Acta 1987; 928:297-304.
42. Ginsberg BH, Jabour J, Spector AA. Effect of alterations in membrane lipid unsaturation on the properties of the insulin receptor of Ehrlich ascites cells. Biochim. Biophys. Acta 1982; 690:157-164.
43. Das UN. Long-chain polyunsaturated fatty acids in the growth and development of the brain and memory. Nutrition 2003; 19:62-65.
44. Das UN. Can memory be improved? A discussion on the role of ras, GABA, acetylcholine, NO, insulin, TNF-α, and long-chain polyunsaturated fatty acids in memory formation and consolidation. Brain Devel. 2003; 25:251-261.
45. Calderon F, Kim HY. Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J. Neurochem. 2004; 90:979- 988.
46. Futerman AH, Banker GA. The economics of neurite outgrowth: the addition of new membrane to growing axons. Trends Neurosci. 1996; 19:144-149.
47. Negre-Aminou P, Nemenoff RA, Wood MR, de la Houssaye BA, Pfenninger KH. Characterization of phospholipase A2 activity enriched in the nerve growth cone. J. Neurochem. 1996; 67:2599-2608.
48. Hornfelt M, Ekstrom PA, Edstrom A. Involvement of axonal phospholipase A2 activity in the outgrowth of adult mouse sensory axons in vitro. Neuroscience 1999; 91:153901547.
49. Darios F, Davletov B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature 2006; 440:813-817.
50. Rickman C, Davletov B. Arachidonic acid allows SNARE complex formation in the presence of Munc18. Chem. Biol. 2005; 12:545-553.
51. Neale ML, Fiera RA, Matthews N. Involvement of phospholipase A2 activation in tumour cell killing by tumour necrosis factor. Immunology 1988; 64:81-85.
52. Hepburn A, Boeynaems JM, Fiers W, Dumont JE. Modulation of tumor necrosis factor-alpha cytotoxicity in L929 cells by bacterial toxins, hydrocortisone and inhibitors of arachidonic acid metabolism. Biochem. Biophys. Res. Commun. 1987; 149:815-822.
53. Hayakawa M, Ishida N, Takeuchi K, Shibamoto S, Hori T, Oku N, Ito F, Tsujimoto M. Arachidonic acid-selective cytosolic phospholipase A2 is crucial in the cytotoxic action of tumor necrosis factor. J. Biol. Chem. 1993; 268:11290-11295.
54. Thorne TE, Voelkel-Johnson C, Casey WM, Parks LW, Laster SM. The activity of cytosolic phospholipase A2 is required for the lysis of adenovirus-infected cells by tumor necrosis factor. J. Virol. 1996; 70:8502-8507.
55. Palmantier R, George MD, Akiyama SK, Wolber FM, Olden K, Roberts JD. Cis-polyunsaturated fatty acids stimulate beta1 integrin-mediated adhesion of human breast carcinoma cells to type IV collagen by activating protein kinases C-epsilon and—mu. Cancer Res. 2001; 61:2445-2452.
56. Arita K, Kanno T, Takehara Y, Fujiwara T, Akiyama J, Horton AA, Utsumi T. Effect of n-3 and n-6 polyunsaturated fatty acids and their ethylesters on stimuli-dependent superoxide generation in neutrophils. Physiol Chem Phys Med NMR 2001; 33:121-132.
57. Das UN. Antibiotic-like action of essential fatty acids. Canadian Med. Assoc. J. 1985; 132:1985.
58. Das UN. Do unsaturated fatty acids function as endogenous anti-bacterial and anti-viral molecules? Am. J. Clin. Nutr. 2006; 83:390-391.
59. Kodicek E. The effect of unsaturated fatty acids on gram-positive bacteria. Symposia Soc. Exp. Biol. 1949; 3:217-232.
60. Lacey RW, Lord VL. Sensitivity of staphylococci to fatty acids: novel inactivation of linolenic acid by serum. J. Med. Microbiol. 1981; 14:41-49.
61. Galbraith H, Miller TB, Paton AM, Thompson JK. Antibacterial activity of long chain fatty acids and the reversal with calcium, magnesium, ergocalciferol and cholesterol. J. Appl. Bact. 1971; 34:803-813.
62. McDonald MI, Graham I, Harvey KJ, Sinclair A. Antibacterial activity of hydrolysed linseed oil ad linolenic acid against methicillin-resistant Staphylococcus aureus. Lancet 1981; 2:1056.
63. Kohn A, Gitelman J, Inbar M. Unsaturated free fatty acids inactivate animal enveloped viruses. Arch. Virol. 1980; 66:301-307.
64. Giron DJ. Inhibition of viral replication in cell cultures treated with prostaglandins E1. Proc Soc Exp Biol Med 1982; 170:25-28.
65. Santoro MG, Benedetto A, Carruba G, Garaci E, Jaffe BM. Prostaglandin A compounds as antiviral agents. Science 1980; 209:1032-1034.
66. Iwamoto KS, Bennett LR, Norman A, Villalobos AE, Hutson CA. Linoleate produces remission n canine mycosis fungoides. Cancer Lett. 1992; 64:17-22.
67. Riley EM, MacLennan C, Wiatkowski DK, Greenwood BM. Suppression of in-vitro lymphoproliferative responses in acute malaria patients can be partially reversed by indomethacin. Parasite Immunol. 1989; 11:509-517.
68. Hsiao LL, Howard RJ, Aikawa M, Taraschi TF. Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. Biochem. J. 1991;274:121-132.
69. Sliwa K, Grundmann HJ, Neifer S, Chaves MF, Sahlmuller G, Blitstein-Willinger E, Bienzle U, Kremsner PG. Prevention of murine cerebral malaria by a stable prostacyclin analog. Infect. Immun. 1991; 59:3846-3848.
70. Kumaratilake LM, Robinson BS, Ferrante A, Poulos A. Antimalarial properties of n-3 and n-6 polyunsaturated fatty acids: in vitro effects on Plasmodium falciparum and in vivo effects on P. berghei. J. Clin. Invest. 1992; 89:961-967.
71. Krugliak M, Deharo E, Shalmiev G, Sauvain M, Moretti C, Ginsburg H. Antimalarial effects of C18 fatty acids on Plasmodium falciparum in culture and on Plasmodium vinckei petteri and Plasmodium yoelii nigeriensis in vivo. Exp. Parasitol. 1995; 81:97-105.
72. Taylor DW, Levander OA, Krishna VR, Evans CB, Morris VC, Barta JR. Vitamin E-deficient diets enriched with fish oil suppress lethal Plasmodium yoelii infections in athymic and scid/bg mice. Infect. Immun. 1997; 65:197-202.
73. Kumar CA, Das UN. Lipid peroxides, nitric oxide and essential fatty acids in patients with Plasmodium falciparum malaria. Prostaglandins Leukot Essent Fatty Acids 1999; 61:255-258.
74. Xiao L, Patterson PS, Yang C, Lal AA. Role of eicosanoids in the pathogenesis of murine cerebral malaria. Am. J. Trop. Med. Hyg. 1999; 60:668-673.
75. Perkins DJ, Kremsner PG, Weinberg JB. Inverse relationship of plasma prostaglandin E2 and blood mononuclear cell cyclooxygenase-2 with disease severity in children with Plasmodium falciparum malaria. J. Infect. Dis. 2001; 183:113-118.
76. Chandra S, Ohnishi ST, Dhawan BN. Reversal of chloroquine resistance in murine malaria parasites by prostaglandin derivatives. Am. J. Trop. Med. Hyg. 1993; 48:645-651.
77. Doering TL, Raper J, Buxbaum LU, Adams SP, Gordon JI, Hart GW, Englund PT. An analog of myristic acid with selective toxicity for African Trypanosomes. Science 1991; 252:1851-1854.
78. Sun CQ, O’Connor CJ, Robertson A. Antibacterial actions of fatty acids and monoglycerides against Helicobacter pylori. FEMS Immunol. Med. Microbiol. 2003; 36:9-17.
79. Giamarellos-Bourboulis EJ, Mouktaroudi M, Adamis T, Koussoulas V, Baziaka F, Perrea D, Karavannacos PE, Giamarellou H. n-6 polyunsaturated fatty acids enhance the activities of ceftazidime and amikacin in experimental sepsis caused by multidrug-resistant Pseudomonas aeroginosa. Antimicrob. Agents Chemother. 2004; 48:4713-4717.
80. Das UN. Can essential fatty acid deficiency predispose to AIDS? Canadian Med. Assoc. J. 1985; 132:900.
81. Das UN. Essential fatty acids and acquired immunodeficiency syndrome. Med. Sci. Monit. 2005; 11:RA206-RA211.
82. Pechacek TF, Asma S, Eriksen MP. Tobacco: global burden and community solutions. In: Evidence Based Cardiology. Yusuf S, Calms A, Camm AJ, Fallen EL, Gersh BJ, eds. 1998. BMJ Books, London. pp. 165-178.
83. Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomised trials. Prog. Cardiovasc. Dis. 1985; 27:335-371.
84. Antithrombotic Trialists Collaboration. Collaborative metaanalysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71-86.
85. Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med. 2000; 342:145-153.
86. Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383-1399.
87. The Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N. Engl. J. Med. 1998; 339:1349-1357.
88. Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N. Engl. J. Med. 1996; 336:1001-1009.
89. Plehn JF, Davis BR, Sacks FM, Rouleau JL, Pfeffer V, Cuddy TE, Moye LA, Piller LB, Rutherford J, Simpson LM, Braunwald E. Reduction of stroke incidence after myocardial infarction with pravastatin: the Cholesterol and Recurrent Events (CARE) study. The Care Investigators. Circulation 1999; 99:216-223.
90. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7-22.
91. Yusuf S. Two decades of progress in preventing vascular disease. Lancet 2002; 360:2-3.
92. Wald NJ, Law MR. A strategy to reduce cardiovascular disease by more than 80%. BMJ 2003; 326:1419-1423.
93. Kumar KV, Das UN. Effect of cis-unsaturated fatty acids, prostaglandins, and free radicals on angiotensin-converting enzyme activity in vitro. Proc. Soc. Exp. Biol. Med. 1997; 214:374-379.
94. Okuda Y, Kawashima K, Sawada T, Tsurumaru K, Asano M, Suzuki S, Soma M, Nakajima T, Yamashita K. Eicosapentaenoic acid enhances nitric oxide production by cultured human endothelial cells. Biochem Biophys Res Commun 1997; 232:487-491.
95. Kaergel E, Muller DN, Honeck H, Theuer J, Shagdarsuren E, Mullally A, Luft FC, Schunck W-H. P450-dependent arachidonic acid metabolism and angiotensin-II-induced renal damage. Hypertension 2002; 40:273-279.
96. Gilroy DW. New insights into the anti-inflammatory actions of aspirin- induction of nitric oxide through the generation of epi-lipoxins. Mem Inst Oswaldo Cruz 2005; 100:49-54.
97. Wang W, Diamond SL. Does elevated nitric oxide production enhance the release of prostacyclin from shear stressed aortic endothelial cells? Biochem. Biophys. Res. Commun. 1997; 233:748-751.
98. Das UN. Can COX-2 inhibitors-induced increase in cardiovascular disease risk be modified by essential fatty acids? J. Assoc. Physicians India 2005; 53:623-627.
99. El-Sohemy A, Archer MC. Regulation of mevalonate synthesis in low density lipoprotein receptor knockout mice fed n-3 or n-6 polyunsaturated fatty acids. Lipids 1999; 34:1037-1043.
100. Nakamura N, Hamazaki T, Jokaji H, Minami S, Kobayashi M. Effect of HMG-CoA reductase inhibitors on plasma polyunsaturated fatty acid concentration in patients with hyperlipidemia. Int. J. Clin. Lab. Res. 1998; 28:192-195.
101. Duncan RE, El-Sohemy A, Archer MC. Regulation of HMG-CoA reductase in MCF-7 cells by genistein, EPA, and DHA, alone and in combination with mevastatin. Cancer Lett. 2005; 224:221-228.
102. El-El-Sohemy A, Archer MC. Regulation of mevalonate synthesis in rat mammary glands by dietary n-3 and n-6 polyunsaturated fatty acids. Cancer Res 1997; 57:3685-3687.
103. Das UN. Essential fatty acids as possible mediators of the actions of statins. Prostaglandins Leukot. Essen Fatty Acids 2001; 65:37-40.
104. Sparrow CP, Burton CA, Hernandez M, Mundt S, Hassing H, Patel S, Rosa R, Hermanowski-Vosatka A, Wang PR, Zhang D, Peterson L, Detmers PA, Chao YS, Wright SD. Simvastatin has anti-inflammatory and antiatherosclerotic activities independent of plasma cholesterol lowering. Arterioscler Thromb. Vasc. Biol. 2001; 21:115-121.
105. Vogel G. Cholesterol-lowering drugs may boost bones. Science 1999; 286:1825-1826.
106. Briel M, Struder M, Glass TR, Bucher HC. Effects of statins on stroke prevention in patients with and without coronary heart disease: a meta-analysis of randomized controlled trials. Am. J. Med. 2004; 117:596-606.
107. Riboldi P, Gerosa M, Meroni PL. Statins and autoimmune diseases. Lupus 2005; 14:765-768.
108. Levine L. Statins stimulate arachidonic acid release and prostaglandin I2 production in rat liver cells. Lipids Health Dis. 2003; 2:1.
109. Scott HD, Laake K. Statins for the prevention of Alzheimer’s disease. Cochrane Database Syst. Rev. 2001; 4:CD003160.
110. Endres S, Ghorbani R, Kelley VE, Georgilis K, Lonnemann G, van der Meer JWM, Rogers TS, Klempner MS, Weber PC, Shaefer EJ, Wolff SM, Dinarello LA. The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. N. Engl. J. Med. 1989; 320:265-271.
111. Kumar GS, Das UN. Effect of prostaglandins and their precursors on the proliferation of human lymphocytes and their secretion of tumor necrosis factor and various interleukins. Prostaglandins Leukot. Essen. Fatty Acids 1994; 50:331-334.
112. Kumar SG, Das UN, Kumar KV, Madhavi, Das NP, Tan BKH. Effect of n-6 and n-3 fatty acids on the proliferation and secretion of TNF and IL-2 by human lymphocytes in vitro. Nutrition Res 1992; 12:815-823.
113. Bhattacharya A, Rahman M, Banu J, Lawrence RA, McGuff HS, Garrett IR, Fischbach M, Fernandes G. Inhibition of osteoporosis in autoimmune disease prone MRL/Mpj-Fas(lpr) mice by N-3 fatty acids. J. Am. Coll. Nutr. 2005; 24:200-209.
114. Das UN. Essential fatty acids and osteoporosis. Nutrition 2000; 16:286-290.
115. Hu FB, Bronner L, Willett WC, Stampfer MJ, Rexrode KM, Albert CM, Hunter D, Manson JE. Fish and omega-3 fatty acid intake and risk of coronary heart disease in women. JAMA 2002; 287:1815-1821.
116. van Gelder BM, Tijhuis M, Kalmijn S, Kromhout D. Fish consumption, n-3 fatty acids, and subsequent 5-y cognitive decline in elderly men: the Zutphen Elderly Study. Am. J. Clin. Nutr. 2007; 87:1142-1147.
117. Gamoh S, Hashimoto M, Hossain S, Masumura S. Chronic administration of docosahexaenoic acid improves the performance of radial arm maze task in aged rats. Clin Exp Pharmacol Physiol 2001; 28:266-270.
118. Lim GP, Calon F, Morihara T, Yang F, Teter B, Salem Ubeda O, N Jr, Frautschy SA, Cole GM. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005; 25:3032-3040.
119. Hashimoto M, Hossain S, Shimada T, Shido O. Docosahexaenoic acid-induced protective effect against impaired learning in amyloid beta-infused rats is associated with increased synaptosomal membrane fluidity. Clin. Exp. Pharmacol. Physiol. 2006; 33:934-939.
120. Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahex- aenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 2005; 115:2774-2783.
121. Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronet K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 2003; 278:43807-43817.
122. Nordoy A, Bonaa KH, Sandset PM, Hansen JB, Nilsen H. Effect of omega-3 fatty acids and simvastatin on hemostatic risk factors and postprandial hyperlipemia in patients with combined hyperlipemia. Arterioscler. Thromb. Vasc. Biol. 2000; 20:259-265.
123. Lopez-Coviella I, Berse B, Krauss R, Thies RS, Blusztajn JK. Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science 2000; 289:313-316.
124. Bayorh MA, Ganafa AA, Eatman D, Walton M, Feuerstein GZ. Simvastatin and losartan enhance nitric oxide and reduce oxidative stress in salt-induced hypertension. Am. J. Hypertens. 2005; 18:1496-1502.
125. Hoffman M. A new role for gases: neurotransmission. Science 1991; 252:1788.
126. Armour KE, Armour KJ, Gallagher ME, Godecke A, Helfrich MH, Reid DM, Ralston SH. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology 2001; 142:760-766.
127. Das UN. Nitric oxide as the mediator of the antiosteoporotic actions of estrogen, statins, and essential fatty acids. Exp. Biol. Med. 2002; 227:88-93.
128. Rhoads GG, Gulbrandsen CL, Kagan A. Serum lipoproteins and coronary heart disease in a population study of Hawaii Japanese men. N. Engl. J. Med. 1976; 294:293-298.
129. Ikewaki K, Rader DJ, Sakamoto T, Nishiwaki M, Wakimoto N, Schaefer JR, Ishikawa T, Fairwell T, Zech LA, Nakamura H, Nagano M, Brewer HB. Delayed catabolism of high density lipoprotein apolipoprotein A-I and A-II in human cholesteryl ester transfer protein deficiency. J. Clin. Invest. 1993; 92:1650-1658.
130. Barter PJ, Brewer HB Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003; 23:160-167.
131. Inazu I, Koizumi J, Mabuchi H. Cholesteryl ester transfer protein and atherosclerosis. Curr. Opin. Lipidol. 2000; 11:389-396.
132. Watts GF. The yin and yang of cholesteryl ester transfer protein and atherosclerosis. Clin. Sci. 2002; 103:595-597.
133. Jansen S, Lopez-Miranda J, Castro P, Lopez-Segura F, Marin C, Ordovas JM, Paz E, Jimenez-Pereperez J, Fuentes F, Perez-Jimenez F. Low-fat and high-monounsaturated fatty acid diets decrease plasma cholesterol ester transfer protein concentrations in young, healthy, normolipemic men. Am. J. Clin. Nutr. 2000; 72:36-41.
134. Hirano R, Igarashi O, Kondo K, Itakura H, Matsumoto A. Regulation by long-chain fatty acids of the expression of cholesteryl ester transfer protein in HepG2 cells. Lipids 2001; 36:401-406.
135. Smaoui M, Hammami S, Attia N, Chaaba R, Abid N, Kilani N, Kchaou H, Mahjoub S, Abid M, Hammami M. Modulation of plasma cholesteryl ester transfer protein activity by unsaturated fatty acids in Tunisian type 2 diabetic women. Nutr. Metab. Cardiovasc. Dis. 2006; 16:44-53.
136. Boekholdt SM, Kuivenhoven JA, Wareham NJ, Peters RJ, Jukema JW, Luben R, Bingham SA, Day NE, Kastelein JJ, Khaw KT. Plasma levels of cholesteryl ester transfer protein and the risk of future coronary artery disease in apparently healthy men and women: the prospective EPIC (European Prospective Investigation into Cancer and nutrition)-Norfolk population study. Circulation 2004; 110:1418-1423.
137. Kastelein JJP, van Leuven AI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ, Revkin JH, Grobbee DE, Riley WA, Shear CL, Duggan WT, Bots ML, for the RADIANCE 1 Investigators. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N. Engl. J. Med. 2007; 356:1620-1630.
138. Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, Ruzyllo W, Bachinsky WB, Lasala GP, Tuzcu EM, for the ILLUSTRATE Investigators. N. Engl. J. Med. 2007; 356:1304-1316.
139. Prichard BNC, Smith CCT, Ling KLE, Betteridge DJ. Fish oils and cardiovascular disease. Br. Med. J. 1995; 310:819-820.
140. Kagawa Y, Nishizawa M, Suzuki M, et al. Eicosapolyenoic acids of serum lipids of Japanese islanders with low incidence of cardiovascular diseases. J. Nutr. Sci. Vitaminol. 1982;28:441-453.
141. Kromhout D, Bosschieter EB, de Lezenne Coulander C. The inverse relation between fish consumption and 20-year mortality from coronary heart disease. N. Engl. J. Med. 1985; 312:1205- 1209.
142. Erkkila AT, Lehto S, Pyorala K, Uusitupa MI. n-3 fatty acids and 5-y risks of death and cardiovascular disease events in patients with coronary artery disease. Am. J. Clin. Nutr. 2003; 78:65-71.
143. He K, Song Y, Daviglus ML, Liu K, Van Horn L, Dyer AR, Greenland P. Accumulated evidence on fish consumption and coronary heart disease mortality: a meta-analysis of cohort studies.Circulation 2004; 109:2705-2711.
144. Dyerberg J, Eskesen DC, Andersen PW, Astrup A, Buermann B, Christensen JH, Clausen P, Rasmussen BF, Schmidt EB, Thol- strup T, et al. Effects of trans- and n-3 unsaturated fatty acids on cardiovascular risk markers in healthy males: an 8 weeks dietary intervention study. Eur. J. Clin. Nutr. 2004; 58:1062-1070.
145. Hu FB, Bronner LL, Willett WC, Stampfer MJ, Rexrode KM, Albert CM, Hunter D, Manson JE. Fish and omega-3 fatty acid intake and risk of coronary heart disease in women. JAMA 2002; 287:1815-1821.
146. Daviglus ML, Stamler J, Orencia AJ, Dyer AR, Liu K, Greenland P, Walsh MK, Morris D, Shekelle RB. Fish consumption and the 30-year risk of fatal myocardial infarction. N. Engl. J. Med. 1997; 336:1046-1053.
147. Bucher HC, Hengstler P, Schindler C, Meier G. N-3 polyunsaturated fatty acids in coronary heart disease: a meta-analysis of randomized controlled trials. Am. J. Med. 2002; 112:298-304.
148. Psota TL, Gebauer SK, Kris-Etherton P, Dietary omega-3 fatty acid intake and cardiovascular risk. Am. J. Cardiol. 2006; 98(suppl):3i-18i.
149. Das UN. Beneficial effect(s) of n-3 fatty acids in cardiovascular diseases: but, why and how? Prostaglandins Leukot. Essen. Fatty Acids 2000; 63:351-362.
150. Das UN. Beneficial actions of polyunsaturated fatty acids in cardiovascular diseases: but, how and why? Current Nutr. Food Sci. In press.
151. GISSI Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet 1999; 354:447-455.
152. Yoko yama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid (EPA) on major coronary events in hypercholesterolemic patients (JELIS): arandomized open-label blinded endpoint analysis. Lancet 2007; 369:1090-1098.
153. Leaf A, Weber PC. Cardiovascular effects of n-3 fatty acids. N. Engl. J. Med. 1988; 322:697-698.
154. Kristensen SD, Schmidt EB, Dyerberg J. Dietary supplementation with n-3 polyunsaturated fatty acids and human platelet function: a review with particular emphasis on implications for cardiovascular disease. J. Intern. Med. 1989; 225(suppl):141-150.
155. Scheurlen M, Kirchner M, Clemens MR, Jaschonek K. Fish oil preparations rich in docosahexaenoic acid modify platelet responsiveness to prostaglandin-endoperoxide/thromboxane A2 receptor agonists. Biochem. Pharmacol. 1993; 46:245-249.
156. Dusing R, Struck A, Gobel BO, Weisser B, Vetter H. Effects of n-3 fatty acids on renal function and renal prostaglandin E metabolism. Kidney Int. 1990; 38:315-319.
157. Barcelli UO, Weiss M, Beach D, Motz A, Thompson B. High linoleic acid diets ameliorate diabetic nephropathy in rats. Am. J. Kidney Dis. 1990; 16:244-251.
158. Singer P, Berger I, Moritz V, Forster D, Taube C. N-6 and N-3 PUFA in liver lipids, thromboxane formation and blood pressure from SHR during diets supplemented with evening primrose, sunflower seed or fish oil. Prostaglandins Leukot Essen Fatty Acids 1990; 39:207-211.
159. Vaskonen T, Laakso J, Mervaala E, Sievi E, Karppanen H. Interrelationships between salt and fish oil in stroke-prone spontaneously hypertensive rat. Blood Press 1996; 5:178-189.
160. Grande JP, Walker HJ, Holub BJ, Warner GM, Keller DM, Haugen JD, Donadio JV Jr, Dousa TP. Suppressive effects of fish oil on mesangial cell proliferation in vitro and in vivo. Kidney Int. 2000; 57:1027-1040.
161. Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acids in the treatment of IgA nephropathy. Semin Nephrol 2004; 24:225-243.
162. Homan van der Heide JJ, Bilo HJ, Tegzess AM, Donker AJ. The effects of dietary supplementation with fish oil on renal function in cyclosporine-treated renal transplant recipients. Transplantation 1990; 49:523-527.
163. Clark WF, Parbtani A, Philbrick DJ, Holub BJ, Huff MW. Chronic effects of omega-3 fatty acids (fish oil) in a rat 5/6 renal ablation model. J. Am. Soc. Nephrol. 1991; 1:1343-1353.
164. Ingram AJ, Parbtani A, Clark WF, Spanner E, Huff MW, Philbrick DJ, Holub BJ. Effects of flaxseed and flax oil diets in a rat-5/6 renal ablation model. Am. J. Kidney Dis. 1995; 25:320-329.
165. Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, Bonventre JV. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 2006; 177:5902-5911.
166. Wiemer G, Fink E, Linz W, Hropot M, Scholkens BE, Wohlfart P. Furosemide enhances the release of endothelial kinins, nitric oxide and prostacyclin. J. Pharmacol. Exp. Ther. 1994; 271:1611-1615.
167. Harris RC, Zhang MZ, Cheng HF. Cyclooxygenase-2 and the renal renin-angiotensin system. Acta Physiol. Scand. 2004; 181:543-547.
168. Christensen JH, Gustenhoff P, Korup E, Aaroe J, Toft E, Moller JM, Rasmussen K, Dyerberg J, Schmidt EB. n-3 polyunsaturated fatty acids, heart rate variability and ventricular arrhythmias in post-AMI-patients. A clinical controlled trial. Ugeskr Laeger. 1997; 159:5525-5529.
169. Christensen JH, Christensen MS, Dyerberg J, Schmidt EB. Heart rate variability and fatty acid content of blood cell membranes: a dose-response study with n-3 fatty acids. Am. J. Clin. Nutr. 1999; 70:331-337.
170. Minami M, Kimura S, Endo T, Hamaue N, Hirafuji M, Togashi H, Yoshioka M, Saito H, Watanabe S, Kobayashi T, Okuyama H. Dietary docosahexaenoic acid increases cerebral acetylcholine levels and improves passive avoidance performance in stroke-prone spontaneously hypertensive rats. Pharmacol. Biochem. Behav. 1997; 58:1123-1129.
171. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405:458-462.
172. Borovikova LV, Ivanova S, Nardi D, Zhang M, Yang H, Ombrellino M, Tracey KJ. Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton.. Neurosci. 2000; 85:141-147.
173. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey CJ. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J. Exp. Med. 2002; 195:781-788.
174. 174. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susaria S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421:384-388.
175. Das UN. Alcohol consumption and risk of dementia. Lancet 2002; 360:490.
176. Das UN. Free radicals, cytokines and nitric oxide in cardiac failure and myocardial infarction. Mol. Cell Biochem. 2000; 215:145-152.
177. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, Blumberg RS, Serhan CN. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc.Natl.Acad.Sci.U.S.A. 2005; 102:7671-676.
178. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac R-L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J.Exp.Med. 2002; 196:1025-037.
179. Kapoor R, Huang YS. Gamma linolenic acid: an antiinflammatory omega-6 fatty acid. Curr. Pharm. Biotechnol. 2006; 7:531- 534.
180. Iversen L, Fogh K, Bojesen G, Kragballe K. Linoleic acid and di- homogammalinolenic acid inhibit leukotriene B4 formation and stimulate the formation of their 15-lipoxygenase products by human neutrophils in vitro. Evidence of formation of antiinflammatory compounds. Agents Actions 1991; 33:286-291.
181. Kambe T, Murakami M, Kudo I. Polyunsaturated fatty acids potentiate interleukin-1-stimulated arachidonic acid release by cells overexpressing type IIA secretory phospholipase A2. FEBS Lett. 1999; 453:81-84.
182. Gilroy DW, Newson J, Sawmynaden P, Willoughby DA, Croxtall JD. A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. FASEB J. 2004; 18:489-498.
183. Das UN. A defect in the activity of ∆6 and ∆5 desaturases may be a factor in the initiation and progression of atherosclerosis. Prostaglandins Leukot. Essen. Fatty Acids. In press.
184. Bernal-Mizrachi C, Gates AC, Weng S, Imamura T, Knutsen RH, DeSantis P, Coleman T, Townsend RR, Muglia LJ, Semenkovich CF. Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature 2006; 435:502-506.
185. Smith EB. The effects of age and of early atheromata on the intimal lipids in men. Biochem. J. 1962; 84:49.
186. Smith EB. Lipids carried by S10-12 lipoprotein in normal and hypercholestero-laemic serum. Lancet 1962; 2:530-534.
187. Klein PD, Johnson RM. Phosphorous metabolism in unsaturated fatty acid-deficient rats. J. Biol. Chem. 1954; 211:103-110.
188. Hayashida T, Portman OW. Swelling of liver mitochondria from rats fed diets deficient in essential fatty acids. Proc. Soc. Exp. Biol. Med. 1960; 103:656-659.
189. Cornwell DG, Panganamala RV. Atherosclerosis an intracellular deficiency in essential fatty acids. Prog. Lipid Res. 1981; 20:365-376.
190. Peyron- Caso E, Taverna M, Guerre-Millo M, Veronese A, Pacher N, Slama G, Rizkalla SW. Dietary (n-3) polyunsaturated fatty acids up-regulate plasma leptin in insulin-resistant rats. J. Nutr. 2002; 132:2235-2240.
191. Reseland JE, Haugen F, Hollung K, Solvoll K, Halvorsen B, Brude IR, Nenseter MS, Christiansen EN, Drevon CA. Reduction of leptin gene expression by dietary polyunsaturated fatty acids. J. Lipid Res. 2001; 42:743-750.
192. Flachs P, Mohamed-Ali V, Horakova O, Rossmeisl M, Hosseinzadeh-Attar MJ, Hensler M, Ruzickova J, Kopecky J. Polyunsaturated fatty acids of marine origin induce adiponectin in mice fed a high-fat diet. Diabetologia 2006; 49:394-397.
193. Borkman M, Storlien LH, Pan DA, Jenkins AB, Chisholm DJ, Campbell LV. The relation between insulin sensitivity and the fatty-acid composition of skeletal-muscle phospholipids. N. Engl. J. Med. 1993; 328:238-244.
194. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science 2000; 289:2122-2125.
195. Hajnal A, Pothos EN, Lenard L, Hoebel BG. Effects of feeding and insulin on extracellular acetylcholine in the amygdala of freely moving rats. Brain Res. 1998; 785:41-48.
196. Wang G-J, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, Netusil N, Fowler JS. Brain dopamine and obesity. Lancet 2001; 357:354-357.
197. Futerman AH, Banker GA. The economics of neurite outgrowth: the addition of new membrane to growing axons. Trends Neu- rosci. 1996; 19:144-149.
198. Negre-Aminou P, Nemenoff RA, Wood MR, de la Houssaye BA, Pfenninger KH. Characterization of phospholipase A2 activity enriched in the nerve growth cone. J. Neurochem. 1996; 67:2599-2608.
199. Hornfelt M, Ekstrom PA, Edstrom A. Involvement of axonal phospholipase A2 activity in the outgrowth of adult mouse sensory axons in vitro. Neuroscience 1999; 91:1539-1547.
200. Kelly RB. Deconstructing membrane traffic. Trends Cell Biol. 1999; 9:M29-M33.
201. Darios F, Davletov B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature 2006; 440:813-817.
202. Rickman C, Davletov B. Arachidonic acid allows SNARE complex formation in the presence of Munc18. Chem. Biol. 2005; 12:545-553.
203. Kitajka K, Sinclair AJ, Weisinger RS, Weisinger HS, Mathai M, Jayasooriya AP, Halver JE, Puskas LG. Effects of dietary omega-3 polyunsaturated fatty acids on brain gene expression. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:10931-10936.
204. Kitajka K, Puskas LG, Zvara A, Hackler L, Barcelo-Coblijn G, Yeo YK, Farkas T. The role of n-3 polyunsaturated fatty acids in brain: Modulation of rat brain gene expression by dietary n-3 fatty acids. Proc. Natl. Acad. Sci U.S.A. 2002; 99:2619-2624.
205. Barcelo- Coblijn G, Hogyes E, Kitajka LG, Zvara A, Hackler L Jr, Nyakas C, Penke Z, Farkas T. Modification by docosahexaenoic acid of age-induced alterations in gene expression and molecular composition of rat brain phospholipids. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:11321-11326.
206. Das UN. Pathophysiology of metabolic syndrome X and its links to the perinatal period. Nutrition 2005; a21:762-773.
207. Das UN. Can perinatal supplementation of long-chain polyunsaturated fatty acids prevents schizophrenia in adult life? Med. Sci. Monit. 2004; 10:HY33-HY37.
208. Das UN. Can perinatal supplementation of long-chain polyunsaturated fatty acids prevent atopy, bronchial asthma and other inflammatory conditions? Med. Sci. Monit. 2006;12:RA99-RA111.
209. Beydoun MA, Kaufman JS, Satia JA, Rosamond W, Folsom AR. Plasma n-3 fatty acids and the risk of cognitive decline in older adults: the Atherosclerosis Risk in Communities Study. Am. J. Clin. Nutr. 2007; 87:1103-1111.
210. van Gelder BM, Tijhuis M, Kalmijn S, Kromhout D. Fish consumption, n-3 fatty acids, and subsequent 5-y cognitive decline in elderly men: the Zutphen Elderly Study. Am. J. Clin. Nutr. 2007; 87:1142-1147.
211. Connor WE, Connor SL. The importance of fish and do- cosahexaenoic acid in Alzheimer disease. Am. J. Clin. Nutr. 2007; 87:929-930.
212. Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N Jr, Frautschy SA, Cole GM. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005; 25:3032-3040.
213. Hashimoto M, Hossain S, Shimada T, Shido O. Docosahexaenoic acid-induced protective effect against impaired learning in amyloid beta-infused rats is associated with increased synaptosomal membrane fluidity. Clin. Exp. Pharmacol. Physiol. 2006; 33:934-939.
214. Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahex- aenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 2005; 115:2774-2783.
215. Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, Foster DA, Sisodia SS, Xu H, Gorelick FS, Greengard P. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:1941-1946.
216. Kim SY, Ahn BH, Min KJ, Lee YH, Joe EH, Min DS. Phospholipase D isozymes mediate epigallocatechin gallate-induced cyclooxygenase-2 expression in astrocyte cells. J. Biol. Chem. 2004; 279:38125-38133.
217. Kihara T, Shimohama S. Alzheimer’s disease and acetylcholine receptors. Acta Neurobiol. Exp. 2004; 64:99-105.
218. Ieraci A, Herrera DG. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med. 2006; 3:101.
219. Chong ZZ, Lin SH, Maise K. NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J. Cereb. Blood Flow Metab. 2004; 24:728-743.
220. Das UN. Fetal alcohol syndrome and essential fatty acids PLoS Med. 2006; 3:e247.
221. Appleton KM, Hayward RC, Gunnell G, Peters TJ, Rogers PJ, Kessler D, Ness AR. Effects of n-3 long-chain polyunsaturated fatty acids on depressed mood: systematic review of published trials. Am. J. Clin. Nutr. 2006; 84:1308-1316.
222. Das UN. Is depression a low-grade systemic inflammatory condition? Am. J. Clin. Nutr. In press.
223. Colin A, Reggers J, Castronovo V, Anssean M. Lipids, depression, and suicide. Encephale 2003; 29:49-58.
224. Su KP, Huang SY, Chiu CC, Shen WW. Omega-3 fatty acids in major depressive disorder: A preliminary double-blind, placebo-controlled trial. Eur Neuropscychopharmacol 2003; 13:267-271.
225. Ranjekar PK, Hinge A, Hegde MV, Ghate M, Kale A, Sitasawad S, et al. Decreased antioxidant enzymes and membrane essential polyunsaturated fatty acids in schizophrenia and bipolar mood disorder patients. Psychiatry Res. 2003; 121:109-122.
226. Parker G, Gibson NA, Brotchie H, Heruc G, Rees AM, Hadzi-Pavlovic D. Omega-3 fatty acids and mood disorders. Am. J. Psychiatry 2006; 163:969-978.
227. De Vriese SR, Christophe AB, Maes M. In humans, the seasonal variation in poly-unsaturated fatty acids is related to the seasonal variation in violent suicide and serotonergic markers of violent suicide. Prostaglandins Leukot. Essent. Fatty Acids 2004; 71:13-18.
228. Arvindakshan M, Ghate M, Ranjekar PK, Evans DR, Mahadik SP. Supplementation with a combination of omega-3 fatty acids and antioxidants (vitamins E and C) improves the outcome of schizophrenia. Schizophr. Res. 2003; 62:195-204.
229. Das UN, Vaddadi KS. Essential fatty acids in Huntington’s disease. Nutrition 2004; 20:942-947.
230. Das UN, Rao KP. Effect of y-linolenic acid and prostaglandins E1 on gamma-radiation and chemical-induced genetic damage to the bone marrow cells of mice. Prostaglandins Leukot. Essen. Fatty Acids 2006; 74:165-173.
231. Das UN. From bench to the clinic: y-linolenic acid therapy of human gliomas. Prostaglandins Leukot. Essen. Fatty Acids 2004; 70:539-552.