Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
3. Basic Energetics: Intermolecular Interactions
3.2. Electrostatic Interaction
The Coulomb energy w of a charge q1 in the presence of another charge q2 is
(3.4) ![]()
Here, ϕ2 represents the potential associated with the force ![]() between the charges q1 and q2 with r = |r| their distance and ε0 the permittivity of vacuum. Molecule 1 can be represented by a charge distribution4)
between the charges q1 and q2 with r = |r| their distance and ε0 the permittivity of vacuum. Molecule 1 can be represented by a charge distribution4) ![]() , and the potential ϕ2 due to the charge distribution of molecule 2 by
, and the potential ϕ2 due to the charge distribution of molecule 2 by ![]() where sj = |sj| = |r−rj| is the distance of charge qj to position r (Figure 3.2). The total interaction energy
where sj = |sj| = |r−rj| is the distance of charge qj to position r (Figure 3.2). The total interaction energy ![]() with sjk = |sjk| = |rj − rk|. After expanding the function ϕ2, evaluating all the derivatives and collecting terms, a procedure with considerable understatement often called “a lengthy but straightforward calculation” (see Justification 3.1), one obtains the desired result. Note that, since this process takes place at constant T and V, the interaction W12 represents the Helmholtz energy.
with sjk = |sjk| = |rj − rk|. After expanding the function ϕ2, evaluating all the derivatives and collecting terms, a procedure with considerable understatement often called “a lengthy but straightforward calculation” (see Justification 3.1), one obtains the desired result. Note that, since this process takes place at constant T and V, the interaction W12 represents the Helmholtz energy.
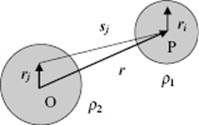
Figure 3.2 The interaction ρ1ϕ2 of two charge distributions ρ1 and ρ2 with common origin O.
Justification 3.1: The multipole expansion*
To find the proper expression for the electrostatic interaction, let us consider an arbitrary charge distribution ρ of point charges qj at position rj from the origin O located at the center of mass (Figure 3.2). The potential energy ϕ at a certain point P located at r outside the sphere containing all charges is
![]()
where sj = |sj| = |r−rj| is the distance of charge qj to P. Developing 1/sj in a Taylor series with respect to rj, we may write 1/sj = 1/r + rj(∇1/r)O + ··· and therefore (using direct notation, Appendix B)
(3.5) 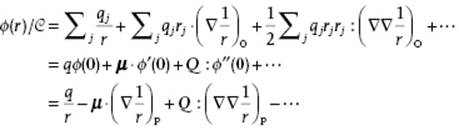
where r = |r| is the distance of the point P to the origin O of ρ. In the second line we defined q = Σjqj as the total charge, μ = Σjqjrj as the dipole moment, and Q = ½Σjqjrjrj as the quadrupole moment. Moreover, we abbreviate 1/r by ϕ(0), [∇(1/r)]O by φ′(0), etc. The minus sign in the last step for the second, fourth, and so on terms arises because the differentiation is now made at P, and not at the origin of the potential O. This follows since for an arbitrary vector x it holds that ∇(1/x) = −x/x3 and therefore [∇(1/r)]P = −[∇(1/r)]O. Equation (3.5) is usually called the multipole expansion of the potential. The interaction W of a point charge q at r in the potential ϕ of a charge distribution is W = qϕ(r). Hence, to calculate the interaction energy between two charge distributions ρ1 and ρ2, separated by r (Figure 3.2), we express this energy as the energy of ![]() in the field of
in the field of ![]() . The interaction energy W12 = ρ1ϕ2 is then
. The interaction energy W12 = ρ1ϕ2 is then
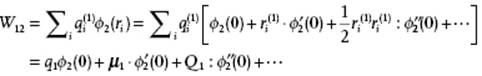
If we take the center of mass P of ρ1 as common origin, the potential ϕ2 due to ρ2 is given by the last line of Eq. (3.5), and substitution in the previous equation results in
(3.6) ![]()
![]()
This is the general expression for the electrostatic interaction energy expressed in terms of multipole moments of the charge distributions with respect to their own center of mass [1]. For further evaluation, we need again ∇(1/r) = −r/r3 and also ∇2(1/r) = 3rr/r5 – I/r3 with I the unit tensor, easily derived using r2 = x2 + y2 + z2, and leading to Eq. (3.7).
The final result reads
(3.7) ![]()
or explicitly (see Figure 3.3 for notation, introducing the abbreviation ω = θ,ϕ)
(3.8) 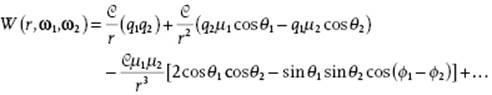
Figure 3.3 The interaction at a distance r of a point charge q1 with a dipole μ2 in which the charges q are separated by a distance δ (a) and the interaction of a dipole with a dipole at distance r (b).
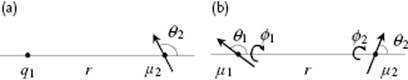
In this expression ![]() is the charge of molecule 1, and
is the charge of molecule 1, and ![]() is the dipole moment with μ1 = |μ1|.
is the dipole moment with μ1 = |μ1|.
The first term (proportional to q1, q2 and 1/r) is just the Coulomb interaction (more precisely the charge–charge Coulomb interaction) between the molecules at distance r. At long distance r this interaction term will dominate, but clearly it is zero if one of the molecules is neutral.
The second term (proportional to q1, μ2, q2, μ1 and 1/r2; Figure 3.3) is the interaction between the charge of molecule 2 with the dipole moment of molecule 1 and the interaction between the charge of molecule 1 with the dipole moment of molecule 2. This is the charge–dipole (Coulomb) interaction. The charge–dipole interaction decreases as 1/r2, as compared to 1/r for the charge–charge Coulomb interaction. A simple way to obtain this result is dealt with in Problem 3.2. Because the charge–dipole interaction is orientation-dependent and molecules move generally rather rapidly, an orientational average (overbar) must be made (we will learn how to do that in Chapter 5). For the moment we just quote the result, valid for sufficiently high temperature,
(3.9) ![]()
The third term (proportional to μ1, μ2 and 1/r3; Figure 3.3) represents the dipole–dipole (Coulomb) interaction between the dipole moments of molecule 1 and molecule 2, and is often addressed as the Keesom interaction (it also includes the charge–quadrupole interaction, but we neglect that interaction). We deal with this interaction in a simplified way in Problem 3.3. This interaction is also orientation-dependent and for the orientational average one obtains, again for sufficiently high temperature and using ω = (θ,ϕ) with θ and ϕ the orientation angles
(3.10) ![]()
For neutral molecules the dipole–dipole interaction is the leading term. Note (again) that the expressions given represent the Helmholtz energies and that the internal energy expressions U are given by U = 2W (see Problem 2.6).
Problem 3.1
Show that for a charged molecule the dipole moment depends on the choice of the origin but is independent of this choice for a neutral molecule.
Problem 3.2: Charge–dipole interaction
A simple model to estimate the charge–dipole interaction is shown in Figure 3.3. The distances between the charge q1 of molecule 1 and the charges of the dipole moment of molecule 2, +q2 and −q2, respectively, are given by
![]()
Show, if it is assumed that the distance r is much larger than the distance δ by using Coulomb's law and the binomial expansion, that the interaction for fixed direction is ![]() where
where ![]() . How large should r be as compared to δ in order to have an error less than 5%?
. How large should r be as compared to δ in order to have an error less than 5%?
Problem 3.3: Dipole–dipole interaction
The dipole–dipole interaction can be estimated in a similar way as for the charge–dipole problem (Figure 3.3). Show that the expression for the in-line orientation energy (orientations parallel to the connection line) is
![]()
Here, ± indicates whether the orientation is in the same way (attraction) or in the opposite way (repulsion). Also show that the expression for the parallel orientation energy (orientations perpendicular to the connection line) is
![]()
again, dependent on whether the orientation is antiparallel (attraction) or parallel (repulsion). How large should r be as compared to δ in order to have an error less than 5%?
Problem 3.4*
Verify Equation (3.6). Alternatively, derive the expression for the electrostatic interaction up to second order using a double Taylor expansion for ![]() with sij = |r + rj−ri| using the complete expression
with sij = |r + rj−ri| using the complete expression ![]() .
.