Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
5. The Transition from Microscopic to Macroscopic: Statistical Thermodynamics
5.3. The Semi-Classical Approximation
The scheme for structureless, independent particles outlined so far comprises the calculation of:
· the quantum energy levels εi,
· the partition function, say Z (Ξ or W may be used as well), either exact via summation or approximate via integration,
· the corresponding thermodynamic potential, say F (or Ω or S), and
· the remaining properties (e.g., S, P …) using phenomenological thermodynamics.
The scheme also applies to other situations, but a few remarks are appropriate. We mentioned “structureless” particles, but molecules do contain internal structure, and the effect of internal structure is discussed in Section 5.5. We also said “independent” particles; however, interaction is important and we deal with the effect of interactions in Section 5.6. The next point relates to the energy. In many cases, the energy EQM as calculated from the Schrödinger equation ![]() has to be approximated by the classical mechanics expression
has to be approximated by the classical mechanics expression ![]() , where
, where ![]() is the Hamilton function. In fact, statistical thermodynamics was largely developed before the introduction of quantum mechanics. The classical development leads to the partition function
is the Hamilton function. In fact, statistical thermodynamics was largely developed before the introduction of quantum mechanics. The classical development leads to the partition function
![]()
where p = p1 … pN = p1xp1yp1zp2x … pNz, q = q1 … qN = q1xq1yq1zq2x … qNz and T(p) and Φ(q) represent the kinetic and potential energy, respectively. Here, pi is the (generalized) momentum and qi the (generalized) coordinate of particle i. The choice of coordinates is to some extent arbitrary, since for conjugated coordinates we have dpdq = dp′dq′. The connection between ZQM and ZCM has been made [4], but the process is rather complex and we illustrate here the result by example only. Since the factor between ZCM and ZQM should depend only on fundamental constants, this is in principle also sufficient. Consider a single particle with three degrees of freedom (DoF) for which the classical energy expression ![]() (p,q) is given by
(p,q) is given by ![]() , so that the classical partition function becomes
, so that the classical partition function becomes
(5.36) ![]()
We see that zCM is similar to zQM except for a factor h−3, which is missing in the classical expression. So, for each DoF an extra factor h−1 is required to match the quantum expression. This result appears to be general: if we calculate the partition function using a classical expression for the energy, we need an extra factor h−1 for every DoF (see Justification 5.3). For a single DoF z1D reads
(5.37) ![]()
The partition function z1D is thus dimensionless. As long as z1D >> 1, this DoF can be described by classical mechanics. As soon as z1D ≅ 1, a full quantum description for that particular DoF is required. This applies also to many particles if l is taken to be the mean particle distance l = (V/N)1/3. To apply classical mechanics to a gas thus requires that Λ3 << V/N. Since μ = kT ln(N/z) with z = V/Λ3, this implies that exp(βμ) >> 1, which is exactly the condition used to arrive at classical statistics. For translation it can be easily shown that a semi-classical description is nearly always sufficient. As will be seen in the next section, this is not generally true for the internal contributions. It is thus quite possible, that classical evaluation is sufficient for one mechanism, while a quantum evaluation is necessary for another. As long as energies are additive, this presents no special problems.
Example 5.2: The harmonic oscillator
A single harmonic oscillator provides a good demonstration for the factor h. In quantum mechanics the energy for an oscillator with spring constant a and mass m is given by ![]() with
with ![]() so that the energy difference ΔE between two successive states is ΔE = ħω. In classical mechanics the total energy can be written as
so that the energy difference ΔE between two successive states is ΔE = ħω. In classical mechanics the total energy can be written as ![]() , where p is the momentum and q is the coordinate. We may also write p2/α2 + q2/β2 = 1 with α = (2mE)1/2 and β = (2E/mω2)1/2. If we plot constant energy curves in μ-space, which is in this case a two-dimensional space, we obtain ellipses. The area enclosed by such an ellipse is given by the integral I = ∮ p dq, where the integration is over one period, or I = παβ = 2πE/ω. Alternatively, we have q = q0 sin(ωt),
, where p is the momentum and q is the coordinate. We may also write p2/α2 + q2/β2 = 1 with α = (2mE)1/2 and β = (2E/mω2)1/2. If we plot constant energy curves in μ-space, which is in this case a two-dimensional space, we obtain ellipses. The area enclosed by such an ellipse is given by the integral I = ∮ p dq, where the integration is over one period, or I = παβ = 2πE/ω. Alternatively, we have q = q0 sin(ωt), ![]() and
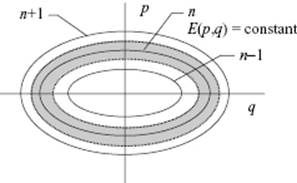
and ![]() . Let us draw ellipses with energies corresponding to n − 1, n, and n + 1 (Figure 5.1). The area between two successive ellipses is the area in classical phase space associated with one quantum state, and this area corresponds to 2πΔE/ω = 2πħω/ω = h. For large n, the classical energy is almost constant in the phase region between n − ½ and n + ½, so that the approximation
. Let us draw ellipses with energies corresponding to n − 1, n, and n + 1 (Figure 5.1). The area between two successive ellipses is the area in classical phase space associated with one quantum state, and this area corresponds to 2πΔE/ω = 2πħω/ω = h. For large n, the classical energy is almost constant in the phase region between n − ½ and n + ½, so that the approximation ![]() can be made. For T→ ∞ and n → ∞ the argument becomes exact.
can be made. For T→ ∞ and n → ∞ the argument becomes exact.
Figure 5.1 Phase space for a 1D oscillator.
Overall, the result for N particles leads to the introduction of the factor (h3NN!)−1 so that we have in total, using the Hamilton function ![]() and potential energy Φ,
and potential energy Φ,
(5.38) ![]()
This result is the most frequently encountered expression for the partition function, also indicated as the semi-classical partition function. We have seen that in Eq. (5.36) for the single-particle partition function the integration over the momenta and position coordinates separate. The factor V, due to the integration over the position coordinates, is denoted as the configurational part while the factor Λ = (h2/2πmkT)1/2, due to the integration over the momenta, is denoted as the kinetic part. The latter part leads to ½kT for the kinetic energy of a classical DoF. Because in classical mechanics energy expressions the kinetic energy is generally only dependent on p and the potential energy is generally only dependent on q, this separation is also generally possible and it can be concluded that in classical models the kinetic energy per DoF is (nearly) always ½kT. For many particles this separation is shown in the last part of Eq. (5.38). For N independent particles the configurational part is deceptively simple (QN = VN), but we will see that this integration becomes the major problem for interacting particles11) (see Section 5.6).
Finally, we note that the expectation value for the canonical average value of a property X(p,q) in the semi-classical approximation becomes
(5.39) ![]()
where the last step can be made if X depends only on the coordinates q.
Justification 5.3: The introduction of h and N!*
First we note that for the Hamilton operator ![]() operating on wave function Ψ and corresponding to the classical Hamilton function
operating on wave function Ψ and corresponding to the classical Hamilton function ![]() we have
we have
![]()
Second, a state |Ψ⟩ for N particles can be expanded in any complete set of the proper symmetry and we use here the antisymmetrized eigenstates of the momentum operator p using the δ-function normalization (see Section 2.2)
![]()
with the permutation operator ![]() for coordinates r (see Section 2.2) and A(p) the corresponding coefficient. Note that |exp(ipTr)⟩ denotes the product function
for coordinates r (see Section 2.2) and A(p) the corresponding coefficient. Note that |exp(ipTr)⟩ denotes the product function ![]() of the individual eigenfunctions
of the individual eigenfunctions ![]() for particle i. Third, we will need for an arbitrary function F(p) of p the relation
for particle i. Third, we will need for an arbitrary function F(p) of p the relation
![]()
Using bra-ket notation the partition function ZN = Σj exp(−βεj) is written as
![]()
with Ψj the energy eigenfunction corresponding to state j. We see that this result can be interpreted as the sum of the diagonal elements of a matrix with as elements ![]() . We expand Ψj in the aforementioned antisymmetrized eigenfunctions of the momentum operator p to obtain
. We expand Ψj in the aforementioned antisymmetrized eigenfunctions of the momentum operator p to obtain
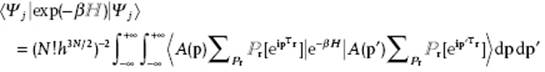
The partition function ZN becomes ![]()
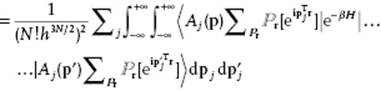
Now, using ![]() for exp(ip′Tr),
for exp(ip′Tr), ![]() ,
, ![]() and
and ![]() , we obtain
, we obtain
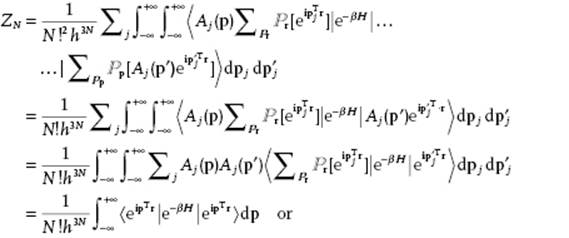
![]()
The classical partition function ZCM is thus (N!h3N)−1 times the phase integral over ![]() . This analysis not only shows the origin of the factor h−3N but also of the factor (N!)−1. Extending the analysis further – in principle a little bit but in practice considerably [5] – by introducing second-order terms in
. This analysis not only shows the origin of the factor h−3N but also of the factor (N!)−1. Extending the analysis further – in principle a little bit but in practice considerably [5] – by introducing second-order terms in ![]() leads to the first-order quantum correction on the classical result which reads, using ⟨ … ⟩ as the canonical average,
leads to the first-order quantum correction on the classical result which reads, using ⟨ … ⟩ as the canonical average,
![]()
so that the Helmholtz energy becomes
![]()
The quantity Φaa is an effective force constant so that ωa = (Φaa/ma)1/2 is the corresponding angular frequency. At room temperature for vibrations with wave number12) ![]() or more, the correction becomes significant.
or more, the correction becomes significant.
Problem 5.11: The thermal wavelength
Calculate Λ for He and Ar and compare these values with the average spacing between the atoms as estimated from the densities at the triple point. Is the classical approximation valid?
Problem 5.12: Angle-averaged potentials
For arbitrary molecules the interaction is dependent on their distance r and both their orientations, indicated by ω = (θ,ϕ) where θ and ϕ are the angles for the polar co-ordinate system (0 ≤ θ ≤ π and 0 ≤ ϕ ≤ 2π). The differential element dω is dω = sinθ dθdϕ. Using the multipole expression for W(r) for fixed distance r, expanding the exponentials and integrating term-by-term, show that,
![]()
![]()
Discuss in physical terms why the terms with n odd are identically zero. Show also that the angle-averaged internal energy
![]()
where the subscript ω indicates averaging over ω only, and that this result is consistent with Problem 2.6.
Problem 5.13: Charge–dipole interaction
Show that, by using the result of Problem 5.12, the charge–dipole contribution to the intermolecular interaction is given by
(3.9) ![]()
Problem 5.14: Dipole-induced dipole interaction
Show that the dipole-induced dipole contribution is given by
(3.12) ![]()
Problem 5.15: Dipole–dipole interaction*
Similarly, show that the dipole–dipole contribution is given by
(3.10) ![]()