Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
5. The Transition from Microscopic to Macroscopic: Statistical Thermodynamics
5.5. Internal Contributions
So far, we have limited the discussion to molecules without internal degrees of freedom, that is, molecules without any other motion than translation. In Section 5.2 we saw that the contributions from internal mechanisms in molecules, as long as their energy is additive, can be incorporated in the overall partition function Z as a factor, for example, ![]() , where ztra, zvib, zrot and zele denote the translation, vibration, rotation and electronic partition function, respectively. In this section we will address the vibration, the (external and internal) rotation, and (briefly) the electronic transitions that can occur in molecules. The pattern of how to calculate thermodynamic properties will be clear by now: find the energy expression, evaluate the partition function; and calculate from the partition function the thermodynamic properties.
, where ztra, zvib, zrot and zele denote the translation, vibration, rotation and electronic partition function, respectively. In this section we will address the vibration, the (external and internal) rotation, and (briefly) the electronic transitions that can occur in molecules. The pattern of how to calculate thermodynamic properties will be clear by now: find the energy expression, evaluate the partition function; and calculate from the partition function the thermodynamic properties.
5.5.1 Vibrations
The simplest molecule with internal structure is a diatomic molecule with m1 and m2 as the masses. The vibrations of a diatomic molecule can be described by the harmonic oscillator model with spring constant a and using the reduced mass μ = m1m2/(m1 + m2) of the two atoms as the mass in the model. The energy of the oscillator using the circular frequency ω, as given in Section 2.2, is
![]()
Hence, for the average energy U = ⟨ε⟩ we find
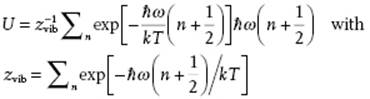
If we use β = (kT)−1, the partition function zvib for a single vibrator can be written as
![]()
and, since this is a geometric series, we can evaluate the sum as
(5.41) ![]()
This expression cannot be used for a too-high temperature for two reasons. First, at higher temperature T, anharmonicity occurs and the harmonic oscillator model no longer applies. Second, in summing zvib we assumed that n → ∞, but in reality n is finite. Knox [8] estimated that one should have kT < 0.1D, where D is the dissociation energy. For a typical value of D = 4 eV, this results in T < 4600 K.
Since zvib⟨ε⟩ = ∑n exp[−ħωβ(n + ½)] ħω (n + ½) = −∂zvib/∂β, we can write
(5.42) ![]()
The Helmholtz energy F is given by
(5.43) ![]()
so that the entropy S is given by
(5.44) ![]()
The energy can also be calculated from U = F + TS. The heat capacity CV reads
(5.45) ![]()
At low and high temperature the expression for CV expands to, respectively,
![]()
The behavior of the harmonic oscillator is characterized by ω. Equivalently, we use the characteristic (vibration) temperature θvib, given as θvib = ħω /k. When T >> θvib, the behavior can be classified as classical and the energy becomes U ≅ kT. When T << θvib, expansion of the expression results in U ≅ ½ħω. When T ≅ θvib, the full expression must be used. Since the characteristic temperature for vibration is typically hundreds of degrees, this implies that in most cases the quantum expression for the partition function must be applied. Values for θvib as well as the equilibrium distance req and dissociation energy Deq for some molecules are given in Table 5.1.
Table 5.1 Vibration, rotation, and dissociation data for several diatomic molecules.a)
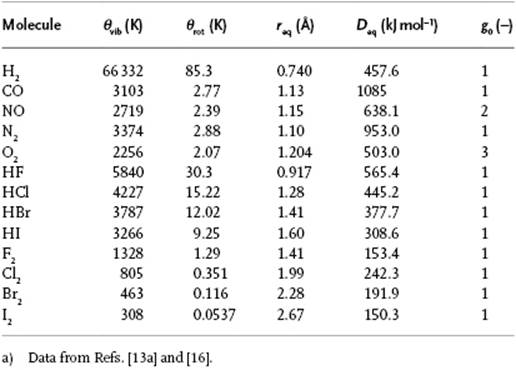
For a N-atomic molecule we can describe the vibrational behavior by 3N − 6 normal coordinates (3N − 5 for a linear molecule), each of which can be modeled as a harmonic oscillator13). These normal coordinates are independent, and hence the behavior can be described as the sum of the behaviors of the individual normal coordinates. In fact, they are an example of generalized coordinates. For example, for H2O the three vibrational temperatures are 2294, 5262, and 5404 K, respectively, while they read 960, 960, 132, and 3380 K for CO2.
As an aside, we note that the harmonic oscillator results are directly applicable to the Einstein model for solids in which each atom vibrates independently of the others with the same (Einstein) frequency ωE. In this case, we have for N atoms ![]() , that is, without the factor N!−1 because, although the atoms are indistinguishable, the lattice sites distinguishable and the total number of configurations is N!.
, that is, without the factor N!−1 because, although the atoms are indistinguishable, the lattice sites distinguishable and the total number of configurations is N!.
Problem 5.17
Show that the entropy S for the harmonic oscillator is given by Eq. (5.44).
Problem 5.18
Show that the specific heat CV for the harmonic oscillator is given by Eq. (5.45). Show also that for T >> θvib, U ≅ kT and CV ≅ k.
Problem 5.19
Show that at 300 K most molecules are in the vibrational ground state.
Problem 5.20: The anharmonic oscillator*
Anharmonicity changes the energy expression and selection rule for the oscillator to
![]()
where εn/hc is the energy in cm−1, ![]() the vibrational wave number (see footnote 11; typically 100 to 4000 cm−1), and x is the anharmonicity constant (typically ∼0.01). Estimate the contribution to CV from anharmonicity.
the vibrational wave number (see footnote 11; typically 100 to 4000 cm−1), and x is the anharmonicity constant (typically ∼0.01). Estimate the contribution to CV from anharmonicity.
5.5.2 Rotations
The rotation behavior of molecules is somewhat more complex than their vibration behavior. We start with homonuclear and heteronuclear diatomic molecules, and thereafter deal briefly with polyatomic molecules and internal rotation.
The energy of the diatomic rigid rotator, as given in Section 2.2, is
(5.46) ![]()
Here, the moment of inertia I = μr2 with the reduced mass μ = m1m2/(m1 + m2) and m1 and m2 the masses of the particles. The constant B is the rotational constant12, alternatively expressed as the characteristic (rotation) temperature θrot = B/k. Each of the energy levels J has a (2J + 1) degeneracy. At low temperature only the first few terms are contributing, and we have
![]()
At high temperature the summation can be approximated by integration and thus
(5.47) 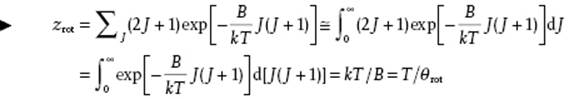
Actually, we have to divide zrot by the symmetry number σ, denoting the number of ways the molecule can be rotated into a configuration indistinguishable from the original configuration. Obviously, σ = 1 for AB and σ = 2 for AA molecules. The origin of this factor lies in the symmetry of the overall wave function and for the details of the derivation we refer to the literature (e.g., McQuarrie [9]). A more complete analysis using the Euler–McLaurin summation expression results in
![]()
which is good to within 1% for θrot < T. Replacing the summation by integration is thus only justified at sufficiently elevated temperature, say for T > 5θrot. For a lower T, one must sum zrot term by term. Typical values for θrot are given in Table 5.1.
If a polyatomic molecule is linear, the same expression as for diatomic molecules applies, of course, with adjusted moments of inertia. For a nonlinear polyatomic molecule a similar, but more complex, reasoning leads to
(5.48) ![]()
where Ix, Iy and Iz denote the principal moments of inertia or, equivalently, θx, θy and θz the rotational temperatures. The symmetry number has the same meaning as before. As examples we quote σ(H2O) = 2, σ(NH3) = 3, σ(CH4) = 12, σ(CH3Cl) = 3, σ(C6H6) = 12, and σ(C6H5Cl) = 2. For polyatomic molecules the semi-classical partition function is generally valid for T > 100 K.
Internal rotations, such as the transition from the staggered to eclipsed conformation in CH3–CH3, also contribute. In the case of molecules for which the associated barrier is relatively large, say > 10kT, the two groups only vibrate with respect to each other and the harmonic oscillator approximation can be used. If the barrier to internal rotation is relatively low, say < kT, one could consider the internal rotation as a free rotation for which the partition function is
(5.49) ![]()
where I′ is the moment of inertia for the internal rotation. In the case of ethane, however, the barrier is ∼12.1 kJ mol−1, while NAkT at 300 K is ∼2.5 kJ mol−1 and thus the neither the harmonic oscillator nor the free rotation approximation may be used. The internal rotation contribution is often the main source of error in calculating thermodynamic data (see, e.g., Ref. [10] for a more detailed discussion).
5.5.3 Electronic Transitions
Generally, the spacing between electronic states in molecules is large as compared with kT. This implies that transitions from the ground state to an excited state are rare, and only the electronic ground state has to be taken into account. Hence the electronic partition function is generally closely approximated by
(5.50) ![]()
where gi indicates the electronic state degeneracy, for the ground state often g0 = 1. Taking the zero level at the electronic ground state the approximation zele = g0 is in most cases sufficiently accurate for both atoms and molecules. One exception is formed by the systems atoms where the electronic ground state is split by spin-orbit coupling. For example, for the F atom the ground state is split in a lower 3P3/2 level with g0 = 4 and an upper 2P1/2 level with g1 = 2. The energy gap between these levels is 404 cm−1, while the gap with the next electronic level is much larger than kT = 207 cm−1 at 298 K. Another example is the NO molecule with an unpaired electron in a π* orbital giving rise to a 2Π1/2 (g0 = 2) and a 2Π3/2 (g1 = 2) level with a gap of 121 cm−1. Again, the gap to the first excited (in this case vibration) level is much larger than kT.
Problem 5.21
Estimate the temperature above which the replacement of the summation in the calculation of the rotational partition function by integration is justified. Take typical numbers from Table 5.1.
Problem 5.22
Calculate the contribution of the 3P3/2 and 2P1/2 levels for the F atom to CV at room temperature and compare the value obtained with the one for translation.
Problem 5.23
Show that for a diatomic molecule the contribution of rotation to the heat capacity CV is given by ![]() and to the entropy S by
and to the entropy S by ![]() .
.
Problem 5.24
Show that the most probable rotational state for a diatomic molecule is given by Jmax ≅ (T/2σθrot)1/2 − 1/2 by treating J as a continuous variable (high temperature approximation).
Problem 5.25: The non-rigid rotor*
Diatomic molecules are not rigid rotors, and the energy is more appropriately described by
![]()
and ![]() the vibrational wave number. For the HCl molecule the rotational constant B and centrifugal distortion constant D are 10.59 cm−1 and 5.28 × 10−4 cm−1, respectively. Calculate the CV according the rigid rotor energy expression, Eq. 5.47. Estimate the contribution of the centrifugal distortion on the energy levels and compare the associated CV with the total CV.
the vibrational wave number. For the HCl molecule the rotational constant B and centrifugal distortion constant D are 10.59 cm−1 and 5.28 × 10−4 cm−1, respectively. Calculate the CV according the rigid rotor energy expression, Eq. 5.47. Estimate the contribution of the centrifugal distortion on the energy levels and compare the associated CV with the total CV.