Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
8. Modeling the Structure of Liquids: The Physical Model Approach
8.5. Scaled-Particle Theory*
In Chapter 7 we showed that using the PY approximation for hard-spheres with diameter σ the pressure P could be described by Eq. (7.43) reading
(8.75) ![]()
with the background correlation function y(r) = g(r)/e(r), direct correlation function c(r), pair correlation function g(r), Boltzmann function e(r) = exp[−βϕ(r)], pair potential ϕ(r), packing fraction η = πσ3ρ/6, and number density ρ. We already mentioned there that y(r) is also denoted as the cavity function, so in this model we need only the value of the correlation function at r = σ and this observation led to scaled-particle theory.
Consider a spherical cavity of radius r inside a fluid and let p0(r) be the probability that no molecular center lies inside this sphere. We take the value of the number density of molecules (hard-spheres) just outside this cavity as ρG(r). Note that a cavity plays exactly the same role as a hard-sphere of diameter 2r − σ. When r = σ, the cavity behaves as a typical molecule and ρG(σ) can be regarded as the number density at a distance σ from a specified particle, that is, we take G(σ) = g(σ) leading to
(8.76) ![]()
Consequently, instead of calculating g(r), we evaluate G(r) at r = σ.
Now, the probability particle of finding particle in a spherical shell between r and r + dr is 4πr2ρG(r)dr. Hence, the joint probability of finding an empty sphere of radius r and no particle in a shell between r and r + dr is given by
(8.77) ![]()
where in the last step the limit dr → 0 is taken. For r < ½σ, at most one sphere can lie in the cavity of radius r, implying that the probability 1 − p0(r) is 4πr3ρ/3. The solution of Eq. (8.77) is thus
(8.78) ![]()
One can show that G(r) and its first derivative are continuous at r = ½σ, but that the second derivative has a finite discontinuity due to the possible introduction of a second sphere. At ![]() , three spheres can be included and a discontinuity occurs in the fourth derivative. With increasing r, more and more spheres can be accommodated and it appears that at the transition from accommodating L spheres to L + 1 spheres a discontinuity occurs in the 2Lth derivative of G(r).
, three spheres can be included and a discontinuity occurs in the fourth derivative. With increasing r, more and more spheres can be accommodated and it appears that at the transition from accommodating L spheres to L + 1 spheres a discontinuity occurs in the 2Lth derivative of G(r).
Considering next the behavior for large r, we recall first that
(8.79) ![]()
where W(r) is the work to create a cavity of radius r, that is, the potential of mean force for this process. From Eqs (8.77) and (8.79) we obtain
(8.80) ![]()
From thermodynamics we learn that
(8.81) ![]()
with volume increment dV = 4πr2dr, surface increment dV = 8πrdr, pressure P, surface tension γ and Tolman length δ for a cavity of radius r (see Chapter 15). Combining Eqs (8.80) and (8.81) leads to
(8.82) ![]()
Although G(r) is not an analytic function, it is still continuous because the discontinuities occur in the high-order derivatives. Hence, we use G(r) as an interpolation function for the complete range ½σ < r < ∞ and determine the quantities P, γ and δ from Eq. (8.76) and the conditions that G(r) and its first derivative should be continuous at r = ½σ. This results, using η = πρσ3/6, in
(8.83) ![]()
Note that the expression for P is identical to that derived from the compressibility equation for hard-spheres in the PY approximation. Reiss [25] applied this model, using a temperature-dependent hard-sphere diameter, also to estimate the surface tension (actually the interface tension with a hard smooth wall). In view of the simplicity of the model the results were surprisingly in agreement with experiment (Table 8.4).
Table 8.4 Scaled-particle and experimental values for γ for various compounds.
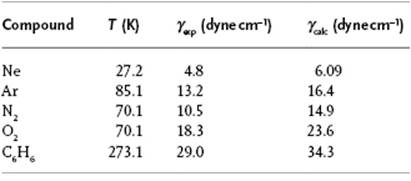
The above-described information presents only the basics of this model. It appears possible to generate systematically relations [26] which improve G(r), and other refinements, in particular relaxing the hard-sphere approximation, are also possible. The model has been successfully applied to estimate hydrophobic interactions of solvent molecules in hydrophobic solvents.
Notes
1) The label “free volume” can easily mean different things to different people. The present definition refers to what is also denoted as fluctuation volume [27].
2) For more details see, e.g., Fowler and Guggenheim (1939), Hirschfelder et al. [3], Barker [4] or McQuarrie [7].
3) In a lattice sum the total potential energy is calculated as the sum of the interactions over all pairs of molecules, arranged according to the lattice specified.
4) Normally we would indicate the number of molecules of type 1 by N1, the number of molecules of type 2 by N2, the total number by N = N1 + N2, and the number of pairs by N11 (N22, N12). To avoid the use of many subscripts for a pure fluid, we use for the total number of sites M and for the number of molecules N. For the number of pairs for we choose N12, relabeled as zX.
5) This is equivalent to using the canonical partition function right away.
6) The adjective “significant” is, of course, rather subjective. Eyring always chose particularly challenging names for his theories. Another example is the theory of ‘absolute’ reaction rates in which there is no element that is ‘absolute’ and which nowadays is often called transition state theory.
7) Different authors provide slightly different ‘best’ estimates. Authors even provide different estimates in the same text [22]: Pcri = 0.119 and 0.141 ε /σ3, Vcri = 3.15 and 3.36 Nσ3, Tcri = 1.28 and 1.31 ε /k).
References
1 Dasannacharya, B.A. and Rao, K.R. (1965) Phys. Rev., 137, A417.
2 See Pryde (1966).
3 See Hirschfelder et al. (1954).
4 See Barker (1963).
5 Eyring H., and Hirschfelder, J.O. (1937) J. Chem. Phys., 41, 249.
6 (a)Lennard-Jones, J.E. and Devonshire, A.F. (1937) Proc. R. Soc., A163, 53; (b)Lennard-Jones, J.E. and Devonshire, A.F. (1938) Proc. R. Soc., A165, 1.
7 See McQuarrie (1973).
8 Wentorf, R.H., Buehler, R.J., Hirschfelder, J.O., and Curtiss, C.F. (1950) J. Chem. Phys., 18, 1484.
9 Buehler, R.J., Wentorf, R.H., Hirschfelder, J.O., and Curtiss, C.F. (1951) J. Chem. Phys., 19, 61.
10 Pople, J. (1951) Philos. Mag., 12, 459.
11 Prigogine, I. and Raulier, S. (1942) Physica, 9, 396.
12 Adams, D.J. and Matheson, A.J. (1972) J. Chem. Soc. Faraday Trans. II, 68.
13 Adams, D.J. and Matheson, A.J. (1973) Chem. Phys. Lett., 22, 484.
14 Hirschfelder, J.O., Stevenson, D.P., and Eyring, H. (1937) J. Chem. Phys., 5, 896.
15 Hoover, W.G. and Ree, F.H. (1968) J. Chem. Phys., 49, 3609.
16 Kirkwood, J.G. (1950) J. Chem. Phys., 18, 380.
17 Cernuschi, F. and Eyring, H. (1939) J. Chem. Phys., 7, 547.
18 Rowlinson, J.S. and Curtiss, C.F. (1951) J. Chem. Phys., 19, 1519.
19 (a) Henderson, D. (1962) J. Chem. Phys., 37, 631; (b) Henderson, D. (1963) J. Chem. Phys., 39, 54.
20 Wentorff, R.H., Buehler, R.J., Hirschfelder, J.O., and Curtiss, C.F. (1950) J. Chem. Phys., 18, 1484.
21 Fuller, E.J., Ree, T., and Eyring, H. (1962) Proc. Natl Acad. Sci. USA, 48, 501.
22 Eyring H., and Jhon, M.S. (1969) Significant Liquid Structures, John Wiley & Sons, Ltd, London.
23 (a) Jhon, M.S. and Eyring, H. (1971) The significant structure theory of liquids, in Physical Chemistry, Vol. VIIIA (ed. D. Henderson), Academic Press, New York, Ch. 5, pp. 335–373; (b) A brief introduction is Eyring, H. and Marchi, R.P. (1963) J. Chem. Educ., 40, 562.
24 Yoon, B.J., Jhon, M.S., and Eyring, H. (1981) Proc. Natl Acad. Sci. USA, 78, 6588.
25 Reiss, H. (1965) Adv. Chem. Phys., 9, 1.
26 Stillinger, F.H., Debenedetti, P.G., and Chatterjee, S. (2006) J. Chem. Phys., 125, 204504–204505.
27 Bondi, A. (1968) Physical Properties of Molecular Crystals, Liquids and Glasses, John Wiley & Sons, London.
Further Reading
Barker, J.A. (1963) Lattice Theories of the Liquid State, Pergamon, London.
Fowler, R.H. and Guggenheim, E.A. (1939) Statistical Thermodynamics, Cambridge University Press, London.
Hirschfelder, J.O., Curtiss, C.F., and Bird, R.B. (1954) Molecular Theory of Gases and Liquids, John Wiley & Sons, Inc., New York.
McQuarrie, D.A. (1973) Statistical Thermodynamics, Harper and Row, New York.
Pryde, J.A. (1966) The Liquid State, Hutchinson University Library, London.