Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
2. Basic Macroscopic and Microscopic Concepts: Thermodynamics, Classical, and Quantum Mechanics
In the description of liquids and solutions, we will need macroscopic and microscopic concepts that we do not consider as part of the subject proper but still are prerequisite to our topic. For macroscopic considerations, we will need phenomenological thermodynamics, and Section 2.1 presents a brief review of this. For microscopic considerations, we introduce in Section 2.2 some concepts from classical mechanics, and in Section 2.3 basic quantum mechanics and a few model system solutions. For further details of these matters, reference should be made to the books in the section “Further Reading” of this chapter, of which free use has been made.
2.1. Thermodynamics
Thermodynamic considerations are basic to many models and theories in science and technology, and in this section the phenomenological aspects of thermodynamics are briefly described but less briefly as in the summary given by Clausius [1]:
· Die Energie der Welt is constant.
· Die Entropie der Welt strebt einem Maximum zu1).
2.1.1 The Four Laws
Thermodynamics is based on four well-known basic laws2). In order to describe these, we start with a few concepts. In thermodynamics the part of the physical world that is under consideration (the system) is for the sake of analysis considered to be separated from the rest (the surroundings). The thermodynamic state of the system is assumed to be determined completely by a set of macroscopic, independent coordinates ai (also labeled a)3) and one “extra” parameter related to the thermal condition of the system. For this parameter one has several choices, one of which is the most appropriate and to which we return later. Together, these constitute the set of state variables. Intensive(extensive) variables are independent of (dependent on) the size and/or quantity of the matter contained in the system. When a new set of state variable values is applied, the system adapts itself via internal processes, collectively characterized by the internal variables ξi (or ξ). When the properties of a system do not change with time at an observable rate given certain constraints, the system is said to be in equilibrium. Thermodynamics is concerned with the equilibrium states available to systems, the transitions between them, and the effect of external influences upon the systems and transitions [2, 3].
Depending on the problem, one assumes a specific type of wall between the system and the surroundings, that is, a diathermal (contrary adiabatic) wall allowing influence from outside on the system in the absence of forces, a flexible (contrary rigid) wall allowing work exchange, or a permeable (contrary impermeable) wall allowing matter exchange. Systems separated by an adiabatic wall are isolated, while systems separated by a diathermal wall are said to be closed, but in thermal contact. Finally, systems separated by diathermal permeable walls are designated as open. Sometimes the wall is taken as virtual, for example, in continuum considerations.
If any two separate systems, each in equilibrium, are brought into thermal contact, the two systems will gradually adjust themselves until they do reach mutual equilibrium. The zeroth law4) states that if two systems are both in thermal equilibrium with a third system, they are also in thermal equilibrium with each other – that is, they have a common value for a state variable T, called (empirical) temperature. If we consider two systems in thermal contact, one of which is much smaller than the other, the state of the larger one will only change negligibly in comparison to the state of the smaller one if heat is transferred from one system to the other. If we are primarily interested in the small system, the larger one is usually known as a temperature bath or thermostat. If, on the other hand, we are primarily interested in the large system, we regard the small system as a measuring device for registering the temperature and refer to it as a thermometer.
With each independent variable ai, often indicated as displacement, a dependent variable is associated, generally denoted as force5) Ai. A force is a quantity in the surroundings that, when multiplied with a change in displacement, yields the associated work. Displacement and force are often denoted as conjugated variables. Work done on the system is counted positive and depends on the path between the initial and the final state. For infinitesimal changes daithe work δW6) is
(2.1) ![]()
The most familiar example of work is the mechanical work δWmec = −P dV, where P and V denote the pressure and volume, respectively7). Other examples are the chemical work δWche = Σαμαdnα with the chemical potential μαand number of moles nα of component α and the electrical work δWele = ϕ dq with the electric potential ϕ and charge q. In general, the integral over δW yields ∫δW = W, where W is not a state function, that is, a function of state variables only, contrary to the integral over a state variable a yielding ∫da = Δa. Hence, δW is not a total differential and consequently we use the notation δW instead of dW.
When work is done on an adiabatically enclosed system it appears that the final state is independent of the type of work and its process of delivery. This leads to the state function U for which we thus have dU = δW. Using other enclosures as well, the first law states that there exists a state function U, called internal energy, such that the change in the internal energy dU from one state to another is given by
(2.2) ![]()
where δQ is identified as heat entering the system8) and interpreted as work done by the microscopic forces associated with the internal variables ξ in the system. Heat entering the system is counted as positive and also depends on the path between the two states. Although δQ and δW are dependent on the path between the initial and final states, dU is independent of the path and depends only on the initial and final states. Hence, dU is a total differential and the internal energy U a state variable which is the proper choice for the “extra”, thermal parameter of state.
Each state is thus characterized by a set of state variables a and the internal energy U. If for a process δQ = 0, it is called adiabatic. If δW = 0 we have isochoric conditions and we refer to the process involved as pure heating or cooling. If both δQ = 0 and δW = 0, the system is isolated. The first law, Eq. (2.2), can thus be stated succinctly as follows: for an isolated system, the energy is constant.
The second law states that there exists another state function S = S(r) + S(i), called entropy and composed of a reversible part S(r) and irreversible part S(i), such that for a transition between two states
(2.3) ![]()
where TdS(r) = δQ and T is the temperature external, that is, just outside, to the volume element considered. The second law, Eq. (2.3), thus expresses that for an isolated system the entropy can increase only or remain constant at most. Since for any system the energy U is a characteristic of the state, the entropy S is a function of U and a and we write S = S(U,a). The entropy of a composite system is usually taken to be additive, that is, the sum of the entropies of the constituent subsystems, to be analytical, that is, continuously differentiable (see Appendix B), and monotonically increasing with energy. Additivity is true as long as the range of interactions between particles is small as compared to the size of the system. Conventionally, additivity is taken to imply extensivity; that is, if the extensive parameters are multiplied by λ, the entropy will obey S(λU,λa) = λS(U,a), and the entropy is thus taken as a homogeneous function of degree 1 of the extensive parameters (see Appendix B). However, this is not necessarily true but only if the effect of the interface of the system with the walls of the enclosure can be neglected. Analyticity and continuity are also not always true. For example, for a phase transition it may be impossible to express the entropy in a Taylor series around the transition temperature and it may show a jump (discontinuous transition) or a discontinuity (continuous transition) in derivatives at that temperature. Normally, we accept analyticity and continuity.
According to the second law, for a given U and a, the equilibrium state is obtained when S is maximized by the microscopic processes associated with the internal variables ξ of the system or dS(U,a) = 0 and d2S(U,a) < 0 (Figure 2.1). If the equality in Eq. (2.3) holds, the process is reversible (dS(i) = 0), otherwise it is irreversible (dS(i) > 0). If for a process dS = 0, it is called isentropic. If dT = 0, we refer to it as isothermal. A natural process (contrary unnatural process) is a process occurring spontaneously in Nature, which proceeds towards equilibrium and in this sense a reversible process can be seen as the limiting case between natural and unnatural processes9).
Figure 2.1 (a) Entropy S as a function of U and V and a quasi-static process from an initial to a final state. (b) The establishment of equilibrium for an isolated system consisting of a cylinder with a frictionless piston moving from initial position i to final position f.
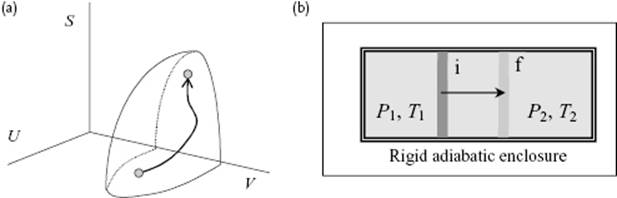
The entropy expressed as S = S(U,a) is called a fundamental equation. Once it is known as a function of its natural variables U and a, all other thermodynamic properties can be calculated. The description S = S(U,a) is called the entropy representation. Since S is a single valued continuously increasing function of U, the equation S = S(U,a) can be inverted to U = U(S,a) without ambiguity. For a given entropy S the equilibrium state is reached when dU(S,a) = 0. From stability considerations it follows that U is minimized by the internal processes of the system or dU(S,a) = 0 and d2U(S,a) > 0. The description U = U(S,a) is the energy representation with natural variables S and aand is also a fundamental equation. In equilibrium we have from10) dU = (∂U/∂S)dS + (∂U/∂a)Tda the (thermodynamic) temperature T = ∂U/∂S and the forces A = ∂U/∂a. From T = ∂U/∂S we easily obtain the relation T−1 = ∂S/∂U. One can show that the temperature T is independent of the properties of the thermometer used, and is consistent with the intuitive concept of (empirical) temperature. The units are kelvins, abbreviated as K and related to the Centigrade or Celsius scale, using °C as unit with the same size but different origin:
(2.4) ![]()
The origin of the absolute scale, 0 K = −273.150 °C, is referred to as absolute zero.
For completeness we mention the third law, stating that for any isothermal process involving only phases in internal equilibrium
(2.5) ![]()
This relation also holds if, instead of being in internal equilibrium, such a phase is in frozen metastable equilibrium, provided that the process does not disturb this frozen equilibrium. According to the third law, absolute zero cannot be reached. The discussion of the third law contains subtleties for which we refer to the literature.
2.1.2 Quasi-Conservative and Dissipative Forces
Only for reversible systems we can associate TdS with δQ and in this case δW = ΣiAidai = ATda is the reversible work. For irreversible systems, TdS > δQ and the work δW contains also an irreversible or dissipative part. To show this, note that the elementary work can be written as
(2.6) ![]()
where dS = dS(r) + dS(i). Since U = U(S,a) we have dU = (∂U/∂S)dS + (∂U/∂a)Tda = TdS + (∂U/∂a)Tda. So we can also write Eq. (2.6) as
(2.7) ![]()
Therefore, the expression TdS(i) has also the form of elementary work and we introduce forces A(d) by writing
(2.8) ![]()
The quantity A(d) is the dissipative force and we refer to ![]() as the dissipative work. Writing for the total force
as the dissipative work. Writing for the total force
(2.9) ![]()
we define the quasi-conservative force A(q) by
(2.10) ![]()
which is the variable conjugated to a. Summarizing, we have
(2.11) 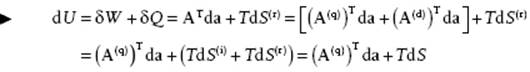
known as the Gibbs equation. The adjective “quasi-” stems from the fact that U is still dependent on the temperature while the adjective “conservative” indicates that U acts as a potential. Generally the adjective “quasi-conservative” is not added; using only the volume V for a, one refers to A = −P(q) as just pressure and labels it as −P. We will do so likewise hereafter. Hence, in the remainder we use Ai = −P, ai = V and write U = U(S,V) so that the pressure becomes P = −∂U/∂V. The Gibbs equation then becomes
(2.12) ![]()
However, note that in a nonequilibrium state and for dissipative processes, P = P(q) is not the total pressure, and that in general there are more independent variables. Since for a reversible process one assumes that the system is at any moment in equilibrium, the rate ![]() must be “infinitely” slow. If this is the case the process is quasi-static. It will be clear that reversible processes are quasi-static, although the reverse is not necessarily true.
must be “infinitely” slow. If this is the case the process is quasi-static. It will be clear that reversible processes are quasi-static, although the reverse is not necessarily true.
2.1.3 Equation of State
Expressed in its natural variables S and V, U(S,V) is a fundamental equation, just like S(U,V). Again, once it is known, all other thermodynamic properties of the system can be calculated. If U is not expressed in its natural variables but, for example, as U = U(T,V), we have slightly less information. In fact it follows from T = ∂U/∂S that U = U(T,V) represents a differential equation for U(S,V). Functional relationships such as
![]()
expressing a certain intensive state variable as a single-valued function of the remaining extensive state variables are called equations of state (EoS) and the intensive variable is thus described by a state function. If all the equations of state are known, the fundamental equation can be inferred to within an arbitrary constant. To that purpose we note that, since U is an extensive quantity, U(S,V) can be taken as homogeneous of degree 1 and thus, using Euler's theorem (see Appendix B), that
(2.13) ![]()
Given T = T(S,V) and P = P(S,V) the energy U can be calculated. A similar argumentation for S leads to S = (1/T)U + (P/T)V. Only in some special cases, for example, if dS(U,V) = f(U)dU + g(V)dV, the differential can be integrated directly to obtain the fundamental equation. Equation (2.13) is often referred to as an Euler equation.
Problem 2.1
The pressure is given by −P = ∂U/∂V = [∂U(S,V)/∂V]S. Show that if one uses the equation of state U = U(T,V), P becomes
![]()
Problem 2.2
A perfect gas is defined by U = cnRT and PV = nRT, where c is a constant, n the number of moles and the other symbols have their usual meaning. Show that the fundamental equation for the entropy S is given by S = S0 + nR ln[(U/U0)c(V/V0)] with U0 and V0 a reference energy and reference volume.
Problem 2.3
Show, using a similar argumentation as for U, that the relation S = (1/T)U + (P/T)V holds.
2.1.4 Equilibrium
Consider an isolated system consisting of two subsystems only capable of exchanging heat (dVi = 0). We now ask ourselves under what conditions thermal equilibrium would occur, and to this purpose we consider arbitrary but small variations in energy in both subsystems. From the first law we have dU = dU1 + dU2 = 0. If the system is in thermal equilibrium, since S is maximal, we must also have dS = dS1 + dS2 = 0. Because
(2.14) ![]()
and since dU1 is arbitrary, we obtain T1 = T2 in agreement with the zeroth law. Moreover, starting from a nonequilibrium state one can show that heat flows from high to low temperature, again in agreement with intuition [3].
The procedure outlined above is in fact the general method to deal with basic equilibrium thermodynamic problems: one considers an isolated system consisting of two subsystems of which one is the object of interest and the other represents the environment. For the total, isolated system we have dU = 0. Moreover, in equilibrium we have dS = 0. Together with the closure relations – that is, the coupling relations between the displacements ai of the system of interest and of the environment – a solution can be obtained.
Let us now consider hydrostatic equilibrium along these lines. Consider therefore again an isolated system consisting of two homogeneous subsystems but in this case capable of exchanging heat and work (Figure 2.1). For this system to be in equilibrium we write again dS = dS1 + dS2 = 0. However, the entropy S is now a function of U and the various values of ai. As we are interested in hydrostatic equilibrium we take just the volume V for each subsystem. From Eq. (2.12) we obtain
(2.15) ![]()
and thus for the equilibrium configuration
(2.16) ![]()
Because the total system is isolated we have the closure relations dU = dU1 + dU2 = 0 and dV = dV1 + dV2 = 0. Therefore, since dU1 and dV1 are arbitrary, we obtain
(2.17) ![]()
regaining thermal equilibrium and
(2.18) ![]()
The latter condition corresponds to hydrostatic equilibrium. Hence, in hydrostatic equilibrium the temperature and the pressure of the subsystems are equal.
2.1.5 Auxiliary Functions
The Gibbs equation for a closed system is given by
(2.19) ![]()
which gives the dependent variable U as a function of the independent, natural variables S and V. From consideration of the second law, the criterion for equilibrium for a closed system with fixed composition is that dS(U,V) = 0 or Sis a maximum at constant U. Equivalently, as stated earlier, dU(S,V) = 0 or U is a minimum at constant S. Both descriptions are complete but not very practical, since it is difficult to keep the entropy constant and keeping the energy constant excludes interference from outside. Therefore, auxiliary functions having more practical natural variables are introduced.
If we write the energy as U(S,V), the enthalpy H is the Legendre transform11) of U with respect to P = −∂U/∂V, which is obtained from
(2.20) ![]()
After differentiation this yields
(2.21) ![]()
Combining with dU = TdS − PdV results in
(2.22) ![]()
The natural variables for the enthalpy H are thus S and P and the equilibrium condition becomes dH(S,P) = 0.
Similarly, writing H(S,P), the Gibbs energy G is the Legendre transform of H with respect to T = ∂H/∂S and given by
(2.23) ![]()
On differentiation and combination with dH = T dS + V dP this yields
(2.24) ![]()
of which the natural variables are T and P. Consequently, the equilibrium condition becomes dG(T,P) = 0. A third transform is the Helmholtz energy F, defined as
(2.25) ![]()
with natural variables T and V and the corresponding equilibrium condition dF(T,V) = 0. It is easily shown that, for example,
(2.26) ![]()
(2.27) ![]()
The functions U(S,V), H(S,P), F(T,V) and G(T,P) thus act as a potential, similar to the potential energy in mechanics, and are called thermodynamic potentials. The derivatives with respect to their natural, independent variables yield the conjugate, dependent variables. These potentials are also fundamental equations. A stable equilibrium state is reached when these potentials are minimized for a given set of their natural variables, that is, when dX = 0 and when d2X > 0, where X is any of the four functions U, H, F, or G. The advantage of using F and G is obvious: While it is possible to control either the set (V,T) or (P,T) experimentally, control of either the set (V,S) or (P,S) is virtually impossible. As an aside we note that the Legendre transform can be also applied to the entropy yielding the so-called Massieu functions. These also act as potentials, for example, X(V,T) = S − U/T or Y(P,T) = S − U/T − PV/T. It will be clear that X is related to F and Y to G. In practice, these functions are but limitedly used, although they were invented before the transforms of the energy.
2.1.6 Some Derivatives and Their Relationships
Apart from the potentials, we also need some of their derivatives, sometimes denotes as response functions, and their mutual relationships. Consider the relations
![]()
The heat capacities are defined by
![]()
It thus follows that
![]()
Three other derivatives that occur frequently are the compressibilities κX (X = T or S) and the (thermal) expansion coefficient or (thermal) expansivity α ≡ αP.
![]()
Moreover, if dϕ = Xdx + Ydy is a total differential, we can use the Maxwell relations (∂X/∂y)x = (∂Y/∂x)y. From the expressions for dU, dH, dF and dG we obtain
![]()
which can be used to reduce any thermodynamic quantity to a set of measurable quantities. Only three of the quantities CV, CP, κT, κS and α are independent because
(2.28) ![]()
Using the third law, it can be shown that at T = 0, CP = CV = 0 and α = 0. Using the relations S = −∂F/∂T and S = −∂G/∂T, we have the Gibbs–Helmholtz equations
(2.29) 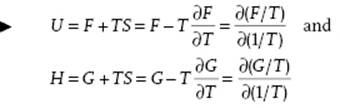
Problem 2.4
Show for a perfect gas (see Problem 2.2) that CV = cNR, CP = (c + 1)NR, κT = 1/P and α = 1/T.
Problem 2.5
Prove Eq. (2.28) and show that CP/CV = κT/κS.
Problem 2.6
Show that, if F = C/T where C is a constant, F = ½U.
Problem 2.7: Blackbody radiation
For blackbody radiation in a volume V the pressure P is given P = u/3 with energy density u = U/V and U(T) the total energy being a function of temperature T only. Using dU = TdS − PdV, show that u = aT4 where the constant a is called the Stefan–Boltzmann constant. Using (∂S/∂V)T = (∂P/∂T)V, show that s = S/V = 4aT3/3 yielding T and P as functions of S. Finally, show that S = CU3/4/4V1/4 with C = 4a1/3/3 a constant.
2.1.7 Chemical Content
The content of a system is defined by the amount of moles nα of a chemical species α in the system which can be varied independently, often denoted as components. A mixture is a system with more than one component. A homogeneous part of a mixture, that is, with uniform properties throughout that part, is addressed as phase while a multicomponent phase is labeled solution. A system consisting of a single phase is called homogeneous, while one consisting of more than one phase is labeled heterogeneous. Moreover, a phase can be isotropic and anisotropic (properties independent or dependent on direction within a phase). The majority component of a solution is the solventwhile solute refers to the minority component. For an arbitrary extensive quantity Z of a mixture, we have the molar quantities ![]() or
or ![]() for a pure component α (sometimes also indicated by Zm(α), zα or even z when no confusion is possible). For mixtures, we also define for any extensive property Z the associated partial (molar) property Zα as the partial derivative with respect to the number of moles nα at constant T and P. For example, the partial volume Vα given by
for a pure component α (sometimes also indicated by Zm(α), zα or even z when no confusion is possible). For mixtures, we also define for any extensive property Z the associated partial (molar) property Zα as the partial derivative with respect to the number of moles nα at constant T and P. For example, the partial volume Vα given by
(2.30) ![]()
and similarly for Uα, Sα and so on. At constant T and P we thus have
(2.31) ![]()
Since Z is homogeneous of the first degree in nα, we have by the Euler theorem
(2.32) ![]()
Hence, we may regard Z as the sum of the contributions Zα for each of the species α.
We note that, besides the number of moles nα, various other quantities are used for amount of substance. We will also use the number of molecules Nα = NAnα with NA as Avogadro's number. Frequently, one is only interested in relative changes in composition, and to that purpose one uses the mole fraction defined by ![]() , using
, using ![]() . In the sequel, the expressions for a binary system are given in parentheses with index 1 referring to the solvent, and index 2 to the solute.
. In the sequel, the expressions for a binary system are given in parentheses with index 1 referring to the solvent, and index 2 to the solute.
Indicating the molar mass of the solvent by M1, one defines the molality mα ≡ nα/n1M1 and since the mole fraction xα = nα/Σαnα = nα/n we have
(2.33) ![]()
For dilute solutions, nα≠1 → 0 and we obtain mα/xα = 1/M1 (m2/x2 = 1/M1). If M1 is expressed in kg mol−1, the unit of molality becomes mol kg−1, often labeled as m, for example, for 0.1 mol kg−1 one writes 0.1 m.
In practice, molarity cα ≡ nα/V with V the total volume of the solution, is also used. Since ρV = ΣαnαMα with ρ the mass density12) of the solution, we have
(2.34) 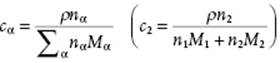
Using again the mole fraction xα, the result
(2.35) 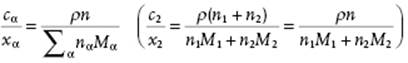
is easily obtained. For dilute solutions, nα≠1 → 0 and we obtain cα/xα = ρ/M1 (c2/x2 = ρ/M1). If cα is expressed in mol l−1, labeled as M, one often speaks of concentration, for example, for cα = 0.1 mol l−1, one writes 0.1 M. Whatever measurement for composition is used, it is expedient to use as other independent variables P and T so that differentiation with respect to T implies constant P, and vice versa. From cα we obtain the relation ∂cα/∂T = −cααP with αP the expansion coefficient, and therefore cα is not independent of temperature. This renders theoretical calculations using cα as independent variable often cumbersome.
For chemical problems, that is, where a change in chemical composition is involved, the fundamental equation becomes U = U(S,V,nα) or S = S(U,V,nα). This leads to
(2.36) ![]()
The partial derivative μα = ∂U/∂nα is called the chemical potential and is the conjugate intensive variable associated with the extensive variable nα in the chemical work dWche = Σαμαdnα. Equilibrium is now obtained when dU(S,V,nα) = 0.
For obtaining a potential in terms of T and V, we use a Legendre transformation of U with respect to T = ∂U/∂S using the Helmholtz energy F = U − TS and obtain
(2.37) ![]()
so that F = F(T,V,nα) and the equilibrium condition reads dF(T,V,nα) = 0.
Applying a Legendre transformation of U with respect to T = ∂U/∂S and P = −∂U/∂V using the Gibbs energy G = U − TS + PV leads to
(2.38) ![]()
The Gibbs energy is thus given by G = G(T,P,nα) and the equilibrium condition becomes dG(T,P,nα) = 0. Because the chemical potential μα of the component α is defined as μα = ∂G/∂nα, it is equal to the partial Gibbs energy Gα. Since G is homogeneous of the first degree in nα, we have by Euler's theorem ![]() . On the one hand, we thus have
. On the one hand, we thus have
![]()
while on the other hand we know that G = G(T,P,nα) and thus that
![]()
Therefore, we obtain by subtraction the so-called Gibbs–Duhem relation
(2.39) ![]()
implying a relation between the various differentials. This relation is here derived for T, P, and nα as the only independent variables, but the extension to any number of variables is obvious. Moreover, although derived here for μα = Gα, it will be clear that a similar relation can be derived for all partial quantities.
Finally, we can also apply a Legendre transformation to U with respect to T = ∂U/∂S and μα = ∂U/∂nα by using Ω = U − TS − Σαμαnα = F − Σαμαnα leading to
![]()
This so-called grand potential Ω is mainly used in statistical thermodynamics (Chapter 5). Since we showed that Σαμαnα = G, we identify the grand potential as
![]()
Problem 2.8
Show that for the perfect gas (see Problem 2.2) the fundamental equation S = S(U,V,n) is given by
![]()
where u and v denote molar quantities and the subscript 0 refers to reference values. First, use the Gibbs–Duhem equation for dμ to show that the chemical potential μ is given by
![]()
Thereafter, use the Euler equation S = (1/T)(U + PV − μN).
2.1.8 Chemical Equilibrium
At constant P and constant T the Gibbs energy in a chemically reacting system varies with composition as dG = Σαμαdnα. For a reaction to occur spontaneously dG = Σαμαdnα < 0 while at equilibrium13) dG = Σαμαdnα = 0. Let us consider the reaction
(2.40) ![]()
where a, … , d denote the stoichiometric coefficients of components A, … , D. It will be convenient to rearrange this expression to
![]()
with a positive value for the coefficient να when α is a product (C, D) and a negative value when α is a reactant (A, B). We define a factor of proportionality dξ(t) in such a way that dnα = ναdξ . Starting at time zero with initially nα(0) moles of each species the changes of the number of moles of each species in the time interval dt are
![]()
where ![]() is the rate of reaction. This leads to
is the rate of reaction. This leads to
![]()
where we introduced the affinity D = −Σα(ναμα). From dG ≤ 0 we conclude that
(2.41) ![]()
as the condition for a reaction to occur. So, D and ![]() must have the same sign or be zero. At equilibrium D = 0. Since να = dnα/dξ, the affinity D is related to the fundamental equations, the most important ones being, using μα = ∂U/∂nα = ∂G/∂nα = −T∂S/∂nα,
must have the same sign or be zero. At equilibrium D = 0. Since να = dnα/dξ, the affinity D is related to the fundamental equations, the most important ones being, using μα = ∂U/∂nα = ∂G/∂nα = −T∂S/∂nα,
(2.42) ![]()
All nα must be positive or zero and the reaction goes to completion if one of the components is exhausted. This implies a lower and upper value for Δξ. Therefore, the factor Δξ is sometimes normalized according to
![]()
where ε is the degree of reaction. Writing ![]() for the chemical rate of work or power mimics the expression for the mechanical power
for the chemical rate of work or power mimics the expression for the mechanical power ![]() with force A and rate of displacement
with force A and rate of displacement ![]() . In both cases the power is positive semi-definite, that is,
. In both cases the power is positive semi-definite, that is, ![]() .
.
Example 2.1
A vessel contains a ½ mole of H2S, ¾ mole of H2O, 2 moles of H2, and 1 mole of SO2 and is kept at constant T and P. The equilibrium condition is
![]()
![]()
If the chemical potentials are known as a function of T, P and the nα's, the solution for dξ can be obtained. Suppose that the solution is dξ = ¼. If dξ = ⅔, ![]() and therefore this is dξmax. If dξ = −⅜,
and therefore this is dξmax. If dξ = −⅜, ![]() and therefore this is dξmin. Therefore, the degree of reaction ε = [¼ − (−⅜)]/[⅔ − (−⅜)] = 3/5.
and therefore this is dξmin. Therefore, the degree of reaction ε = [¼ − (−⅜)]/[⅔ − (−⅜)] = 3/5.
The above formulation leads immediately to the conventional chemical description. Let us introduce for component α the absolute activity λα = exp(μα/RT) and define, considering again the reaction given by Eq. (2.40), the reaction product
(2.43) ![]()
Using the more compact notation introduced before we write more briefly ![]() . The equilibrium condition D = −Σα(ναμα) = 0 can then be written as
. The equilibrium condition D = −Σα(ναμα) = 0 can then be written as ![]() .
.
For solid–gas reactions we distinguish between gases (α) and solids (β). This allows us to write ![]() , where the first product contains all the terms relating to gaseous species and the second to the solid species. Now note that the chemical potential μα of a gaseous component α is given by
, where the first product contains all the terms relating to gaseous species and the second to the solid species. Now note that the chemical potential μα of a gaseous component α is given by ![]() , where
, where ![]() is the chemical potential in the standard state, R is the gas constant, yα = fx,αxαP is the activity (for gases often denoted as fugacity), fx,α is the (mole fraction) activity coefficient, P is the total pressure, and P° is the standard pressure (1 bar). Hence for a gas
is the chemical potential in the standard state, R is the gas constant, yα = fx,αxαP is the activity (for gases often denoted as fugacity), fx,α is the (mole fraction) activity coefficient, P is the total pressure, and P° is the standard pressure (1 bar). Hence for a gas ![]() , where
, where ![]() is the value of λα for P = P°. For gases at low pressures (hence activity coefficient fx,α = 1) the activity becomes the partial pressure Pα given by Pα = xαP. For solids, on the other hand, we have
is the value of λα for P = P°. For gases at low pressures (hence activity coefficient fx,α = 1) the activity becomes the partial pressure Pα given by Pα = xαP. For solids, on the other hand, we have ![]() , only weakly dependent on the pressure. In total we have
, only weakly dependent on the pressure. In total we have
![]()
![]()
The (pressure) equilibrium constant KP is related to the standard Gibbs energy
(2.44) ![]()
via Δμ = −RTln KP. Since Δμ is constant at constant temperature, the value of KP is constant at constant temperature, which explains the name. If activities are used throughout, we refer to the activity equilibrium constant Ky.
For reactions in solution we start again with ![]() and use
and use ![]() with fm,α the (molality) activity coefficient for molality mα and
with fm,α the (molality) activity coefficient for molality mα and ![]() the value for
the value for ![]() . This leads to
. This leads to
(2.45) ![]()
where Km is the (molality) equilibrium constant. For ideal solutions or ideally dilute solutions mα → 0, fm,α → 1 and thus in that case ![]() . Similarly using
. Similarly using ![]() with fc,α the (molarity) activity coefficient for component α for molarity cα and
with fc,α the (molarity) activity coefficient for component α for molarity cα and ![]() the value for
the value for ![]() leads to
leads to
(2.46) ![]()
where Kc is the (molarity) equilibrium constant. If cα → 0, fc,α → 1.
Problem 2.9
Derive the chemical equilibrium condition as done along the lines of hydrostatic and thermal equilibrium.