Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
13. Mixing Liquids: Polymeric Solutions
In discussing the behavior of molecular solutions, although we kept track of the differences in the size of the molecules, the entropy was (nearly) always assumed to behave ideally. This is, however, no longer the case for polymeric solutions, in which the difference in size between the components becomes appreciable. In this chapter we discuss the extension leading to the famous Flory–Huggins theory.1) We first discuss entropy and then deal with energy modifications; thereafter, the essence of some alternative theories is indicated. A brief review of polymer structural characteristics precedes all of this.
13.1. Polymer Configurations
Polymers consist of long molecular chains of covalently bonded atoms. Typically, the molecule is constructed from a set of repeating units, the monomers. To a good approximation the energy of a single molecule can be estimated by the adding bond energies (Table 13.1).
Table 13.1 Bond energy Ubon and bond length d for various bonds.a)
|
Bond |
Ubon (eV) |
d (Å) |
|
C–H |
4.3 |
1.08 |
|
C–C |
3.6 |
1.54 |
|
C=C |
6.3 |
1.35 |
|
C≡C |
8.7 |
1.21 |
|
C–O |
3.6 |
1.43 |
|
C=O |
7.6 |
1.22 |
|
C–F |
5.0 |
1.36 |
|
C–Cl |
2.8 |
1.76 |
|
C–Si |
3.1 |
1.93 |
|
Si–H |
3.0 |
1.45 |
|
Si–Si |
1.8 |
2.34 |
|
Si–F |
5.6 |
1.81 |
|
Si–Cl |
3.7 |
2.16 |
|
Si–O |
3.8 |
1.83 |
a) Data from Ref. [36].
1 eV = 96.48 kJ mol−1.
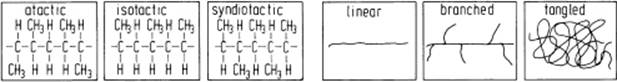
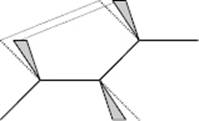
The chemical structure of the chains is complicated somewhat by isomerism, a phenomenon whereby one overall composition can have different geometric structures. A simple example of chemical isomerism is provided by the vinyl polymers for which one may have either head-to-head (–CH2–CHX–CHX–CH2–) or head-to-tail (–CH2–CHX–CH2–CHX–) addition. A somewhat more complex case involves steric isomerism. Consider again the case of vinyl polymers in which a side group is added to every alternate carbon atom. If the groups are all added in an identical way, we obtain an isotactic polymer (Figure 13.1). However, if there is an inversion for each monomer unit, we obtain a syndiotactic polymer. Finally, an irregular addition sequence leads to an atactic polymer. Of course, a chain can be branched and, generally, the chains are entangled.
Figure 13.1 Tacticity and chain structure of polymers.
Each sample of polymer will consist of molecular chains of varying length and, consequently, of varying molecular mass. The molecular mass distribution is important for many properties. One can distinguish between the number average Mn and weight average Mw, defined by
(13.1) 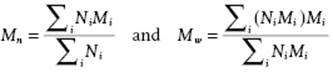
respectively, where Ni is the number of molecules with molecular mass Mi and the summation is over all molecular masses. Since the degree of polymerization (DP) involved is generally very high, the difference between a discrete and a continuous distribution is usually negligible, and we can write
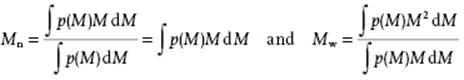
where the second step for Mn can be made if p(M) is assumed to be normalized. Since the second central moment or variance of the distribution function is given by
![]()
we have
![]()
and the polydispersity index (PDI) Mw/Mn describes the width of the distribution. The shape of p(M) can vary widely, dependent on the polymerization process.
Mixtures of various types of polymers are possible. A blend is a mixture of two or more polymers. In a graft, a chain of a second polymer is attached to the base polymer. If in the main chain a chemical combination exists between two monomers [A] and [B], then the material is a copolymer. In the latter case, we can distinguish between: (i) a block copolymer, where the monomer A is followed first by a sequence of other monomers A such as AAA and subsequently by a series of B monomers; and (ii) random copolymers, where a monomer A is followed randomly by either monomer A or B. This results in the absence of long sequences of A and B monomers.
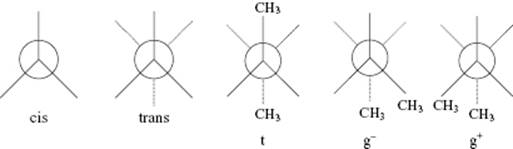
We now turn to the molecular conformations, the most important of which are gauche and trans conformations. Consider first the central bond between two C atoms as in ethane, C2H6. Figure 13.2 shows the two extremes in conformation, namely cis and trans, in a view along the C–C bond axis. In ethane, three equivalent minimum energy or trans conformations are present. To rotate the two CH3 groups with respect to each other, energy must be spent and an energy barrier exists between the two trans states. Substituting on each C atom one H atom by a CH3 group, to produce butane, C4H10, the equivalence between the trans states is lost and we obtain one trans (t) conformation and two equivalent (g+, g−) gauche conformations with dihedral angles ϕ = 0 ° and ϕ = +120 ° and ϕ = –120 °, respectively, for the minimum energy conformations (Figure 13.2). Continuing with the substitution of end H atoms with CH3 groups results in polyethylene (PE). Although the details for each C–C bond for this molecule may slightly differ, one trans and two gauche conformations are present for each C–C bond. They must all be specified for a complete description of the molecule. For PE, the lowest energy conformation is the all-trans conformation with a zig-zag structure of the C–C bonds.
Figure 13.2 The cis and trans conformations in ethane and the t, g+, and g− conformations in butane, as shown by the Newman projection.
This is no longer true for other polymers, where the H atoms have been replaced by other atoms or groups. Consider for example polytetrafluorethylene (PTFE), where all H atoms have been replaced by F atoms. As the F atoms are larger than the H atoms, the nonbonded repulsive interactions between CF2 groups of the second-nearest carbon atoms become much more important (Figure 13.3), and repulsive energy can be decreased by rotating slightly along the C–C axis of each bond. Of course, this increases the bond rotation energy, and by balancing the two contributions an equilibrium is reached. In the case of PTFE, an optimum dihedral angle of ϕ ≅ 16.5 ° is obtained. The result of all this is that the molecule forms a helix along its axis in which the positions of the side groups (the F atoms in the case of the PTFE) rotate along the molecular axis. After n screws along the axis the position of the mth monomer regains the position of the first monomer, apart from a shift along the axis. When described in this way, we refer to these as m/n helices. For example, PE has a 2/1 helix, while PTFE has a 13/6 helix below 19 °C and a 15/7 helix above 19 °C. This description for the (semi-)crystalline state is not as exact as it appears however, as the “periodicity” along the chain may vary slightly [1].
Figure 13.3 Nonbonded interactions between CX2 groups of the second-nearest C atoms.
Focusing on the chains themselves, a first estimate of the end-to-end distance2) X is made via the freely jointed chain model: n bonds, each of length l, connected without any restriction. The probability distribution of the end-to-end vectors for long chain molecules is described by the random walk model resulting in
(13.2) ![]()
For such a model chain one obtains, in the limit of a large number of atoms,
(13.3) ![]()
where ⟨r2⟩ = ⟨x2⟩ + ⟨y2⟩ + ⟨z2⟩ is the mean square end-to-end distance of the chains. The end-to-end distance X = ⟨r2⟩1/2 is thus proportional to n1/2.
However, we know that the bonds are not freely connected but have a certain bond angle τ. Leaving the bonds otherwise unrestricted, we obtain the freely rotating chain model for which it holds in the limit of a large number of bonds that
(13.4) ![]()
As expected, the square-root dependence on n is preserved but the proportionality factor is changed. In the case of sp3-hybridized carbon atoms, for example, in a PE chain, with a bond angle of τ = 109.5 °, we have approximately ⟨r2⟩ = 2.0nl2.
A further improvement is obtained by using the independent hindered rotation model, that is, a rotating chain but with preferential orientation for the dihedral (bond rotation) angle ϕ between two groups connected by a bond. In this model one has
(13.5) ![]()
Again, the square-root dependence on n is preserved and the proportionality factor changes. For the PE chain we have one trans (t) configuration with a dihedral angle ϕ = 0 ° and two equivalent gauche (g+, g−) configurations with a dihedral angle ϕ = 120 ° and ϕ = –120 °, respectively (Figure 13.2). The latter have a higher energy by an amount Egau. Denoting the Boltzmann factor by σ = exp(–Egau/RT), we obtain for the average dihedral angle
(13.6) ![]()
For the end-to-end distance we thus have
(13.7) ![]()
For PE at 140 °C, using Egau = 2.1 kJ mol−1, we find σ = 0.54 leading to ⟨r2⟩ ≅ 3.4nl2.
Finally, we recognize that the hindered rotation around a bond is correlated, and this is taken into account in the correlated hindered rotation model. The final expression becomes
(13.8) ![]()
where the characteristic ratio C is a function of the correlation of the rotations along the chain and is therefore a measure of the stiffness of the chain. For PE, Flory calculated, taking into account the correlation up to two bonds away, that C = 6.7 ± 0.2, in good agreement with experiment; for other polymers, different values of C are obtained (see Table 13.2). Actually the solvent used should be indicated if non-theta conditions are present (see Section 13.2).
Table 13.2 Values for characteristic ratio C for various polymers.
|
Material |
C |
|
PEO |
4.0/4.1 |
|
PE |
6.7/6.8 |
|
a-PS |
10.0 |
|
i-PS |
10.7 |
|
a-PP |
5.5 |
|
i-PP |
5.8 |
|
s-PP |
5.9 |
|
a-PMMA |
8.4 |
|
i-PMMA |
10 |
|
s-PMMA |
7 |
|
PVC |
13 |
|
a-PVAc |
8.9/9.4 |
|
PDMS |
6.2 |
|
a-PiB |
6.6 |
|
PC |
2.4 |
PE, polyethylene; PEO, polyoxyethylene; PS, polystyrene; PP, polypropylene; PMMA, poly(methyl methacrylate); PVC, poly(vinyl chloride); PVAc, poly(vinyl acetate); PDMS, poly(dimethylsiloxane); PiB, poly(isobutylene); PC, poly(carbonate); a, atactic; i, isotactic; s, syndiotactic.
The above-described considerations led to the introduction of the equivalent chain (Figure 13.4), in which a real chain, containing n correlated and rotation-hindered bonds of length l, is described as a freely jointed chain of nKsegments of length b. Each of the segments thus represents a number of real bonds; however, as the correlation along the chain is limited to a few bonds, these segments can be considered as freely jointed. For this description we use the end-to-end distance X and the contour length L, which is the length of the fully extended real chain with all valence angles and bond lengths at their equilibrium value. In fact, we match X = ⟨r2⟩ = Cnl2 with nKb2 and L with nKb. This can be done in a unique way, leading to b = ⟨r2⟩/L and nK = L2/⟨r2⟩. Let us take again PE as an example. For the PE chain with a bond angle τ = 109 °, L = nl sin(τ/2) ≅ 0.83 nl, while ⟨r2⟩ = 6.7nl2. This leads to b ≅ 8l and nK ≅ 0.1n. The segment thus contains about 10 (real) bonds, and its length b is often addressed as the Kuhn length. For other polymers, of course, various Kuhn lengths are obtained, with higher values reflecting a greater stiffness of the molecular chain. In discussing the properties of polymers, frequent use is made of the equivalent chain model since, for this model many mathematical results are known. In the polymer literature, mention is often made of Kuhn monomers or even monomers when addressing the entities that we have labeled here as segments. In order to avoid confusion, we systematically use the term “monomer” for a repetitive chemical unit, and the term “segment” when speaking about a unit for the equivalent chain. The results obtained for the equivalent chain are then translated to real chain via the characteristic ratio C.
Figure 13.4 The equivalent chain for polyethylene with segments containing approximately one-tenth of the bonds of the real polyethylene chain, and each segment having a length approximately eightfold that of a C–C bond length. This results in the same end-to-end distance X and contour length L as the real polyethylene chain.
Justification 13.1
A simple justification for the distribution function P(n,r) runs as follows. We consider a freely jointed (ideal) chain of n bonds or segments of length l. If we add to the chain an extra segment with vector l (components li) we have P(n + 1,r) = P(n,r + l). Assuming that l ≡ |l| << r ≡ |r|, we may expand P(n + 1,r) in a Taylor series in l obtaining
![]()
Averaging over all possible orientations of l, taking into account that ⟨l⟩ = 0, the result is
![]()
where ⟨lilj⟩ = ⅓l2δij has been used. We further write ⟨P(n,r)⟩ = P(n,r) for the average over all orientations of r. Using ⟨P(n + 1,r)⟩ = P(n + 1,r) we also have for n >> 1
![]()
and equating P(n + 1,r) with ⟨P(n,r + l)⟩ we have approximately
![]()
The relevant solution depends only on r, since the undisturbed molecule has a spherical shape and reads P ∼ n−3/2exp(–3r2/2nl2). Using the normalization condition ∫P(n,r)4πr2 dr = 1, we have for the complete solution
![]()
Note that for r > nl, P(n,r) ≠ 0, though this condition should be obeyed for a realistic solution. However, for n >> 1, P(n,r) ≅ 0 when r > nl.
The possibility of having trans or gauche states at each bond renders polymer molecules relatively flexible, so that for polymers in good solvents (and in polymer melts as well as in glassy amorphous polymers) the chains exhibit a random configuration and the polymer molecule appears as a coil. The configuration of a polymer molecule, whether in solution or in the melt, can be (partially) characterized by the end-to-end distance3) X. For the freely jointed (and therefore the equivalent) chain the probability analysis shows that the molecules have a Gaussian end-to-end distribution, also indicative of the fact that polymer molecules normally are coiled, and one often speaks of a Gaussian chain. In a good solvent, polymer–solvent attractions prevail, the coil expands, and X increases. In a poor solvent, polymer–polymer attraction prevails, irrespective of whether they are due to parts from the same or from a different chain. The coil shrinks and X decreases until the effective monomer–monomer (segment–segment) repulsion due to excluded volume forces sets in. Under certain conditions the intramolecular interactions are similar in magnitude to the intermolecular interactions. In other words, the enthalpy and entropy contributions from solvent–monomer (solvent–segment) and monomer–monomer (segment–segment) interactions to the Helmholtz energy of the assembly of molecules under consideration compensate each other, and one part of the molecule seems not to “notice” other parts of the same molecule, nor other molecules. The molecules behave like “phantoms,” and are indeed sometimes referred to as phantom chains. The temperature at which this occurs is termed the Flory temperature θ, and one speaks of theta conditions, under which the coil neither shrinks nor expands, and has unperturbed dimensions. The influence of the solvent can be described by
(13.9) ![]()
where the subscript denotes the theta conditions and α a parameter dependent on solvent, temperature, and molecular mass. At the Flory temperature T = θ, theta conditions hold and α = 1. We will discuss the behavior of α somewhat further in the next paragraph. In addition to the main topic, we note here that for solids the Flory theorem is important: in a dense polymeric system theta conditions prevail. Describing theta conditions as the configuration where intramolecular and intermolecular interactions compensate, and since the “solvent” is the polymer melt itself, the theorem is highly plausible. Rephrasing, on the one hand, the monomers of a certain reference chain is subject to a repulsive potential due to the excluded volume effect of its own monomers and this leads to an expansion of the coil. On the other hand, the other chains, which interpenetrate the reference chain, generate a counteracting attractive potential acting inwards on the reference chain such that, under theta conditions, the two effects cancel leading to (pseudo)-unperturbed chains. The results of small-angle neutron-scattering experiments have supported this theorem.
Finally, we note it is not difficult to calculate the Helmholtz energy F for a Gaussian chain of n bonds with length l. We use Boltzmann's estimate for the entropy S = klnW, where W is the number of configurations (to be precise, with the same energy). The value of W can be estimated from P(n,r), realizing that P(n,r) = W(n,r)/∫W(n,r)dr. By using Eq. (13.2) we find
(13.10) ![]()
Since we assume that all conformations have the energy, F = U − TS becomes F = −TS = 3kTr2/2nl2, so that for the force to extend this chain we obtain f = ∂F/∂r = 3kTr/nl2.
Problem 13.1
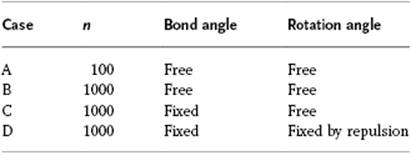
Consider a polymer with the main chain carbon atoms in sp2 conformation (bond length l = 0.1 nm) and with a strong preference for a rotation angle of 45 ° deviating from the zig-zag conformation. Calculate the contour length L and end-to-end distance X for the conditions, as given in the accompanying table. Compare the results and explain the differences qualitatively.
Problem 13.2
Suppose that two adjacent bonds have a correlated orientation due to the (fixed) bond angle τ, but that no correlation exists between bonds with at least one bond in between. Calculate the end-to-end distance for a polymer with 1000 bonds in the main chain and a bond length of 0.1 nm.