Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
14. Some Special Topics: Reactions in Solutions
14.2. Transition State Theory
Transition state theory (TST), also known as “activated complex theory”, is an important follow-up from statistical mechanics, used throughout in chemistry and physics, which has been developed mainly by Eyring and coworkers. In this section we will illustrate this theory for chemical reactions in the gas phase.
14.2.1 The Equilibrium Constant
The first result we need is the equilibrium condition. Associating (as in Chapter 5) the macroscopic internal energy U with the statistical mechanics expression for the average energy ⟨E⟩, the macroscopic number of molecules N with the average number of molecules ⟨N⟩ and recalling that the grand canonical partition function reads kT ln Ξ = PV, we conclude from the expression for the grand canonical entropy
(14.12) ![]()
that ∂S/∂N = –μ/T. The equilibrium condition at constant U and V for a system of several components thus becomes dS = ∑j (∂Sj/∂Nj) dNj = –∑j (μj/T) dNj = 0, where the index j denotes the various reactants and products. If we have a reaction
![]()
where R and P denote reactants and products, respectively, we obtain
![]()
Using the relation μj = kT ln(Nj/zj), obtained from μj = ∂F/∂Nj with zj denoting the partition function of component j, the result is
(14.13) 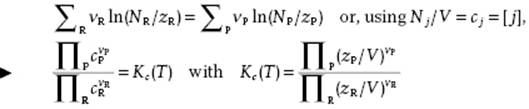
where Kc(T) is the equilibrium constant. This relationship is known as the law of mass action. The reference level of energy for each factor in Eq. (14.13) is the same. However, for the evaluation of the various partition functions it is more convenient to use the ground state of each species as a reference. Let us take the gas-phase chemical reaction AB + C ↔ A + BC as a simple example with K(T) = zAzBC/zABzC. Shifting the reference level of all species to an arbitrary level and denoting the partition function with respect to the ground state for each species by z′, we obtain
(14.14) 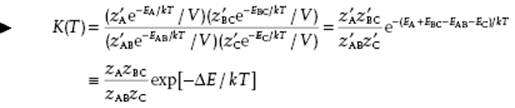
where in the last step the primes have been removed (using, from now on, as reference level for each of the species the ground state) and ΔE ≡ (EA + EBC − EAB − EC) represents the difference in ground-state energies of the reactants and products.
14.2.2 Potential Energy Surfaces
The second concept we need is the potential energy surface. We note that the potential energy of a system containing atoms or molecules can be written as a function of special combinations of the nuclear spatial coordinates, usually referred as generalized or normal coordinates. Generally, the pictorial representation of the potential energy hypersurface is difficult. The concept can be grasped from a simple example, for which we take the collinear reaction between three atoms A, B, and C. In Figure 14.1 a map is shown for this reaction with, as axes, the distances AB and BC, respectively. The map shows two valleys separated by a col, and in general, thermal fluctuation creates continuous attempts to pass from one valley to another. In order to calculate the rate constant for the chemical reaction A + BC ↔ AB + C, we must calculate all trajectories on the potential energy surface. With this in mind we need the concept of a dividing surface, defined as a surface which cannot be passed without passing a barrier. In Figure 14.1, for example, one of the dividing surfaces is given by RAB = RBC. When calculating the rate constant, we must take into account only those trajectories that do pass the dividing surface. An upper limit to the reaction rate r(T) is then given by
(14.15) ![]()
where j(T) represents the total amount of reactant systems crossing a dividing surface per unit volume and per unit time at a temperature T – that is, the flux. Equation (14.15) is known as the Wigner variational theorem. All statistical methods for computing reaction rates are based on this principle. The calculation of all allowed trajectories to estimate the flux j(T) provides an enormous task if performed in a rigorous fashion, and therefore approximate methods are normally introduced. The variation theorem provides the opportunity to make an optimum choice for the dividing surface – that is, to select the surface that provides the lowest flux j(T). This may be achieved by using a surface depending on several parameters and choosing these parameters in such a way that a minimum j(T) is obtained.
Figure 14.1 The potential energy surface for the reaction A + BC ↔ AB + C, showing a col between two valleys.
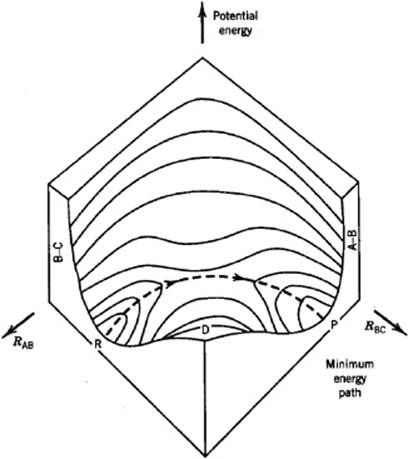
14.2.3 The Activated Complex
The simplest approach along the above lines is to take into account only the trajectory following the minimum energy path from one valley to the other valley. In the case of the chemical reaction A + BC ↔ AB + C, this means the path from the configuration represented by A + BC (point R in Figure 14.1) to the configuration represented by AB + C (point P in Figure 14.1). The coordinate along this path is called the reaction coordinate. The top of the col between the two valleys, which actually is a saddle point of the potential energy surface, is known as the transition state. A configuration in the neighborhood of the transition state is addressed as an activated complex. Transition state theory is based on a number of assumptions. The first assumption is that the optimum dividing surface passes through the transition state and is perpendicular to the reaction coordinate. The second assumption is that activated complexes are at all times in equilibrium with both the reactants and the products. For the forward reaction A + BC ↔ (ABC)‡, where (ABC)‡ denotes the activated complex, this implies that the equilibrium constant Kfor is given by
(14.16) ![]()
where, as usual, [X] denotes the concentration of species X. The equilibrium constant Krev for the reverse reaction AB + C ↔ (ABC)‡ is
(14.17) ![]()
Since in Eq. (14.16) and (14.17) [(ABC)‡]for and [(ABC)‡]rev are equal, we have [(ABC)‡]for = [(ABC)‡]rev = ½[(ABC)‡]. The combination of these two conditions together is called the quasi-equilibrium assumption. Finally, it is assumed that once the transition state is reached the reaction completes – that is, the reactants do not return to the nonreacted state.
Here, we consider the forward reaction in some detail. Equation (14.14) shows that the equilibrium constant for the reaction A + BC ↔ (ABC)‡ can be written as
(14.18) ![]()
We also assume that the partition function of each species X can be (approximately) factorized as
![]()
where zele, ztra, zvib, and zrot represent the electronic, translation, vibration, and rotation partition functions, respectively. The contribution of zele reduces to the degeneracy number of the ground state, usually 1, since we assume the reaction to proceed on a potential energy surface, which exists only by virtue of the adiabatic assumption. Therefore, no electronic transitions are allowed.
If we have sufficient information on the transition state – that is, we know the structure and relevant force constants – we can construct its partition function. The translation and rotation partition function do not provide a problem in principle, as they can be constructed as for normal molecules. However, the vibration partition function requires some care. The usual normal coordinate analysis of an N-atom molecule can be made, and from that we obtain 3N – 6 normal coordinates for a nonlinear molecule or 3N – 5 normal coordinates for a linear molecule. Of these vibration coordinates, all but one has a positive coefficient in the second-order terms in the potential energy expansion. The last, negative coefficient corresponds to an imaginary frequency for this vibration coordinate, and represents the reaction coordinate. This implies that, in the transition state, a small fluctuation in the reaction coordinate will lead to an unstable configuration with respect to this coordinate. It is customary [4]3) to treat this coordinate as a translation coordinate over a small length δ at the top of the potential energy barrier with a partition function z = (δ /h)(2πm‡kT)1/2. The complete partition function z(ABC)‡ for the activated complex is then
(14.19) ![]()
where z‡ represents the partition function for the remaining coordinates – that is, the true vibration coordinates, the translation and rotation coordinates. Further, m‡ denotes the mass of the activated complex and k, T and h have their usual meanings.
The concentration of the activated complexes due to the forward reaction is given by Eq. (14.16), where the forward equilibrium constant is given by Eq. (14.18). Since there are [(ABC)‡]for complexes per unit volume which populate the length δ and which are moving forward, the forward reaction rate rfor is rfor = [(ABC)‡]for/τ, where τ is the average time to traverse the length δ, given by τ = δ /vave. Thus, we need the average velocity vave in one direction over the length δ. Using the same approximation of a free translatory motion again, we borrow from kinetic gas theory [5]
(14.20) ![]()
Combining Eq. (14.16), (14.18), (14.19) and (14.20) with rfor = [(ABC)‡]vave/δ, we obtain
(14.21) ![]()
A similar expression is obtained for the reverse reaction, and it is easily verified that the forward and reverse reactions are in equilibrium. The forward rate constant kfor can thus be calculated given the relevant information. However, for a first-principles calculation many pieces of information are required: the energy barrier for the reaction; the structure; and the force constants associated with the dynamics of the reactants, products, and activated complex. Typically, this information is incompletely available. This is also true for other mechanisms, although of course for mechanisms other than gas reactions different arguments apply. The exponential dependence on the barrier energy is generally valid, however, and rationalizes the generally observed Arrhenius-type behavior.
It remains to be discussed how to connect the results to experimental data. Experimentally, it is often observed that, if the logarithm of the rate constant is plotted against the reciprocal temperature, a straight line is obtained. The gradient of this line is used to define the (empirical) activation energy Eact by
(14.22) ![]()
Substituting Eq. (14.21), taking logarithms, and differentiating with respect to T yields
![]()
Using Eq. (14.22) and the van't Hoff relation ![]() , where ΔU‡ is the change in energy, we obtain
, where ΔU‡ is the change in energy, we obtain
![]()
This equation thus provides a link between the experimentally observed Eact and ΔU‡. It should be noted that neither Eact nor ΔU‡ is identical to ΔE, which appears in Eq. (14.18) because of the temperature dependence of the various partition functions. For the various partition functions, the temperature dependencies read
![]()
For the vibrations, however, the behavior is less simple. For kT << ħω, Zvib ≈ T0, while for kT >> ħω, Zvib ≈ T, where n is the number of vibrational modes of the molecule. At intermediate temperature, Zvib ≈ Ta with 0 ≤ a ≤ n. For a restricted (intermediate) temperature range a is approximately constant. In total, this yields
![]()
explaining the modified Arrhenius equation. As noted above, typically |m| < 2.
Finally, we note that formalism can also be interpreted thermodynamically, and to this purpose we write Eq. (14.21) in terms of molar quantities
(14.23) ![]()
(14.24) ![]()
with ΔS‡, ΔH‡, ΔU‡ and ΔV‡ the molar entropy, enthalpy, energy, and volume of activation, respectively. In a gas reaction, the term PΔV‡ may be put equal to RTΣjνj, where Σjνj is the stoichiometric sum for activated complex formation. For a unimolecular reaction the stoichiometric sum Σjνj = 0 and ΔH‡ = ΔU‡ = Eact − RT, or Eact = ΔH‡ + RT. Consequently, A = e(kT/h)exp(ΔS‡/R). For a bimolecular reaction Σjνj = −1 and ΔH‡ = ΔU‡ − RT = Eact − 2RT, or Eact = ΔH‡ + 2RT. Consequently, A = e2(kT/h)exp(ΔS‡/R). In solution, the volume term is nearly always negligible, and therefore ΔH‡ ≅ ΔU‡. We see that the pre-exponential factor in the empirical expression krea = A exp(−Eact/RT) corresponds approximately to the entropy.
Here, our brief overview on gas-phase TST ends, and in the next sections we apply TST to reactions in liquids. It must be re-emphasized that, for a detailed calculation, a considerable amount of information is required.
Problem 14.1: The dissociation of I2
Consider the reaction I2 ↔ 2I at 300 K. Note that the ground state for the I atom is 2P2/3 (hence fourfold degenerate), while that for the I2 molecule is ![]() (hence nondegenerate). For I2 the rotational temperature θrot = 0.054 K, the vibrational temperature θvib = 308 K, the dissociation energy from the ground state is D = 1.5417 eV, and the molecular mass mI = 127 g mol−1.
(hence nondegenerate). For I2 the rotational temperature θrot = 0.054 K, the vibrational temperature θvib = 308 K, the dissociation energy from the ground state is D = 1.5417 eV, and the molecular mass mI = 127 g mol−1.
a) Show that the translational and electronic partition function for the I atom are ztra = Λ−3V and zele = 4 exp[−½(D/kT + θvib/2T)], respectively.
b) Show that the translational, rotational, vibrational and electronic partition function for the I2 molecule are ztra = 2−3/2Λ−3V, zrot = T/2θrot, zvib = [exp(θvib/2T) − exp(−θvib/2T)]−1, and zele = 1, respectively.
c) Show that Kc = 32(πmkT/h2)3/2(θrot/T)[1 − exp(−θvib/T)]exp(−D/kT), and that KP = kTKc.
d) Calculate the numerical value for KP at 300 K.