MCAT General Chemistry Review - Steven A. Leduc 2015
Thermodynamics
6.1 SYSTEM AND SURROUNDINGS
Why does anything happen? Why does a creek flow downhill, a volcano erupt, a chemical reaction proceed? It’s all thermodynamics: the transformation of energy from one form to another. The laws of thermodynamics underlie any event in which energy is transformed.
The Zeroth Law of Thermodynamics
The Zeroth Law is often conceptually described as follows: If two systems are both in thermal equilibrium with a third system, then the two initial systems are in thermal equilibrium with one another.
Thus, the Zeroth Law establishes a definition of thermal equilibrium. When systems are in thermal equilibrium with one another, their temperatures must be the same. When bodies of different temperatures are brought into contact with one another, heat will flow from the body with the higher temperature into the body with lower temperature in order to achieve equilibrium at the same temperature value. This means that devices (thermometers) may be designed to achieve thermal equilibrium with their surroundings, and give a quantified, relative value of the temperature at this equilibrium.
In this way, the Zeroth Law defines what we call temperature, and is the logical basis for the subsequent thermodynamic laws that rely on it. It also establishes the link between heat and temperature. An important practical application of the Zeroth Law is calorimetry, which will be discussed in more detail in Chapter 7.
The First Law of Thermodynamics
The First Law states that the total energy of the universe is constant. Energy may be transformed from one form to another, but it cannot be created or destroyed.
An important result of the First Law is that an isolated system has a constant energy—no transformation of the energy is possible. When systems are in contact, however, energy is allowed to flow, and thermal equilibrium can be attained. In addition, the First Law also establishes that work can be put into a system to increase its overall energy. This may or may not occur with a corresponding change in temperature. The concept of work and its effects on physical thermodynamics can be examined more closely in the MCAT Physics and Math Review.
Conventions Used in Thermodynamics
In thermodynamics we have to designate a “starting line” and a “finish line” to be able to describe how energy flows in chemical reactions and physical changes. To do this we use three distinct designations to describe energy flow: the system, the surroundings, and the thermodynamic universe (or just universe).
The system is the thing we’re looking at: a melting ice cube, a solid dissolving into water, a beating heart, anything we want to study. Everything else: the table the ice cube sits on and the surrounding air, the beaker that holds the solid and the water, the chest cavity holding the heart, is known collectively as the surroundings. The system and the surroundings taken together form the thermodynamic universe.
We need to define these terms so that we can assign a direction—and therefore a sign, either (+) or (—)—to energy flow. For chemistry (and for physics), we define everything in terms of what’s happening to the system.
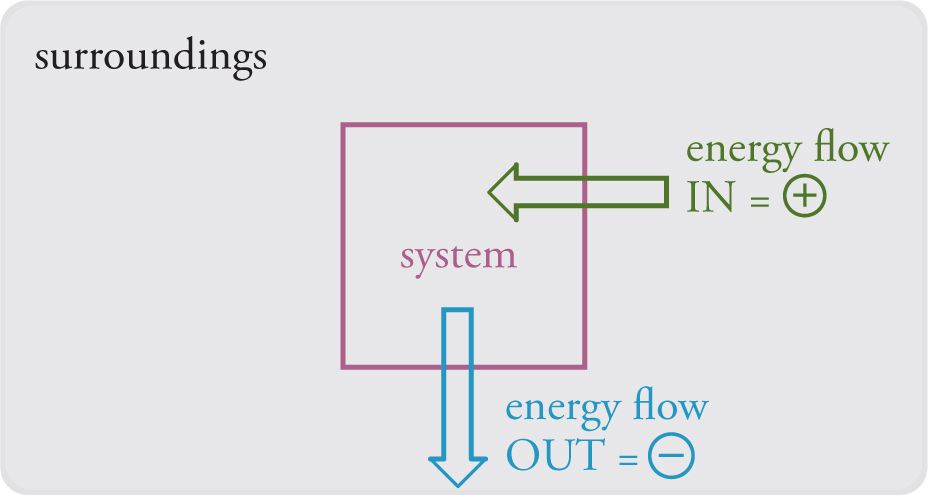
Consider energy flowing from the surroundings into the system, like the heat flowing from the table to the ice cube that’s sitting on it. What is happening in the system? As energy flows in (here it’s heat), the molecules in the system absorb it and start to jiggle faster. Eventually enough energy is absorbed to cause the ice to melt. Overall, the energy of the system increased, and we therefore give it a (+) sign. What about water when it freezes? Here energy (once again, heat) leaves the water (our system), and the water molecules’ jiggling slows down. The energy of the system has decreased, and we therefore assign a (—) sign to energy flow. Finally, energy that flows into the system flows out of the surroundings, and energy that flows out of the system flows into the surroundings. Therefore, we can make these statements:
1) When energy flows into a system from the surroundings, the energy of the system increases and the energy of the surroundings decreases.
2) When energy flows out of a system into the surroundings, the energy of the system decreases and the energy of the surroundings increases.
Keep this duality in mind when dealing with energy.
6.2 ENTHALPY
Enthalpy is a measure of the heat energy that is released or absorbed when bonds are broken and formed during a reaction that’s run at constant pressure. The symbol for enthalpy is H. Some general principles about enthalpy prevail over all reactions:
• When a bond is formed, energy is released. ∆H < 0.
• Energy must be put into a bond in order to break it. ∆H > 0.
In a chemical reaction, energy must be put into the reactants to break their bonds. Once the reactant bonds are broken, the atoms rearrange to form products. As the product bonds form, energy is released. The enthalpy of a reaction is given by the difference between the enthalpy of the products and the enthalpy of the reactants.
∆H = Hproducts — Hreactants
The enthalpy change, ∆H, is also known as the heat of reaction.
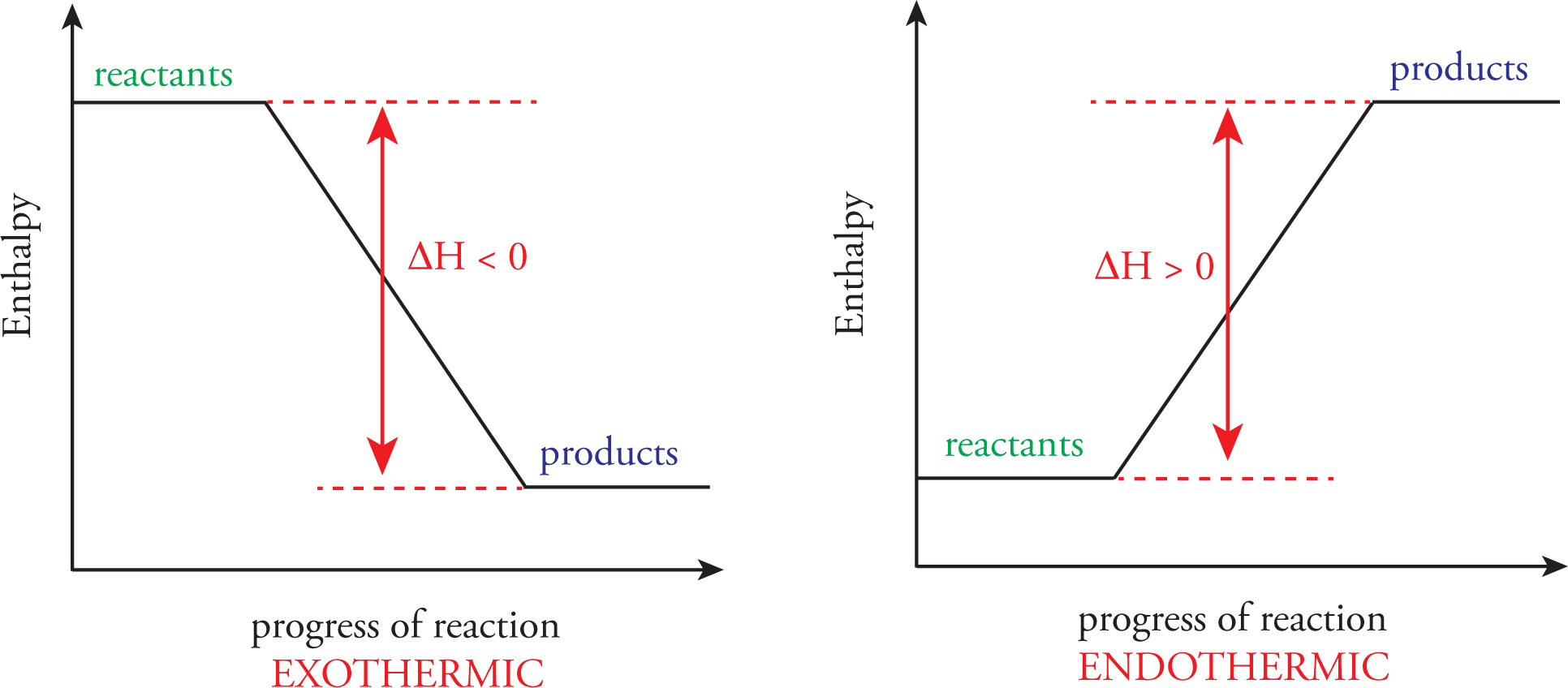
If the products of a chemical reaction have stronger bonds than the reactants, then more energy is released in the making of product bonds than was put in to break the reactant bonds. In this case, energy is released overall from the system, and the reaction is exothermic. The products are in a lower energy state than the reactants, and the change in enthalpy, ∆H, is negative, since heat flows out of the system. If the products of a chemical reaction have weaker bonds than the reactants, then more energy is put in during the breaking of reactant bonds than is released in the making of product bonds. In this case, energy is absorbed overall and the reaction is endothermic. The products are in a higher energy state than the reactants, and the change in enthalpy, ∆H, is positive, since heat had to be added to the system from the surroundings.
Example 6-1: The combustion of methanol is given by this reaction:
2 CH3OH(g) + 3 O2(g) → 2 CO2(g) + 4 H2O(g), ∆H = —1352 kJ
a) How much heat is produced when 16 g of oxygen gas reacts with excess methanol?
b) Is the reaction exothermic or endothermic?
c) How many moles of carbon dioxide are produced when 676 kJ of heat is produced?
Solution:
a) The molecular weight of O2 is 2(16) = 32 g/mol, so 16 g represents one-half mole. If 1352 kJ of heat is released when 3 moles of O2 react, then just (1/6)(1352 kJ) = 225 kJ of heat will be released when one-half mole of O2 reacts.
b) Because ∆H is negative, the reaction is exothermic. And since 6 moles of gaseous products are being formed from just 5 moles of gaseous reactants, the disorder (entropy) has increased.
c) The stoichiometry of the given balanced reaction tells us that 2 moles of CO2 are produced when 1352 kJ of heat is produced. So, half as much CO2 (that is, 1 mole) is produced when half as much heat, 676 kJ, is produced.
Example 6-2: Which one of the following processes does NOT contribute to the change in enthalpy, ∆Hrxn, of a chemical reaction?
A) Phase change
B) Formation of stronger intermolecular forces
C) Breaking covalent bonds
D) The presence of a heterogeneous catalyst
Solution: A catalyst lowers the activation energy, but does not affect an equilibrium constant, enthalpy, entropy, or free energy in any way. Choice D is the answer. The other choices do fall under the umbrella of enthalpy.
6.3 Calculation of ∆Hrxn
The heat of reaction (∆Hrxn) can be calculated in a number of ways. Each of these will lead to the same answer given accurate starting values. The three most important methods to be familiar with are the use of heats of formation (∆Hf), Hess’s law of heat summation, and the summation of individual bond enthalpies.
Standard Conditions
Essentially every process is affected by temperature and pressure, so scientists have a convention called standard conditions for which most constants, heats of formation, enthalpies, and so on are determined. Under standard conditions, the temperature is 298 K (25°C) and the pressure is 1 atm. All solids and liquids are assumed to be pure, and solutions are considered to be at a concentration of 1 M. Values that have been determined under standard conditions are designated by a ° superscript: ∆H°, for example. Be careful not to confuse standard conditions with standard temperature and pressure (STP). STP is 0°C, while standard conditions means 25°C.
Heat of Formation
The standard heat of formation, ∆H°f , is the amount of energy required to make one mole of a compound from its constituent elements in their natural or standard state, which is the way the element exists under standard conditions. The convention is to assign elements in their standard state forms a ∆H°f of zero. For example, the ∆H°f of C(s) (as graphite) is zero. Diatomic elements, such as O2, H2, Cl2 and so on are also defined as zero, rather than their atomic forms (such as O, Cl, etc.), because the diatomic state is the natural state for these elements at standard conditions. For example, ∆H°f = 0 for O2, but for O, ∆H°f = 249 kJ/mol at standard conditions, because it takes energy to break the O=O double bond.
When the ∆H°f of a compound is positive, then an input of heat is required to make that compound from its constituent elements. When ∆H°f is negative, making the compound from its elements gives off energy.
You can calculate the ∆H° of a reaction if you know the heats of formation of the reactants and products:
∆H°rxn = (Σn × ∆H°f, products) — (Σn × ∆H°f, reactants)
In the above equation “n” denotes the stoichiometric coefficient applied to each species in a chemical reaction as written. ∆H°f of a given compound is the heat needed to form one mole, and as such if two moles of a molecule are needed to balance a reaction one must double the corresponding ∆H°f in the enthalpy equation. If only half a mole is required one must divide the ∆H°f by 2.
Example 6-3: Which of the following substances does NOT have a heat of formation equal to zero at standard conditions?
A) F2(g)
B) Cl2(g)
C) Br2(g)
D) I2(s)
Solution: Heat of formation, ∆H°f , is zero for a pure element in its natural phase at standard conditions. All of the choices are in their standard state, except for bromine, which is a liquid, not a gas, at standard conditions. The correct answer is C.
Example 6-4: What is ∆H° for the following reaction under standard conditions if the ∆H°f of CH4(g) = —75 kJ/mol, ∆H°f of CO2(g) = —393 kJ/mol, and ∆H°f of H2O(l) = —286 kJ/mol?
CH4(g) + 2 O2(g) → CO2(g) + 2 H2O(l)
Solution: Using the equation for ∆H°rxn, we find that
∆H°rxn = (∆H°f CO2 + 2 ∆H°f H2O) — (∆H°f CH4 + 2 ∆H°f O2)
= (—393 kJ/mol + 2(—286) kJ/mol) — (—75 kJ/mol + 0 kJ/mol)
= —890 kJ/mol
Hess’s Law of Heat Summation
Hess’s law states that if a reaction occurs in several steps, then the sum of the energies absorbed or given off in all the steps will be the same as that for the overall reaction. This is due to the fact that enthalpy is a state function, which means that changes are independent of the pathway of the reaction. Therefore, ∆H is independent of the pathway of the reaction.
For example, we can consider the combustion of carbon to form carbon monoxide to proceed by a two-step process:
1) |
C(s) + O2(g) → CO2(g) |
∆H1 = —394 kJ |
2) |
CO2(g) → CO(g) + 1/2 O2(g) |
∆H2 = +283 kJ |
To get the overall reaction, we add the two steps:
C(s) + 1/2 O2(g) → CO(g)
So, to find ∆H for the overall reaction, we just add the enthalpies of each of the steps:
∆Hrxn = ∆H1 + ∆H2 = —394 kJ + 283 kJ = —111 kJ
It’s important to remember the following two rules when using Hess’s law:
1) If a reaction is reversed, the sign of ∆H is reversed too.
For example, for the reaction CO2(g) → C(s) + O2(g), we’d have ∆H = +394 kJ.
2) If an equation is multiplied by a coefficient, then ∆H must be multiplied by that same value.
For example, for 1/2 C(s) + 1/2 O2(g) → 1/2 CO2(g), we’d have ∆H = —197 kJ.
Summation of Average Bond Enthalpies
Enthalpy itself can be viewed as the energy stored in the chemical bonds of a compound. Bonds have characteristic enthalpies that denote how much energy is required to break them homolytically (often called the bond dissociation energy, or BDE; see Section 5.2).
As indicated at the start of this section, an important distinction should be made here in the difference in sign of ∆H for making a bond versus breaking a bond. One must, necessarily, infuse energy into a system to break a chemical bond. As such the ∆H for this process is positive; it is endothermic. On the other hand, creating a bond between two atoms must have a negative value of ∆H. It therefore gives off heat and is exothermic. If this weren’t the case it would indicate that the bonded atoms were higher in energy than they were when unbound; such a bond would be unstable and immediately dissociate.
Therefore we have a very important relation that can help you on the MCAT:
Energy is needed to break a bond.
Energy is released in making a bond.
From this we come to the third method of determining ∆Hrxn. If a question provides a list of bond enthalpies, ∆Hrxn can be determined through the following equation:
∆Hrxn = Σ (BDE bonds broken) — Σ (BDE bonds formed)
One can see that if stronger bonds are being formed than those being broken, then ∆Hrxn will be negative. More energy is released than supplied and the reaction is exothermic. If the opposite is true and breaking strong bonds takes more energy than is regained through the making of weaker product bonds, then the reaction is endothermic.
Example 6-5: Given the table of average bond dissociation energies below, calculate ∆Hrxn for the combustion of methane given in Example 6-4.
Bond |
Average Bond Dissociation Energy (kJ/mol) |
C—H |
413 |
O—H |
467 |
C=O |
799 |
C=N |
615 |
H—Cl |
427 |
O=O |
495 |
A) 824 kJ/mol
B) 110 kJ/mol
C) —824 kJ/mol
D) —110 kJ/mol
Solution: First determine how many of each type of bond are broken in the reactants and formed in the products based on the stoichiometry of the balanced equation. Then using the bond dissociation energies we can calculate the enthalpy change:
∆Hrxn = ∑ (BDE bonds broken) — ∑ (BDE bonds formed)
∆Hrxn = (4(C—H) + 2(O=O)) — (2(C=O) + 4(O—H))
= (4(413) + 2(495)) — (2(799) + 4(467))
= —824 kJ/mol
The correct answer is C. You may notice that the two methods of calculating the reaction enthalpy for the same reaction did not produce exactly the same answer. This is due to the fact that bond energies are reported as the average of many examples of that type of bond, whereas heats of formation are determined for each individual chemical compound. The exact energy of a bond will be dependent not only on the two atoms bonded together but also the chemical environment in which they reside. The average bond energy gives an approximation of the strength of an individual bond, and as such, the summation of bond energies give an approximation of ∆Hrxn.
6.4 ENTROPY
The Second Law of Thermodynamics
There are several different ways to state the second law of thermodynamics, each appropriate to the particular system under study, but they’re all equivalent. One way to state this law is that the disorder of the universe increases in a spontaneous process. For this to make sense, let’s examine what we mean by the term spontaneous. For example, water will spontaneously splash and flow down a waterfall, but it will not spontaneously collect itself at the bottom and flow up the cliff. A bouncing ball will come to rest, but a ball at rest will not suddenly start bouncing. If the ball is warm enough, it’s got the energy to start moving, but heat—the disorganized, random kinetic energy of the constituent atoms—will not spontaneously organize itself and give the ball an overall kinetic energy to start it moving. From another perspective, heat will spontaneously flow from a plate of hot food to its cooler surroundings, but thermal energy in the cool surroundings will not spontaneously concentrate itself and flow into the food. None of these processes would violate the first law, but they do violate the second law.
Nature has a tendency to become increasingly disorganized, and another way to state the second law is that all processes tend to run in a direction that leads to maximum disorder. Think about spilling milk from a glass. Does the milk ever collect itself together and refill the glass? No, it spreads out randomly over the table and floor. In fact, it needed the glass in the first place just to have any shape at all. Likewise, think about the helium in a balloon: It expands to fill its container, and if we empty the balloon, the helium diffuses randomly throughout the room. The reverse doesn’t happen. Helium atoms don’t collect themselves from the atmosphere and move into a closed container. The natural tendency of all things is to increase their disorder.
We measure disorder or randomness as entropy. The greater the disorder of a system, the greater is its entropy. Entropy is represented by the symbol S, and the change in entropy during a reaction is represented by the symbol ∆S. The change in entropy is determined by the equation
∆S = Sproducts — Sreactants
If randomness increases—or order decreases—during the reaction, then ∆S is positive for the reaction. If randomness decreases—or order increases—then ∆S is negative. For example, let’s look at the decomposition reaction for carbonic acid:
H2CO3 ![]() H2O + CO2
H2O + CO2
In this case, one molecule breaks into two molecules, and disorder is increased. That is, the atoms are less organized in the water and carbon dioxide molecules than they are in the carbonic acid molecule. The entropy is increasing for the forward reaction. Let’s look at the reverse process: If CO2 and H2O come together to form H2CO3, we’ve decreased entropy because the atoms in two molecules have become more organized by forming one molecule.
In general, entropy is predictable in many cases:
• Liquids have more entropy than solids.
• Gases have more entropy than solids or liquids.
• Particles in solution have more entropy than undissolved solids.
• Two moles of a substance have more entropy than one mole.
• The value of ∆S for a reverse reaction has the same magnitude as that of the forward reaction, but with opposite sign: ∆Sreverse = —∆Sforward.
While the overall drive of nature is to increase entropy, reactions can occur in which entropy decreases, but we must either put in energy or gain energy from making more stable bonds. (We’ll explore this further when we discuss Gibbs free energy.)
Example 6-6: Which of the following processes would have a negative ∆S?
A) The evaporation of a liquid.
B) The freezing of a liquid.
C) The melting of a solid.
D) The sublimation of a solid.
Solution: Only the change described in choice B involves a decrease in randomness—the molecules of a solid are more ordered and organized than those in a liquid. So this process would have a negative change in entropy.
Example 6-7: Of the following reactions, which would have the greatest positive entropy change?
A) 2 NO(g) + O2(g) → 2 NO2(g)
B) 2 HCl(aq) + Mg(s) → MgCl2(aq) + H2(g)
C) 2 H2O (g) + Br2(g) + SO2(g) → 2 HBr (g) + H2SO4(aq)
D) 2 I− (aq) + Cl2(g) → I2(s) + 2 Cl− (aq)
Solution: The reactions in choices A, C, and D all describe processes involving a decrease in randomness, that is, an increase in order. However, the process in choice B has a highly ordered solid on the left, but a highly disordered gas on the right, so we’d expect this reaction to have a positive entropy change.
Example 6-8: For the endothermic reaction
2 CO2(g) → 2 CO(g) + O2(g)
which of the following is true?
A) ∆H is positive, and ∆S is positive.
B) ∆H is positive, and ∆S is negative.
C) ∆H is negative, and ∆S is positive.
D) ∆H is negative, and ∆S is negative.
Solution: Since we’re told that the reaction is endothermic, we know that ∆H is positive. This eliminates choices C and D. Now, what about ∆S? Has the disorder increased or decreased? On the reactant side, we have one type of gas molecule, while on the right we have two. The reaction increases the numbers of gas molecules, so this describes an increase in disorder. ∆S is positive, and the answer is A.
Example 6-9: A gas is observed to undergo condensation. Which of the following is true about the process?
A) ∆H is positive, and ∆S is positive.
B) ∆H is positive, and ∆S is negative.
C) ∆H is negative, and ∆S is positive.
D) ∆H is negative, and ∆S is negative.
Solution: Condensation is the phase change from gas to liquid, which releases heat (since the reverse process, vaporization, requires an input of heat). Therefore, ∆H is negative, and choices A and B are eliminated. Now, because the change from gas to liquid represents an increase in order—since gases are so highly disordered—this process will have a negative change in entropy. The answer is therefore D.
The Third Law of Thermodynamics
The Third Law defines absolute zero to be a state of zero-entropy. At absolute zero, thermal energy is absent and only the least energetic thermodynamic state is available to the system in question. If only one state is possible, then there is no randomness to the system and S = 0. In this way, the Third Law describes the least thermally energetic state, and therefore the lowest achievable temperature. Kelvin defined the temperature at this state as 0 on his temperature scale.
6.5 GIBBS FREE ENERGY
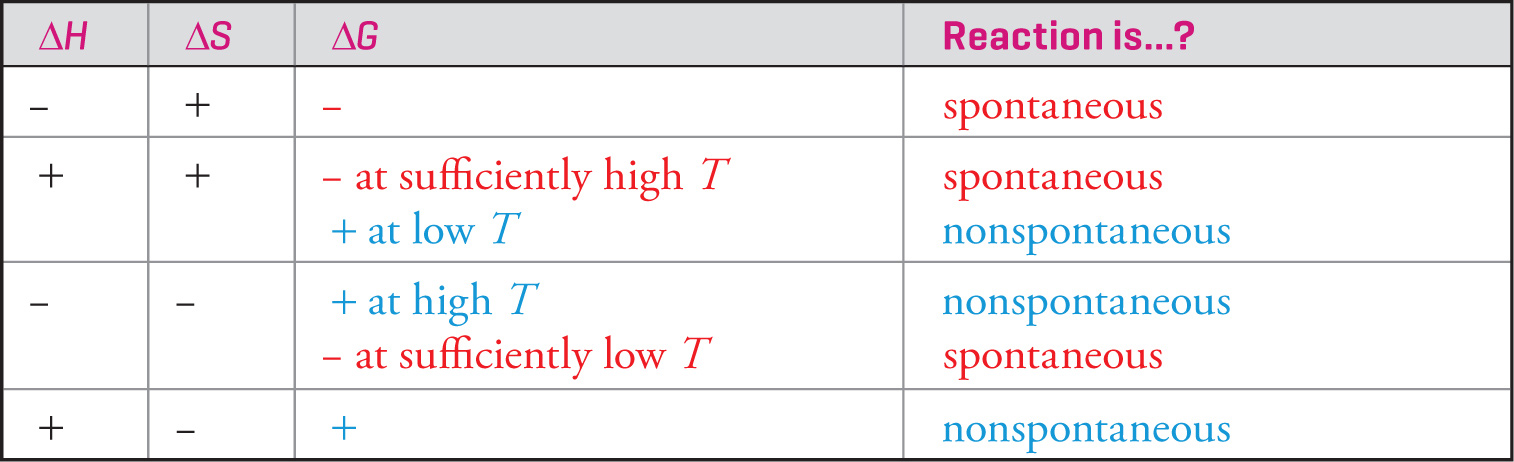
The magnitude of the change in Gibbs free energy, ∆G, is the energy that’s available (free) to do useful work from a chemical reaction. The spontaneity of a reaction is determined by changes in enthalpy and in entropy, and G includes both of these quantities. Now we have a way to determine whether a given reaction will be spontaneous. In some cases—namely, when ∆H and ∆S have different signs—it’s easy. For example, if ∆H is negative and ∆S is positive, then the reaction will certainly be spontaneous (because the products have less energy and more disorder than the reactants; there are two tendencies for a spontaneous reaction: to decrease enthalpy and/or to increase entropy). If ∆H is positive and ∆S is negative, then the reaction will certainly be nonspontaneous (because the products would have more energy and less disorder than the reactants).
But what happens when ∆H and ∆S have the same sign? Which factor—enthalpy or entropy—will dominate and determine the spontaneity of the reaction? The sign of the single quantity ∆G will dictate whether or not a process is spontaneous, and we calculate ∆G from this equation:
Change in Gibbs Free Energy
∆G = ∆H — T∆S
where T is the absolute temperature (in kelvins). And now, we can then say this:
• ∆G < 0 → spontaneous in the forward direction
• ∆G = 0 → reaction is at equilibrium
• ∆G > 0 → nonspontaneous in the forward direction
If ∆G for a reaction is positive, then the value of ∆G for the reverse reaction has the same magnitude but is negative. Therefore, the reverse reaction is spontaneous.
∆G and Temperature
The equation for ∆G shows us that the entropy (T∆S) term depends directly on temperature. At low temperatures, the entropy doesn’t have much influence on the free energy, and ∆H is the dominant factor in determining spontaneity. But as the temperature increases, the entropy term becomes more significant relative to ∆H and can dominate the value for ∆G. In general, the universe tends towards increasing disorder (positive ∆S) and stable bonds (negative ∆H), and a favorable combination of these will make a process spontaneous. The following chart summarizes the combinations of ∆H and ∆S that determine ∆G and spontaneity.
Important note: While values of ∆H are usually reported in terms of kJ, values of ∆S are usually given in terms of J. When using the equation ∆G = ∆H − T∆S, make sure that your ∆H and ∆S are expressed both in kJ or both in J.
Example 6-10: What must be true about a spontaneous, endothermic reaction?
A) ∆H is negative.
B) ∆G is positive.
C) ∆S is positive.
D) ∆S is negative.
Solution: Since the reaction is spontaneous, we know that ∆G is negative, and since we know the reaction is endothermic, we also know that ∆H is positive. The equation ∆G = ∆H − T∆S then tells us that ∆S must be positive, choice C.
Example 6-11: If it’s discovered that a certain nonspontaneous reaction becomes spontaneous if the temperature is lowered, then which of the following must be true?
A) ∆S is negative and ∆H is positive.
B) ∆S is negative and ∆H is negative.
C) ∆S is positive and ∆H is positive.
D) ∆S is positive and ∆H is negative.
Solution: Here’s one (rather mathematical) approach. The nonspontaneous reaction has ∆G1 = ∆H −T1∆S, and the spontaneous reaction has ∆G2 = ∆H − T2∆S. Subtracting these equations, we find that ∆G1 — ∆G2 = (T2 − T1)∆S. Since ∆G1 > 0 and ∆G2 < 0, we know that the left-hand side is positive, so the right-hand side must be positive also. Because T2 < T1, the term (T2 — T1) is negative. This implies that ∆S must be negative, and we eliminate choices C and D. Now let’s figure out the sign of ∆H. From the equation ∆G2 =∆H − T2∆S, we get ∆H = ∆G2 + T2∆S. Because ∆G2 and T2∆S are both negative, so is ∆H, and the answer is B.
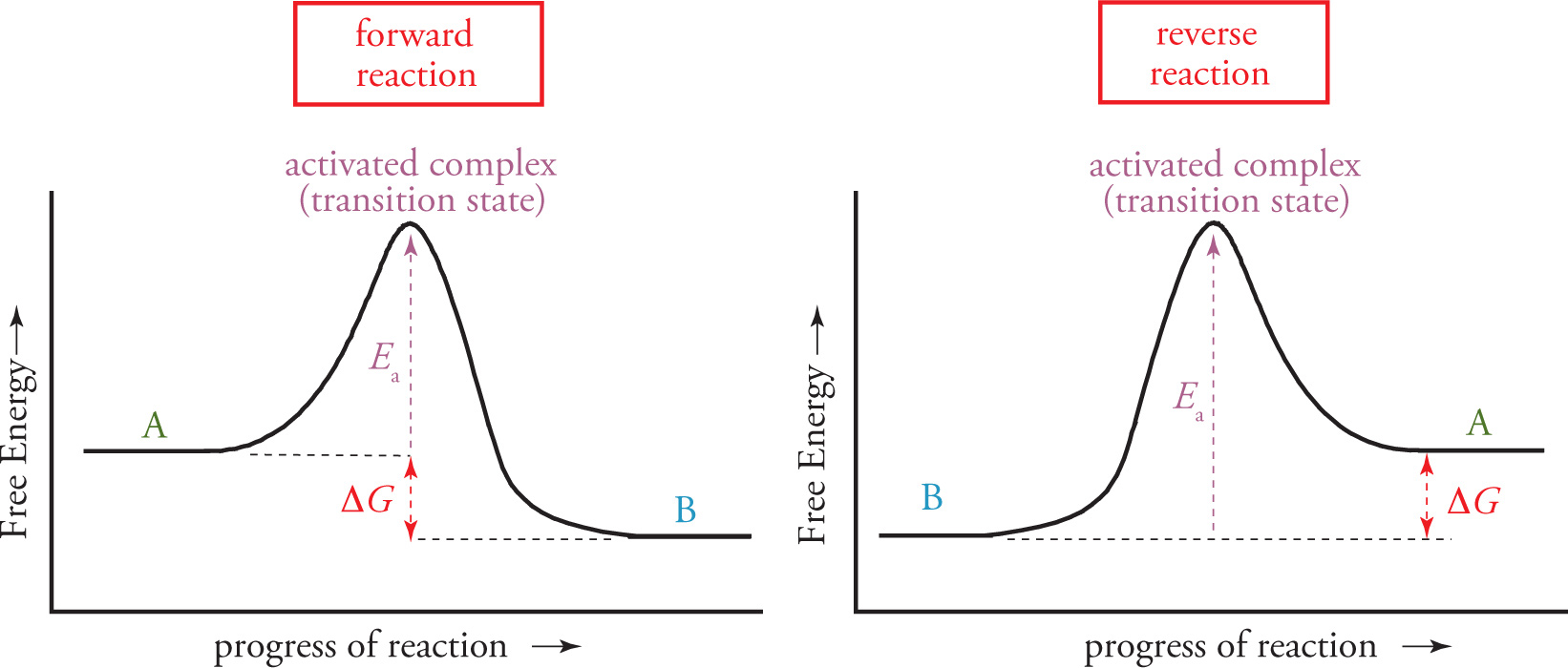
6.6 REACTION ENERGY DIAGRAMS
A chemical reaction can be graphed as it progresses in a reaction energy diagram. True to its name, a reaction energy diagram plots the free energy of the total reactions versus the conversion of reactants to products.
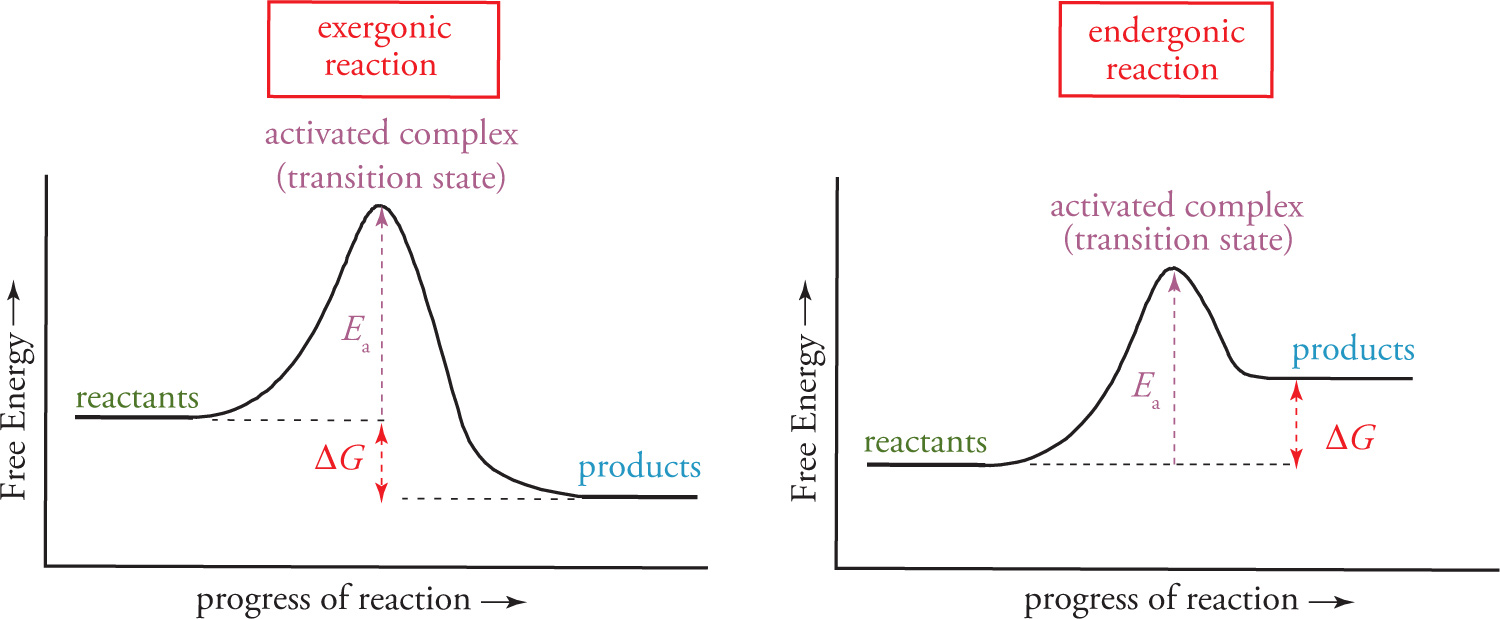
The ∆G of the overall reaction is the difference between the energy of the products and the energy of the reactants: ∆Grxn = Gproducts − Greactants. When the value of T∆S is very small, then ∆G can approximate ∆H, with the difference between the energy of products and reactants being very close to the heat of reaction, ∆H.
The activation energy, Ea, is the extra energy the reactants required to overcome the activation barrier, and determines the kinetics of the reaction. The higher the barrier, the slower the reaction proceeds towards equilibrium; the lower the barrier, the faster the reaction proceeds towards equilibrium. However, Ea does not determine the equilibrium, and an eternally slow reaction (very big Ea) can have a very favorable (large) Keq. Many more details of both kinetics and equilibrium will be discussed in Chapters 9 and 10, respectively.
Kinetics vs. Thermodynamics
Just because a reaction is thermodynamically favorable (i.e., spontaneous), does not automatically mean that it will be taking place rapidly. Do not confuse kinetics with thermodynamics (this is something the MCAT will try to get you to do many times!). They are separate realms. Thermodynamics predicts the spontaneity (and the equilibrium) of reactions, not their rates. If you had a starting line and a finish line, thermodynamics tells you how far you will go, while kinetics tells you how quickly you will get there. A classic example to illustrate this is the formation of graphite from diamond. Graphite and diamond are two of the several different forms (allotropes) of carbon, and the value of ∆Gο for the reaction C(diamond) → C(graphite) is about —2900 J/mol. Because ∆Gο is negative, this reaction is spontaneous under standard conditions. But it’s extremely slow. Even diamond heirlooms passed down through many generations are still in diamond form.
Reversibility
Reactions follow the principle of microscopic reversibility: The reverse reaction has the same magnitude for all thermodynamic values (∆G, ∆H, and ∆S) but of the opposite sign, and the same reaction pathway, but in reverse. This means that the reaction energy diagram for the reverse reaction can be drawn by simply using the mirror image of the forward reaction. The incongruity you should notice is that Ea is different for the forward and reverse reactions. Coming from the products side towards the reactants, the energy barrier will be the difference between Gproducts and the energy of the activated complex.
Chapter 6 Summary
• The Zeroth Law of Thermodynamics states that energy will flow from a body at a higher temperature to a body at a lower temperature until both bodies have the same temperature.
• Energy flow into a system has a positive sign. Energy flow out of a system has a negative sign.
• The First Law of Thermodynamics states that energy cannot be created or destroyed.
• The internal energy of an object is proportional to its temperature.
• The Second Law of Thermodynamics states that all processes tend toward maximum disorder, or entropy (S).
• Enthalpy (H) is a measure of the energy stored in the bonds of molecules.
• Breaking bonds requires energy (+∆H), while forming bonds releases energy (−∆H).
• Standard conditions are 1 atm and 298 K, 1 M concentrations. Standard conditions are denoted by a superscript “°”.
• An endothermic reaction has a ∆H > 0. An exothermic reaction has a ∆H < 0.
• ∆Hreaction = Hproducts − Hreactants. This equation can also be applied to ∆G and ∆S.
• ∆G, the Gibbs free energy, is the amount of energy in a reaction available to do chemical work.
• For a reaction under any set of conditions, ∆G = ∆H −T∆S.
• If ∆G < 0, the reaction is spontaneous in the forward direction. If ∆G > 0, the reaction is nonspontaneous in the forward direction. If ∆G = 0, the reaction is at equilibrium.
CHAPTER 6 FREESTANDING PRACTICE QUESTIONS
1. During the electrolysis of liquid water into hydrogen and oxygen gas at standard temperature and pressure, energy is:
A) absorbed during the breaking of H—H bonds and the reaction is spontaneous.
B) released during the formation of H—H bonds and the reaction is nonspontaneous.
C) absorbed during the formation of O=O bonds and the reaction is spontaneous.
D) released during the breaking of O—H bonds and the reaction is nonspontaneous.
2. What could make the following nonspontaneous endothermic reaction spontaneous?
2 H2O(l) → 2 H2(g) + O2(g)
A) Decreasing volume
B) Increasing temperature
C) Decreasing temperature
D) The reaction will always be nonspontaneous.
3. Which of the following should have the highest enthalpy of vaporization?
A) N2
B) Br2
C) Hg
D) Al
4. A 36 gram sample of water requires 93.4 kJ to sublime. What are the heats of fusion (DHfus) and vaporization (DHvap) for water?
A) ∆Hfus = —20 kJ/mol, DHvap = 66.7 kJ/mol
B) ∆Hfus = 40.7 kJ/mol, DHvap = 6.0 kJ/mol
C) ∆Hfus = 6.0 kJ/mol, DHvap = 40.7 kJ/mol
D) ∆Hfus = 12.0 kJ/mol, DHvap = 81.4 kJ/mol
5. Given the standard enthalpies of formation (∆Hf˚) at 298 K for the compounds below, all of the following reactions are exothermic EXCEPT:
Compound |
∆Hf° (kJ/mol) |
C2H5OH(l) |
—238.86 |
CH3CHO(l) |
—77.80 |
CH3COOH(l) |
—484.50 |
H2O(l) |
—285.83 |
A) CH3COOH(l) → CH3CHO(l) + ½ O2(g)
B) C2H5OH(l) + ½ O2(g) → CH3CHO(l) + H2O(l)
C) CH3CHO(l) + ½ O2(g) → CH3COOH(l)
D) C2H5OH(l) + O2(g) → CH3COOH(l) + H2O(l)
6. The citric acid cycle consists of reactions that break down acetate into carbon dioxide. Given that some steps are thermodynamically unfavorable, why does the cycle proceed in the forward direction overall?
A) The rate constant for the unfavorable reactions is very large.
B) The cycle contains exergonic reactions that drive the endergonic reactions forward.
C) The endothermically unfavorable reactions also have a negative entropy change.
D) The activation energies of the unfavorable reactions are lowered by catalysts.
CHAPTER 6 PRACTICE PASSAGE
The extent to which a salt dissolves in water can be quantified by its solubility product constant, (Ksp) which is defined, for a hypothetical salt XaYb, as shown in Equation 2. The greater the value of Ksp, the more soluble the compound. The Ksp of a salt is related to the free energy of dissolution by the equation ∆Godiss = —RT ln(Ksp). Table 1 lists the Ksp values for some insoluble salts.
XaYb(s) ![]() a Xb+(aq) + b Ya—(aq)
a Xb+(aq) + b Ya—(aq)
Equation 1
Ksp = [Xb+]a [Ya—]b
Equation 2
Salt |
Ksp |
PbCl2 |
1.2 × 10−5 |
MgCO3 |
6.8 × 10−6 |
BaSO4 |
1.1 × 10−10 |
AgCl |
5.4 × 10−13 |
Table 1 Ksp values for select insoluble salts
When a solid completely dissolves, solute particles are separated and encapsulated by solvent molecules. This process requires several steps: 1) breaking all solute-solute interactions, 2) disrupting some solvent-solvent interactions, and 3) forming new solute-solvent interactions. The combination of these processes determines the overall enthalpy change for the dissolution, which can be either exothermic or endothermic regardless of the solubility of the salt. Table 2 shows the enthalpies of dissolution for several soluble salts.
Salt |
∆Hdiss (kJ/mol) |
LiCl |
—37.03 |
KCH3CO2 |
—15.33 |
NaCl |
3.87 |
NH4NO3 |
25.69 |
KClO4 |
41.38 |
Table 2 Dissolution enthalpies for some soluble salts
As solids are low entropy materials, their dissolution entails an increase in entropy. The size of ∆Sdiss is dependent on the organization of solvent molecules in the solvation sphere of the dissolved ions.
1. Which of the following species is isoelectronic with the silver ion in AgCl?
A) Rh+
B) Pd
C) Cd2+
D) In−
2. Given that the dissolution of sodium chloride is endothermic and spontaneous below the saturation concentration, which of the following statements must be true?
A) Forming solute-solvent interactions requires energy, while breaking solute-solute and solvent-solvent interactions releases energy.
B) The increase in entropy must outweigh the endothermic process to create a negative Gibbs free energy.
C) Sodium chloride is only soluble at high temperatures.
D) All ionically-bound materials are substantially soluble in water.
3. Which one of the salts in Table 1 has the smallest value of ∆Godiss?
A) PbCl2
B) MgCO3
C) BaSO4
D) AgCl
4. Which of the following is consistent with the differences in ∆Hodiss for NaCl and LiCl?
A) The electrostatic forces in solid LiCl are much stronger than in solid NaCl, while coordination of water is equivalent for both salts.
B) The electrostatic forces in the two solids are approximately equivalent, while water molecules coordinate much more effectively to Na+ than Li+.
C) The electrostatic forces in solid LiCl are weaker than in solid NaCl, while water cannot effectively coordinate to the very small Li+ cation.
D) The electrostatic forces in solid NaCl are slightly weaker than in solid LiCl, while water far more efficiently coordinates Li+ than Na+.
5. The transfer of heat to or from a solution changes the temperature of the solution according to the equation q = mc∆T where q is the heat transferred, m is the mass of solvent, and c is the specific heat of the solvent. If a 1 g sample of a salt was dissolved in 20 mL of water (specific heat = 4.18 J/g°C) in an insulated beaker and the temperature was found to decrease by 4°C, which of the following salts was used? Assume no phase change for the water.
A) LiCl
B) KCH3CO2
C) NH4NO3
D) NaCl
SOLUTIONS TO CHAPTER 6 FREESTANDING PRACTICE QUESTIONS
1. B Electrolysis requires energy. Water will not split into hydrogen and oxygen gas spontaneously at standard temperature and pressure which eliminates choices A and C. When bonds are broken, energy is absorbed (eliminates choice D). Energy is released when bonds are formed.
2. B Choice A is eliminated because decreasing volume would cause an increase in pressure, which would inhibit the transformation of a liquid to a gas. The question alludes that the process has a positive ∆H. Since the reaction involves changing two moles of liquid to three moles of gas, entropy increases so it will have a positive ∆S. Using the equation ∆G = ∆H T∆S, a reaction with a positive ∆H and ∆S will be spontaneous only at high enough temperatures. Therefore, choices C and D can be eliminated, making choice B correct.
3. D Enthalpy of vaporization is the heat energy required per mole to change from the liquid to gas phase. N2 is a gas at room temperature, Br2 and Hg are both liquids at room temperature, and Al is a solid at room temperature. Therefore, it is expected that Al will have the highest enthalpy of vaporization, making choice D correct.
4. C Both fusion (melting) and vaporization (boiling) require energy and are endothermic, eliminating choice A. Comparing both processes, vaporization takes substantially more energy. During vaporization, intermolecular forces are essentially completely overcome, and gaseous molecules separate widely due to their increased kinetic energy. Choice B is therefore eliminated. A 36 gram sample of water is 2 moles, so the heat of sublimation of 1 mole is half of 93.4 kJ, or 46.7 kJ/mol. This eliminates choice D. Examining the fusion and vaporization of water and adding their enthalpies by Hess’s law gives choice C as the correct answer:
H2O(s) → H2O(l) |
∆Hfus = X (6.0 kJ/mol) |
H2O(l) → H2O(g) |
∆Hvap = Y (40.7 kJ/mol) |
H2O(s) → H2O(g) |
∆Hvap = X + Y = 46.7 kJ/mol |
5. A The change in enthalpy (∆H) for a reaction is equal to the sum of ∆Hf° for products minus the sum of ∆Hf° for reactants: ∆H = Σ ∆Hf (products) — Σ ∆Hf (reactants). For choice A, the lone reduction reaction, ∆H = (—77.8 kJ/mol) — (—484.50 kJ/mol). This is a positive value indicating that enthalpy is absorbed and the reaction is endothermic. Note that the heat of formation of diatomic oxygen gas or any element in its naturally occurring form is defined as 0. The other answer choices, which are all oxidation reactions, will yield a negative value indicating they are exothermic.
6. B The question is asking about thermodynamic principles, so answer choices A and D that involve kinetics can be eliminated. Reactions with a positive ∆H and a negative ∆S are never spontaneous according to ∆G = ∆H — T∆S, eliminating choice C. If the sum of all reactions is more exergonic (—∆G) than endergonic (+∆G), the net release of free energy will drive the cycle forward, making choice B the best answer.
SOLUTIONS TO CHAPTER 6 PRACTICE PASSAGE
1. C Isoelectronic species have the same electron configurations, and hence the same number of electrons. Ag+ has 46 electrons, eliminating choices A and D because they have 44 and 50 electrons, respectively. The electron configuration of Ag+ is [Kr] 4d10. The electron configuration of Pd is [Kr] 5s23d 7 (eliminate choice B). The electron configuration of Cd2+ is [Kr] 4d10, which is isoelectronic with Ag+.
2. B Similar to bond formation, forming solute-solvent interactions is exothermic, meaning energy is released, not required; similarly breaking solute-solute or solvent-solvent interactions is endothermic (requires energy), like bond breaking (eliminate choice A). In addition, sodium chloride is soluble at room temperature (eliminate choice C). Salts are held together by ionic bonds, but Table 1 shows through the small Ksp values that not all of them are substantially soluble (eliminate choice D). For the dissolution of sodium chloride to be spontaneous, the increase in entropy must outweigh the endothermic process, yielding a negative Gibbs free energy.
3. A The important relationship between the standard state Gibbs free energy of dissolution and the solubility product constant is given in the passage:
∆Godiss = —RTlnKsp
Since the question asks for an extreme, first eliminate the two choices for Ksp in Table 1 that are the middle values (choices B and C). The ln function is similar to the log function, and can be thought of in the same way when judging relative magnitudes of ∆Godiss in the equation above. For values of Ksp > 1, the ∆Go value will be negative, and for values of Ksp < 1, the ∆Go value will be positive. Therefore, the larger value of Ksp for PbCl2 will give the smallest value for ∆Go.
4. D Effective dissolution involves the endothermic step of overcoming the electrostatic charges holding the solid salts together and the exothermic step of coordinating solvents to the separated ions. A negative value of ∆Hodiss likely indicates relatively weak electrostatic forces in the solid (small endothermic step), and effective solvation by water (large exothermic step). Table 2 shows that LiCl has a much more negative value of ∆Hodiss than NaCl. Choice A, stronger electrostatic forces in LiCl and no difference in solvation, would lead to a more negative ∆Hodiss for NaCl. Choice B would also result in a more negative value of ∆Hodiss for NaCl, as it indicates that electrostatics are equivalent while Na+ has stronger interactions with water. Choice C is incorrect because a large negative value of ∆Hodiss would be difficult to achieve if water were unable to coordinate Li+. Choice D includes a viable combination of slightly weaker attractive forces in NaCl but much better solvation for Li+.
5. C Since the question states that the temperature of the solution decreased, the salts with exothermic dissolution enthalpies (choices A and B) can be eliminated. Using the calorimetry equation given in the question stem (q = mc∆T), we can estimate:
20 g × ≈ 4 J/g°C × 4°C = ≈ 320 J = ≈ 0.32 kJ = q
Since this heat is associated with 1 g of salt, in order to compare to the ∆Hodiss in Table 2, convert this energy to a per mole basis by multiplying by the molar mass of the salt.
For choice C (NH4NO3, MW = 80 g/mol), this yields:
≈ 0.3 kJ/1g NH4NO3 × 80 g NH4NO3/mol = 24 kJ/mol
which is close to the given 25.69 kJ/mol in the table. The comparable calculation for NaCl yields:
≈ 0.3 kJ/1g NaCl × ≈ 60 g NaCl/mol = 18 kJ/mol
so choice D can be eliminated.