Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
5. Homogeneous Reactions
Takahide Fukuyama, Md. Taifur Rahman, and Ilhyong Ryu
5.1. Acid-Promoted Reactions
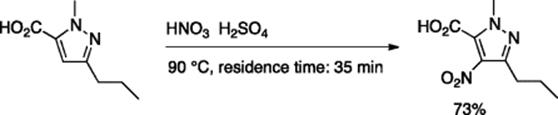
The nitration reaction is one of the fundamental reactions in organic synthesis and is industrially important. Standard batch processes frequently employ a combination of sulfuric and nitric acids, which often become violent and hazardous when scaled-up. Because nitration reactions are highly exothermic, they are temperature sensitive. This led chemists to examine the feasibility of running nitration reactions in a continuous flow manner using microreactors, which would obviate the safety issue with excellent temperature control in a tiny reaction space. Taghavi-Moghadam and coworkers reported that continuous-flow nitration using microreactors represents a safe and controllable method for the nitration of aromatic compounds [1]. For example, the nitration of pyrazole-5-carboxylic acid was carried out using microchannels 100 μm in width (CYTOS Lab System), which allowed strict temperature control at 90 °C. This process resulted in the formation of the desired nitration product in good yield (Scheme 5.1). In the batch system, the nitration of pyrazole- 5-carboxylic acids suffers from decarboxylation by heat evolution; however, this was not the case for this microflow system as the reaction temperature could be kept strictly below the decarboxylation limit of 100 °C.
Scheme 5.1
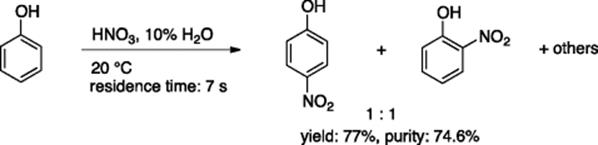
Durcy and Roberge reported controlled nitration of phenol in a glass microreactor with a channel width of 500 μm and an internal volume of 2.0 ml [2]. Nitration was most efficient and controlled under nearly solvent-free conditions at 20 °C without addition of sulfuric acid or acetic acid (Scheme 5.2). Under these concentrated conditions, autocatalysis spontaneously started in the mixing zone, allowing safe control of the reaction. Undesirable polymer formation, which is significant in batch reactions, was effectively suppressed by a factor of 10.
Scheme 5.2
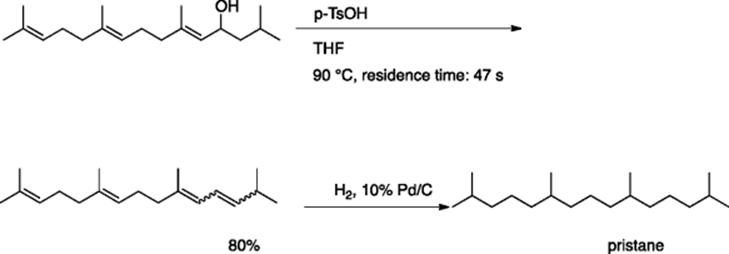
Synthetic-organic chemists have now recognized the potential of microreactors to increase reaction efficiency in organic synthesis. Fukase and coworkers reported that p-toluenesulfonic acid-catalyzed dehydration of allylic alcohols proceeds effectively in a microreactors system consisting of IMMs micromixer (channel width = 40 μm) and an additional residence time unit (diameter =1 mm, length = 1 m) [3]. Thus, dehydration of an allylic alcohol, which was prepared by two steps from farnesol, produced the corresponding conjugate diene in 80% yield (Scheme 5.3). The corresponding batch reaction gave lower yields, especially when the reaction was conducted on a larger scale. A multi-kilogram synthesis of pristane, an adjuvant for monoclonal production, was attained using a continuous method to reduce the dehydration product. A parallel system was created using 10 micromixers (Comet X-01), and the continuous flow reactions occurred over 3–4 days. Hydrogenation of the product, using 10% Pd/C, provided about 5 kg of the desired pristane.
Scheme 5.3
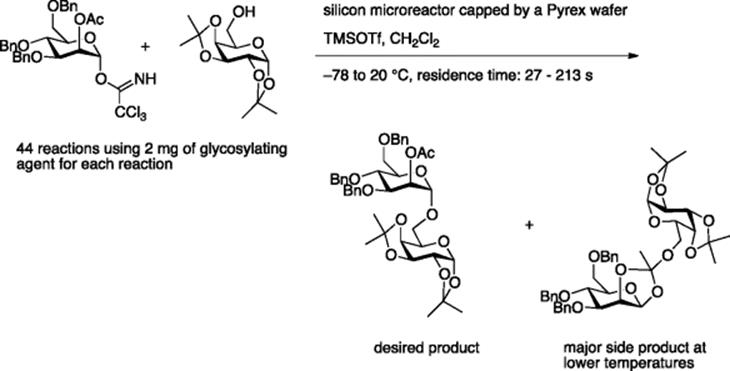
In general, method for optimization frequently requires large quantities of valuable starting materials. In this regard, microreactor-based optimization is advantageous, even for glycosylation, which notoriously lacks reliable general conditions. Seeberger, Jensen, and coworkers used microreactors to optimize glycosylation reactions [4]. With a single preparation of reagents, 44 reactions were completed at varying temperatures and reaction times, requiring just over 2 mg of glycosylating agent for each reaction (Scheme 5.4).
Scheme 5.4
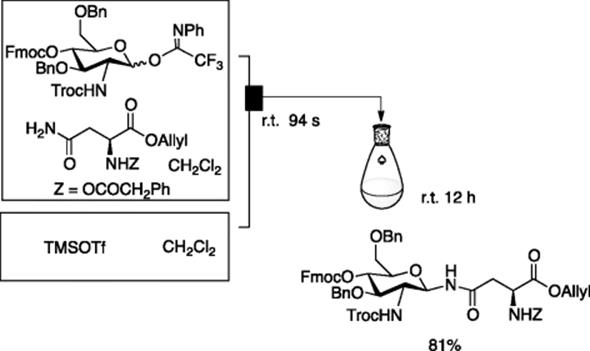
The N-glycosylation of asparagine amide is catalyzed by a Lewis acid such as TMSOTf. However, the batch reaction suffers from variable yields, reflecting on a slight change of the reaction conditions. Tanaka, Miyagawa, and Fukase employed an IMM micromixer (channel width = 40 μm) at the stage of mixing of the substrates with the Lewis acid. The resulting reaction mixture was kept in the batch flask for 12 h at room temperature [5]. This combined protocol gave constantly good yields of N-glycosylated products. It is assumed that efficient mixing and rapid heat transfer associated with the use of micromixer merits the reaction (Scheme 5.5).
Scheme 5.5
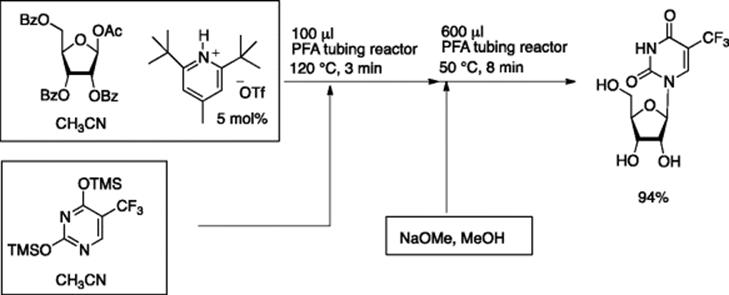
Jamison and coworkers reported on the one-flow, two-step synthesis of nucleosides by Br![]() nsted acid catalyzed glycosylation and base-promoted deprotection [6]. The first glycosylation was carried out by treating protected ribofuranose with silyl-protected thymine in the presence of 5 mol% of pyridinium triflate in acetonitrile. The formed glycosylated product was then treated with methanolic sodium methoxide (0.15 M) for deprotection, yielding the desired nucleoside in 94% yield (Scheme 5.6).
nsted acid catalyzed glycosylation and base-promoted deprotection [6]. The first glycosylation was carried out by treating protected ribofuranose with silyl-protected thymine in the presence of 5 mol% of pyridinium triflate in acetonitrile. The formed glycosylated product was then treated with methanolic sodium methoxide (0.15 M) for deprotection, yielding the desired nucleoside in 94% yield (Scheme 5.6).
Scheme 5.6
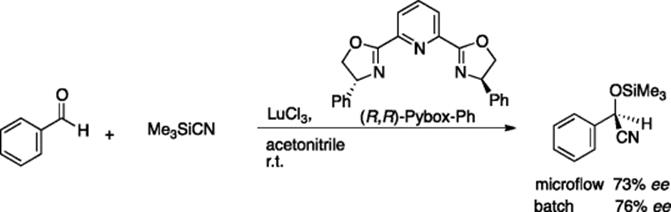
Asymmetric cyanosilylation of ketones and aldehydes is highly important, since the cyanohydrin product can easily be converted into optically active aminoalcohols by reduction. Moberg, Haswell, and coworkers reported on a microflow version of the catalytic cyanosilylation of aldehydes using Pybox [7]/lanthanoid triflates as the catalyst for chiral induction. A T-shaped borosilicate microreactor with channel dimensions of 100 μm × 50 μm was used in this study [8]. Electro-osmotic flow (EOF) was employed to pump an acetonitrile solution of phenyl-PYBOX, LuCl3, and benzaldehyde (reservoir A) and an acetonitrile solution of TMSCN (reservoir B). LnCl3-catalyzed microflow reactions gave similar enantioselectivity to that observed in analogous batch reactions, whereas lower enantioselectivity was observed for the YbCl3-catalyzed microflow reactions than that observed for the batch reaction (Scheme 5.7).
Scheme 5.7
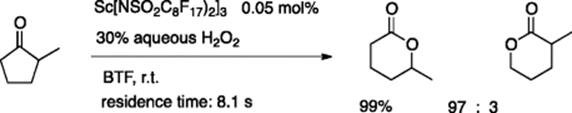
Mikami and coworkers examined the Baeyer–Villiger reaction of cyclic ketones with aqueous hydrogen peroxide, using a borosilicate microreactor that was 3 cm in length, 30 μm in depth, and 30 μm in width [9]. A BTF (benzotrifluoride) solution of a fluorous lanthanide catalyst, Sc[N(SO2C8F17)2]3 was used (Scheme 5.8). Extremely low flow rates (25–200 nl/min), which the authors termed, “nanoflow,” were well suited for the short length of the channel (3 cm). The residence time under optimal conditions was only 8.1 s. In several cases, regioisomeric ratios observed using a microreactor were superior to those observed using a batch reactor. Effective precomplexation in microchannels of the scandium catalyst with hydrogen peroxide to generate Sc-peroxide species seems to account for the better selectivity.
Scheme 5.8
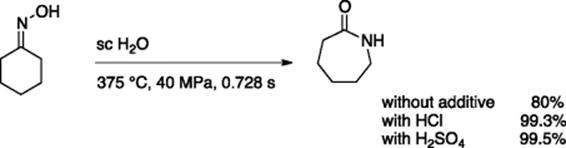
The Beckman rearrangement of cyclohexanone oxime into ε-caprolactam is an industrially important process, since ring-opening polymerization of ε-caprolactam produces nylon 6. Ikushima and coworkers invented a new continuous-microflow process for effective Beckman rearrangement of cyclohexanone oxime using supercritical water [10]. Using a flow device consisting of a Hastelloy C-276 alloy, the reaction proceeded well without any additives, whereas addition of a small amount of hydrochloric or sulfuric acid gave higher yields with the reaction time <1 s (Scheme 5.9).
Scheme 5.9
Kim and coworkers prepared polyimide (PI)-based film microreactors by ArF excimer laser or UV laser treatment. The PI microreactor (ArF-SH type) was successfully operated in extremely strong acidic conditions without any leaking and swelling problems [11]. The Beckman rearrangement of cyclohexanone oxime to ε-caprolactam by sulfuric acid at 130 °C proceeded in 0.9 s residence time with 46% yield, which is much more efficient than the results obtained from the corresponding batch reaction (28%, 1 h).
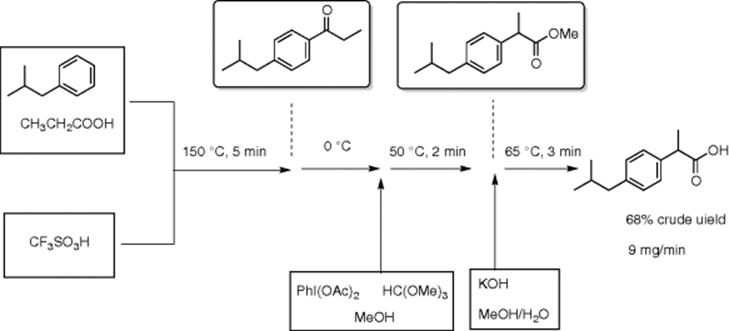
The McQuade group developed a continuous flow synthesis of ibuprofen using a consecutive three-step flow process [12]. The first process is the Friedel–Crafts acylation to prepare 4-isobutylpropiophenone from isobutylbenzene and propionic acid using triflic acid (5 equiv.), which proceeded at 150 °C for 5 min residence time (Scheme 5.10). Then, the product ketone was cooled to 0 °C and subjected to oxidation with one equiv of PhI(OAc)2 and four equiv of trimethyl orthoformate at 50 °C for 2 min. A concurrent 1,2-aryl migration produced methyl 2-aryl-propanoate. The third step is flow saponification using KOH solution in MeOH/H2O at 65 °C for 3 min, which gave ibuprofen in 68% crude yield. The system can generate 9 mg/min of crude ibuprofen.
Scheme 5.10