Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
5. Homogeneous Reactions
5.4. Condensation Reactions
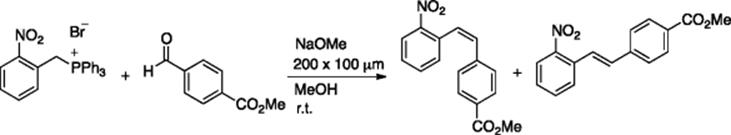
Haswell and coworkers carried out the Wittig reaction of 2-nitrobenzyl triphenylphosphonium bromide with methyl 4-formylbenzoate in a microflow system [24, 25]. They used a borosilicate glass microreactor, with T-shaped channels (width = 200 μm, depth = 100 μm), and the reagents were added via electro-osmotic flow (EOF) by applying a constant and controlled voltage. The Z/E ratio varied between 0.57 and 5.2 in the microflow system (Scheme 5.19), whereas the batch reaction using varying reactant ratios (phosphonium bromide:aldehyde = 2: 1 to 1: 10) gave Z/E ratios in the range of 2.8–3.0 [25]. Haswell et al. also reported enamine [26] and ester synthesis [27] using a similar microreaction system.
Scheme 5.19
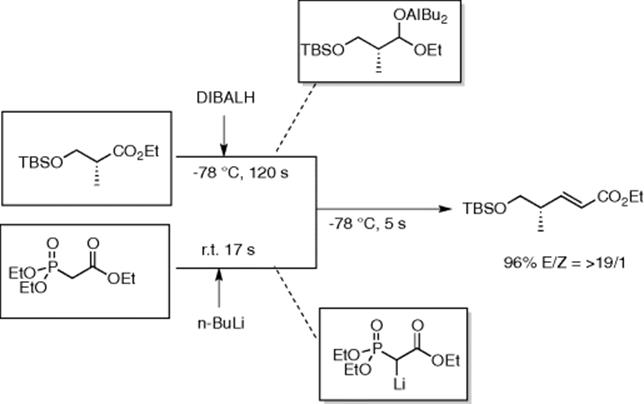
The Jamison group reported that a cryogenically controlled microreactor setup enabled diisobutylaluminum hydride (DIBALH) reduction of esters to give corresponding aldehydes in high yield and superior selectivity (trace amount of over-reduction products: alcohol) within a few seconds of reaction time [28]. They extended their work to combine three highly exothermic reactions in a single microreactor setup. They carried out Horner–Wadsworth–Emmons olefination by rapid reaction between two in situ-formed unstable intermediates, aluminated hemiacetal (from DIBALH-ester reaction) and lithiated-phosphonate (by deprotonation of phosphonate ester by n-BuLi) to furnish α,β-unsaturated ester in good yields and excellent E/Z-selectivity (Scheme 5.20) [29]. Since this integrated in-line protocol did not require any isolation of the intermediates, success with highly volatile and racemization-prone aldehyde precursor (e.g., silyl ether of ethyl lactate) manifested the operational superiority of this microreactor system over the existing batch protocols. In both reports, microreactor setup was constructed from readily available PFA (perfluoroalkoxy perfluoroalkanes) tubings and T-mixers.
Scheme 5.20
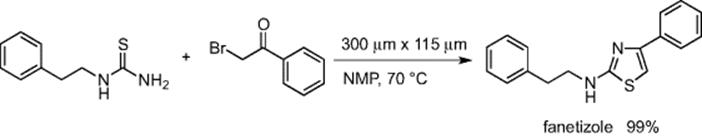
N-Heterocyclic compounds are found in many drug molecules and have potent medicinal activity. Hence, there is a growing interest in the utilization of microreactors as a tool for the synthesis of N-heteocylces. Garcia-Egido and coworkers reported on the Hantzsch synthesis of 2-aminothiazoles using a microflow system (width = 300 μm, depth = 115 μm) under EOF-driven flow using N-methyl-2-pyrrolidinone (NMP) as the solvent [30]. The reaction temperature was kept at 70 °C using a Peltier heater. Fanetizole, a pharmacological agent with activity for the treatment of rheumatoid arthritis, was prepared with excellent yield (Scheme 5.21).
Scheme 5.21
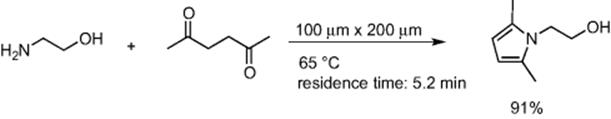
Condensation of 1,3-diketones with hydrazines or hydroxyamines was conducted in a microflow system to give pyrazoles and isoxazoles in good yields [31]. High throughput synthesis of pyrrole by the Paal–Knorr condensation of ethanolamine and acetonylacetone was achieved using the CPC CYTOS Lab System [32]. Running the system for 165 min resulted in 714 g of the pyrrole (Scheme 5.22).
Scheme 5.22
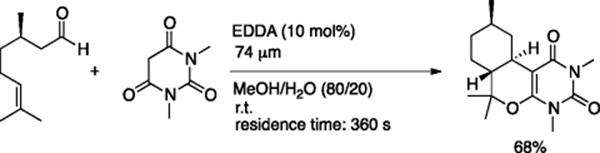
Fernandez-Suarez and coworkers investigated a domino reaction using a glass microreactor with a channel 74 μm in width [33]. The reaction of citronellal with 1,3-dimethylbarbituric acid, in the presence of ethylenediamine acetate (EDDA) and a residence time of 360 s, gave the tricyclic compound in 68% yield. Knoevenagel condensation and intramolecular Diels–Alder reaction took place subsequently (Scheme 5.23), and the syntheses of three different cycloadducts in parallel were also demonstrated.
Scheme 5.23
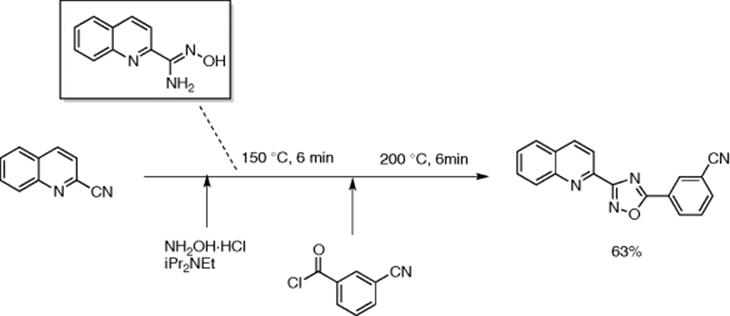
Cosford and coworkers presented a simple microreactor setup for enabling multistep synthesis of bis-substituted 1,2,4-oxadiazoles that required differential and controlled thermal treatments (between 0 and 200 °C) (Scheme 5.24) [34]. A base-assisted reaction between arylnitrile and hydroxylamine hydrochloride at 150 °C in the first reactor produced amidoxime, which was quickly cooled to 0 °C before it was mixed with the acid chloride. This mixture was then warmed and maintained at room temperature for 2 min in the connected tube before it entered a superheated chip-microreactor where the high temperature (200 °C) and pressure (7.5–9.0 bar) accelerated the reaction leading to 40–63% of differently functionalized oxadiazoles within 30 min of total process time. Relatively inferior yields were obtained from over three-day-long reaction in a sealed tube for the same products.
Scheme 5.24
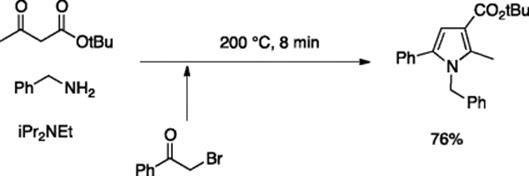
In a similar manner, this group accomplished rapid and high yielding three-component Hantzsch Pyrrole syntheses [35]. A DMF solution of alkyl acetoacetate, benzylamine, and N,N-diisopropylethylamine (DIPEA) was mixed with α-bromoacetophenone in a chip-reactor and allowed to react at above solvent's boiling point (200 °C) and at 5.0 bar pressure for 8 min reaction time, thereby furnishing 1H-pyrrole-3-carboxylate ester (Scheme 5.25). 1H-pyrrole-3-carboxylic acids were obtained when the byproduct HBr was not fully neutralized to intentionally allow hydrolysis of the primarily formed ester. This carboxylic acid can be further converted to amides by off-line carboxydiimide amidation chemistry. A multi-chip variant of the above concept was explored for the synthesis of imidazo[1,2-a]pyridine-2-carboxamides from amino pyridines by integrating an in-line amidation step [36].
Scheme 5.25
Microwave irradiation has been proven useful in accelerating chemical reactions. A unique approach to multi-component reactions – the combination of microwave irradiation and microreactors – was developed by Organ and Bremner [37]. The three-component coupling reaction of amino pyrazole with aldehyde and diketone in a glass capillary tube microflow system (1180 μm i.d.) under microwave irradiation (170 W) proceeded smoothly to give the desired quinolinone in high yield (Scheme 5.26). Without microwave irradiation, the reaction efficiency was very low.
Scheme 5.26
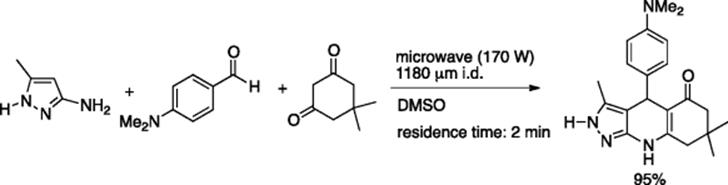
Kappe et al. generated a library of potential drug candidates, namely 4-(pyrazol-1-yl)carboxanilides, by employing a commercially available microreactor that is capable of recreating the high-temperature, high-pressure conditions of a microwave reactor (Scheme 5.27) [38]. Cyclocondensation of 4-nitrophenylhydrazine with appropriate 1,3-dicarbonyl compounds was carried out in X-Cube Flash reactor (Thales nanotechnology Inc.) at 175–230 °C producing 1-(4-nitrophenyl) 1H-pyrazoles that were subsequently reduced, in-line, via Pd/C-hydrogenation to corresponding aniline-derivatives by combining a gas-liquid microreactor (H-Cube) in the downstream. Finally, amidation of the free amine was achieved by a microwave protocol. They also explored the alternative route of instilling the amido group by a microflow high-temperature (100–230 °C) condensation of 4-bromophenylhydrazine with 1,3-dicarbonyl compounds followed by microwave-assisted (150 °C) substitution of bromine-atom by Buchwald–Hartwig amidation reaction.
Scheme 5.27
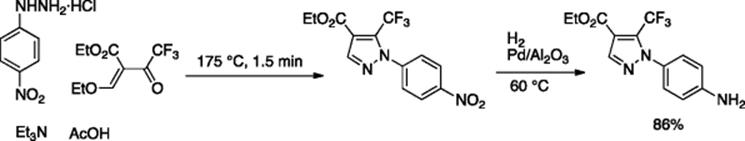
The Seeberger group reported a microflow synthesis of amides that used a highly pyrophoric reagent: trimethylaluminum (Scheme 5.28) [39]. A simple microreactor system, made of PTFE-tubing residence time unit and T-mixer, was used to perform the amidation reactions with a through-put of up to one mole of amide per day. They employed the trimethylaluminum-based amidation as the key step for the synthesis of two pharmaceutically active substrates: rimonabant and efaproxiral.
Scheme 5.28
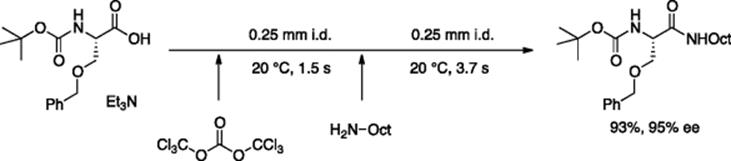
Using phosgene is a very useful and straightforward method to assist the synthesis of amides from amines and carboxylic acids, via the formation of an acid chloride. However, the inherent hazardous and toxic nature of phosgene inhibits its wide application. Moreover, the intermediate acid chloride is a highly activated specie and can undergo epimerization in case the parent acid is chiral. Takahashi et al. presented a facile way of in situ generation of phosgene from its less hazardous precursor; triphosgene in a microfluidic reactor that immediately reacted with the N-Boc-protected O-benzyl-l-serine acid to produce acid chloride in a Teflon tube reactor in 1.5 s (Scheme 5.29) [40]. As the acid chloride was rapidly produced and immediately coupled with amine in the second microtube reactor, epimerization was significantly suppressed (<3%). By controllably neutralizing the released HCl by-product, N-Boc protecting group remained intact throughout the process.
Scheme 5.29
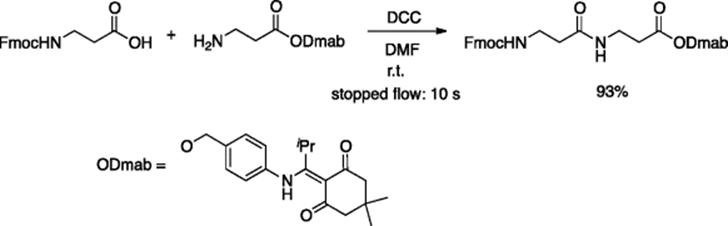
Microreaction technology has also been applied to peptide synthesis. Haswell and coworkers demonstrated that, using a borosilicate glass microreactor, the desired dipeptide was obtained by the reaction of N-Fmoc-β-alanine with β-alanine Dmab ester in the presence of DCC (1,3-dicyclohexylcarbodiimide) (Scheme 5.30) [41, 42]. It also was demonstrated that the Fmoc group could be removed by DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), using a microreactor, to yield the free amine. A multi-step synthesis of a tripeptide also was carried out using a microflow system, although the overall conversion was rather modest [42]. Using a microreactor, the degree of racemization for the α-peptide synthesis was slightly less than that observed in bulk reactions [43].
Scheme 5.30
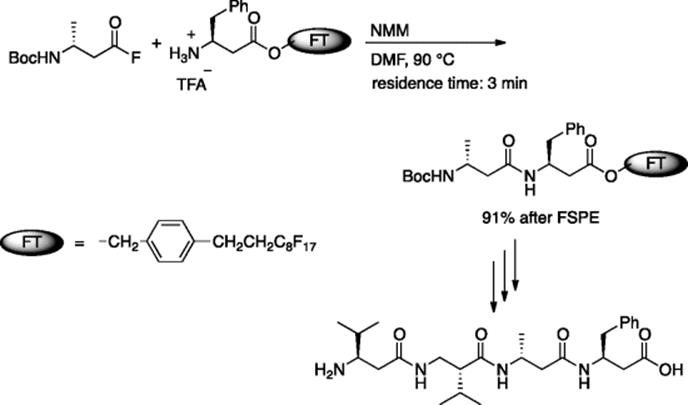
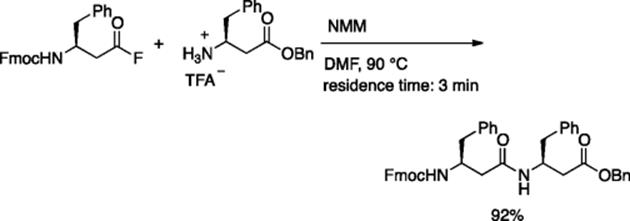
Seeberger and coworkers prepared synthetically useful amounts of β-peptide (0.2–0.6 mmol) using a microreactor (reactor volume = 78.3 μl) [44]. The reaction of acid fluoride and the TFA salt of amino acid benzyl ester in the presence of N-methylmorpholine (NMM) at 90 °C (3 min residence time) gave the dipeptide in 92% yield (Scheme 5.31). A fluorous tag method was utilized for efficient synthesis of tetrapeptide. Amino acid esters having fluorous tags were used to facilitate purification by fluorous solid-phase extraction (FSPE) (Scheme 5.32).
Scheme 5.31
Scheme 5.32