Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
6. Homogeneous Reactions II: Photochemistry and Electrochemistry and Radiopharmaceutical Synthesis
6.3. Radiopharmaceutical Synthesis in Microreactors
Positron emission tomography (PET) [32, 33] is a very powerful diagnostic technique, which can be used in a variety of medical applications including oncology, neurology, and cardiology. The basic principle of PET relies on the incorporation of short-lived positron emitting radioisotopes, most commonly ![]() (half-life of 110 min) or
(half-life of 110 min) or ![]() (half-life of 20 min) into the radiopharmaceutical. The fact that the
(half-life of 20 min) into the radiopharmaceutical. The fact that the ![]() and
and ![]() radioisotopes have such short half-lives is a significant challenge for the synthetic chemist, as the organic reactions used to prepare the molecule of interest must be relatively fast chemical transformations, with the goal being to prepare, purify, and isolate the desired product in 1–2 half-lives. Hence, in the case of
radioisotopes have such short half-lives is a significant challenge for the synthetic chemist, as the organic reactions used to prepare the molecule of interest must be relatively fast chemical transformations, with the goal being to prepare, purify, and isolate the desired product in 1–2 half-lives. Hence, in the case of ![]() this means that the chemist must synthesize and isolate the pure target in <30 min. As a result, although a large number of PET radiopharmaceuticals have been discovered, the complexity of the syntheses means that only a few of these PET radiopharmaceuticals are routinely used; the most common being 2-deoxy-2-[
this means that the chemist must synthesize and isolate the pure target in <30 min. As a result, although a large number of PET radiopharmaceuticals have been discovered, the complexity of the syntheses means that only a few of these PET radiopharmaceuticals are routinely used; the most common being 2-deoxy-2-[![]() ]fluoro-d-glucose ([
]fluoro-d-glucose ([![]() ]FDG). For a more detailed analysis of other PET radiopharmaceuticals, readers are directed to a review by Elizarov [34].
]FDG). For a more detailed analysis of other PET radiopharmaceuticals, readers are directed to a review by Elizarov [34].
It is now well established that microreactor technology enables the residence time of chemical reactions to be substantially reduced [35–37]. Fundamentally, this improvement in reaction efficiency is because the mixing [38, 39] within the microreactors is highly efficient and furthermore the reaction itself may be intensified by application of high-temperature conditions [40]. Furthermore for PET imaging since only nanogram quantities of radiopharmaceutical are actually needed, consequently these volumes of product are very easily made within even the smallest microreactors on the market.
Consequently for this application, classic “lab-on-a-chip” type microreactors are commonly used. They typically have internal volumes ranging from nanoliters to microliters. As with any synthetic chemistry application, the most important consideration is chemical compatibility of the reagents and solvents with the reactor material and consequently glass reactors are the most widely reported, however, polymeric reactors have also been reported in some applications. It should be noted that Future Chemistry (The Netherlands) have recently launched a product in collaboration with Veenstra Instruments [41], specifically aimed at the radiochemistry market (Figure 6.2).
Figure 6.2 FlowSafe by Future Chemistry.

In summary, microreactors enable reaction times to be reduced, which is ideal for short-lived isotope chemistry and the volume of these are perfectly aligned with the amount of product needed for imaging applications. In the following section, a summary of the literature based around ![]() and
and ![]() are highlighted.
are highlighted.
6.3.1 Fluorinations in Microreactors
Within the field of microreactor and continuous flow synthesis, a wide variety of fluorination reactions have been performed. For example, Seeberger et al. [42] demonstrated the efficient fluorination of carboxylic acids, aldehydes, and alcohols using diethylamino sulfur trifluoride (DAST) in a PTFE tube reactor. An alternative method for fluorination is the use of F2 gas; where Chambers et al. [43, 44] demonstrated the use of a nickel flow reactor for the selective fluorination of a range of 1,3-dicarbonyl compounds employing 10% elemental fluorine in nitrogen carrier gas. Jensen et al. [45] further demonstrated the direct fluorination of aromatic compounds within a silicon/Pyrex reactor, with nickel-coated microchannels. Consequently, at first glance, one would think that conducting radiofluorinations would be incremental, however, it needs to be very clearly emphasized that the challenges associated with ![]() chemistry are substantially more difficult. Unlike the traditional fluorine chemistry above, the radiolabeled fluorine cannot be purchased and must be manufactured as part of the total synthesis, which is itself a complex process.
chemistry are substantially more difficult. Unlike the traditional fluorine chemistry above, the radiolabeled fluorine cannot be purchased and must be manufactured as part of the total synthesis, which is itself a complex process.
First, [![]() ]F− is produced within a cyclotron from oxygen enriched H218O by proton bombardment. The fluoride anion is produced as a very dilute aqueous species, and is inserted into the pharmaceuticals most commonly by nucleophilic substitution reactions under appropriate activation conditions (Figure 6.3). This activation is generally done by dehydrating the solution, replacing it with an organic aprotic solvent (usually acetonitrile or DMF) and adding alkali salts with or without the use of kryptands. The most commonly used system for [
]F− is produced within a cyclotron from oxygen enriched H218O by proton bombardment. The fluoride anion is produced as a very dilute aqueous species, and is inserted into the pharmaceuticals most commonly by nucleophilic substitution reactions under appropriate activation conditions (Figure 6.3). This activation is generally done by dehydrating the solution, replacing it with an organic aprotic solvent (usually acetonitrile or DMF) and adding alkali salts with or without the use of kryptands. The most commonly used system for [![]() ]F− incorporation utilizes the CH3CN/H2O azeotrope for drying the fluoride in presence of K2CO3 and Kryptofix 2.2.2 (K2.2.2) (Figure 6.4) as the kryptand. The nucleophilicity of the fluoride anion of such complexes is high enough to react with a variety of substrates. A detailed protocol for this process is reported by Elizarov [34].
]F− incorporation utilizes the CH3CN/H2O azeotrope for drying the fluoride in presence of K2CO3 and Kryptofix 2.2.2 (K2.2.2) (Figure 6.4) as the kryptand. The nucleophilicity of the fluoride anion of such complexes is high enough to react with a variety of substrates. A detailed protocol for this process is reported by Elizarov [34].
Figure 6.3 Process for generating and reacting ![]() .
.
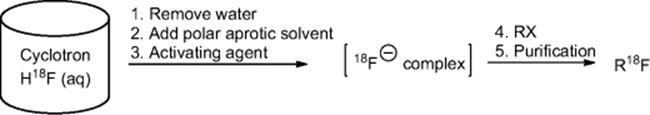
Figure 6.4 Structure of Kryptofix 2.2.2.
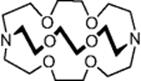
One of the first reports was published by Lu and coworkers [46], who demonstrated the use of a glass microreactor [channel dimensions, 220 μm (wide) × 60 μm (deep) × 1.4 cm (length)] for the rapid synthesis of a series of radiolabeled compounds (Scheme 6.14). A premixed solution of 3-pyridin-3-yl-propionic acid (41) (0.01 M) and tetra-n-butylammonium hydroxide (0.01 M in DMF) was introduced from one inlet of the reactor and a solution of premade radioactive tosylate (42) (0.01 M in DMF) was added from the other inlet. A residence time of 12 s afforded the respective labeled ester (43) (Scheme 6.14) with a radiochemical yield (RCY) of 10%. Although, in this first example, the yield was not significantly better than other techniques, the advantage was that the overall processing time was reduced to just 10 min, which is ideal for PET tracer synthesis.
Scheme 6.14 Tagging of ![]() label to 3-pyridin-3-yl-propionic acid in a microreactor.
label to 3-pyridin-3-yl-propionic acid in a microreactor.
As discussed, [![]() ]FDG is the most widely used radiotracer, consequently this has been the most studied reaction when benchmarking the efficiency of microreactor technology for this application. In this synthesis, mannose triflate (44) is first reacted with the activated fluoride complex to give (45), followed by hydrolysis under acidic or basic conditions to give [
]FDG is the most widely used radiotracer, consequently this has been the most studied reaction when benchmarking the efficiency of microreactor technology for this application. In this synthesis, mannose triflate (44) is first reacted with the activated fluoride complex to give (45), followed by hydrolysis under acidic or basic conditions to give [![]() ]fluorodeoxyglucose ([
]fluorodeoxyglucose ([![]() ]FDG) (46) (Scheme 6.15).
]FDG) (46) (Scheme 6.15).
Scheme 6.15 Synthesis of 2-[![]() ]fluorodeoxyglucose ([
]fluorodeoxyglucose ([![]() ]FDG).
]FDG).
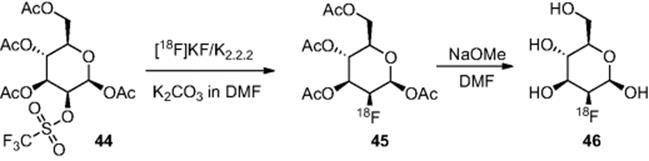
In the first instance, research focused on investigating if the fluorination step could be conducted within microreactors. Steel [47] used a microfluidic chip [channel dimensions, 300 μm (wide) × 50 μm (deep)], where a solution of mannose triflate (44) in anhydrous acetonitrile is reacted with a premade complex from [![]() ]KF, Kryptofix 2.2.2 and K2CO3 in acetonitrile. They reported a 40% conversion for the radiolabeling reaction [(44) to (45)] when a residence time of 2 min was used.
]KF, Kryptofix 2.2.2 and K2CO3 in acetonitrile. They reported a 40% conversion for the radiolabeling reaction [(44) to (45)] when a residence time of 2 min was used.
Gillies and coworkers [48, 49] subsequently used a polycarbonate microreactor, in which a chamber was designed to effect very rapid turbulent mixing. The microreactor was used to conduct the fluorination of mannose triflate (44), followed by acid hydrolysis of the intermediate (45) to synthesize [![]() ]fluorodeoxyglucose ([
]fluorodeoxyglucose ([![]() ]FDG) (46) (Scheme 6.15), whereby 50% overall incorporation of the radiolabel was achieved with a residence time of just 4 s. However, the polymeric material did limit what solvents could be used within the system.
]FDG) (46) (Scheme 6.15), whereby 50% overall incorporation of the radiolabel was achieved with a residence time of just 4 s. However, the polymeric material did limit what solvents could be used within the system.
Similarly Wester and coworkers more recently used a capillary reactor (inner diameter 300 μm, length 0.7 m) for the production of [![]() ]FDG (46) [50]. They reported that the reaction had optimum performance at a temperature of 105 °C and residence time of 40 s. The group subsequently conducted a NaOH hydrolysis to afford [
]FDG (46) [50]. They reported that the reaction had optimum performance at a temperature of 105 °C and residence time of 40 s. The group subsequently conducted a NaOH hydrolysis to afford [![]() ]FDG (46) in 88% radiochemical yield within a processing time of 7 min.
]FDG (46) in 88% radiochemical yield within a processing time of 7 min.
Undoubtedly, the most sophisticated synthesis of [![]() ]FDG was reported by Cheng-Lee et al. [51]. They used a PDMS microreactor consisting of a complex array of channels typically 200 μm wide and 45 μm deep. They employed a sequence of five steps, comprising (i) fluoride concentration (500 μCi), (ii) solvent exchange from water to acetonitrile, (iii) nucleophilic substitution of the mannose triflate (44) (324 ng), (iv) solvent exchange back from acetonitrile to water, and finally (v) acid hydrolysis to afford [
]FDG was reported by Cheng-Lee et al. [51]. They used a PDMS microreactor consisting of a complex array of channels typically 200 μm wide and 45 μm deep. They employed a sequence of five steps, comprising (i) fluoride concentration (500 μCi), (ii) solvent exchange from water to acetonitrile, (iii) nucleophilic substitution of the mannose triflate (44) (324 ng), (iv) solvent exchange back from acetonitrile to water, and finally (v) acid hydrolysis to afford [![]() ]FDG (46). The reactor was designed in order to have different temperature zones, reporting that the fluorination reaction was conducted at 100 °C for 30 s followed by 120 °C for 50 s, while the acid hydrolysis was conducted at 60 °C. Using this approach, [
]FDG (46). The reactor was designed in order to have different temperature zones, reporting that the fluorination reaction was conducted at 100 °C for 30 s followed by 120 °C for 50 s, while the acid hydrolysis was conducted at 60 °C. Using this approach, [![]() ]FDG (46) was obtained in 38% radiochemical yield but in very high purity (97%). Overall the total processing time was reduced to 14 min, compared with typically 50 min for the current batch protocol.
]FDG (46) was obtained in 38% radiochemical yield but in very high purity (97%). Overall the total processing time was reduced to 14 min, compared with typically 50 min for the current batch protocol.
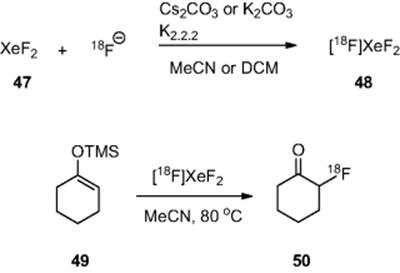
With the advantages of microreactor chemistry becoming more obvious, other reactions have recently been developed. For instance, Lu and coworkers [52] have reported the synthesis and reaction of [![]() ]XeF2 in a microreactor (inner diameter 100 μm, length 4.0 m, volume 31.4 μl). The authors report that, although XeF2 chemistry has been acknowledged as a very efficient electrophilic fluorinating agent, there was previously no practical means of efficiently preparing the radiolabeled derivative. The authors report that the optimum method for the synthesis was to react xenon difluoride (47) with cyclotron produced [
]XeF2 in a microreactor (inner diameter 100 μm, length 4.0 m, volume 31.4 μl). The authors report that, although XeF2 chemistry has been acknowledged as a very efficient electrophilic fluorinating agent, there was previously no practical means of efficiently preparing the radiolabeled derivative. The authors report that the optimum method for the synthesis was to react xenon difluoride (47) with cyclotron produced [![]() ]fluoride in either dichloromethane at room temperature or acetonitrile at 80 °C, with a residence time of ~1.5 min. With the optimum conditions for the generation of [
]fluoride in either dichloromethane at room temperature or acetonitrile at 80 °C, with a residence time of ~1.5 min. With the optimum conditions for the generation of [![]() ]XeF2 (48) in hand, the authors subsequently investigated the reaction of the complex. Reaction of a solution of the trimethyl silyl enol ether (49) in acetonitrile with [
]XeF2 (48) in hand, the authors subsequently investigated the reaction of the complex. Reaction of a solution of the trimethyl silyl enol ether (49) in acetonitrile with [![]() ]XeF2 (48) at 80 °C for 3.1 min afforded the radiolabeled ketone (50) in 78% radiochemical yield (Scheme 6.16).
]XeF2 (48) at 80 °C for 3.1 min afforded the radiolabeled ketone (50) in 78% radiochemical yield (Scheme 6.16).
Scheme 6.16 Synthesis and reaction of electrophilic [![]() ]XeF2.
]XeF2.
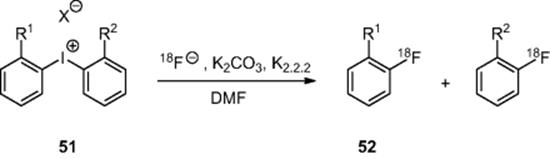
Chun and coworkers [53] have used a microreactor (inner diameter 100 μm, length 4.0 m, volume 31.4 μl) to convert diaryliodonium salts (51) into labeled fluoroarenes (52) (Scheme 6.17). Using an Advion Nanotek system, they reacted a solution of fluoride with a variety of diaryliodonium salts in DMF as solvent. In the case of symmetrical diaryliodonium salts, the products were obtained in moderate to high radiochemical yields (51–85%) with residence times <4 min. The authors went on to study the effect of unsymmetrical diaryliodonium salts, reporting that yields were more variable and that the selectivity was predominantly governed by the nature of the ortho-substituent.
Scheme 6.17 Reaction of diaryliodonium salts in a microreactor.
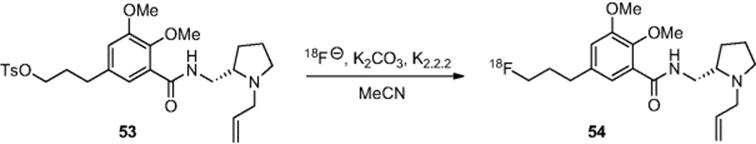
Using the Advion Nanotek microreactor platform, the authors also report the synthesis of [![]() ]fallypride, whereby they reacted tosylate (53) with anhydrous fluoride in acetonitrile to give [
]fallypride, whereby they reacted tosylate (53) with anhydrous fluoride in acetonitrile to give [![]() ]fallypride (54) (Scheme 6.18) in 88% radiochemical yield at a temperature of 170 °C in total run times of 4 min [54]. Using the similar methodology, separate reports on the synthesis of [
]fallypride (54) (Scheme 6.18) in 88% radiochemical yield at a temperature of 170 °C in total run times of 4 min [54]. Using the similar methodology, separate reports on the synthesis of [![]() ]setoperone [55], [
]setoperone [55], [![]() ]FLT [56], and [
]FLT [56], and [![]() ]fluoromisonidazole [57] have also been described.
]fluoromisonidazole [57] have also been described.
Scheme 6.18 Synthesis of [![]() ]fallypride in a microreactor.
]fallypride in a microreactor.
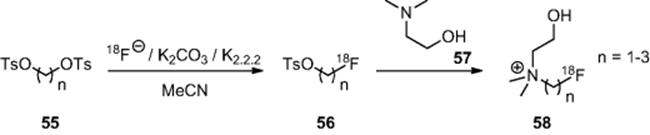
Pascali and coworkers recently reported the concept of using microreactor technology (inner diameter 100 μm, length 2.0 m, volume 15.6 μl) to effect “dose-on-demand” synthesis of PET molecules [58]. Using the Advion system, they reacted ditosylates (55) with anhydrous fluoride in acetonitrile to prepare the [![]() ]tosylates (56) in situ before introduction of a neat solution of choline (57) to synthesize the [
]tosylates (56) in situ before introduction of a neat solution of choline (57) to synthesize the [![]() ]fluorocholine derivatives (58) in 22–54% radiochemical yield within 15 min total processing time (Scheme 6.19).
]fluorocholine derivatives (58) in 22–54% radiochemical yield within 15 min total processing time (Scheme 6.19).
Scheme 6.19 Synthesis of [![]() ]fluorocholine derivatives in a microreactor.
]fluorocholine derivatives in a microreactor.
6.3.2 Synthesis of 11C-Labeled PET Radiopharmaceuticals in Microreactors
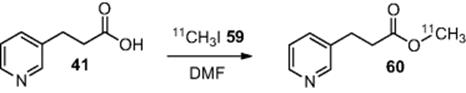
As an extension of their fluorination chemistry, Lu and coworkers [46] also demonstrated the use of their glass microreactor system for the rapid synthesis of a series of ![]() radiolabeled compounds (Scheme 6.20). A premixed solution of 3-pyridin-3-yl-propionic acid (41) (0.01 M) and tetra-n-butylammonium hydroxide (0.01 M in DMF) was introduced from one inlet of the reactor and a solution of radioactive
radiolabeled compounds (Scheme 6.20). A premixed solution of 3-pyridin-3-yl-propionic acid (41) (0.01 M) and tetra-n-butylammonium hydroxide (0.01 M in DMF) was introduced from one inlet of the reactor and a solution of radioactive ![]() (59) (0.01 M in DMF) was added from the other inlet. A residence time of 12 s afforded the respective labeled ester (60) (Scheme 6.9) with a radiochemical yield (RCY) of 88%.
(59) (0.01 M in DMF) was added from the other inlet. A residence time of 12 s afforded the respective labeled ester (60) (Scheme 6.9) with a radiochemical yield (RCY) of 88%.
Scheme 6.20 Methylation of 3-pyridin-3-yl-propionic acid in a microreactor.
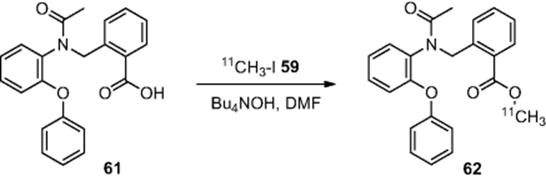
Among other examples, the authors also demonstrated the synthesis of a peripheral benzodiazepine receptor (PBR) ligand (62) via the ![]() -methylation of carboxylic acid (61) (Scheme 6.21), obtaining the desired PBR ligand (62) in a RCY of 65% [46].
-methylation of carboxylic acid (61) (Scheme 6.21), obtaining the desired PBR ligand (62) in a RCY of 65% [46].
Scheme 6.21 Synthesis of ![]() -labeled peripheral benzodiazepine ligand in a microreactor.
-labeled peripheral benzodiazepine ligand in a microreactor.
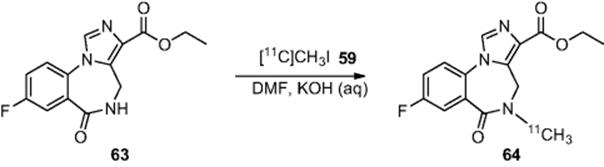
Cleij and coworkers [59] used a homemade PEEK lined stainless steel loop reactor constructed from HPLC fittings to effect the synthesis of [![]() ]flumazenil (Scheme 6.22). Reaction of amine (63) with labeled methyl iodide (59) afforded [
]flumazenil (Scheme 6.22). Reaction of amine (63) with labeled methyl iodide (59) afforded [![]() ]flumazenil (64) in 80% radiochemical yield at room temperature. The radiosynthesis itself took 5 min, while total processing was reported to be completed in 20 min.
]flumazenil (64) in 80% radiochemical yield at room temperature. The radiosynthesis itself took 5 min, while total processing was reported to be completed in 20 min.
Scheme 6.22 Synthesis of [![]() ]flumazenil in a microreactor.
]flumazenil in a microreactor.
[![]() ]Carbon monoxide is another important
]Carbon monoxide is another important ![]() -labeling reagent for radiotracer synthesis as it can provide access to a range of
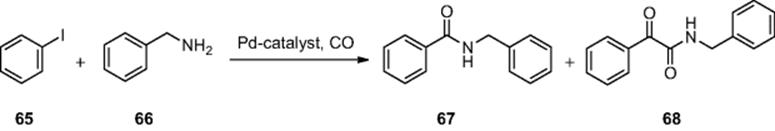
-labeling reagent for radiotracer synthesis as it can provide access to a range of ![]() -labeled molecules. De Mello et al. [60] demonstrated the continuous synthesis of a series of secondary amides via a carbonylative coupling reaction within a glass microreactor [channel dimensions = 200 μm (wide) × 75 μm (deep) × 5 m (length)]. Employing a biphasic reaction system, comprising gaseous carbon monoxide and a solution of iodobenzene (65), benzylamine (66), and a palladium-phosphine catalyst, the effect of liquid flow rate under a constant gas flow was investigated. Using the synthesis of N-benzylbenzamide (67) as a model reaction (Scheme 6.23), the authors found that annular flow dominated when flow rates of 5.0–20.0 μl/min were employed. Conducting the microreactions for 10 min, an increase in conversion as a function of increased reagent residence time was reported, an observation that the authors attribute to the formation of a stable flow regime within the reactor. Using the optimal flow rate of 5.0 μl/min, 46% conversion to the respective amide (67) was achieved along with 9% α-ketoamide (68). The study was extended to use
-labeled molecules. De Mello et al. [60] demonstrated the continuous synthesis of a series of secondary amides via a carbonylative coupling reaction within a glass microreactor [channel dimensions = 200 μm (wide) × 75 μm (deep) × 5 m (length)]. Employing a biphasic reaction system, comprising gaseous carbon monoxide and a solution of iodobenzene (65), benzylamine (66), and a palladium-phosphine catalyst, the effect of liquid flow rate under a constant gas flow was investigated. Using the synthesis of N-benzylbenzamide (67) as a model reaction (Scheme 6.23), the authors found that annular flow dominated when flow rates of 5.0–20.0 μl/min were employed. Conducting the microreactions for 10 min, an increase in conversion as a function of increased reagent residence time was reported, an observation that the authors attribute to the formation of a stable flow regime within the reactor. Using the optimal flow rate of 5.0 μl/min, 46% conversion to the respective amide (67) was achieved along with 9% α-ketoamide (68). The study was extended to use ![]() as the reagent and using the same methodology produced amide (67) in 79% radiochemicals yield and 96% purity. They extended the study to produce a series of other
as the reagent and using the same methodology produced amide (67) in 79% radiochemicals yield and 96% purity. They extended the study to produce a series of other ![]() labeled derivatives in 45–67% yield.
labeled derivatives in 45–67% yield.
Scheme 6.23 Microscale carbonylative synthesis of N-benzylbenzamide.
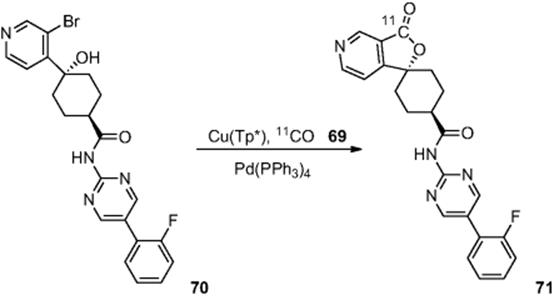
In a more sophisticated extension, Kealey et al. [61] reported the use of a copper [![]() ]carbonyl complex (Cu(Tp*)[
]carbonyl complex (Cu(Tp*)[![]() ]CO) (69) in a NanoTek reactor, which the authors report as a convenient reagent to handle in solution, which opens up a new radiolabeling techniques. Initially, the authors used the same reaction as shown in Scheme 6.23 to develop the methodology, whereby they report that the amide (67) was produced in a 73% yield at 100–125 °C. With the basic methodology in hand, the group used the technique to prepare neuropeptide YY5 receptor antagonist [
]CO) (69) in a NanoTek reactor, which the authors report as a convenient reagent to handle in solution, which opens up a new radiolabeling techniques. Initially, the authors used the same reaction as shown in Scheme 6.23 to develop the methodology, whereby they report that the amide (67) was produced in a 73% yield at 100–125 °C. With the basic methodology in hand, the group used the technique to prepare neuropeptide YY5 receptor antagonist [![]() ]MK-0233 (71). Optimization established that alcohol (70) could be reacted at a temperature of 160 °C to afford [
]MK-0233 (71). Optimization established that alcohol (70) could be reacted at a temperature of 160 °C to afford [![]() ]MK-0233 (71) in 81% yield in a residence time of just 15 s (Scheme 6.24).
]MK-0233 (71) in 81% yield in a residence time of just 15 s (Scheme 6.24).
Scheme 6.24 Synthesis of neuropeptide YY5 receptor antagonist [![]() ]MK-0233 in a microreactor.
]MK-0233 in a microreactor.