Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
7. Heterogeneous Reactions
7.6. Flow Reaction with an Immobilized Catalyst: Metal Catalysts Coordinated by a Polymer-Supported Ligand
Metal and metalloid atoms can be coordinated with, generally, an ionic or electron-negative molecule to form metal-organic complexes. Ligand selection is a critical consideration in Lewis acid and transition metal catalysis because ligands can dictate the reactivity of the central metal atom. Furthermore, metal catalysts bound to a chiral ligand allow production of enantiomerically enriched molecules [117]. Chiral ligands are often expensive, and the separation of products from the ligands may sometimes be troublesome. Consequently, preparation of polymer-supported ligands and evaluation of their reactivity in a flow stream have been extensively studied. Because the degradation of immobilized ligands and leaching of central metal can be problematic under the flow conditions, the recyclability of the polymer-supported ligands even with longer-term use remains a challenging issue.
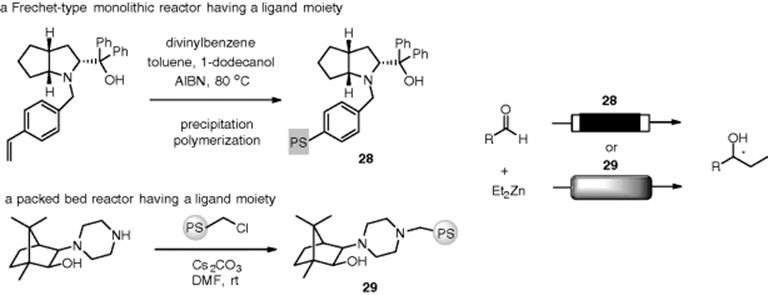
Alkylation of aldehydes by dialkylzinc is accelerated by a catalytic amount of ligands, such as β-amino alcohols. The use of chiral ligands may afford chiral sec-alcohols in an enantioselective manner. Luis, Martens, and coworkers synthesized monoliths (28) containing an amino alcohol moiety, which achieve an enantioselective addition of ZnEt2 to benzaldehyde (99% ee) [118]. This reactor could be reused at least four times, with equivalent yields and no loss of its catalytic efficiency. Pericàs and coworkers developed polystyrene-supported chiral amino alcohols, such as (29), for the enantioselective alkylation of aldehydes by diethylzinc (Scheme 7.27) [119–121]. The functionalized polymer (29) turned out to be highly enantioselective for a broad scope of substrates, allowing repeated recycling without any loss of activity or enantioselectivity. These authors also achieved an enantioselective arylation of aldehydes with triarylboroxin in the presence of diethylzinc under flow conditions [122].
Scheme 7.27 Representative methods for the preparation of polymer-supported ligands and their use for enantioselective alkylation under flow conditions.
Asymmetric cyanation of imines, a modified Strecker synthesis, affords enantiomerically enriched α-aminonitriles, which can be converted into α-amino acids. Seayad and Ramalingam reported a homogeneous catalyst system that was derived from partially hydrolyzed titanium alkoxide and chiral β-amino alcohol ligands as a highly enantioselective catalyst for the asymmetric Strecker reaction [123]. In these extensive investigations, a self-supported chiral titanium cluster (SCTC) catalyst (30) was used as a heterogeneous catalyst. The research group investigated a continuous-flow asymmetric Strecker reaction using a packed-bed reactor, in which SCTC was placed. The system showed high enantioselectivity (up to 98% ee) with complete conversion of imine substrates. They demonstrated three-component Strecker reaction from aldehydes, amines, and TMSCN under continuous flow. Interestingly, in the case of the more-challenging unbranched aldehydes (31), enantiomeric excess of (32) under flow conditions (87% ee) was improved compared with that of the batch reaction (70% ee) (Scheme 7.28) [124].
Scheme 7.28 Flow asymmetric Strecker reaction with a single reactor and successive reactors.

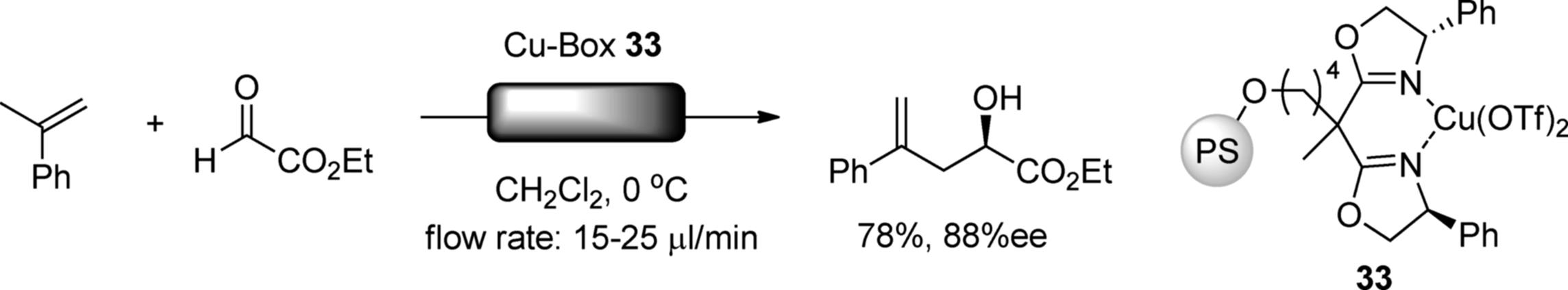
Asymmetric CߝC bond-forming reactions have also been accomplished in flow. Chiral bisoxazolines (Box) are utilized in many asymmetric catalytic reactions as nitrogen-containing bidantate ligands for Lewis acidic metals as well as transition metals. Chiral Cu-Box can be utilized as a Lewis acid catalyst. Salvadori et al. investigated the enantioselective ene reaction using a polystyrene-bound Cu-Box catalyst (33) under flow conditions (Scheme 7.29) [125].
Scheme 7.29 Asymmetric ene reaction in flow.
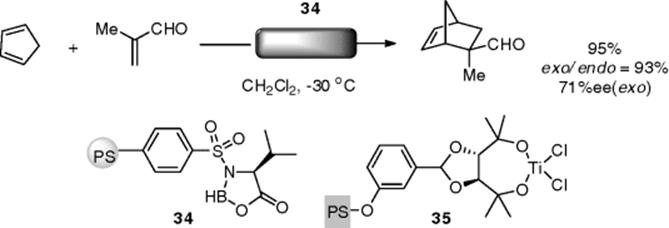
The enantioselective Diels–Alder reaction is another main motif in chiral Lewis acid catalysis. In 1996, Itsuno and coworkers reported an asymmetric Diels–Alder reaction using polymer-supported catalysts under flow conditions. Immobilized chiral oxazoboloridune (34) was prepared from a copolymer of N-sulfonylvaline and borane having styrene moiety, affording the Diels–Alder adduct in an enantioselective manner (up to 71% yield) [126]. The authors used a gravity-fed-type column for the flow reaction. Ti-TADDOL-functionalized monolithic resins (35) were developed by Altava and Luis for the asymmetric Diels–Alder reaction (Scheme 7.30).
Scheme 7.30 Polymer-supported Lewis acids for a flow asymmetric Diels–Alder reaction.
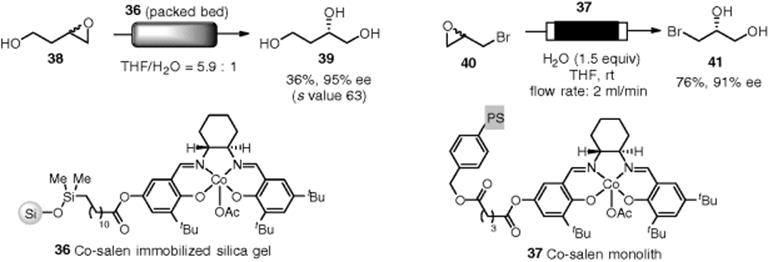
The Salen motif has been widely utilized as a ligand for transition metals. Jacobsen et al. reported that chiral salen–cobalt complex (Co-salen) could be utilized as a Lewis acid catalyst for hydrolytic kinetic optical resolution of racemic epoxides with H2O to give the corresponding diols in an enantioselective manner. Silica bound chiral Co-salen complex (36) was synthesized and adapted to a continuous-flow reaction. The optical kinetic resolution of racemic epoxide (38) was successful to yield the desired triol (39) in good yield (36% conversion) and high enantiomeric excess (Scheme 7.31) [127]. A PASSflow microreactor consisting of Co-salen monolith (37) was used for the dynamic kinetic resolution of epibromohydrin (40). Three runs performed on the 1-mmol scale were completed and afforded (R)-(41) in 76–87% yield with constant enantiomeric purity of 91–93% ee [128].
Scheme 7.31 Hydrolytic kinetic resolution of racemic epoxides.
Delueze and coworkers reported a polymer-supported titanium alkoxide catalyst for the transesterification. They evaluated the efficiency and stability of the catalyst and tested it in a laboratory-scale continuous-flow reactor under equilibrium conditions [129]. The average metal leaching was estimated to less than 1% of the total amount of titanium engaged.
7.6.1 Flow Reactions Using Immobilized Ligands with a Transition Metal Catalyst
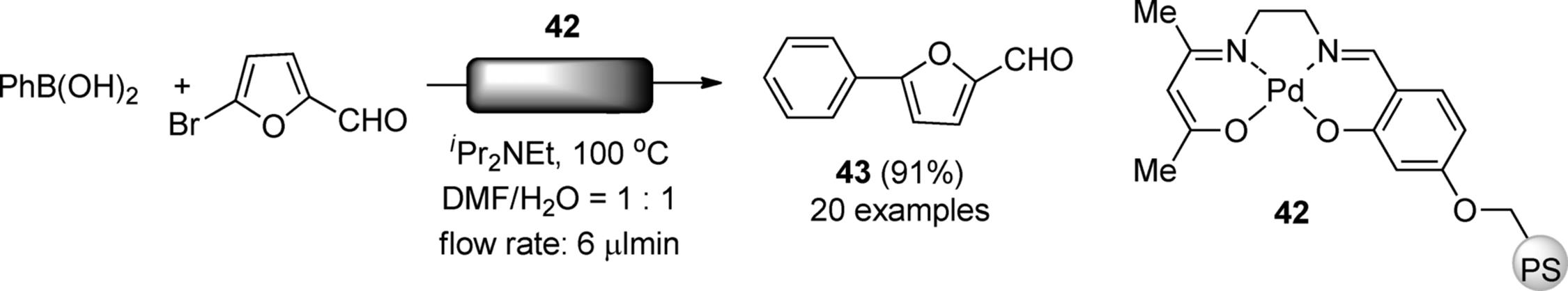
The nature of ligands, such as Boxs, phosphines, and N-heterocyclic carbenes (NHC), bound to a transition metal center usually affects the selectivities and reaction rates in the organic reactions. Styring et al. described the preparation of a Pd(II)–salen complex immobilized on Merrifield resin (42). The polymer catalyst facilitated the Suzuki-Miyaura cross-coupling reaction in a continuous-flow system (Scheme 7.32) [130]. The reactor continuously afforded biaryl adducts (43) from aryl and heteroaryl bromides in good yields. The intensification of the process over the stirred batch reaction was estimated to represent a 20-fold increase in the reaction rate due to increased reagent–catalyst contact. The residence/space time on the reactor was 10.5 min, compared with 24 h in batch. Flow Kumada-Tamao Corriu coupling of aryl magnesium bromide was also achieved by using Ni-salen-functionalized catalysts [131, 132].
Scheme 7.32 Immobilized Pd-salen complex for Suzuki–Miyaura coupling.
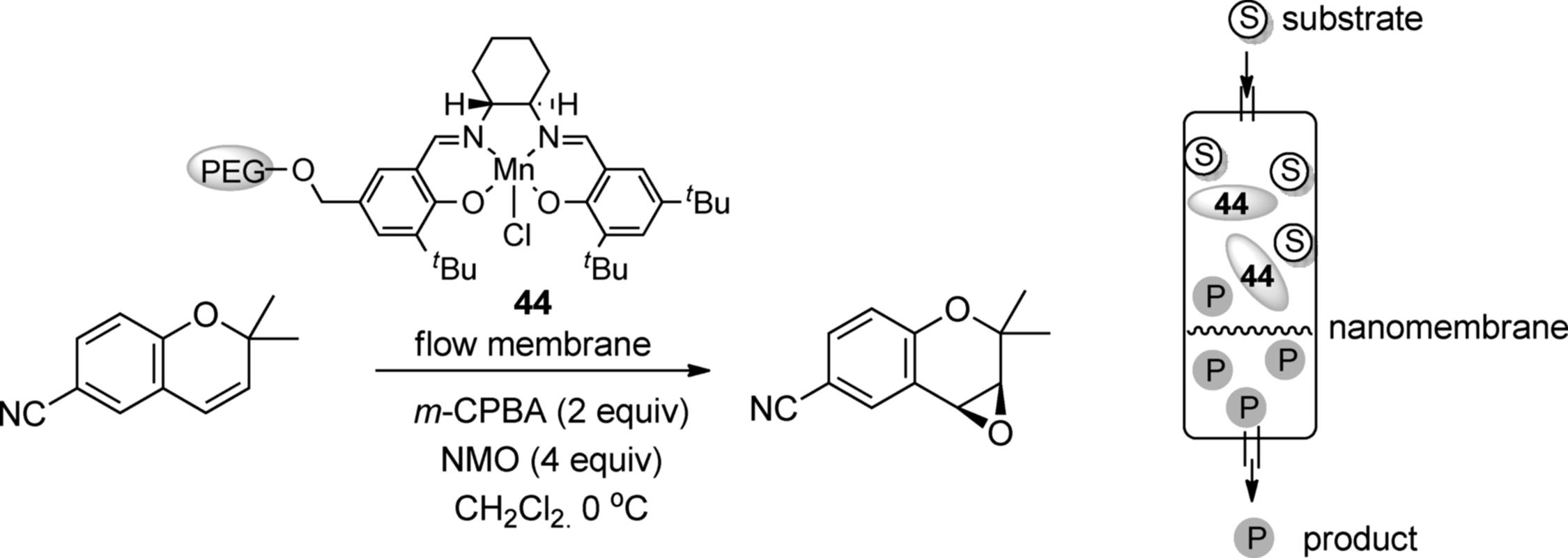
Chiral manganese salen catalysts have been widely used for the asymmetric oxidation of unactivated olefins. The dendritic polyglycerol-supported Mn-salen catalyst (44) was developed for the asymmetric epoxidation of the chromene derivative in a continuous membrane flow reactor. This flow system involves the continuous removal of the product (and unreacted substrate) from the high-molecular-weight dendritic catalyst (44) by filtration through a nanomembrane (Scheme 7.33). Under optimal conditions, 70% conversion with up to 92% ee was achieved [133]. In this system, however, the dendritic catalyst (44) worked as a homogeneous catalyst rather than a heterogeneous one.
Scheme 7.33 Asymmetric epoxidation using a dendritic polymer catalyst and a nanomembrane flow system.
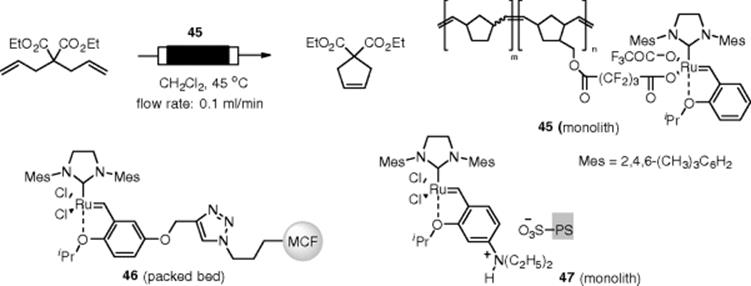
Grubbs olefin metathesis, which allows the exchange of substituents between different olefins (transalkylidenation), has become one of the most useful CߝC bond formations in synthetic chemistry. A variety of Grubbs' catalysts and related catalysts have been developed (Scheme 7.34). Nuyken and Buchmeiser reported a ring closing metathesis (RCM) under flow conditions using an immobilized Grubbs' catalyst [134]. For immobilization, a monolithic support was synthesized by a ring-opening metathesis polymerization reaction (ROMP) by applying a Grubbs' first generation catalyst. A Grubbs-Hoveyda catalyst was installed into the carboxylic side chain to give the porous monolithic catalyst (45). Although the catalyst achieved a turnover number TON) of up to 500 in the flow RCM of 1,6-diene, its catalytic activity fell to about one-fifth after 140 min. Lim et al. reported a siliceous mesocellular foam microparticle (MCF)-supported Grubbs-Hoveyda catalyst for RCM. Due to the inhibition of (46) by the generated by-product ethylene, its catalytic activity decreased rapidly. To overcome this problem, a circulating flow reactor that allows the in situ removal of ethylene was developed [135]. Based on a different concept, a non-covalent immobilization of a Grubbs catalyst (47) was prepared by Kirschning and Grela [136]. The Hoveyda-Grubbs catalyst was modified by an additional amino group, and immobilization was achieved by treatment with sulfonated polystyrene to form the corresponding ammonium salt. The amino group plays a twofold role, being first an active anchor for immobilization and second, after protonation, activating the catalysts.
Scheme 7.34 Flow metathesis catalysts.
The asymmetric reduction of ketones under continuous-flow conditions was investigated using polymer-supported ligand-metal complexes. A ruthenium catalyst bound to a chiral ligand (48) immobilized onto modified silica gel was employed to catalyze an asymmetric hydride transfer (Meerwein-Ponndorf-Verley reduction: MPV reduction). Under optimal flow conditions, the reduction of acetophenone in isopropanol provided phenetylalcohol in 95% yield with 90% ee (Scheme 7.35) [137]. A Ru-salen complex fixed on polysiloxane for asymmetric MPV reduction was prepared by Liese's group. The catalyst continuously provided high ee of chiral alcohols by using a flow membrane reactor [138]. Polystyrene-supported Ru-diarylethylenediamine (DPEN) achieved an asymmetric reduction of alkyl aryl ketones with formic acid-triethylamine [139].
Scheme 7.35 Asymmetric Meerwein-Ponndorf-Varley (MPV) reduction under flow conditions.

As a different immobilization strategy for the heterogeneous asymmetric catalyst, Ding et al. developed a “self-supported” polymer catalyst for the flow asymmetric reduction [140]. Self-assembly of chiral multitopic ligands and reactive metal ions afforded homochiral metal–organic coordination polymers, which exhibited extremely low solubility in common organic solvents. A self-supported Rh-BINOL catalyst (49) showed high catalytic performance for the asymmetric hydrogenation of α-dehydroamino acids with excellent enantioselectivity under flow conditions (Scheme 7.36). The leaching of Rh from the system was quite low (0.13 ppm).
Scheme 7.36 A self-supported heterogeneous catalyst for flow asymmetric hydrogenation.
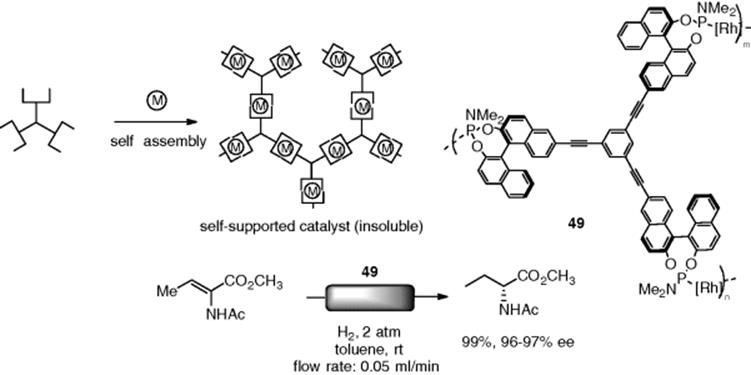
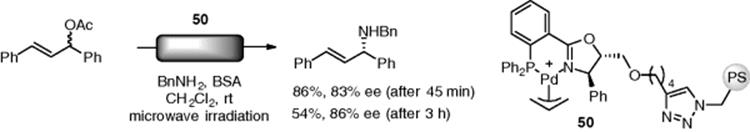
Pericàs et al. prepared polystyrene-supported chiral phosphinooxazoline (PHOX) ligands having a 1,2,3-triazole tether, which was constructed by a 1,3 dipolar cycloaddition. The catalyst (50) gave allylamine from racemic allyl acetate in high yield with excellent enantioselectivity under microwave-assisted continuous-flow conditions (Scheme 7.37) [141]. Although the enantioselectivity was not changed, the catalytic activity of the polymer catalyst was decreased after 3 h.
Scheme 7.37 A flow asymmetric allylic amination reaction.
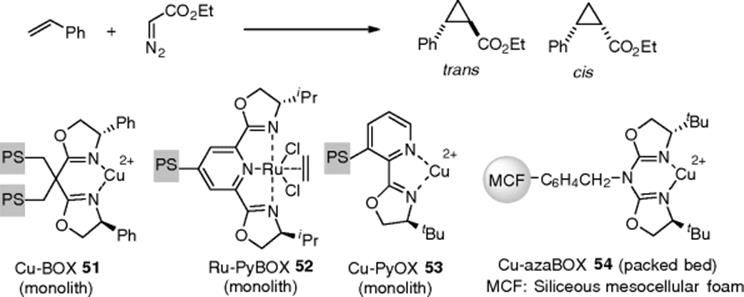
A Cu-Box complex supported on monolith (51) was developed for the enantioselective cyclopropanation of ethyl diazoacetate. The flow reactions using (51) provided an increase in enantioselectivities of about 20% relative to those for the homogenous batch process (Scheme 7.38) [142]. Pyridine-oxazolidine based monoliths (52) and (53), whose central metals were Ru and Cu, respectively, were also developed [143, 144]. Mesoporous silica was utilized as a support for the Cu-Box complex for asymmetric cyclopropanation in a flow reactor. Aza(bisoxazoline) was easily immobilized on siliceous mesocellular foam MCF) microparticles, which are generally useful for fixation of bulky complexes. A flow circulating system through a packed-bed reactor consisting of the MCF-supported catalyst (54) offered an enantioselectivity and a yield as high as those in cyclopropanation as the homogeneous counterpart, and excellent recyclability [145].
Scheme 7.38 Immobilized Box and related ligands for asymmetric cyclopropanation.
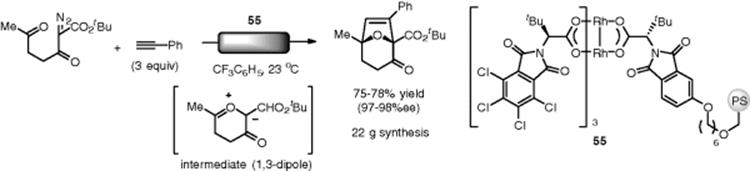
Hashimoto and coworkers accomplished the immobilization of chiral dirhodium(II) catalyst on a polystyrene-based copolymer. The polymer catalyst (55), which was packed in a gravity fed column, was successfully applied in a domino carbonyl ylide formation – dipolar cycloaddition under continuous-flow conditions (Scheme 7.39). The desired bicyclic adduct was obtained in high yield and high levels of asymmetric induction (up to 99% ee). The flow reactor was demonstrated by the retention of activity and enantioselectivity even after 60 h with a low metal leaching level (2.1 ppm) [146].
Scheme 7.39 Enantioselective carbonyl ylide cycloaddition using a continuous-flow system.