Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
9. Gas–Liquid Reactions
9.3. Gas–Liquid Reactions
Details on gas–liquid applications in microstructured reactors can be found as well in the book [4] and in the reviews [26–28].
9.3.1 Direct Fluorination of Aromatics
9.3.1.1 Direct Fluorination of Aromatics
Fluorinated aromatics are particularly important as intermediates in drug synthesis for pharmaceutical industry. Fluorinations of aromatics are typically carried out as multistep reactions via the Balz–Schiemann route introducing the fluorine moiety through the diazonium BF4− precursor. In selected cases, the Halex process is applied which is a nucleophilic aromatic substitution and only works with selected aromatic compounds having a special substituent pattern. For several decades, attempts were made to use highly reactive elemental fluorine in a direct route and to avoid the circumventing chemistry in solution. However, this route proved to be unselective and harmful [107, 108]. This is due to the radical nature of the direct process (under most conditions) being so fast and exothermic that large amounts of heat are released, which have an autoaccelerating effect on the formation of radicals. As a consequence of this ever increasing conversion, explosions happened frequently in direct fluorinations [108, 110]. Even when carried out safely, fluorine radicals react via nonselective pathways yielding a broad spectrum of side and consecutive products [109, 110]. Even chain growth to oligomerization is not uncommon, coating and blocking reactors by precipitation. Furthermore, the insolubility of fluorine in most solvents challenges reactant dosing, because the gas–liquid interface, typically not well defined, is now the means to determine and control mass transfer and reaction rate and selectivity.
Operation under extreme dilutions and/or extremely reduced temperatures (cryogenic) [109, 111–114] are possible ways for reaction control. However, not really far-fetching ones since these measures also hamper practical exploitation and are limited to mechanistical and analytical studies. In addition, a few modern concepts make use of clever combinations of solvents and processing and demonstrate increased selectivity with safe operation at ambient temperature on laboratory scale [114]. Both the low- and the high-temperature studies confirm that an electrophilic pathway is possible with direct fluorination, as given for the other halogenations such as chlorinations and brominations and being much more selective than radical chemistry. Still, a completely different approach not fixed to certain boundary conditions is desired.
The issues to be solved for direct fluorinations are heat release and mass transfer via the gas–liquid interface. Multiphase microstructured reactors enable process intensification [4, 26–28, 115–117]. Often geometrically well-defined interfaces are formed with large specific values, for example, up to 20 000 m2/m3 and even more. These areas can be easily accessible, as flow conditions are often highly periodic and transparent microreactors are available. For the nondispersing microreactors, the specific interfaces are quasi constant for the whole operation time. This enables to have high local fluorine concentrations at the interfaces and to have control over them. The large heats released can be transferred by virtue of the small fluid layers and integration of micro heat exchangers. As last, microreactors allow to set defined and short reaction time which are necessary as opposing measure to the aggressive conditions deliberately faced.
Direct fluorination of toluene was achieved using the falling film microreactor and the micro bubble column [115, 116, 118, 119], dual- or multichannel microreactor [49, 120, 121]. Even extreme conditions were realized such as operation at high substrate concentration (typically about 0.1 M, but also up to 1.0 M), large fluorine gas contents (up to 50%), and at comparatively high temperatures (−10 °C up to room temperature instead of cryogenic conditions). Mono-fluoro toluenes were generated with yields up to 28%. Conversions of 44–77% and yields of 60–78% were obtained in a dual-channel microreactor using varying contents of a special solvent mixture (formic acid–acetonitrile) [65].
The distribution of isomers is consistent to an electrophilic mechanism with fluorine cation intermediates.
(9.2) 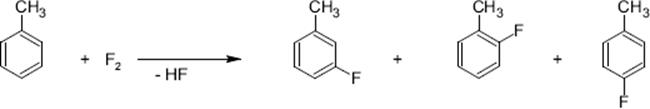
A small amount of side-chain fluorination was observed [65]. Benzyl fluoride was formed in about the same extent as the meta-isomer. Small amounts of difluoro- and trifluorotoluenes as well as some unidentified high-boiling compounds were found as well [65]. Addition products were not detected [119].
The reaction cannot be performed in fully fluorinated apolar solvents such as octafluorotoluene, which is probably because of missing polarization of fluorine that is necessary to attack the aromatic system. Polar solvents such as methanol or acetonitrile provide better environments for fluorination [119]. Formic acid as a protic solvent gives even better results and is also quite inert as this small molecule has no labile site for fluorine attack [65].
Conversion rises, as expected, with increasing temperature, as to be expected [119]. For the falling film microreactor, conversion is increased from 15% to 30% when heating up from −40 °C to −15 °C. The selectivity varies largely and exhibits no clear trend.
Using the falling film microreactor or a microbubble column, yields of up to 28% were obtained with acetonitrile as solvent at conversions ranging from 7% to 76% and selectivities from 31 to 43% with regards to the monofluorinated product [119]. Using the dual-channel reactor, conversions from 17% to 95% and selectivities from 37% to 10% were achieved using methanol as solvent [65]. The conversion of a laboratory bubble column, taken for comparison, ranged from 6% to 34% with selectivities of 17–50%, which is equivalent to yields of 2–8% [119].
The most striking point about the fluorination results is the high intrinsic speed of the reaction (Figure 9.29). The falling film microreactor was operated at seconds scale and the microbubble column even at microseconds scale [119]. This is in contrast to fluorinations in laboratory flasks taking hours.
Figure 9.29 Reduction of reaction times by several orders of magnitude using a falling film microreactor (FFMR) or micro bubble columns (MBC I and II, denoting different dimensions, as given in Ref. [119]) as compared to standard organic laboratory processing with a laboratory bubble column (LBC). τ: residence time. Source: By courtesy of IMM.
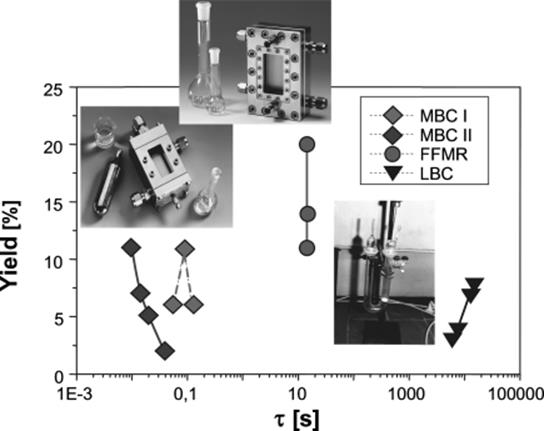
Accordingly, the respective space-time yields are higher by orders of magnitude [119]. The space-time yields for these microreactors ranged from about 20 000 to 110 000 mol mono-fluorinated product/(m3 h). The falling film microreactor had two times higher space-time yields as compared to the microbubble column. The performance of the laboratory bubble column was in the order of 40–60 mol mono-fluorinated product/(m3 h).
The fluorine content in the gas phase of a falling film microreactor was varied between 10%, 25%, and 50% [119]. A nearly linear increase in conversion results at constant selectivity. The substitution pattern, but the ratio of ortho- to para-isomers is strongly affected.
(9.3) 
Toluene fluorination with acetonitrile as a solvent in a single-, dual-, and 20-channel microreactor was performed at high superficial gas and liquid velocities (between 2.9 and 14.6 m/s) [121]. With bigger velocities, the toluene conversion increased from 63% to 76%, while the combined selectivity of ortho-, meta-, and para-fluorotoluene isomers remained constant at 26%. 20-channel reactor operating at jG = 14.6 m/s and jL = 6.1 × 10−2 m/s would provide 14 g of ring monofluorinated toluenes per day. These results suggest that for fast reactions, careful design of the gas–liquid contacting in the microchannels may lead to microreactors with high productivity.
Scale-up of the direct fluorination of a range of benzaldehyde derivatives is achieved using microstructured flow devices [121]. The results are collected in Figure 9.30.
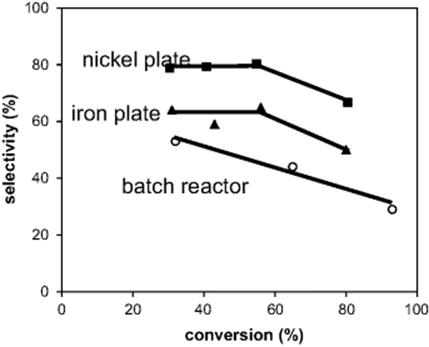
Figure 9.30 Comparison of selectivity for 1-chloromethyl-2,4-diisocyanotobenzene depending on toluene-2,4-diisocyanate using different reactor plate materials. Source: By courtesy of the Swiss Chemical Society [130].
Very large quantities of appropriate fluorinated products could be produced with the use of continuous flow process techniques [120].
9.3.1.2 Direct Fluorination of Aliphatics and Non-C-Moieties
The basic limitations of direct fluorinations are similar for aliphatic compounds as discussed already using the example of aromatic derviatives and so is the potential of microreactors. The fluorination of ethyl acetoacetate was carried out in an annular-flow microreactor.
(9.4) 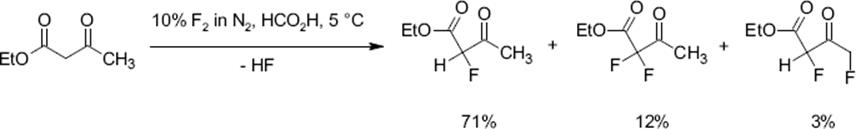
The microreactor contained a single channel single-channel microreactor with two feeds for gas and liquid [123, 124]. The gas flows were set so high that an annular flow regime was reached with a central gas core surrounded by a liquid film wetting the channel. This flow pattern has a very high interface and low liquid-side resistance due to the thin film. Formic acid was used as solvent.
High yields of the mono-fluorinated product at short reaction times were obtained which exceeded the performance of standard batch laboratory processing [122]. Yields of 72% were achieved at 99% conversion [122, 123]. The metallic construction material interacts with the reaction and impacts the keto/enol equilibrium to the advantage of the enol species which is fluorinated faster.
This microflow processing was demonstrated also fluorination of β-ketoesters such as ethyl 2-chloro-3-oxobutanoate [122, 123] or ethyl 2-methyl-3-oxobutanoate [123]. Five- and six-ring β-ketoester derivatives such as 3-acetyl-3,4,5-trihydrofuran-2-one (1) [123], 2-acetylcyclohexan-one [123], and ethyl 2-oxocyclohexane carboxylate (2) [123] were directly fluorinated as well.
(9.5) 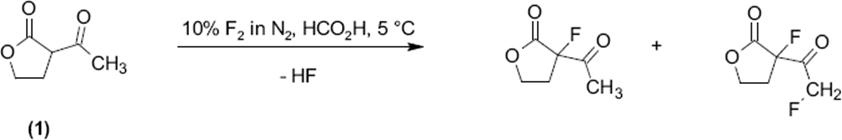
(9.6) 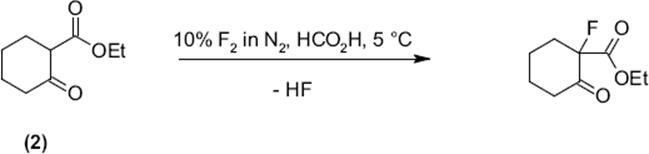
The performance of a single-channel and a numbered up three-channel microreactor was similar [123]. Some differences were found which probably origin more from fluctuations in the hydrodynamics rather than from the numbering up.
Hazardous perfluorination processes with high yields were carried out safely in microreactors such as the perfluorination of tetrahydrofurane and cyclohexane derivatives [122]
(9.7) 
(9.8) 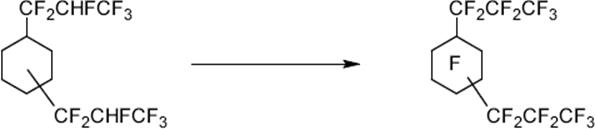
A pilot fluorination reactor was built for direct fluorination of ethyl acetoacetate and is described in Ref. [124] and reported in more detail in Section 6.3.1, page 141. A numbered up reactor with 30 microchannels was used.
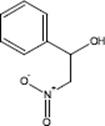
Still, many fluorination reactions are performed in a liquid phase. The safe and convenient series of fluorination methods including nucleophilic fluorination, electrophilic fluorination, and trifluoromethylation facilitated using a modular flow reactor is described in Refs [125, 126]. Nucleophilic fluorination is achieved using diethylaminosulfur trifluoride (DAST) as a commonly used reagent to lead the conversion of alcohols and carbonyl compounds to their corresponding fluoro derivatives. Its use in continuous-flow reactor is safer and more flexible than in corresponding batch reactors because of to its volatility and dismutation. A popular alternative reagent to DAST for the fluorination of organic substrates is (1-chloromethyl-4-fluoro-1,4-diazoniabicyclo-[2.2.2]octane) bis(tetrafluoroborate (Selectfluor®) that was used for electrophilic fluorination [125].
Although some of the reagents employed are known to be toxic and difficult to work with in batch mode, the flow setup proved to be very reliable and easy to use. Some of the successfully performed reactions are shown in Table 9.1.
Table 9.1 Some fluorination reactions described in Ref. [125]
|
Starting material |
Fluorination product |
Yield (%) |
|
|
|
73 |
|
|
|
97 |
|
|
|
65 |
|
|
|
87 |
|
|
|
83 |
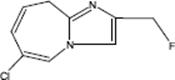
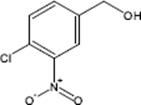
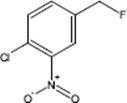
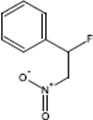
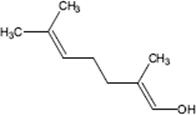
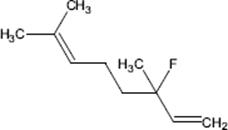
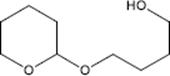
9.3.1.3 Direct Fluorination of Heterocyclic Aromatics
Selective fluorination of quinoline aromatics leads to various commercially important products such as 5-fluorouracil, 5-fluoroprimaquine, and ciprofloxacin with the fluorine moiety being decisive for their chemical and biological properties.
The fluorination of quinoline was performed in a microstructured reactor operated in the annular flow regime, which contained single-channel microreactor with two consecutive feeds for gas and liquid [127, 128]. The role of the solvent was large. The reaction was totally unselective in acetonitrile and gave only tar-like products. With formic acid, a mixture of mono- and poly-fluorinated products besides tar was formed. No tar formation was observed with concentrated sulfuric acid as solvent at 0–5 °C. In this way, a high selectivity of about 91% at medium conversion was achieved. Substitution was effective only in the electron-rich benzenoid core, and not in the electron-poor pyridine-type core. The reactivity at the various positions in the quinoline molecule is 5 > 8 > 6 and thus driven by the vicinity to the heteroatom nitrogen which corresponds to the electrophile reactivity known from proton/deuterium exchange studies in strong acid media.
(9.9) 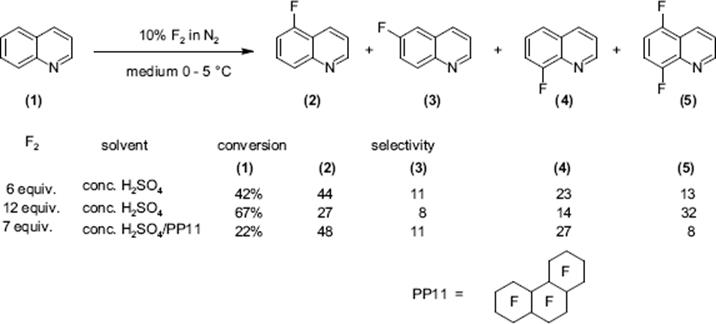
(9.10) 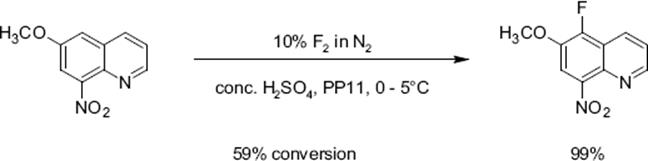
Mixtures obtained by diluting sulfuric acid by an inert perfluorocarbon fluid, acting as heat-transfer medium, were also used without loss of performance [127, 128]. Substituted quinolines allow to study the impact of electron-donating and electron-withdrawing groups on regioselectivity. Electron-donating groups such as —CH3 and —OCH3 direct the fluorine substituent into an ortho and para pattern, consistent with an electrophilic substitution path, and electron withdrawing groups such as —NO2 leads to a meta-substitution pattern.
(9.11) 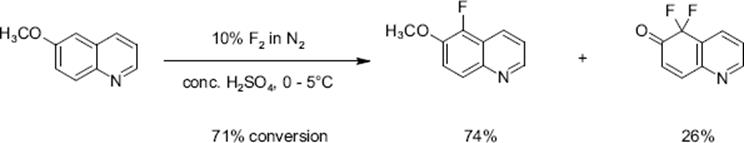
(9.12) 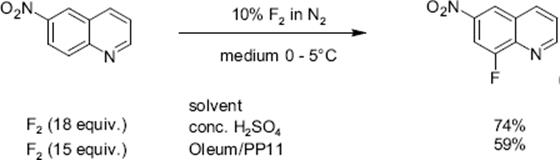
9.3.2 Oxidations of Alcohols, Diols, and Ketones with Fluorine
For some oxidations, toxic heavy metal oxidants are used [128]. This can be circumvented by use of microreactor technology due to safe handling of elemental fluorine, which can be used to mediate oxidation reactions [129]. This is possible in a direct way via fluorine introduction into the substrate and subsequent replacement by an oxygen moiety. In an indirect manner, intermediate oxygen transfer reagents such as HOF·MeCN can be generated by reaction of aqueous acetonitrile with elemental fluorine, which then attack the substrate. The only byproduct is hydrogen fluoride, which could be recycled by electrolysis.
The oxidation of cyclohexanol to cyclohexanone with fluorine and aqueous acetonitrile was performed in a single-channel microreactor operated under annular flow at room temperature. A conversion of 84% and a selectivity of 74% was observed [129]. In a similar way, diols such as oxidation of 1,2-cyclohexandiol were partly or fully oxidized. A total of 53% selectivity to the mono-oxidation product was obtained at a conversion of 87%; the di-oxidation product was obtained with 30% yield.
(9.13) 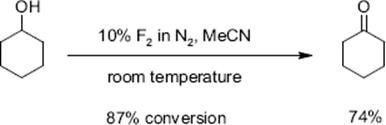
(9.14) 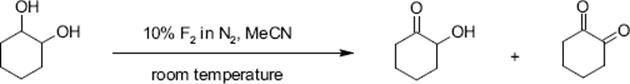
The Baeyer–Villiger oxidation of cyclohexanone to the seven-membered lactone used aqueous formic acid (5% water) as medium and a 60% conversion at 88% selectivity was found [129].
(9.15) 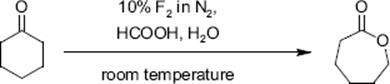
9.3.3 Photochlorination of Aromatic Isocyanates
Side-chain photo-chlorination of toluene isocyanates leads to important industrial intermediates for polyurethane synthesis, one of the most important classes of polymers [130]. Irradiated thin liquid layers in microchannels should have much higher photon efficiency (quantum yield) than given for conventional processing.
The reaction of toluene-2,4-diisocyanate with chlorine to 1-chloromethyl-2,4-diisocyanatobenzene was carried out in a falling film microreactor with a transparent window for irradiation [130]. There are two modes of reaction. The desired radical process proceeds with the photoinduced homolytic cleavage of the chlorine molecules and the chlorine radical reacts with the side chain of the aromatic compound. At too high chlorine concentrations, radical recombination becomes dominant and consecutive processes such as chlorinations of the side chain may occur as well. Another undesired pathway is the electrophilic ring substitution to toluene-5-chloro-2,4-diisocyanate, promoted by Lewis acidic catalysts in polar solvents at low temperature. Even small metallic impurities probably from corrosion of the reactor material can enhance the formation of electrophilic byproducts.
(9.16) 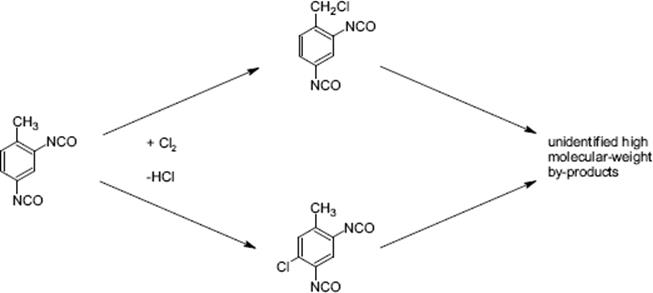
The yield of the product 1-chloromethyl-2,4-diisocyanatobenzene ranged from 24% to 54% at 130 °C (conversion: 30%–81%; respective selectivity: 79%–67%) [130]. The content of the ring-substituted product decreases with increasing conversion (12–5%), while simultaneously the other by-products were formed in much larger amount (8–29%) (Figure 9.31). This is indicative of consecutive reaction steps, that is, multiple chlorination. The superior performance of the microreactor is explained by the better photon yield because of the very thin liquid films. Owing to the low penetration of light into the conventional batch reactor, a large part of the reaction volume is actually not irradiated. Here, thermal rather than photo-induced pathways are followed which favor ring-chlorination.
Figure 9.31 Microplant for toluene sulfonation with falling film microreactor as central apparatus and unitized backbone as fluidic bus system. Source: By courtesy of Elsevier [133].

Reaction time can also be reduced by means of microprocess technology [130]. A reaction time of 30 min was necessary for a 30 ml-batch reactor to achieve a conversion of 65% at a selectivity of 45%, whereas with the falling film microreactor only ~14 s for the same performance was required. This leads also to a large space-time yield with 401 mol/(l h) for the falling film microreactor as compared to 1.3 mol/(l h) for the batch reactor. The impact of Lewis-acid formation on the reaction course was investigated using an iron microreactor instead of the usually used nickel microreactor. The plate surface is converted to iron chloride under the reaction conditions. The selectivity of the target product then drops from 67% to 50%.
9.3.4 Photoradical Chlorination of Cycloalkenes
Photo-initiated radical chlorination of cycloalkanes was investigated using microflow reactors [131]. Under natural light, microflow chlorination of cyclohexane with molecular chlorine gives chlorocyclohexane with high selectivity (>95%) and yield of 20% (Table 9.2).
Table 9.2 Microflow chlorination of cyclohexane with sulfuryl chloride [131]

The continuous microflow system was composed of a T-shaped micromixer (i.d. = 500 μm) and the reactor, two gas/liquid feed stainless tube and an outlet.
Single-chlorination reaction of cycloalkanes with sulfuryl chloride can also be successfully carried out using a microflow reactor under irradiation using a 15 W black light.
Also chlorination using sulfuryl chloride as a chlorination reagent is examined [131]. The Mikroglas Dwel Device (Foturan glass, 1000 μm width, 500 μm depth, and 1.9 m length, and total hold-up volume of 0.95 ml) was used as a microreactor.
When a solution containing sulfuryl chloride and 40 molar equivalents of cyclohexane was introduced to a microreactor with a residence time 19 min at room temperature, the reaction gave chlorocyclohexane selectively in 22% yield (entry 1). While increasing molar ratio of cyclohexane to 80 equiv. did not affect the yield of the product (20%, entry 2), extending residence time (57 min, flow rate: 1.0 ml/h) raised product yield to 35% (entry 3).
9.3.5 Mono-Chlorination of Acetic Acid
Selectivity is a major issue for the mono-chlorination of acetic acid to chloroacetic acid since second chlorination gives dichloroacetic acid and other chlorinated species including acid chloride formation [132]. The removal of these impurities, especially of the dichloroacetic acid is laborious and costly, either by crystallization or by reduction with a palladium catalyst.
(9.17) 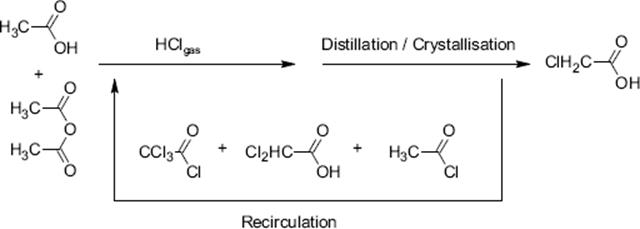
A yield of 85% was obtained with a falling film microreactor, which outperforms large-scale bubble column processing [132]. Selectivity was also superior with <0.05% dichloroacetic acid formed. Conventional processing has much higher levels of impurity (~3.5%). When both, temperature and pressure, were slightly increased, the yield raised from 85% to 90%. The content of dichloroacetic acid was still neglegible (<0.05%).
9.3.6 Sulfonation of Toluene
The sulfonation of toluene proceeds via a complex scheme of elemental reactions with numerous side and consecutive reactions. Toluene sulfonic acid and sulfur trioxide can react in a consecutive process to give toluene pyrosulfonic acid (2), which can undergo further reactions [133, 134]. Reaction of (2) with another molecule of toluene yields two molecules toluene sulfonic acid (3) or di-tolyl sulfone (4). In addition, toluene sulfonic anhydride may be formed via reaction of toluene pyrosulfonic acid with toluene sulfonic acid (5).
Precise control of concentration and residence time can increase the selectivity of the sulfonation of toluene, as this allows to optimally set the interplay between the reactions (1) to (4) [133, 134]. The highly exothermic nature of the reaction demands for good temperature control. A single microreactor is not suited to conduct the various reaction steps with all their different needs on temperature and residence time. Thus, a continuously operated plant with many microflow tools was developed. The plant design was based on a fluidic backbone providing unitized ports and plant unit sites in order to facilitate the connection of microstructured components from different suppliers (Figure 9.32).
(9.18) 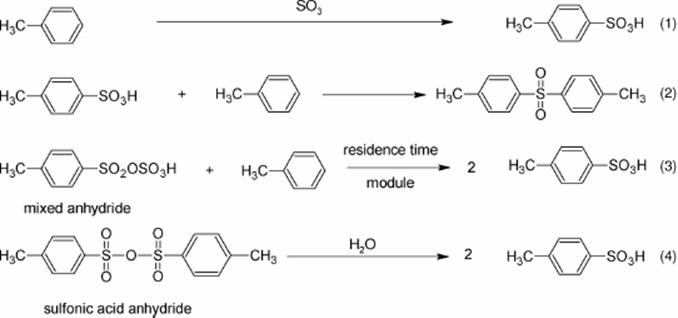
Figure 9.32 Process flow sheet for the microplant used for toluene sulfonation. Source: By courtesy of Elsevier [133].
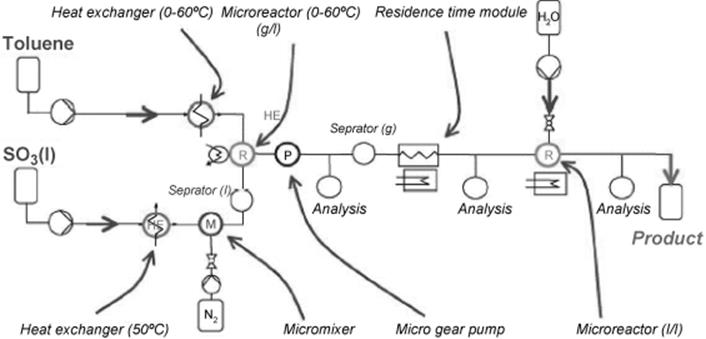
The process flow sheet of the microreactor plant is shown in Figure 9.33 [133, 134]. Toluene is heated to 40 °C using a microstructured heat exchanger while, at the same time, liquid sulfur trioxide is vaporized by heating to 60 °C. Nitrogen is added to the sulfur trioxide gas in a micromixer for dilution. This stream is then passed into a separator to remove liquid contents before entering the microstructured falling film reactor where it reacts with the liquid toluene. Following the discussion on the chemistry given above, a delay-loop reactor had to be added to complete toluene pyrosulfonic acid conversion. This was achieved in a heated wound tube. The need for anhydride hydrolysis demands for a further processing step, which is performed in a tempered liquid–liquid microstructured reactor by mixing with water.
Figure 9.33 Selectivity for toluene sulfonation in a special microplant with falling film microreactor, when increasing contents of sulfur trioxide are fed. Source: By courtesy of Elsevier [133].
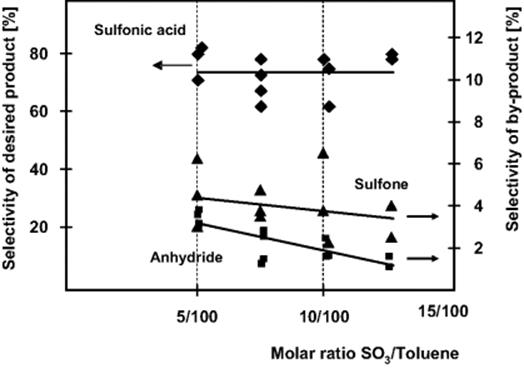
As the distribution of the products depends largely on reaction time and on the type of operations performed, several sampling stations were inserted at various positions of the microreactor plant [133, 134]. Analysis was performed directly after the falling film microreactor, where the ratio of sulfur trioxide to toluene widely varied. The selectivity for the formation of the toluene sulfonic acid is constant at about 73% for molar ratios of sulfur trioxide/toluene ranging from 5/100 to 15/100, while the selectivity for anhydride formation decreases from 8% to 2% (Figure 9.34). The selectivity for the pathway to di-tolyl sulfone is low at about 3%. If sampling is done at the stage behind the delay loop, the selectivity of toluene sulfonic acid increases to 82% because of the conversion of toluene pyrosulfonic acid with toluene. The anhydride selectivity is further decreased to 2%, while that of di-tolyl sulfone is not altered. After hydrolysis with water, a nearly quantitative conversion of the anhydride to the acid is achieved, if mixing conditions and the amount of water were optimized. Totally, a selectivity of 82% for the target product toluene sulfonic acid was achieved at nearly complete conversion. Side products are, for example, the sulfones given by reaction path (2) in the scheme above.
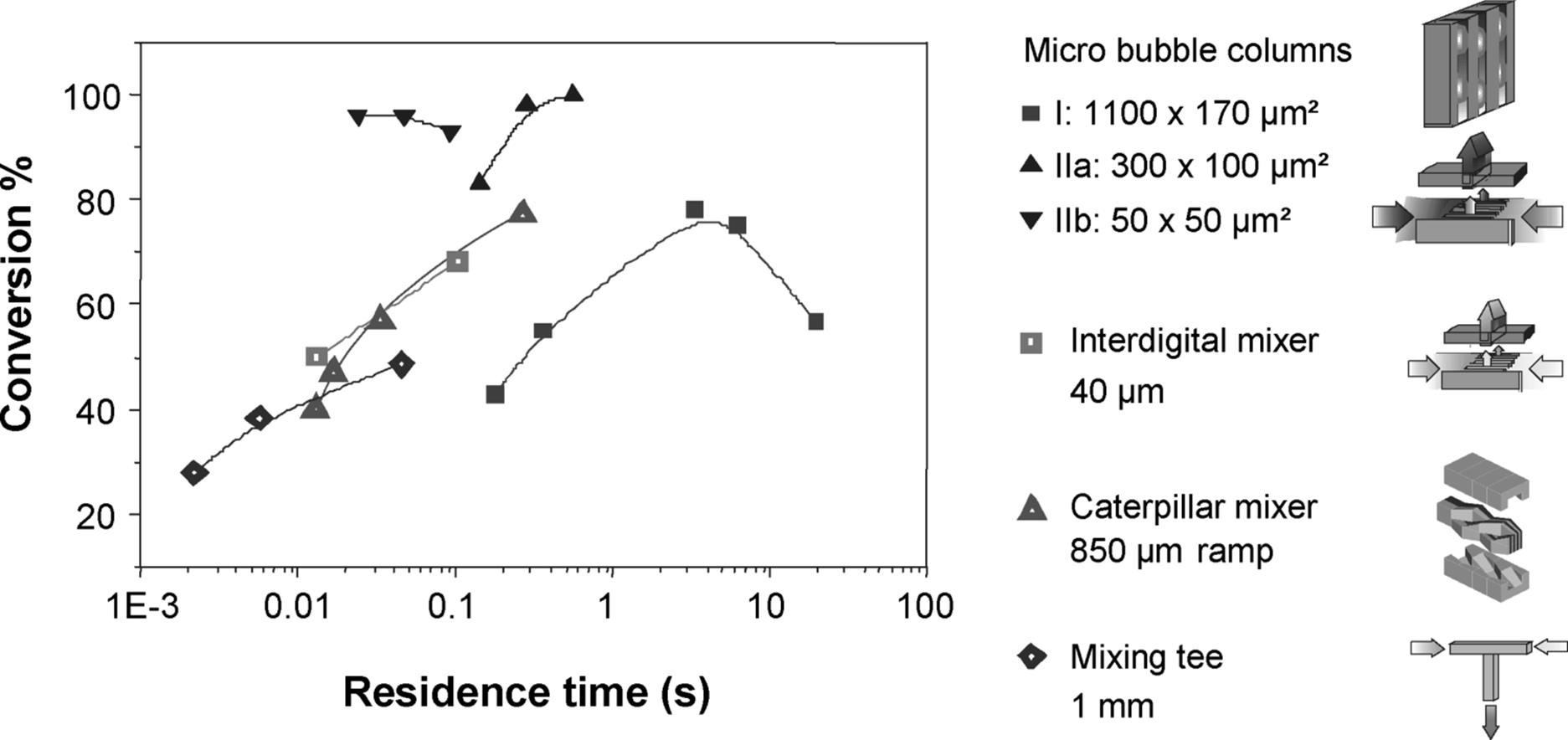
Figure 9.34 Special-type multipurpose microdevices and mixing tee used for investigation of CO2 absorption. Comparison of their reactor performance in dependence of the residence time. Micro bubble column (![]() ), (1100 μm × 170 μm); micro bubble column (
), (1100 μm × 170 μm); micro bubble column (![]() ), (300 μm × 100 μm); micro bubble column (
), (300 μm × 100 μm); micro bubble column (![]() ), (50 μm × 50 μm); interdigital mixer (
), (50 μm × 50 μm); interdigital mixer (![]() ), (40 μm); caterpillar mixer (
), (40 μm); caterpillar mixer (![]() ), (850 μm ramp); mixing tee (◊), (1 mm). Source: By courtesy of IMM [141].
), (850 μm ramp); mixing tee (◊), (1 mm). Source: By courtesy of IMM [141].
9.3.7 Photooxidation Reactions
A [4 + 2]-cycloaddition of singlet oxygen to α-terpinene in methanol yields ascaridole in the presence of catalytic amounts of Rose Bengal as photosensitizer. The reaction was carried out in one-channel chip microreactor made from glass [73].
(9.19) 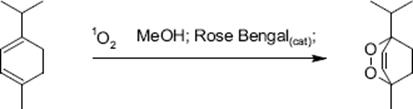
This enabled the use of singlet oxygen without need for preparation of large quantities of oxygenated solutions [73]. Further benefits of microreactor processing are to facilitate scale-up and avoid unwanted sample heating, which results in system simplification by eliminating the need for collimators and refrigeration to control the tungsten lamp power. In addition, safety issues which arise for aerated, oxygenated organic solvents can be addressed differently. Owing to the good mass transfer, no pre-saturation of the α-terpinene with oxygen is required, and after reaction the oxygenated solution can be instantly degassed with nitrogen.
The quantum yield should also be high, as the efficiency of the use of light for the reaction can be kept high through the thin liquid layer (the losses by absorption are low, respectively) [73]. Albeit this is generic to all photochemical applications, it is here of special importance because Rose Bengal has a large extinction coefficient which leads to a substantial increase in absorption. Radiation is not effective anymore after a short path length. In turn, the chip microreactor allowed the use of high sensitizer concentrations of up to 5 × 10−3 M at large optical transmittance of 95%. Conversions of about 80% were obtained at short irradiation times (5 s).
The photooxygenation of cyclopentadiene with Rose Bengal as photosensitizer was performed using a falling film microreactor in methanol [135]. The endoperoxide is first generated and then reduced to 2-cyclopenten-1,4-diol, which is used as an intermediate in pharmaceutical drug synthesis. This route is not easily possible by batch processing since the explosive endoperoxide intermediate is formed in substantial amounts.
(9.20) 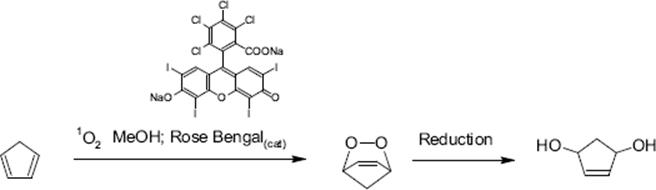
A xenon lamp was used as light source, which irradiated the thin falling films flowing through a quartz glass window in the reactor [135]. Thiourea in methanol was used to reduce the labile endoperoxide directly to the stable diol product. The yield of cis-2-cyclopenten-1,4-diol was 20% at 10–15 °C.
Another singlet oxygen photochemical oxygenation of α-terpinene was performed in a microstructured reactor [136]. A solid supported [60] fullerene photosensitizer that was grafted to Tentagel® beads was used as singlet oxygen sensitizer. Oxidation of α-terpinene was achieved within 50 s.
(9.21) 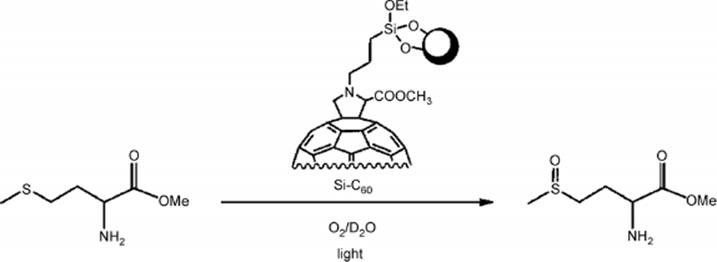
With the same fullerene-method, a quantitative conversion of methionine to the corresponding sulfoxides was achieved in about 40 s, using low power, white LED illumination. The reaction time is considerably shorter when compared to the batch procedure that requires, for the same process, about 1 h illumination and the use of a 300 W tungsten halogen lamp. This may be due to the narrow channels space in a microreactor, which favors singlet oxygen diffusion, thus determining a faster reaction.
The excellent performance of a triple-channel microreactor fabricated by means of a soft-lithography technique was demonstrated by carrying out photosensitized oxygenations of α-terpinene, citronellol, and allyl alcohols [137]. The reaction solution containing the reactant and a photosensitizer was delivered through the middle channel, whereas the outer two channels were used for oxygen supply.
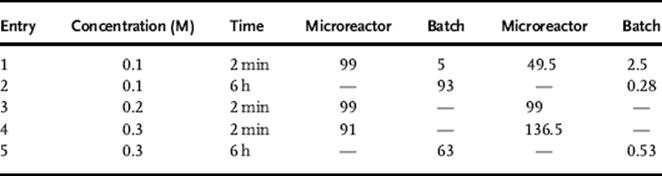
The summary of photosensitized oxygenation of (−)-citronellol and allyl alcohols in batch and in a microreactor is shown in Tables 9.3 and 9.4.
(9.22) 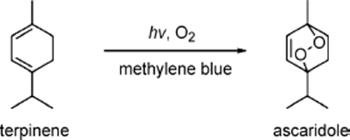
(9.23) 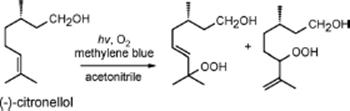
(9.24) 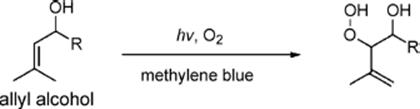
Table 9.3 Photosensitized oxygenation of (−)-citronellol [137] conversion (%)a) STY (mmol/l/min)b)
a. Conversions were determined by ![]() NMR using an internal standard.
NMR using an internal standard.
b. Space-time yield (STY) = mmol of products/(reactor volume × time).
Table 9.4 Photosensitized oxygenation of allyl alcohols [137] conversion (%)a) STY (mmol/l/min)b)
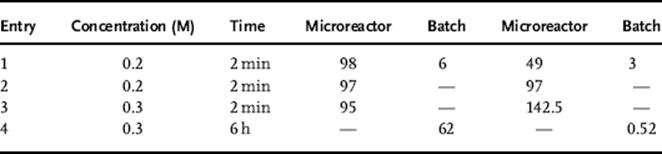
a. Conversions were determined by ![]() NMR using an internal standard.
NMR using an internal standard.
b. Space-time yield (STY) = mmol of products/(Reactor volume × time).
As a result of the increased illumination as well as the increased gas–liquid contact area per unit volume, the triple-channel microreactor exhibited better performance compared to the batch reactor, or even compared to a typical dual-channel microreactor [138, 139]. Moreover, the scaleup process using the microreactor revealed higher productivity than the batch reactor, which would be valuable for the practical applications in a broad range of gas–liquid chemical reactions.
Another interesting photo reaction performed in a microreactor is the conversion of dihydroartemisinic acid to artemisinin, an important drug used for malaria treatment [140].
(9.25) 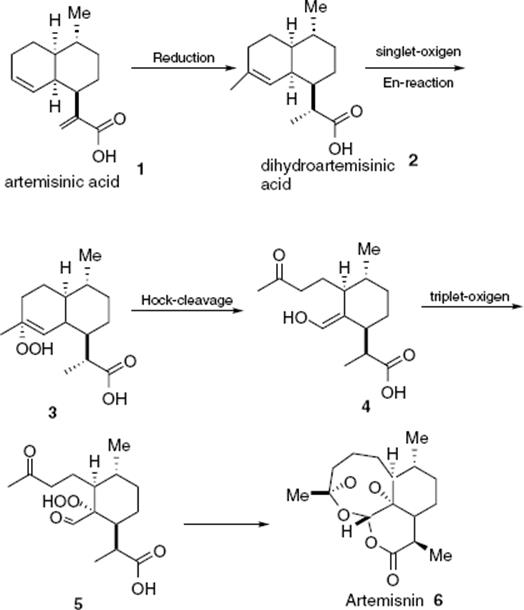
Complete reaction took about 4.5 min and gave 39% yield of final product (6). This cost efficient, continuous, and scalable process opens the possibility for continuous and affordable supply of this important drug.
9.3.8 Reactive Carbon Dioxide Absorption
This reaction serves as literature known model reaction to characterize mass transfer efficiency in microreactors [141]. As it is a very fast reaction, solely mass transfer can be analyzed. The analysis can be done simply by titration; the reactants are inexpensive, and not toxic.
(9.26) ![]()
The mass transfer efficiency of different gas–liquid contactors as a function of residence time was compared, including an interdigital micromixer, a caterpillar minimixer, a mixing tee, and three microbubble column with microchannels of varying diameter (Figure 9.35) [141]. The two microbubble columns comprising the smaller microchannels reached almost 100% conversion. The microbubble column with the largest hydraulic diameter reached at best 75% conversion. The respective curve passes over a maximum due to the antagonistic interplay between residence time and specific interfacial area.
Figure 9.35 Comparison of nitrobenzene conversion and aniline selectivity as a function of reaction time for the incipient wetness catalyst. NB: nitro benzene and AN: aniline. Source: By courtesy of Elsevier [150].
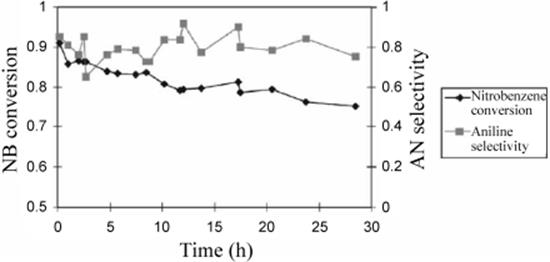
All other devices showed only the increasing part of such dependency, that is, the highest performance was obtained at the longest residence time [141]. The best conversions of interdigital micro and caterpillar minimixers of ~78% and ~70%, respectively, still exceed notably the performance of a conventional mixing tee (1 mm inner diameter).
The mass-transfer efficiency of the falling film microreactor was determined at various carbon dioxide volume contents (0.1; 1.0; and 2.0 M) [141]. The molar ratio of carbon dioxide to sodium hydroxide was constant at 0.4 for all experiments, that is, the liquid reactant was in light excess. The higher the base concentration, the higher was the conversion of carbon dioxide. For all concentrations, complete absorption was achieved; however, at different carbon dioxide contents in the gas mixture. The results show the interdependency of the carbon dioxide content, the gas flow velocity, and the sodium hydroxide concentration.
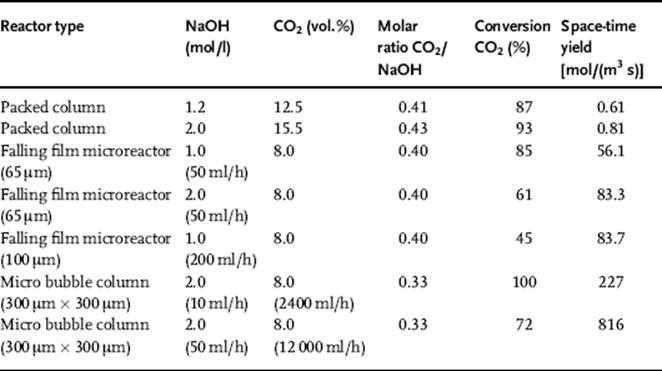
The mass transfer efficiency of the falling film microreactor and the micro bubble column was compared quantitatively to literature reports on conventional packed columns (see Table 9.5) [141]. The process conditions were chosen as similar as possible for the different devices. The conversion of the packed columns was 87–93%; the microdevices had conversions of 45–100%. Furthermore, the space-time yield was compared. Here, the microdevices resulted in larger values by orders of magnitude. The best results for falling film microreactors and the microbubble columns were 84 and 816 mol/(m3 s), respectively, and are higher than for conventional packed bed reactors of about 0.8 mol/(m3 s).
Table 9.5 Comparison of space–time yields for CO2 absorption when using microdevices or conventional packed columns.
Source: By courtesy of IMM [141].
A more detailed mass-transfer study on the carbon dioxide absorption in sodium hydroxide solution was performed using a falling film microreactor, both experimentally and numerically [142, 143]. Experimental investigations were made at a liquid flow of 50 ml/h, with three NaOH concentrations (0.1, 1, and 2 M), at a fixed inlet molar ratio CO2:NaOH of 0.4, and for a range of CO2 concentration of 0.8–100%. A two-dimensional reactor model was developed and the results are similar to the experimental data at low NaOH concentrations (0.1 and 1 M). The agreement is less pronounced for higher concentrations such as 2 M NaOH, which could be explained by either maldistribution of the liquid through the reactor channels or model simplifications. The model indicates that carbon dioxide is consumed within a short distance from the gas–liquid interface.
A numerical model accounts for the variation in liquid film thickness and back mixing effects. It can be used for a wide range of process conditions in microstructured falling-film contactors. It enables also investigation of physical and chemical absorption systems with slower reaction kinetics, in which the mass-transfer resistance is concentrated in the liquid phase [143].