Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
10. Bioorganic and Biocatalytic Reactions
10.2. Bioorganic Syntheses Performed in Microreactors
Many reactions for biomolecular synthesis were performed using microfluidic reactors. Microreactors offer several advantages over conventional reaction systems, one of which is the control of mixing reagents and reaction time, which is simply achieved by controlling the length of the microchannel or by integrating a micromixing device. Such control of fluid allows regulation of reaction by mixing. Basically, syntheses of biomolecules in microreactors are performed by organic synthesis or by biochemical conversion. The reactions are basically carried out in organic media hence, generally, silicon or glass as material for the microreactors has been widely used.
10.2.1 Biomolecular Syntheses in Microreactors: Peptide, Sugar and Oligosaccharide, and Oligonucleotide
10.2.1.1 Peptide Synthesis
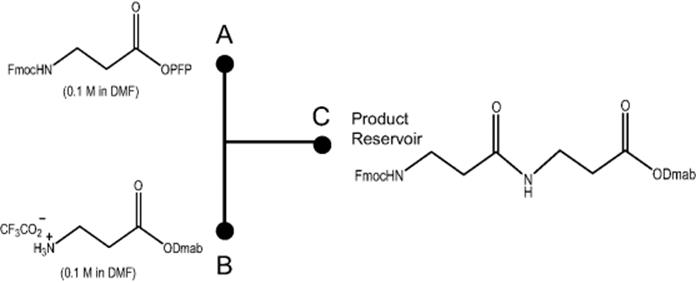
Peptide synthesis that was realized by multistep synthesis by utilizing a microfluidic reactor was demonstrated by Watts and his group [17–19]. The authors reported the synthesis of β-peptides using a borosilicate glass microreactor, which was produced by photolithographic and wet etching methods. The reagents were immobilized by electroosmotic flow (EOF). They initially prepared dipeptide from preactivated carboxylic acids. The methodology has been extended such that the peptides may also be produced via the pentafluorophenyl ester derivatives of amino acids using a microreactor as illustrated in Scheme 10.1. They demonstrated that performing such reactions in a microreactor operating under electrokinetic control represented a significant increase in yield over the traditional batch method. They postulated that the enhancement in the rate of reaction is an electrochemical phenomenon due to the reaction being performed in an electric field. It was also demonstrated that selective deprotection of the resultant dipeptides can be achieved. The authors used the methodology to consecutively react pentafluorophenyl esters and amines to produce a library of peptides within a computer controlled microreactor system operating under EOF [18].
Scheme 10.1 Schematic of microreactor manifold. Source: Reprinted with permission from Ref. [19]. Copyright 2004 Elsevier.
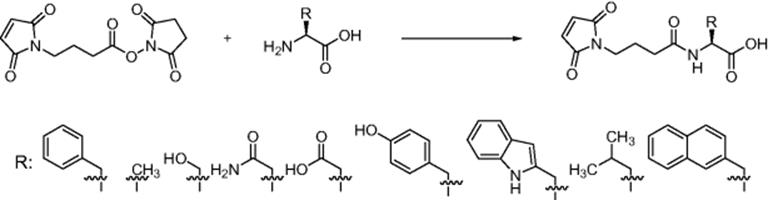
Biomolecular synthesis depends on the molecular conformation, steric hindrance, and electronics of both reaction partners. Predicting the outcome of the reaction is complicated by the difficulty in predicting the reactivity of the coupling partners. Moreover, reaction variables such as stoichiometry, concentration, temperature, reaction time, and activator play an undisputable role in the outcome of a given synthesis. Given such requirements, microfluidic system serves as an ideal tool to achieve successful synthesis. For instance, Miyazaki's group [20] demonstrated the utility of microreactor as an active reaction vessel for performing biochemical synthesis. An important feature of the microfluidic systems is their superior controllability of fluids, which cannot be achieved in the conventional large-scale batch reactions. The effects of a microfluidic system on chemical reactions of two different miscible solutions were examined using amino acid substitution as a model reaction. A simple condensation reaction of the amino group with an active ester using a dimethylformamide (DMF) solution of N-(4-maleimidobutyloxy) succinimide (GMBS) and an aqueous solution of phenylalanine (Scheme 10.2) was performed in a microfluidic system. Phenylalanine substitution in a microreaction system using separate solutions was more efficient than batch reaction with reaction yields at slower flow rate exceeding 90% in a 10 min reaction time compared with 80% yield after 12 h in batchwise reaction. Comparison between microchannel reactor using separate solutions and microchannel reactor using premixed homogenous mixture showed that while yield exceeded 90% for separate solutions, the reaction did not proceed in a premixed homogenous solution even with slower flow rate (<5 μl/min). Such results show that formation of interface between the water and DMF solutions in a microchannel affects the reaction. The microchannel used in the experiment is composed of straight channels and several turns. Secondary flow occurs at the turn structure of the microreactor, which disrupts the solution interface in a water–water system. The same effects also exist in DMF–water systems and enable efficient mixing at higher flow rates while at lower flow rates, diffusion mainly affects mixing. Substitution of other amino acids showed that reaction enhancement is caused by localization of hydrophobic amino acids at the interface of DMF and water. Using the rapid Michael addition reaction of the SH group of cysteine to a maleimide group, it was also demonstrated that such reaction involving hydrophilic compounds diminished in a microfluidic system. The results are proofs that microreactors are novel apparatus for regulating chemical reactions, depending on the structure of the reactant molecules, via control of fluid that enables regulation of reaction by mixing.
Scheme 10.2 Reaction of amino acids with active ester [20]. Source: Copyright 2004 The Royal Society of Chemistry.
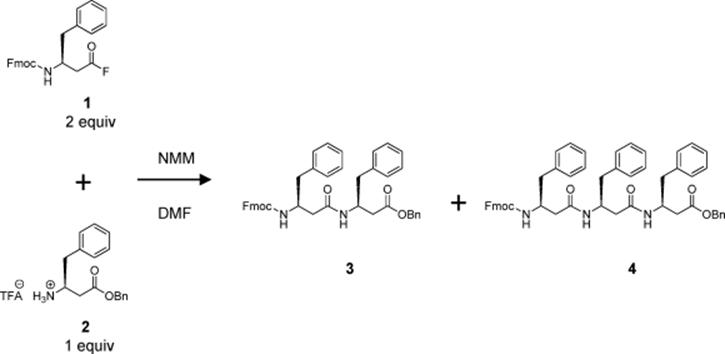
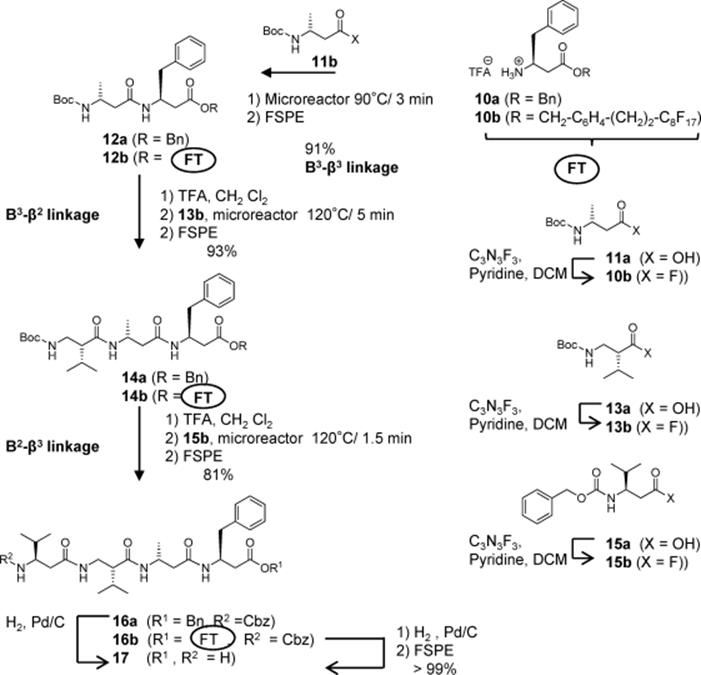
Seeberger's group described the first application of a silicon microreactor for the synthesis of β-peptides [21]. They demonstrated that reaction conditions can be rapidly optimized and larger amounts of peptide can be produced using a microreactor. The microreactor allowed for detailed investigation and optimization of reaction parameters using minimal amount of reagents. Moreover, it paved the way for the use of unprecedented high reaction temperatures, leading to homogenous reaction mixtures and significantly reduced reaction times. The authors described the first peptide coupling reactions of Boc- and Fmoc-protected amino acids within 1–5 min at an unusually high temperature of 120 °C, the use of β2- (Scheme 10.3) and β3- (Scheme 10.4) homo amino acid fluorides for the peptide coupling reaction, and the successful application of a C10H4F17-substituted benzylic ester protecting group (Scheme 10.5) for peptide synthesis in solution. The method is capable of large-scale production.
Scheme 10.3 Synthesis of dipeptide (3) in the microreactor.
Scheme 10.4 Synthesis of the β3 dipeptides (7) and (9) in a continuous flow microreactor. 2-ClZ = 2-chlorobenzyloxycarbonyl.
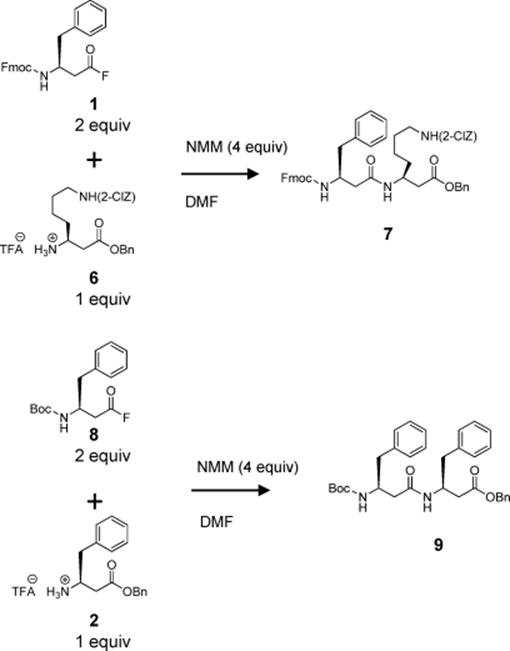
Scheme 10.5 Synthesis of the β-tetrapeptide in a continuous flow microreactor.
Cell-free protein synthesis was performed using polydimethylsiloxane (PDMS)-based microreactor arrays [22]. The microreactor array chip comprised a temperature control chip made of glass and a disposable reaction chamber chip made of PDMS. To evaluate the performance of this microreactor array, rat adipose-type fatty acid binding protein, glyceraldehyde-3-phosphate dehydrogenase, cyclophilin, and firefly luciferase were synthesized from their respective DNA templates using a cell-free extract prepared from Escherichia coli.
10.2.1.2 Sugar and Oligosaccharide Synthesis
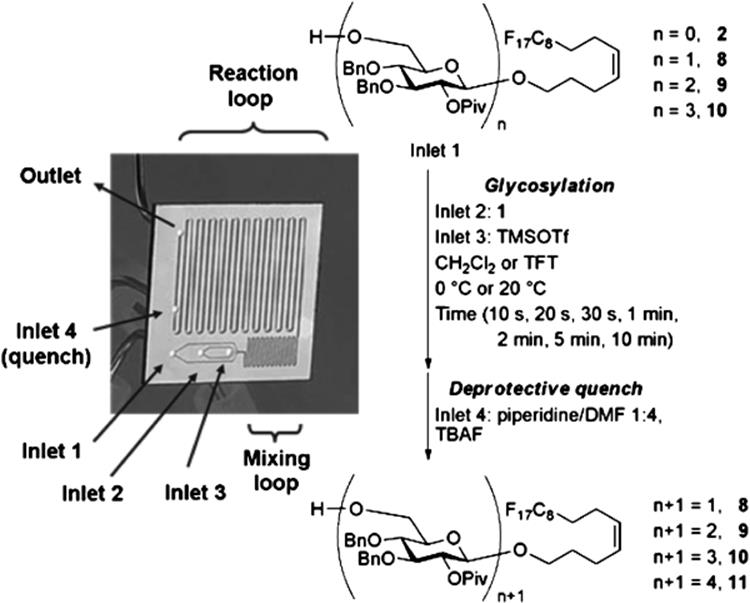
Synthesis of oligosaccharides is one of the challenges in the field of synthetic organic chemistry. Glycosylation reaction is difficult to carry out due to its dependence on a wide variety of factors such as nature of coupling partners, concentration and solvent, stoichiometry, temperature, and reaction time. Moreover, the building blocks used for oligosaccharide assembly are precious synthetic intermediates and often require multistep syntheses. Slow batch-wise reaction was overcome using the microreactor. The silicon microreactor from Jensen was used by the Seeberger's group to systematically study glycosylations (Scheme 10.6) [23]. The silicon-glass microreactor was chosen for its excellent thermal conductivity and stability to a broad range of organic solvents and reagents. A five-port silicon microreactor was designed with three primary inlets to mix and react glycosylating agent, nucleophilic (acceptor), and activator. Glycosylation reactions were performed rapidly over a wide range of conditions using this microreaction system (Figure 10.1). They monitored the reaction progress of the coupling between protected mannoside and galactoside and found it to be a function of temperature and time, wherein, low temperatures (−80 to −70 °C) and short reaction times of <1 min favored orthoester formation while higher temperature (−40 °C) and longer times of ~4 min favored formation of the desired α-linked product. Glycosylation reactions were performed rapidly over a wide range of conditions using the microreactor within a day. This microfluidic system enables easy handling of hazardous reagents such as trimethylsilyl trifluoromethanesulfonate at microliter scale. In addition to reaction optimization, the internal volume of 78.3 μl renders it suitable for reaction screening and larger scale production.
Figure 10.1 Optimization of glycosylation reactions with the use of a silicon-based microreactor [27]. Source: Copyright 2007 American Chemical Society.
Scheme 10.6 (a) Disaccharide formation by glycosylation reaction of protected mannoside (glycosyl donor) and galactoside (acceptor). (b) Glycosylation reaction involving mannosyl donor and acceptor to form dissacharide. Orthoester was also formed.
Another experiment reported by Fukase's group described continuous flow synthesis by integration of a microreactor and a synthesis based on affinity separation (SAS) system for high throughput synthesis of oligosaccharide [24]. In their synthetic methodology, SAS, the desired tagged compound was separated from the reaction mixture by solid-phase extraction using specific molecular recognition of the tag. In their experiment, a new SAS method using a podand-type tag, a pseudo-benzo-31-crown-10 structure was used for oligosaccharide synthesis. After glycosylation of the tagged monosaccharide with a glycosyl donor, the reaction mixture was subjected to the affinity separation. Separation of the desired compounds possessing the podand tag was achieved by the affinity between the podand and the ammonium ion.
In a different approach, transgalactosylation, which is the reverse reaction of the hydrolysis reaction by glycosidase, was carried out in organic solvent–buffer system as a reaction solvent to reduce the water concentration, and consequently the equilibrium of the reverse reaction was shifted [25]. The reaction was performed in a poly(methyl methacrylate) (PMMA) microreactor with two different inlets. Through simple syringe pumping, the enzyme and reagents were loaded from these inlets, and the reaction was terminated by heating. The reaction gave three kinds of stereoisomers, that is, p-nitrophenyl-2-acetamide-2-deoxy-3-O-(β-d-galactopyranosyl)-β-d-glucopyranoside, p-nitrophenyl-2-acetamide-2-deoxy-4-O-(β-d-galactopyranosyl)-β-d-glucopyranoside, and p-nitrophenyl-2acetamide-2-deoxy-6-O-(β-d-galactopyranosyl)-β-d-glucopyranoside (Scheme 10.7). The ratios between those isomers in the products were not determined, but the total amount of the disaccharide mixture (galactosylated p-nitrophenyl-2-acetamide-2-deoxy-6-O-(β-d-galactopyranysol)-β-d-glycopyranoside; Gal-GlcNAcPNP) gave better yield when the reaction was conducted in the microreaction channel compared with that in the micro-test tube. This result suggests that the microreactor is an effective device for enzymatic glycosylation.
Scheme 10.7 Synthesis of disaccharide by transgalactosylation that yielded three kinds of isomers.
Multistep syntheses that employ interconnected microreactors hold great promise in creating complex molecules in a continuous flow mode. An example of optimization of synthetic efficiency by multistep syntheses performed in a single microreactor made of PDMS is the rapid synthesis of [![]() ]fluoride radiolabeled molecular imaging probe, 2-deoxy-2-[
]fluoride radiolabeled molecular imaging probe, 2-deoxy-2-[![]() ]fluoro-d-glucose ([
]fluoro-d-glucose ([![]() ]FDG). Sequentially, five steps have been combined in a single microdevice; [
]FDG). Sequentially, five steps have been combined in a single microdevice; [![]() ] fluoride concentration, water evaporation, radiofluorination, solvent exchange, and hydrolytic deprotection [26]. The five sequential processes yielded 2-desoxy-2-fluoro-d-glucose, [
] fluoride concentration, water evaporation, radiofluorination, solvent exchange, and hydrolytic deprotection [26]. The five sequential processes yielded 2-desoxy-2-fluoro-d-glucose, [![]() ]FDG, a widely used radiolabeled molecular probe. Higher radiochemical yield and purity was obtained within shorter synthesis time relative to conventional automated synthesis. The entire process was automated and required only 14 min in comparison to the conventional batch process that took 50 min. This acceleration is significant considering that the half-life of [
]FDG, a widely used radiolabeled molecular probe. Higher radiochemical yield and purity was obtained within shorter synthesis time relative to conventional automated synthesis. The entire process was automated and required only 14 min in comparison to the conventional batch process that took 50 min. This acceleration is significant considering that the half-life of [![]() ] fluorine is only 110 min and rapid synthetic steps are required to obtain the imaging probes.
] fluorine is only 110 min and rapid synthetic steps are required to obtain the imaging probes.
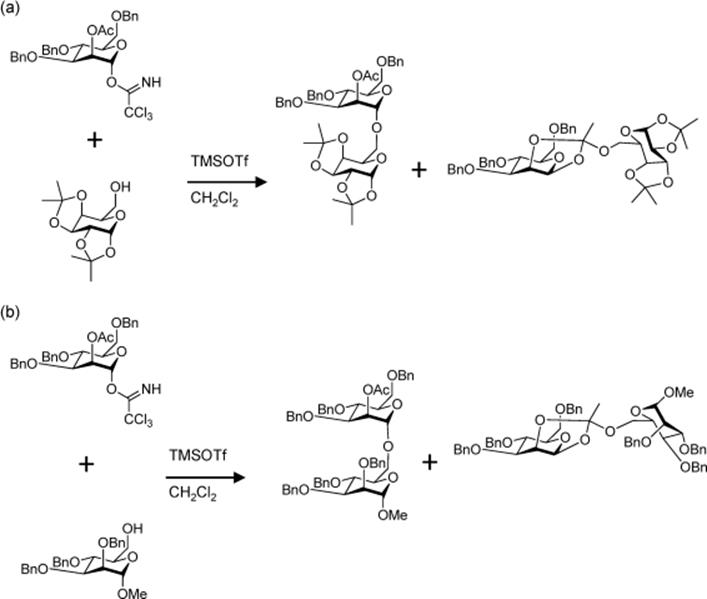
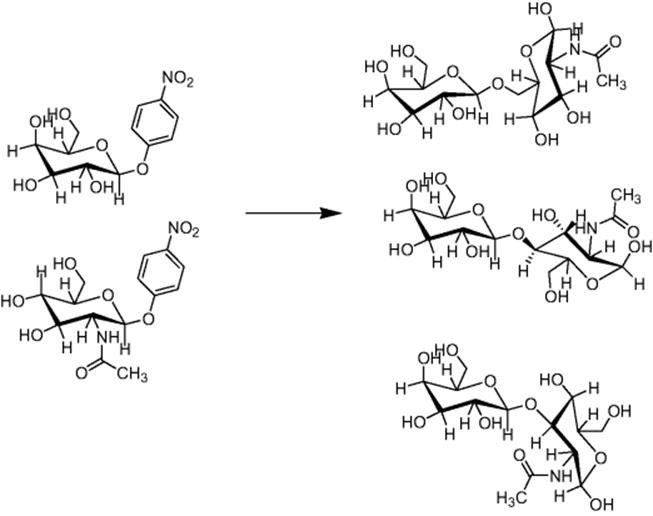
In another study by Seeberger's group, after establishing the reaction conditions for single glycosylation reactions in silicon-glass microreactors [23], the synthesis of an oligosaccharide in continuous flow was explored [27]. A combination of continuous flow microreactors and fluorous phase chemistry was employed to assemble oligosaccharides as illustrated in Scheme 10.8. Synthesis of the β-(1–6) linked d-glucopyranoside homotetramer was performed in a silicon-glass microreactor by iteratively coupling glycosylphosphate to a growing carbohydrate chain on a perfluorinated support linker for facile purification by fluorous solid phase extraction (FSPE). Each glycosylation was optimized and scaled out in the microfluidic device to obtain the desired oligosaccharides in excellent yield.
Scheme 10.8 Synthesis of β-(1 → 6) linked d-glucopyranoside homotetramer via iterative glycosylation in a combined system of microreactor and fluorous phase chemistry [27]. Source: Copyright 2007 American Chemical Society.
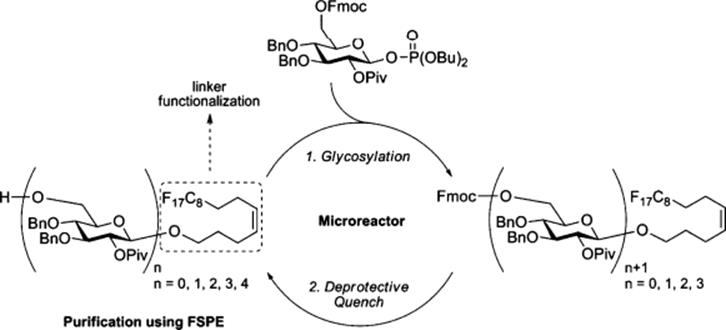
Another challenging topic in the field of oligosaccharide synthesis is the inherent reactivity of the sialyl donor. In another study by Fukase's group, they successfully achieved highly α-selective sialylation of sialic acid N-phenyltrifluoroacetamide toward the synthesis of the α-(2–6) and α-(2–3)-NeuAc-Gal units with various galactose and lactose acceptors by tuning the electronic properties of the C-5 nitrogen-protecting groups [28]. The microfluidic system was applied to the α-sialylation (Scheme 10.9) and an efficient route to large-scale synthesis was established.
Scheme 10.9 Optimization of α-(2–6)-sialylation between donor and acceptor using microreactor [28]. Source: Copyright 2007 Taylor and Francis.
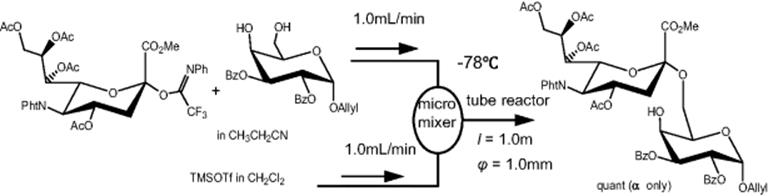
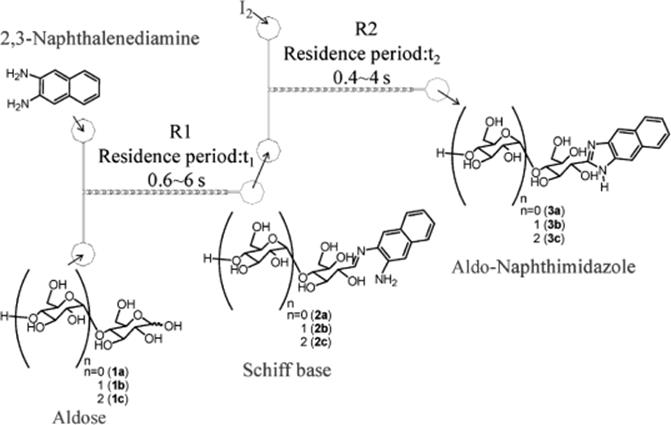
Recently, an efficient and rapid synthesis of carbohydrate derivatives such as aldo-napththimidazoles formed by linking naphthalenediamine with mono-, di-, and trisaccharides was accomplished using a split-and-recombine (SAR)-microreactor (Figure 10.2) [29]. The SAR-microreactors combine mixing mechanisms of both diffusion and chaotic advection [29]. The 3-D structure enables the two fluids to generate sequential stretching and folding, which consequently increases the interfacial area between two fluids and thus facilitates mixing [30–32]. A solution of aldose (d-glucose, d-maltose, or d-maltotriose) in AcOH and a solution of 2,3-naphthalenediamine in AcOH were loaded into reactor 1 (R1) through a Teflon tube using syringe pump, and the product from R1 passed into reactor 2 (R2) was mixed with a solution of iodine in AcOH that was introduced from another inlet of R2 (Scheme 10.10). Using two-step reaction process in SAR-microreactors, the carbohydrate derivatives were generated by linking naphthalenediamine with mono-, di-, and trisaccharides requiring just subseconds to seconds, much less than for the flask, with comparable yields. With the same duration, the yield of the product formed in SAR-microreactors was much enhanced than in T-shaped microreactors. The SAR-microreactors were suggested to be suitable for studying the reactive parameters (e.g., reaction duration, temperature) and developing drugs. Increasing the total amount of carbohydrate derivatives yield can be achieved by concurrent use of multiple microreactors.
Figure 10.2 Illustration of the experiment and geometric design of a SAR-microreactor. SEM: scanning electron microscope; OM: optical microscope [29]. Source: Copyright 2011 Elsevier.
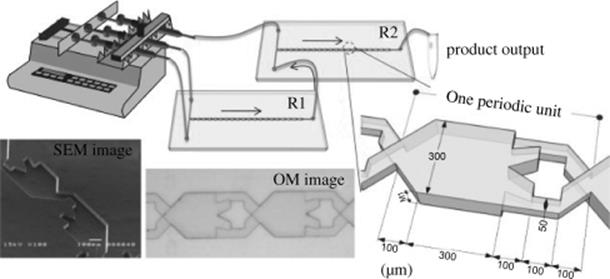
Scheme 10.10 Schematic diagram of the microreaction system for the synthesis of carbohydrate derivatives [29]. Source: Copyright 2011 Elsevier.
Several researchers from the field of synthetic biology have extensively studied significant progress toward gene synthesis using microfluidic systems. The polymerase chain reaction is the most widely used technique for oligonucleotide synthesis. The two general strategies for microchip-based PCR are flow through and stationary chamber formats. We just raise recent examples of oligonucleotide synthesis within microreactors in this subsection. A discussion on microreactors used for genetic analysis will be covered in Section 10.3.1.1.
10.2.1.3 Oligonucleotide Synthesis
Chemical synthesis of oligonucleotide in microfluidic platform made of perfluoropolyether (PFPE) has been reported by Huang and other [33]. The microfluidic reactor was used to perform reaction cycles adopted from the widely used phosphoramidite method [33]. PfPE is an elastomer with excellent chemical compatibility thus making it an ideal material for organic chemical reactions. The device demonstrated capability of synthesizing 60 pmol of DNA oligonucleotides while consuming <500 nl of 0.1 mol/l phosphoramidite solution in each reaction cycle. A 60-fold reduction of reagent consumption over the conventional automated method has been reported. This approach demonstrates the utility of integrated micromechanical valves for complicated multistep organic synthetic reactions and enables automated chemical experiments with a broad choice of solvents.
The first integrated microfluidic device capable of performing two-step gene synthesis to assemble a pool of oligonucleotides into genes with the desired coding sequence was presented [34]. The device composed of two PCR, temperature-controlled hydrogel valves, electromagnetic micromixer, shuttle micromixer, volume meters, and magnetic beads based solid-phase PCR purification, fabricated using a fast prototyping method without lithography process. The fabricated device was integrated with a miniaturized thermal cycler to perform gene synthesis. Oligonucleotides were primarily assembled into genes by polymerase chain assembly, and the full-length gene was amplified by a second PCR. The microdevice successfully attained syntheses with low oligonucleotide concentration of 10 nM and primer concentration of 0.4 μm using one-step and two-step PCR-based gene synthesis process. Separation of the product, a green fluorescent protein fragment (GFPuv) (760 bp), was separated from the PCR reaction mixture by magnetic-beads-based solid-phase purification. The system could be used for constructing a more comprehensive system for fully automated gene synthesis.
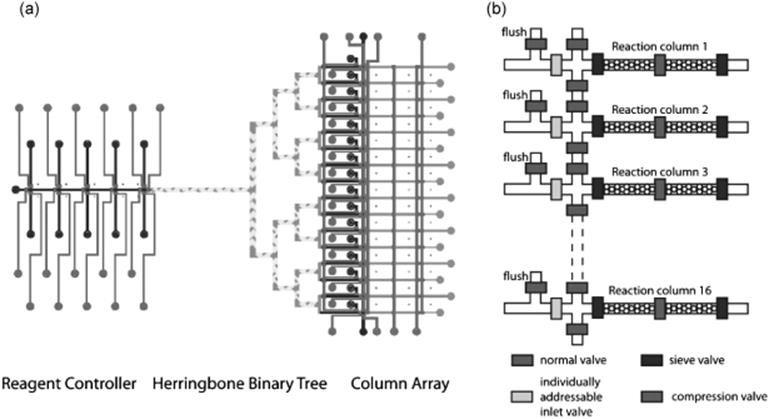
There are many limitations inherent in microarray DNA synthesis technologies such as low yields [35], requirement for oligonucleotides to be cleaved off the chip as a single mixture, and sometimes lower quality compared to the existing solid-phase strategies due to the inefficient de-blocking step. To overcome the mentioned limitations, a programmable microfluidic synthesis platform was developed [36]. The microfluidic platform for parallel solid phase synthesis of oligonucleotides significantly reduced the cost of gene synthesis by reducing reagent consumption (by 100-fold) while maintaining a ~100 pmol synthesis scale that eliminated the need for amplification before the assembly. Sixteen oligonucleotides were synthesized in parallel on this platform and then successfully used in a ligation-mediated assembly method to generate DNA constructs ~200 bp in length (Figure 10.3).
Figure 10.3 (a) Schematic diagram of a 16-column microfluidic DNA synthesizer. The control lines, fluidic lines, herringbone mixers, and the square profiled binary tree and reactor columns are shown. (b) Close up schematic of the column array. The normal valve and the individually addressable inlet valve close fully when activated; the sieve valve and compression valve of the reaction columns allow fluid flow while trapping the CPG beads. Each column has a flush valve that opens and closes in unison to allow residual reagent in the binary tree to be washed away between steps to avoid contamination. The addressable inlet valve controls which columns the reagents enter. The compression valves prevent rearrangement of closely packed spherical beads when a pressure drop occurs between steps [36]. Source: Copyright 2010 Oxford University Press.