Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
11. Industrial Microreactor Process Development up to Production
11.3. Process Development at Laboratory Scale
11.3.1 Nitration of Substituted Benzene Derivatives
BASF in Ludwigshafen, Germany, and university partners reported the nitration of several disubstituted benzene derivatives using a capillary flow microreactor [29]. The exact nature of these species, however, was not disclosed.
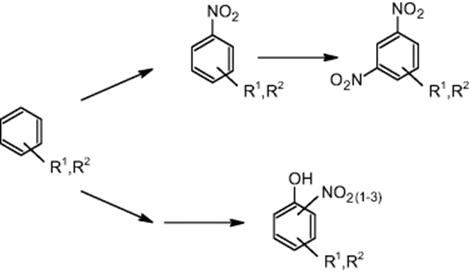
Because the benzene derivative and nitric acid are immiscible, the impact of mixing/distribution on slug formation was investigated. Uniform slugs of the aromatic compound/nitric acid were formed in a Y-piece [29]. The capillary attached has a stabilizing effect on the slug flow. The deviation of slug size distribution is very small (about 5%). Hence, interfacial area is nearly constant for this type of capillary flow.
For the nitration of benzene derivatives, experiments were performed at two temperature levels, 60 and 120 °C [29]. For the 60 °C experiment, very small amounts of by-products such as phenol derivatives and dinitrated species were formed, not exceeding 50 ppm each. At 120 °C, high levels of by-products were found, with 300 ppm dinitrated species and 200 ppm phenol derivatives. The mechanism of by-product formation was also investigated [29]. Dinitrated products were generated by nitration of the mononitrated product. It was concluded that phenolic by-products were formed directly from the aromatic starting material, rather than from the mononitrated product. This proposed reaction mechanism could be confirmed by performing selective nitration of the mononitrated product.
The influence of interphase mass transfer between liquid–liquid slugs was investigated for nitration of aromatic compounds in a capillary flow reactor (see Figure 11.2) [29]. This was achieved by changing flow velocity via volume flow setting, while residence time was kept constant by increasing the capillary length.
Figure 11.2 Interphase mass transfer coefficient obtained from reaction engineering model. Source: Courtesy of Elsevier [29].
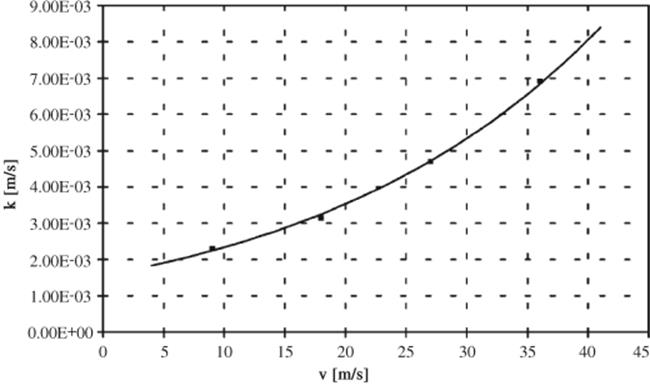
Conversion to the mononitrated benzene derivative increased linearly with increasing flow velocity due to enhanced mass transfer. The formation of phenol by-products increased in the same manner for similar reasons. In turn, consecutive by-products, dinitrated aromatics, were formed in a linear decreasing fashion. This was explained by a mass transfer-induced removal of the mononitrated product from the reacting slug.
In the last step, a reactor model was developed, taking into account both mass transfer of organic components between the two phases and the homogeneous reaction within the aqueous phase, the latter relying on the literature data [29]. From there, an extended kinetic model was developed and applied, considering the kinetics of the homogeneous side reactions as well [29]. Using this, the activation energies of these processes could be derived.
11.3.2 Microflow Azide Syntheses
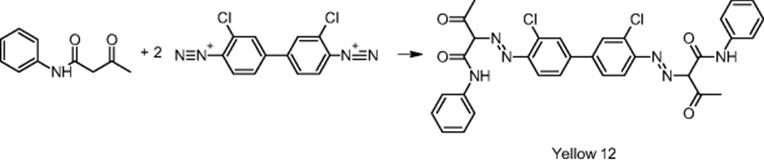
Trust Chem Company in Hangzhou, China, was involved in the synthesis of the commercial azo pigment Yellow 12 [30].
Particle synthesis in general yields products that differ in particle average size and distribution, shape, morphology, selectivity, and many more properties. Mixing is the key step for particle seed formation and growth and can determine the above-mentioned product qualities. Due to uniform concentration profiles and a good correlation between experiment and theory, product properties of particles made in microdevices are often superior and process development and upscaling can be faster and more predictable as compared to conventional technologies. In the ideal case of numbering up, the laboratory-scale performance is maintained during piloting and production, since the fluid dynamics are not changed or at least shift by analogy in a known manner.
The synthesis of the azo pigment Yellow 12 involves a very fast precipitation and, consequently, is largely impacted by mixing in micromixers. Target of an investigation of the Trust Chem Company was to obtain small-sized crystals by microprocess technology. Using a laboratory-scale slit-type interdigital micromixer–reactor [30], smaller particles with a uniform size distribution were obtained for the commercial azo pigment Yellow 12 (see Figure 11.3) [30].
Figure 11.3 Particle size distribution of the batch (a) and the micromixer-based synthesis (b) of Yellow 12. Source: Courtesy of ACS [30].
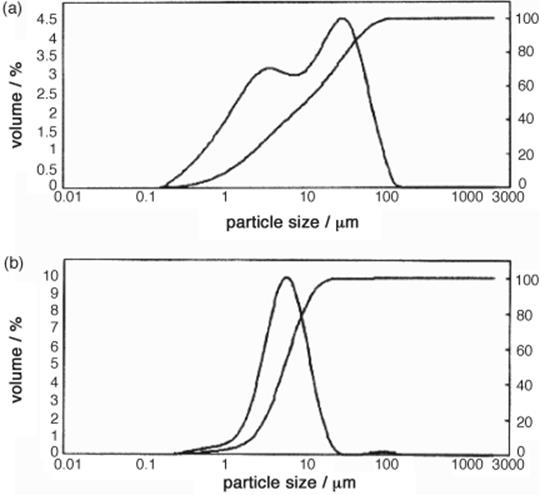
Related product properties are improved, such as optical properties like the glossiness and transparency (see Table 11.1). There are no negative consequences for dye manufacturing with the new microreactor-made crystals, as the tinctorial power is the same as for conventional synthesis. Tinctorial power is a measure for the adhesion of the pigment to wool stuff or similar material. The intensification in coloration properties means that the same amount of material can be treated now with less amount of the Yellow 12 azo pigment, which reduces materials costs and increases the profitability of the pigment manufacture.
Table 11.1 Glossiness of the imprinted color (GU = glossiness units) for micromixer-based and conventional Yellow 12 pigment manufacture.
Source: From Ref. [30].

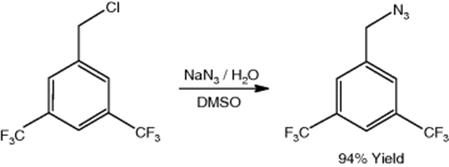
A research at Eli Lilly and Company, Chemical Product Research and Development, Indianapolis, IN, USA, demonstrated the microcapillary tube reactor process for efficient production of azide, 1-(azidomethyl)-3,5-bis-(trifluoromethyl)benzene, which minimizes potential hazards associated with hydrazoic acid condensation in the reactor [31]. This substance is a key starting material for a new phase II investigational drug candidate at Eli Lilly and Company, a potential neurokinin 1 antagonist, used for emesis treatment. The continuous-flow thermal tube reactor allowed for safe operation at higher processing temperatures compared to analogous batch processing systems. The azide was prepared in a simple biphasic solvent system using phase-transfer catalysis, which results in an overall low e-factor.
11.3.3 Vitamin Precursor Synthesis
BASF in Ludwigshafen, Germany, carried out process development for a reaction as part of a multistep process, finally yielding a vitamin [32, 33]. This involved short-temperature processing (<4 s) that simply was not possible using macroscopic bench-scale apparatus. In the latter case, almost 50% of the reaction heat was released already at the mixer unit, rather than after entering the subsequent heat exchanger. The temperature rise led to side reactions, reducing the yield.
The reactants and the product were not disclosed in the open literature [32, 33]. Concentrated sulfuric acid is present in quantitative amounts besides the organic solvent, so a liquid/liquid process results. The reactant quickly forms an intermediate that again quickly reacts to give the product. Thermally induced side reactions occur at each stage.
A maximum yield of 80–85% was obtained at 4 s residence time and a temperature of 50 °C by microreaction system processing [32, 33]. Using ordinary laboratory processing with standard laboratory glassware gave only 25% yield and the continuous industrial process gave a yield of 80–85%; the former employing the semibatch industrial process gave a yield of 70%. The temperature and the residence time of industrial and microreactor continuous processing were identical.
Retinol (Vitamin A form) was synthesized by ester cleavage in a microreactor-based system at Sigma-Aldrich, Switzerland [34].
A total of 800 g/day of product was synthesized and the advantages compared to batch technology were evident on a 500 g scale and are shown in Table 11.2.
Table 11.2 Comparison of batch and microreactor process for retinol production.
|
Batch process |
Microreactor process |
|
|
Reactor |
20 l vessel |
1.5 ml microreactor |
|
Reaction conditions |
KOH in methanol/water |
Feed 1 : 1 M acetate in THF |
|
Feed 2 : 2 M KOH |
||
|
Reaction time |
16 h |
39 min |
|
Hours of work |
26 |
10 |
|
Upscale |
No |
Unrestricted |
|
Yield |
40% |
70% |
By transferring the batch synthesis routine into a flow chemistry process in a microreactor, the yield of the reaction was improved to 70%, now giving a constant output of 70% product each time the synthesis is performed [34]. This improvement is a direct result of the flow process. In the continuous flow, every portion of product is streamed out of the reaction zone instantly after it has formed.
It should be highlighted that the savings achieved with this single new synthesis procedure have already justified all investments into microreactor technology at Sigma-Aldrich.
11.3.4 Ester Hydrolysis to Produce an Alcohol
Sigma-Aldrich performed an ester hydrolysis to produce an alcohol that decomposes quickly. None of these compounds could be disclosed [2]. Scale-up could not satisfy the commercial interest in the labile product, as the yield decreased strongly with batch vessel size. Seventy percent yield was obtained at 5 l scale, 35% at 20 l scale, and 10% at 100 l scale.
The investigation in a microreactor was initially hampered by the presence of an insoluble compound for the ester process [2]. Since this reactant had to be changed, a process development study had to be carried out. A model reaction, an ester hydrolysis yielding a stable alcohol, was used instead of the real one, in order to facile the process development. Twelve different conditions were used. Having acquired the process know-how, the same kind of process development was done for the real reaction in only 2 h with success.
11.3.5 Synthesis of Methylenecyclopentane
Methylenecyclopentane was synthesized by Sigma-Aldrich using an undisclosed route [2].
![]()
The reaction is highly exothermic, which makes scale-up difficult [4]. In addition, 30% of the yield is the thermodynamically stable side product 1-methylcyclopentene. Product and side product are difficult to separate. Both unsolved issues led to a stop in process development using conventional technology.
Using microreactor processing 70% conversion was achieved, which gave solely product and no side product [2]. Actually, the microreactor conversion is lower than that for the batch mode, but the first is now the preferred route, as separation of the product from the reactant can be accomplished, while product, as mentioned, hardly can be purified from the side product. A throughput of 300 g/h was achieved.
11.3.6 Condensation of 2-Trimethylsilylethanol
Sigma-Aldrich performed the condensation of 2-trimethylsilylethanol and p-nitrophenyl chloroformate to give 2-(trimethylsilyl)ethyl-4-nitrophenyl carbonate [2]. Reduction of residence time is required to achieve high selectivity, as the product degrades and side products form. While conventional technology needs 14 h to complete the reaction, only about 18 min are needed when carrying out the reaction in a microreactor, which enhances selectivity. A reason for the better microreactor performance is however not detailed.
11.3.7 Staudinger Hydration
A two-stage microreactor continuous-flow system developed by Sigma-Aldrich allows the handling of organic azides safely in small holdup volumes with immediate conversion in the second microreactor (Figure 11.4) [35].
Figure 11.4 Two-stage microreactor assembly for the safe production and immediate conversion of organic azides [35].
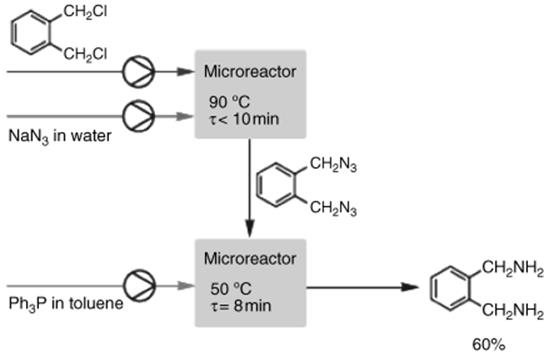
In the first microreactor, the azide intermediate is formed by nucleophilic substitution. The azide intermediate is submitted to Staudinger hydration with triphenylphosphine in a subsequent second microreactor [35]. A phase-transfer catalyst moderates the initial reaction with efficient mixing by the microreactor. Production capacity of o-xylylenediamine in two subsequent glass microreactors exceeds 1 kg/day with an overall yield of 60%. Workup and isolation of the potentially hazardous intermediate are completely avoided between reaction steps, saving time and money and offering scale-independent safety levels.
11.3.8 (S)-2-Acetyl Tetrahydrofuran Synthesis
SK Corporation in Daejeon, Korea, performed the synthesis of (S)-2-acetyl tetrahydrofuran with an alkylating step using a Grignard reaction and a hydrolysis step, finally yielding a ketone moiety in the side chain starting with a cyano group [36].
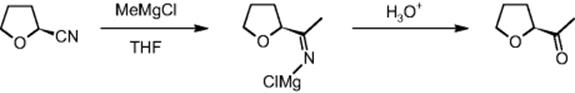
The methylating agent MeMgCl is very reactive, even compared to other Grignard reagents, and thus not easy to handle at large scale, as substantial safety and hazardous issues would result [36]. The microreactor allowed one to minimize moisture and oxidizing effects due to encased processing at low degree of contamination of decomposing species for Grignard reactants.
Overalkylation can lead to tertiary alcohol formation by consecutive reaction [29]. Product quality demands to keep this impurity level <0.2%. Microreactor operation yielded the overalkylated alcohol follow-up product at 0.18%, while level of impurity was 1.56% for the batch process [36]. The reason is probably the lower backmixing in the microflow system, with concentration profiles being less deteriorated from ideal; that is, no excess of alkylating agent is generated locally to promote the follow-up reaction.
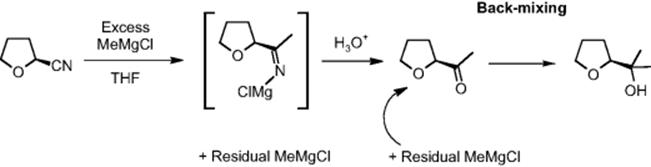
The α-hydrogen of the reactant is unstable under the basic reaction conditions applied, leading to a small degree of racemization [36]. Conservation of stereochemistry was largely achieved by microreactor operation; 98.4% enantiomeric excess (ee) was found as compared to 97.9% ee at batch level (see Table 11.3).
Table 11.3 Impurity and optical purity of the batch and microreactor processes in the (S)-2-acetyl tetrahydrofuran synthesis [36]
|
Impurity (%) |
Optical purity (%) |
|
|
Batch |
1.56 |
97.7 |
|
Microreactor process |
0.18 |
98.4 |
11.3.9 Synthesis of Intermediate for Quinolone Antibiotic Drug
LG Chem in Daejeon, Korea, performed the multistep synthesis of gemifloxacin (Factive™), a quinolone antibiotic drug with enhanced activity against Gram-positive bacteria [37]. This drug has high activity against Gram-negative bacteria, atypical strains, and major respiratory pathogens.
In one reaction step, the enamine moiety is protected by the t-Boc group in a fast and highly exothermic (ΔH = −213 kJ/mol) reaction [37].
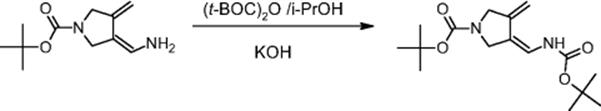
A consecutive reaction occurs with higher rate constant and higher activation energy, thus being more dominant at higher temperatures [37]. The impurity level generated by the consecutive step becomes too high at temperatures >25 °C. This can be minimized by effective heat removal from the exothermic reaction. In addition, the product can react with KOH, so efficient mixing is required that avoids spatially strongly varying concentrations of reactants. For similar reasons, it was speculated that narrower residence time distribution could increase selectivity.
A slit-type interdigital micromixer was used for fast mixing [37] and compared to a tubular reactor and five Kenics mixers connected in series. Details of further components of the micromixer rig were not given, but most likely a capillary reactor was added for efficient heat exchange.
Mixing is essential to achieve dispersion of the two-phase mixture. For the tube reactor (without mixing function), the flow had to be very high to achieve turbulence (Re > 2000) and in this way serve for dispersion [37]. As a result of the high flow rate, residence time was too short to complete reaction and only 27% conversion was achieved. At lower flow rate, phase separation occurs. An extrapolation of the reactor length for complete conversion yields an impractical tube length of about 2 km. The combination of five Kenics static mixers guaranteed good mixing over an extended length, so a conversion of 97% was achieved due to the micromixer. The microreactor used has good mixing and was also able to remove the heat of reaction. No by-products were formed at conversions as high as 96%. Thus, the microreactor performance in terms of selectivity is equal to that of the static mixers, but has the advantage of operation at ambient reaction temperature of 15 °C, whereas the latter need outside cooling of 0 or −20 °C due to the necessary heat removal.
11.3.10 Domino Cycloadditions in Parallel Fashion
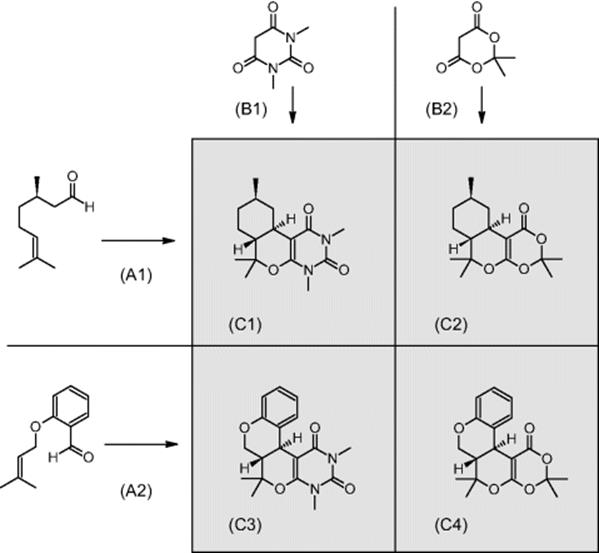
GlaxoSmithKline Pharmaceuticals in Harlow, UK, investigated domino cycloadditions in a commercial chip and extended their process development by operation in parallel fashion in a three-member array, which was one of the first examples of a parallel multireaction [38].
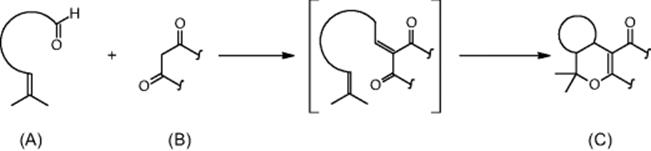
The domino reaction consists of a Knoevenagel condensation giving an intermediate that immediately undergoes an intramolecular hetero-Diels–Alder reaction with inverse electron demand [38]. As aldehydes, rac-citronellal, an aromatic aldehyde, and two commercially available 1,3-diketones, 1,3-dimethylbarbituric acid and Meldrum's acid, were selected. By combinations of these reactants, different cycloadducts were generated.
Process development was accomplished in single-run reactions [38]. The conversion of the microchannel processing amounted typically to about 50–75%, depending on the nature of cycloadduct and the residence time chosen, and was comparable to the results of batch syntheses [38]. By combinations of aldehydes and 1,3-diketones, different cycloadducts were generated simultaneously in one run on one chip, that is, an undesired transfer of solutions from one channel to another by imperfect sealing between these channels. The conversions were comparable to the single runs, with one exception. Also, cross-contamination was observed. It ranged from a few percent to about 50%.
11.3.11 Phase-Transfer Catalysis-Mediated Knoevenagel Condensation
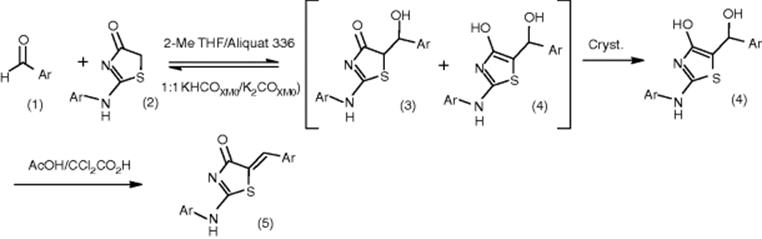
Another example of Knoevenagel condensation performed at GlaxoSmithKline is a reaction between an aromatic aldehyde and thiazalone [39]. Although the Knoevenagel reaction has been applied widely in organic synthesis, it faces problems regarding elimination of tautomers and activation of methylene compound. The reaction time exceeds 30 h and in the best case 84% conversion was obtained. Screening of influence of phase-transfer catalysts and Lewis acids on the efficiency of Knoevenagel condensation was performed in a system of parallel microreactors. Microreactors allowed conditions to be determined both rapidly (within 2 weeks) and with minimal material (~3 g). Intermediate 5 (scheme below) was synthesized in over 92% yield and with a cycle time of less than 6 h, which is a great reduction from >30 h when using traditional conditions.
11.3.12 Ciprofloxazin® Multistep Synthesis
The synthesis of Ciprofloxazin was one among several syntheses being performed in contract research by a microreactor developer for pharmaceutical industry and feasibility was demonstrated [40]. In this multistep synthesis, alkylaminodefluorinations were essential part of the chemistry. Ciprofloxazin, the final product, is an antibiotic compound with a high sales volume.
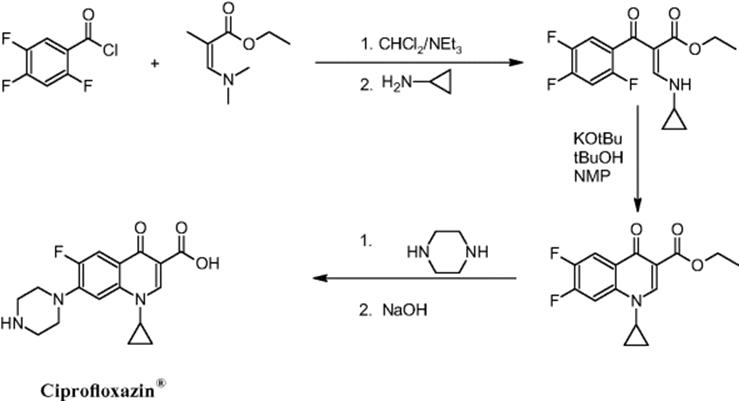
This reaction scheme involves two substitutions of fluorine moieties at the aromatic ring by amines, yielding the final product for pharmaceutical applications [40, 41]. All in all, five synthesis steps are actually required and performed subsequently in a microreactor system to get the target molecule.
11.3.13 Methyl Carbamate Synthesis
The Chemical Development & Drug Evaluation branch of Johnson & Johnson Pharmaceutical Research & Development LLC in Raritan, NJ, USA, performed the exothermic reaction of methyl chloroformate with amines to give methyl carbamates [42]. Due to large heat release hot spots occur. For the reaction of N-methoxycarbonyl-l-tert-leucine with methyl chloroformate to give the amino acid derivative, this is even observed at laboratory scale.
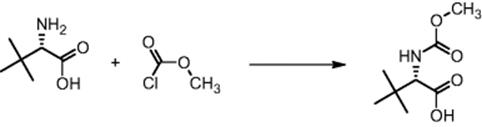
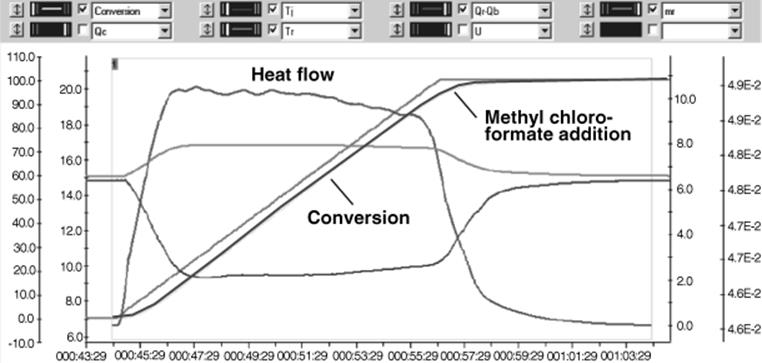
Calorimetric measurements show that the addition is exothermic [42]. The heat release rate is mainly feed controlled, as the square shape of the heat flow curve demonstrates (see Figure 11.5). Whenever feed is added, the heat flow responds without delay.
Figure 11.5 Reaction calorimetry results from the addition of methyl chloroformate (slight excess) to l-tert-leucine. Source: Courtesy of ACS [42].
In case of complete malfunction of cooling and stirring systems, the temperature may exceed the solvent reflux temperature [42]. Accordingly, a slow dosing of the methyl chloroformate is necessary to have control over the heat release. After having determined the reaction parameters at 1 g scale, the reaction was carried out in a microreactor with 91% yield at 7 min residence time. More than 1 kg of N-methoxycarbonyl-l-tert-leucine was prepared within 12 h.
Scale-up by using a microreactor was done also for the amidation of p-tolyl chlorodithionoformate with dimethylamine to give p-tolyl dimethyldithiocarbamate without further safety precautions at 96% yield, which is comparable to the batch process [42]. At 1.4 min residence time, a yield of 155 g/h was achieved.
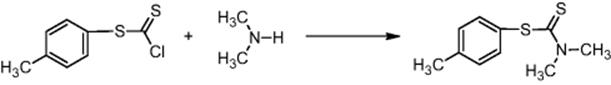
11.3.14 Newman–Kuart Rearrangement
The Chemical Development & Drug Evaluation branch of Johnson & Johnson Pharmaceutical Research & Development LLC in Raritan, NJ, USA, tested microreactors for processes at elevated temperatures above the limit of most multipurpose conventional reactors, which is above ~140 °C [42]. Operation above this limit is possible only by means of special reactors equipped with heat transfer units.
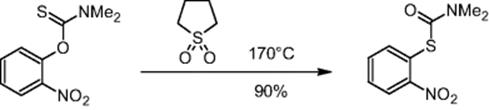
The Newman–Kuart rearrangement is an example of a high-temperature reaction [42]. Using a microreactor, the reaction temperature could be extended up to 200 °C. O-(2-Nitrophenyl)-N,N-dimethylthiocarbamate was converted to S-(2-nitrophenyl)-N,N-dimethylcarbamothioate at 170 °C in 14 min in 90% yield. Quantitative conversion with a throughput of 34 g/h was achieved with sulfolane as solvent at the same temperature and reaction time.
11.3.15 Ring-Expansion Reaction of N-Boc-4-Piperidone
The Chemical Development & Drug Evaluation branch of Johnson & Johnson Pharmaceutical Research & Development LLC in Raritan, NJ, USA, investigated the ring-expansion reaction of N-Boc-4-piperidone with ethyl diazoacetate in a microreactor system as an example of processing hazardous substances [42].
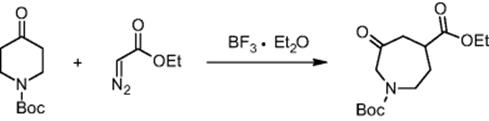
A crude yield of 90% was obtained in ether at −25 °C. When performed in a batch mode on 70 mg scale, no safety issues were taken and an 81% yield was obtained [42]. Reaction calorimetry reveals a very exothermic reaction after feeding and subsequent mixing with an initiation period; that is, the response of the heat flow curve is delayed by about 1 min as compared to the feed curve (see Figure 11.6). Therefore, the heat release rate with this mode of addition is not feed controlled. The reaction is very sluggish, since the reaction occurs at a single blow as soon as 60% of the material has been added. Calculating the worst-case temperature rise shows that the reaction would rapidly increase in temperature and can approach the solvent reflux temperature, possibly throwing out the reaction mixture in case of cooling or stirring malfunction. This would particularly include the full accidental release of all the BF3·Et2O.
Figure 11.6 Addition of BF3·Et2O to N-Boc-4-piperidone and ethyl diazoacetate. Source: Courtesy of ACS [42].
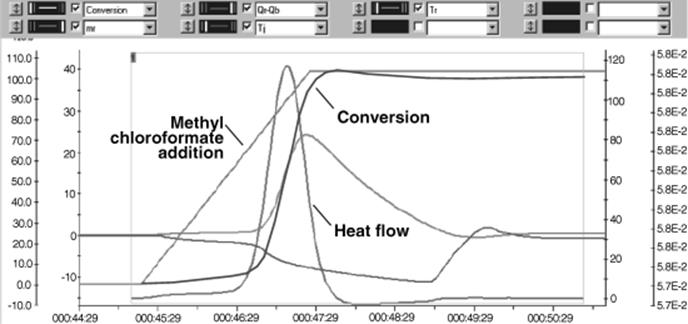
The calculated worst-case temperature of 45.6 °C approaches also the reflux temperature of the solvent (diethyl ether), but does not reach the decomposition temperature of ethyl diazoacetate [42]. Furthermore, the evolution of large amounts of nitrogen gas during the reaction could lead to an overpressurization of the reaction vessel. All these reasons together prevented a scaling of the ring-expansion reaction to kilogram scales due to related safety issues. Operating the microreactor system, however, allowed without any further optimization a precise control of the reaction and 89% yield was obtained. Reaction time was only 1.8 min with a throughput of 91 g/h.
Inspired by the Johnson & Johnson research, this procedure was developed further and is used by Sigma-Aldrich for the catalog production of N-Boc-hexahydro-1H-azepin-4-one. The microreaction is followed by base-induced decarboxylation performed in a conventional batch reactor [43].
11.3.16 Synthesis of Aldehydes
Reduction of nitriles and esters to aldehydes with diisobutylaluminum hydride (DIBAL-H) applying continuous-flow technology was investigated by Janssen Pharmaceuticals, division of Johnson & Johnson [44, 45]. Optimization of reaction conditions is performed by the control of residence time. Pumping, mixing, and quenching continuously the reagents through the microreactor enabled rapid experimentation and scale-up, thus shortening the time from research to development and production.
Reaction conditions were optimized for the reduction of benzonitrile 1a to benzaldehyde 2a. These optimized reaction conditions were used successfully for other aromatic nitriles [44].
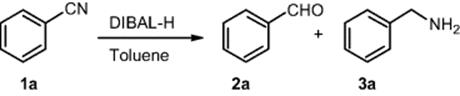
All the explored examples (representatives for aryl, benzyl, alkyl, and heterocyclic aldehydes) are obtained in good to excellent yields and selectivities using a simple and economical setup.
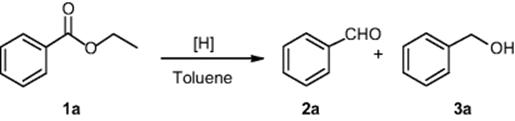
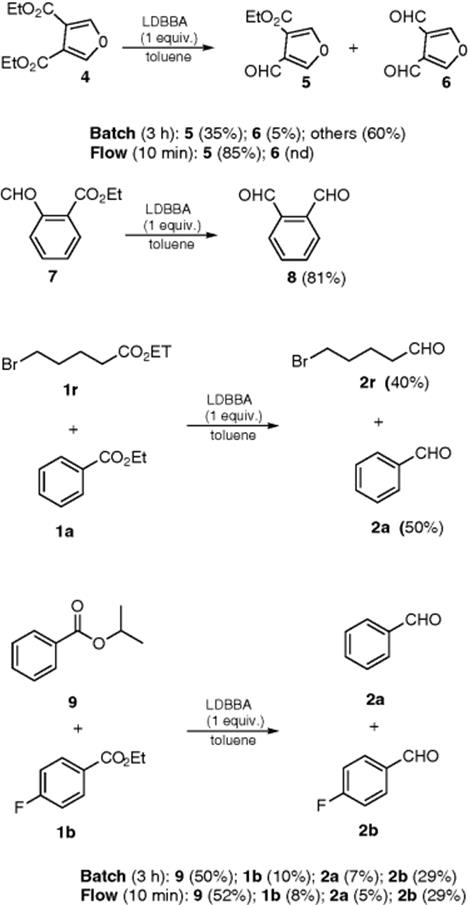
Another important transformation, the reduction of esters to aldehydes with lithium diisobutyl-tert-butoxyaluminum hydride (LDBBA) in flow, was performed in the laboratory of Janssen Pharmaceuticals [45]. DIBAL-H applied in their previous research [44] proved to be unsuccessful in reducing ethyl benzoate (1a) to benzaldehyde (2a). LDBBA appeared to be the most general and effective alternative to DIBAL-H. The selective reduction of an ester group in the presence of an aldehyde, the selective reduction of a single ester group, and the selective reduction of a primary ester in the presence of a secondary ester are achieved with the efficiency that cannot be achieved under traditional batch conditions.
11.3.17 Grignard Reactions and Li–Organic Reactions
Lonza Ltd in Visp, Switzerland, performed the Grignard reaction of an acid chloride with a phenylethyl Grignard reactant in THF in a multiple-injection microreactor [46] (Figure 11.7). Many injection points were chosen to split and delocalize the reaction and release heat, so several small hot spots arise instead of one large hot spot (ΔH = −260 kJ/mol, ΔTadiabatic = ~70 °C). The reaction is quenched at the reactor outlet with water.
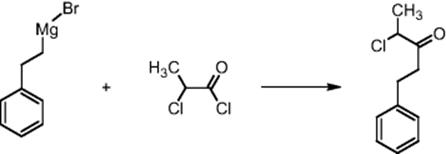
Figure 11.7 Impact of number of injection points and cooling on the yield of the reaction of an acid chloride with a phenylethyl Grignard reactant. Source: Courtesy of D. Roberge, Lonza [46].
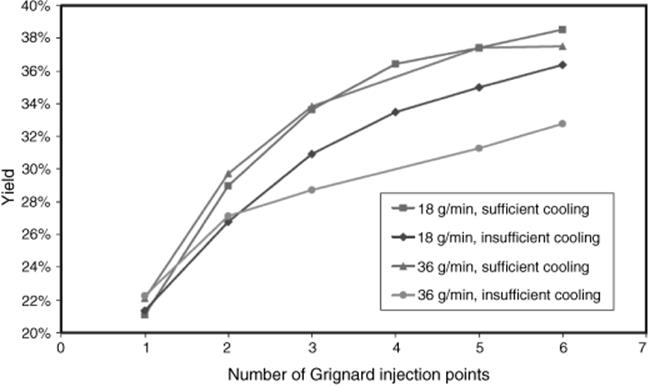
The reaction performance was given for mixers with one to six injection points (see Figure 11.8) [46]. A considerable increase in yield from 21 to 38% can be seen.
Figure 11.8 Impact of improved cooling on the yield of the reaction of an acid chloride with a phenylethyl Grignard reactant. Source: Courtesy of D. Roberge, Lonza [46].
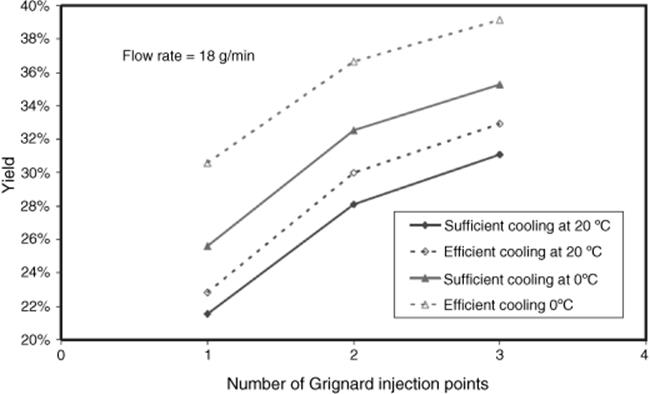
In another process optimization step, the cooling was further improved (from “sufficient” to “efficient”) by using a lower temperature of the cooling liquid, that is, 0 °C instead of 20 °C [46]. In this way, a yield of 39% was obtained with only three injection points, thus reducing the complexity of the microdevice and pressure drop (see Figure 11.8).
The Chemical Development & Drug Evaluation branch of Johnson & Johnson Pharmaceutical Research & Development LLC in Raritan, NJ, USA, used the reaction of the Grignard reagent 3-methoxyphenylmagnesium bromide and a ketone to obtain the product tramadol. cis- and trans-isomers are formed in about 4 : 1 ratio [42]. The organometallic compounds formed are unstable intermediates and are sensitive to moisture.
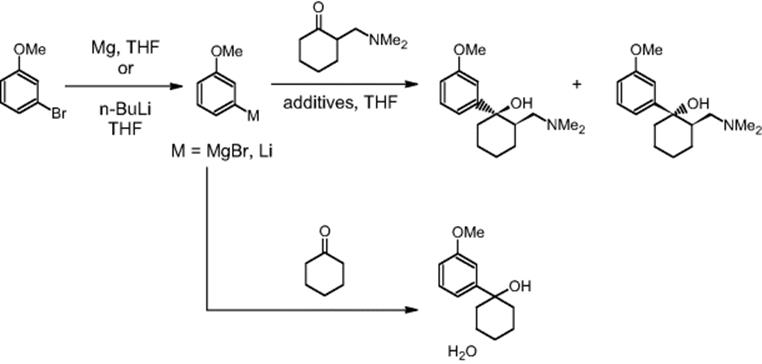
Using aryllithium instead of the Grignard reagent resulted in a higher ratio of cis-isomer formation [42]. In reaction calorimetric studies, it was found that both steps, the formation of 3-methoxyphenyllithium and its addition to ketone, are pretty exothermic with worst-case temperature rises of up to 62 and 133 °C, respectively. The lithium intermediate has to be kept at very low reaction temperatures to prevent decomposition. We concluded then that a continuous reaction may be a good alternative to batch synthesis to improve the reaction yield and to minimize the safety concerns due to the exothermicity of the reaction sequence.
3-Methoxyphenyllithium and cyclohexanone were reacted in a batch mode at −10 and −65 °C to give yields of 32 and 80%, the expected tertiary alcohol, respectively [42]. It was planned to use such temperature effect also in the microreactor. The metal–halogen exchange step could be performed at −14 °C with 17 s residence time and the lithium intermediate was further reacted with cyclohexanone in the batch mode at −40 °C. Lower temperatures were not possible due to chiller limitations and the availability of only one microreactor accounted for the combined continuous-flow batch processing. In this way, a yield of 87% at a throughput of 54 g/h was achieved.
11.3.18 Continuous Synthesis of Disubstituted Triazoles
A library of 1,4-disubstituted 1,2,3-triazoles was synthesized at Cornell University in collaboration with Pfizer Inc. [47]. Six different acetylenes, six different alkyl halides, and sodium azide were used for obtaining the triazoles. Thirty triazoles were synthesized in a continuous-flow Conjure reactor (a copper flow reactor) in good to excellent yields [47] (Figure 11.9).
Figure 11.9 The 1,4-disubstituted 1,2,3-triazoles synthesized by using the Conjure flow reactor.
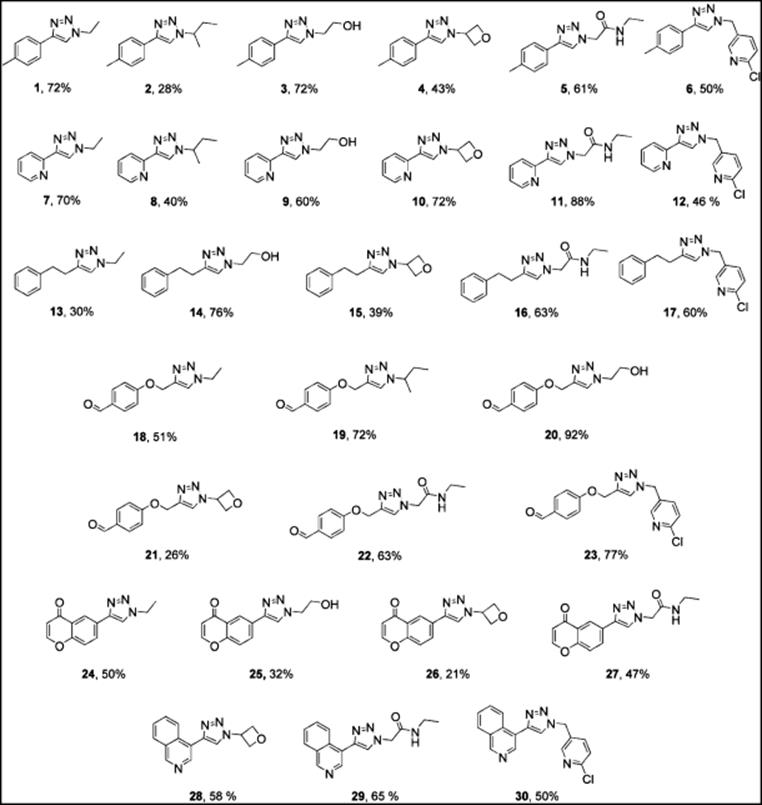
Triazoles were synthesized from simple starting materials without the need for additional catalyst. Organic azides, generated in situ from alkyl halides and sodium azide, were reacted with acetylenes using the copper-catalyzed Huisgen 1,3-dipolar cycloaddition.
Reactions could be easily scaled up with fewer safety concerns than the corresponding batch processes. This one-pot click reaction using a copper flow reactor eliminated both the handling of organic azides and the need for additional copper catalyst and enabled the preparation of numerous triazoles in a continuous-flow process.
11.3.19 Production of 6-Hydroxybuspirone
The complete continuous production process for the hydroxylation of the azapirone psychotropic agent buspirone to give 6-hydroxybuspirone (6-hydroxy-8-[4-(4-pyrimidin-2-ylpiperazin-1-yl)-butyl]-8-aza-spiro[4.5]decane-7,9-dione), including enolization, oxidation, and quenching, is tested at Bristol-Myers Squibb, New Jersey, USA [48].
The preparation scheme including two microreactors is shown in Figure 11.10. Continuous oxidation was considered the most challenging aspect of the process development effort. A single- and a two-stage microreactor were used to study the feasibility of a continuous oxidation process.
Figure 11.10 The preparation of 6-hydroxybuspirone can be completed via a continuous process using at least two microreactors.
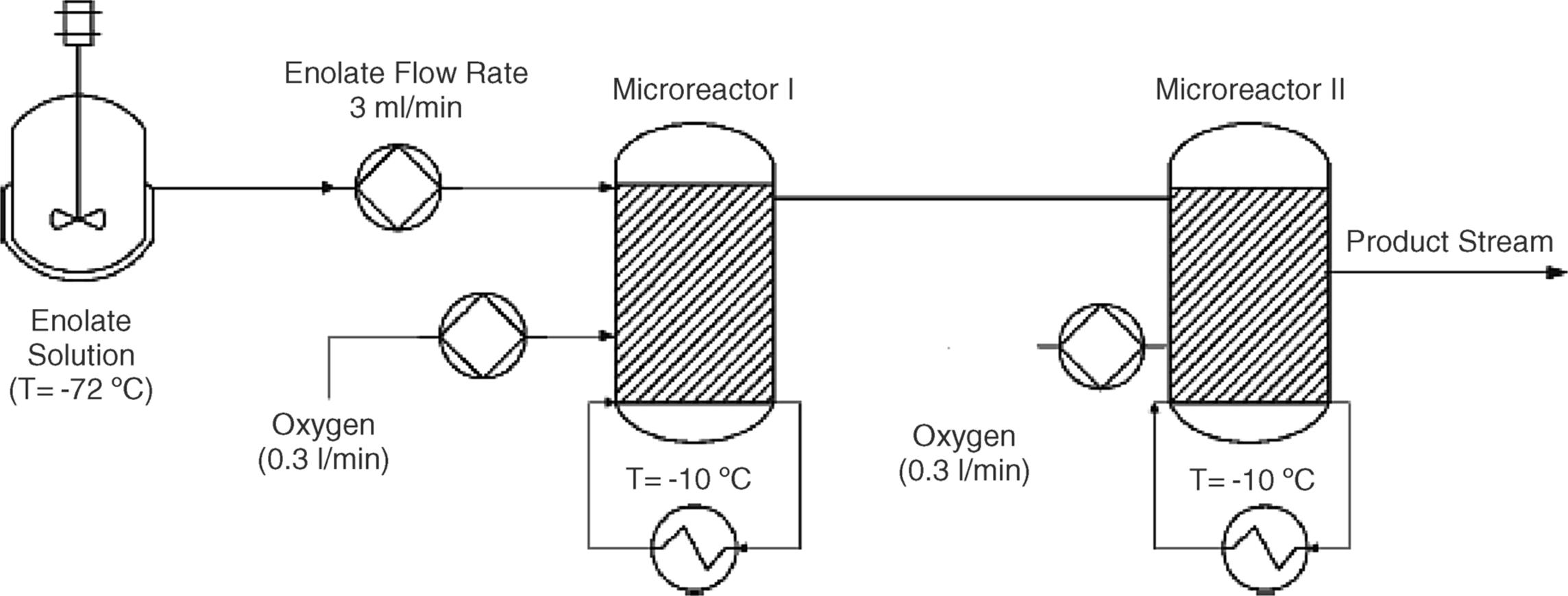
A much faster reaction was enabled due to higher operating temperatures and enhanced mass transfer in the continuous process. Long and variable batch reaction times (from 8 to 24 h) were diminished to less than 4 min in a continuous reaction. The continuous process was inherently safer with a much smaller reaction volume (about three orders of magnitude) of a flammable solvent in the presence of oxygen.
The continuous process developed in the laboratory proved to be a scalable process in the pilot plant. The trickle bed oxidation reactor was scaled by numbering up. Laboratory operating parameters were replicated for each oxidation column operated in parallel. Heat and mass transfer characteristics were identical in each oxidation column. Steady-state operation was demonstrated in the pilot plant for over 50 h. Three continuous reactors were operated in succession for production of a stable quenched product solution. Over 100 kg of API was produced during the pilot plant campaign using this setup. Also, the quality of the product produced by the continuous process was comparable to that of the batch process.
11.3.20 Swern–Moffatt Oxidation
The Swern–Moffatt oxidation in a continuous-flow microreactor system was investigated by Organon N.V., The Netherlands [49].
The Swern–Moffatt oxidation is a versatile metal-free oxidation method that finds application in the transformation of primary and secondary alcohols into aldehydes and ketones, respectively. However, its application in process chemistry is hampered by the low-temperature requirement, namely, −70 °C, and the highly exothermic behavior, which makes temperature control very difficult. The highly efficient heat transfer in the microreactor should solve the problem of limited cooling capacity in a batch reactor.
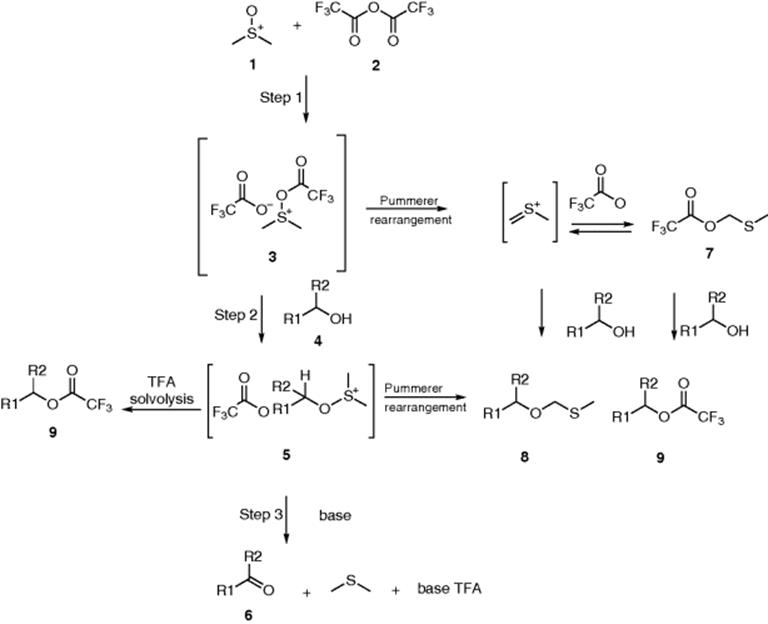
Because of the small reactor volume, accumulation of the labile trifluoroacetoxydimethylsulfonium salt (3) and alkoxydimethylsulfonium salt (5) is minimized in a microreactor. Further, because of the short residence times, which can be applied in the microreactor, the exothermic Pummerer rearrangement of the unstable intermediate 3 is limited. HPLC pumps and the Ehrfeld microreactor system were employed in the system, allowing for a real continuous process. Operation at much higher temperatures was possible, up to 30 °C [49].
The microreactor could also serve as a tool for quick scale-up of Swern–Moffatt oxidations. The scalability and reliability of the microreactor were tested by running the system for several hours. For testosterone, the system was operated for 1.5 h, resulting in an 4-androstene-3,17-dione production rate of 64 g/h.