March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 13. Aromatic Substitution: Nucleophilic and Organometallic
13.C. Reactions
In the first part of this section, reactions are classified according to attacking species, with all leaving groups considered together, except for hydrogen and N2+, which are treated subsequently. Finally, a few rearrangement reactions are discussed.
13.C.i. All Leaving Groups Except Hydrogen and N2+
A. Oxygen Nucleophiles
13-1 Hydroxylation of Aromatic Compounds
Hydroxy-de-halogenation
![]()
Direct hydroxylation of aryl halides to give phenols generally requires the presence of activating groups or exceedingly strenuous reaction conditions.96 When the reaction is carried out at high temperatures, cine substitution is observed, indicating a benzyne mechanism.97 However, phenols are prepared from aryl halides using KOH and a Pd catalyst at 100 °C.98 Formation of phenols is possible using AgNO3 with microwave irradiation,99 or CuI.100 There is a hydrogen peroxide promoted hydroxylation of aryl halides with metal hydroxide salts.101 Other microwave-promoted phenol-forming reactions are known.102
A slightly related reaction involves the amino group of naphthylamines where can be replaced by a hydroxyl group after treatment with aq bisulfite.103 The scope is greatly limited; the amino group, with very few exceptions may be NH2 or NHR and must be on a naphthalene ring. The reaction is reversible (see 13-6), and both the forward and reverse reactions are called the Bucherer reaction.
![]()
An indirect method for conversion of an aryl halide to a phenol involves initial conversion to an organometallic, followed by oxidation to the phenol. For the conversion of aryl Grignard reagents to phenols, a good procedure is the use of trimethyl borate followed by oxidation with H2O2 in acetic acid104 (see Reaction 12-31). Phenols have been obtained from unactivated aryl halides by treatment with borane and a metal (e.g., lithium), followed by oxidation with alkaline H2O2.105 Arylboronic acids [ArB(OH)2] are oxidized by aq H2O2 to give the corresponding phenol.106 The reaction of an aromatic compound with a borane in the presence of an Ir catalyst, followed by oxidation with aq Oxone, gave the corresponding phenol.107 Aryllithium reagents have been converted to phenols by treatment with oxygen.108 In a related indirect method, arylthallium bis(trifluoroacetates) (prepared by Reaction 12-23) can be converted to phenols by treatment with lead tetraacetate followed by triphenylphosphine, and then dilute NaOH.109 Diarylthallium trifluoroacetates undergo the same reaction.110
OS I, 455; II, 451; V, 632. Also see, OS V, 918.
13-2 Alkali Fusion of Sulfonate Salts
Oxido-de-sulfonato-substitution
![]()
Aryl sulfonic acids can be converted, through their salts, to phenols, by alkali fusion. In spite of the extreme conditions, the reaction gives fairly good yields, except when the substrate contains other groups that are attacked by alkali at the fusion temperatures. Milder conditions can be used when the substrate contains activating groups, but the presence of deactivating groups hinders the reaction. The mechanism is not clear, but a benzyne intermediate has been ruled out by the finding that cine substitution does not occur.111
OS I, 175; III, 288.
13-3 Replacement by OR or OAr
Alkoxy-de-halogenation
![]()
This reaction is similar to 13-1 and, like that one, generally requires activated substrates.96,112 With unactivated substrates, side reactions predominate, although aryl methyl ethers have been prepared from unactivated chlorides by treatment with MeO− in HMPA.113 This reaction gives better yields than 13-1 and is used more often. A good solvent is liquid ammonia. Aryl chlorides react with phenol and KOH with microwave irradiation to give the diaryl ether.114 Potassium phenoxide reacts with iodobenzene in an ionic solvent at 100°C with CuCl.115 Sodium methoxide reacted with o- and p-fluoronitrobenzenes ~109 times faster in NH3 at −70°C than in MeOH.116 Phase-transfer catalysis has also been used.117 Phenols reacted with aryl fluorides118 or aryl chlorides119 to give the diaryl ether. Intramolecular versions are known that produce benzofurans.120 Heating aryl iodides and phenols in an ionic liquid forms ethers.121 In addition to halides, leaving groups can be other OR, or even OH.122
For aroxide nucleophiles, the reaction is promoted by Cu salts,123 and activating groups need not be present. This method of preparation of diaryl ethers is called the Ullmann ether synthesis124 and should not be confused with the Ullmann biaryl synthesis (13-11). The reactivity order is typical of nucleophilic substitutions, despite the presence of the Cu salts.125 Copper-catalyzed coupling is known using ligand and additive-free conditions.126 Because aryloxycopper(I) reagents (ArOCu) react with aryl halides to give ethers, it has been suggested that they are intermediates in the Ullmann ether synthesis.127 Indeed, high yields of ethers can be obtained by reaction of ROCu or ArOCu with aryl halides.128 Aryl halides are converted to aryl ethers with aliphatic alcohols in the presence of suitable Cu salts.129
An increasingly important variation of this reaction couples an alkoxide and an aryl halide to give aryl ethers in the presence of a Pd catalyst and a suitable ligand.130 Ligand effects are important in such reactions.131 A Pd catalyzed, intramolecular displacement of an aryl halide with a pendant alkoxide unit leads to dihydrobenzofurans.132 Nickel catalysts have also been used.133 Aryl iodides react with phenols in the presence of K2CO3, CuI, and Raney nickel alloy.134 An Fe catalyzed etherification reaction is known.135
In a related reaction, acid salts (RCOO−) are sometimes used as nucleophiles.136 Unactivated substrates have been converted to carboxylic esters in low-to-moderate yields under oxidizing conditions.137 The following chain mechanism, called the SON2 mechanism,138 has been suggested138:
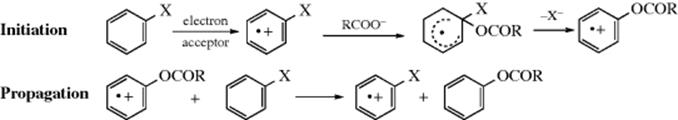
OS I, 219; II, 445; III, 293, 566; V, 926; VI, 150; X, 418.
B. Sulfur Nucleophiles
13-4 Replacement by SH or SR
![]()
Aryl thiols and thioethers can be prepared by reactions that are similar to 13-1 and 13-3.139 Activated aryl halides generally give good results, but side reactions are occasionally important. Some reagents give the thiol directly. 4-Bromonitrobenzene reacts with Na3SPO3, in refluxing methanol, to give 4-nitrothiophenol, for example.140
Diaryl sulfides can be prepared by the use of ![]() .141 Even unactivated aryl halides react with
.141 Even unactivated aryl halides react with ![]() if polar aprotic solvents (e.g., DMF,142 DMSO143 1-methyl-2-pyrrolidinone,144 or HMPA)145 are used, though the mechanisms are still mostly or entirely nucleophilic substitution. 2-Iodothiophene reacts directly with thiophenol to give 2-(phenylthio)thiophene.146
if polar aprotic solvents (e.g., DMF,142 DMSO143 1-methyl-2-pyrrolidinone,144 or HMPA)145 are used, though the mechanisms are still mostly or entirely nucleophilic substitution. 2-Iodothiophene reacts directly with thiophenol to give 2-(phenylthio)thiophene.146
Metal-catalyzed reactions of aryl halides and thiols lead to thioethers. Perhaps the most common metal used nowadays is Pd.147 Copper catalysts have been used,148 including catalysis in aqueous media,149 and also Ni150 or In151catalysts. Copper-catalyzed coupling is known using ligand- and additive-free conditions.152 Aryl iodides react with dialkyl disulfides and a nickel catalyst to give aryl alkyl sulfides.153 Diaryl sulfides can also be prepared (in high yields) by treatment of unactivated aryl iodides with ArS− in liquid ammonia under irradiation.154 The mechanism in this case is probably SRN1. The reaction (with unactivated halides) has also been carried out electrolytically, with a Ni catalyst.155 In the presence of a Pd catalyst, thiophenols react with diaryliodonium salts (Ar2I+−BF4) to give the unsymmetrical diaryl sulfide.156
Other sulfur nucleophiles also react with activated aryl halides:
![]()
Arylboronic acids [ArB(OH)2] react with thiols and copper(II) acetate to give the corresponding alkyl aryl sulfide.157 Arylboronic acids also react with N-methylthiosuccinimide, with a Cu catalyst, to give the aryl methyl sulfide.158
Aryl sulfones have been prepared from sulfinic acid salts, aryl iodides, and CuI.159 A Pd catalyzed arylation of sulfenate anions leads to aryl sulfoxides.160The copper-catalyzed reaction of NaO2SMe and aryl iodides give the aryl methyl sulfone,161 and aryl sulfones have been prepared from arylboronic acids using a Cu catalyst.162 A similar synthesis of diaryl sulfones has been reported using a Pd catalyst.163
Aryl selenides (ArSeAr and ArSeAr') can be prepared by similar methodology. Symmetrical diaryl selenides were prepared by the reaction of iodobenzene with diphenyl diselenide (PhSeSePh), in the presence of Mg and a Cu catalyst.164 Aryl halides react with tin selenides (ArSeSnR3), with a Cu catalyst, to give the diaryl selenide,165 and a CuS–Fe catalyst was also used.166
Thiocyanates have been generated from unactivated aryl halides using charcoal supported copper(I) thiocyanate.167
OS I, 220; III, 86, 239, 667; V, 107, 474; VI, 558, 824. Also see, OS V, 977.
C. Nitrogen Nucleophiles
13-5 Replacement of Halogen by NH2, NHR, or NR2
Amino-de-halogenation
Amido-de-halogenation
![]()
Activated aryl halides react with ammonia and primary and secondary amines to give the corresponding arylamines. Primary and secondary amines usually give better results than ammonia, with piperidine especially reactive. Picryl chloride (2,4,6-trinitrochlorobenzene) is often used to form amine derivatives. 2-Chloronitrobenzene also reacts with aniline derivatives directly with microwave irradiation.168 Other leaving groups in this reaction may be NO2,169 N3, OSO2R, OR, SR, N=NAr (where Ar contains electron-withdrawing groups)170 and even NR2.171 Aryl triflates react directly with secondary amines in N-methylpyrrolidine solvent using microwave irradiation.172 Aniline derivatives react with activated aromatic rings, in the presence of tetrabutylammonium fluoride and under photolysis conditions, to give a N,N-diarylamine.173 Arylation of amines with aryl halides has also been done in ionic liquids174 and in supercritical CO2.175 Flow reactor technology has been used for the direct, uncatalyzed amination of 2-chloropyridine.176
Aryl halides can be converted to amines by the use of NaNH2, NaNHR, or NaNR2.177 Lithium dialkylamides also react with aryl halides to give the N-arylamine.178 With amide base reagents, the benzyne mechanism generally operates, so cine substitution (Sec. 13.A.iii, category 2) is often found. The amide base is usually generated by reaction of the amine with an organolithium reagent, but other bases may be used. The reaction of an amine, an aryl halide, and potassium tert-butoxide generates the N-aryl amine.179 Triarylamines have been prepared in a similar manner from ArI and Ar′2NLi, even with unactivated ArI.180
Aryl fluorides react in the presence of KF–alumina and 18-crown-6 in DMSO.181 Aryl fluorides react with amines in the presence of potassium carbonate/DMSO and ultrasound,182 and aryl chlorides react on basic alumina with microwave irradiation.183 2-Fluoropyridine reacts with R2NBH3Li to give the 2-aminoalkylpyridine.184 2,4-Dinitrofluorobenzene, known as Sanger's reagent, is used to tag the amino end of a peptide or protein chain.185
The reaction of amines with aryl halides can be done under milder conditions using a catalyst to initiate or mediate the reaction. Both amines (aliphatic and aniline derivatives)186 and amide bases187 have been coupled to aryl halides using Pd catalysts and an appropriate ligand.188 A considerable amount of work189 has been done to vary the nature of the ligand and the Pd catalyst, as well as the base.190 Work to discern the mechanism of this reaction has also received considerable attention.191 This amination has come to be called the Buchwald–Hartwig Cross-Coupling Reaction.
Polymer-supported Pd catalysts,192 polymer-bound phosphine ligands used in conjunction with a Pd catalyst,193 and polymer-bound amines194 have been used for N-arylation. These reactions have been done in ionic liquids using a Pd catalyst.195 Palladium-catalyzed amination of aryl halides has been reported using microwave irradiation.196 Catalysts are available for the amination of aryl halides in aqueous media.197 The Pd-catalyzed amination of aryl substrates is not limited to halides, and the reaction with mesylates lead to arylamines.198 Arylamines with chiral substituents on nitrogen can be prepared using a Pd-catalyst with optically active ligands.199
Aniline derivatives have been prepared by the reaction of aryl chlorides with silylamines (Ph3SiNH2) using lithium hexamethyldisilazide and a Pd catalyst.200 Amines react with Ph2I+BF4−, in the presence of Pd catalysts,201 or a CuI catalyst202 to give the N-phenyl amine. Aminoalkylation of heteroaromatic rings is possible, as in the reaction of 3-bromothiophene with a primary amine and a Pd catalyst.203 2-Halopyridines react to give the 2-aminoalkyl pyridine.204
Copper catalysts have been used for the reaction of amines or aniline derivatives with aryl halides.205 The selectivity of O- versus N-arylation for reactions of amino alcohols has been discussed.206 Copper-catalyzed amination reactions can be done in aqueous media using 2-dimethylaminoethanol as a ligand.207 Ammonium salts serve as a nitrogen source in some cases.208 In the Goldberg reaction, an aryl bromide reacts with an acetanilide in the presence of K2CO3 and CuI to give an N-acetyldiarylamine, which can be hydrolyzed to a diarylamine: ArBr + Ar′NHAc → ArAr′NAc.209
Nickel catalysts have been used in the reaction of aryl halides with N-alkyl aniline derivatives210 and also with aliphatic amines.211N-Arylation was also accomplished with butyllithium and a secondary amine using Ni/C-diphenylphosphinoferrocene (dppf).212 An intramolecular reaction of a pendant aminoalkyl unit with an aryl chloride moiety, catalyzed by Ni(0) gave a dihydroindole.213 An arylbismuth reagent reacts with aliphatic amines, in the presence of copper(II) acetate, to give an N-arylamine.214 Diarylzinc reagents give the N-aryl amine in the presence of a Cu215 or a Ni catalyst216. Aryl halides also react with Zn(NTMS2)2 in the presence of a Pd catalyst to give arylamines.217 Iron-catalyzed arylation reactions are known.218 A Mo(CO)6 mediated, Pd catalyzed reaction is known for allylamines and aryl halides.219
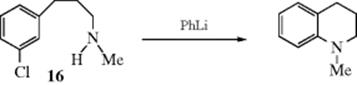
Intramolecular versions of this reaction generate bicyclic or polycyclic amines.220 An example is the conversion of 16 to the tetrahydroquinoline.221 Larger ring amines can be prepared using this approach: 8- and even 12-membered.
Arylboronic acids react with aliphatic amines222 or aq ammonia223 in the presence of a Cu catalyst. N-Aryl imides have been prepared by similar methodology, from arylboronic acids.224 Trifluoroarylboronates react with copper(II) acetate and then an aliphatic amine to give the N-phenylamine.225 Primary aromatic amines (ArNH2) were converted to diaryl amines (ArNHPh) by treatment with Ph3Bi(OAc)2226 and a Cu powder catalyst.227
The metal-catalyzed reaction with ammonia or amines likely proceeds by the SNAr mechanism.228 This reaction, with phase-transfer catalysis, has been used to synthesize triarylamines.229 In certain cases, the SRN1 mechanism has been found (Reaction 10-26). When the substrate is a heterocyclic aromatic nitrogen compound, still a different mechanism [the SN(ANRORC) mechanism], involving opening and reclosing of the aromatic ring, has been shown to take place.230
There are a number of indirect approaches for the preparation of aryl amines. Activated aromatic compounds can be directly converted to the N-aryl amine in high yield with hydroxylamine in the presence of strong bases.231 Aryl halides can be converted to the corresponding Grignard reagent (Reaction 12-38). Subsequent reaction with allyl azide followed by hydrolysis leads to the corresponding aniline derivative.232 Aryl Grignard reagents react with nitroaryl compounds to give, after reduction with FeCl3/NaBH4, a diaryl amine.233 Aryl halides can be converted to the aryllithium via halogen–lithium exchange or hydrogen–lithium exchange (Reactions 12-38 and 12-39).
The use of transition metal catalysts allows aryl halides to react with the nitrogen of amides or carbamates to give the corresponding N-aryl amide or N-aryl carbamate. Amides react with aryl halides in the presence of a Pd234 or a Cu catalyst.235N-Aryl lactams are prepared by the reaction of a lactam with an aryl halide in the presence of a Pd catalyst.236 β-Lactams also react.237 The reaction of 2-oxazolidinones with aryl halides in the presence of a Pd catalyst gave the N-aryl-2-oxazolidinone.238N-Aryl amides are prepared from amides using PhSi(OMe)3/Cu(OAc)2/Bu4NF.239N-Boc hydrazine derivatives (BocNHNH2) gave the N-phenyl derivative [BocN(Ph)NH2] when reacted with iodobenzene and a catalytic amount of CuI and 10% of 1,10-phenanthroline.240 3-Bromothiophene was converted to the 3-amido derivative with an amide and CuI-dimethylethylenediamine.241N-(2-Thiophene)-2-pyrrolidinone was similarly prepared from 2-iodothiophene, the lactam, and a copper catalyst.242N-Arylation of urea is possible using a Cu catalyst.243
The transition metal catalyzed couplings of primary or secondary phosphines with aryl halides or sulfonate esters to give arylphosphines is known.244 The Pd catalyzed conversion of aryl halides to aryl phosphines using (trimethylsilyl)diphenylphosphine tolerates many functional groups (not those that are easily reducible, e.g., aldehydes, because Zn metal245 is often used as a coreagent), but it is mainly limited to aryl iodides.246 Diphenylphosphine reacts with aryl iodides and a copper catalyst to give the triarylphosphine.247 Aryl iodides also react with a secondary phosphine and 5% Pd/C to give the P-arylphosphine.248 Tertiary phosphines can be used via aryl–aryl exchange, as in the reaction of an aryl triflate and triphenylphosphine and a Pd catalyst, for example, gave the arylphosphonium salt (ArPPh3).249 Aryl iodides can also be used.250
OS I, 544; II, 15, 221, 228; III, 53, 307, 573; IV, 336, 364; V, 816, 1067; VII, 15. OS III, 664. OS X, 423.
13-6 Replacement of a Hydroxy Group by an Amino Group
Amino-de-hydroxylation
![]()
The reaction of naphthols with ammonia and sodium bisulfite is called the Bucherer reaction. Primary amines can be used instead of ammonia, in which case N-substituted naphthylamines are obtained. In addition, primary naphthylamines can be converted to secondary (ArNH2 + RNH2 + NaSO3 → ArNHR), by a transamination reaction. The mechanism of the Bucherer reaction amounts to a kind of overall addition–elimination, via 18 and 19.251
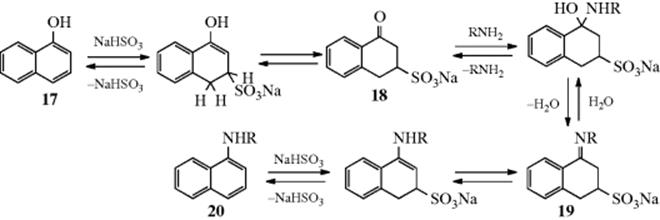
The first step in either direction consists of addition of NaHSO3 to one of the double bonds of the ring, which gives an enol from 17 (or enamine from 20) that tautomerizes to the keto form 18 (or imine form, 19). The conversion of 18 to 19 (or vice versa) is an example of Reaction 16-13 (or 16-2). Evidence for this mechanism was the isolation of 18252 and the demonstration that for β-naphthol treated with ammonia and HSO3−, the rate of the reaction depends only on the substrate and on HSO3−, indicating that ammonia is not involved in the rate-determining step.253 If the starting compound is a β-naphthol, the intermediate is a 2-keto-4-sulfonic acid compound, so the sulfur of the bisulfite in either case attacks meta to the OH or NH2.254
Hydroxy groups on benzene rings can be replaced by NH2 groups if they are first converted to aryl diethyl phosphates. Treatment of these with KNH2 and potassium metal in liquid ammonia gives the corresponding primary aromatic amines.255 The mechanism of the second step is SRN1.256
OS III, 78.
D. Halogen Nucleophiles
13-7 The Introduction of Halogens
Halo-de-halogenation, and so on
![]()
It is possible to replace a halogen on an aromatic ring by another halogen257 if the ring is activated. In such cases, there is equilibrium, which is usually shifted in the desired direction by the use of an excess of added halide ion.258A phenolic hydroxy group can be replaced by chloro with PCl5 or POCl3, but only if the ring is activated. Unactivated phenols give phosphates when treated with POCl3: 3 ArOH + POCl3 → (ArO)3PO. Phenols, even unactivated ones, can be converted to aryl bromides by treatment with Ph3PBr2259 (see Reaction 10-47) and to aryl chlorides by treatment with PhPCl4.260
Halide exchange is particularly useful for putting fluorine into a ring, since there are fewer alternate ways of doing this than for the other halogens. Activated aryl chlorides give fluorides when treated with KF in DMF, DMSO, or dimethyl sulfone.261 Reaction of aryl halides with Bu4PF/HF is also effective for exchanging a halogen with fluorine.262
Halide exchange can also be accomplished with copper halides. Since the leaving-group order in this case is I > Br > Cl ![]() F, which means that iodides cannot normally be made by this method, the SNAr mechanism is probably not operating.263 However, aryl iodides have been prepared from bromides, by the use of Cu supported on charcoal or Al2O3,264 with an excess of NaI and a Cu catalyst,265 and by treatment with excess KI and a Ni catalyst.266Interestingly, aryl chlorides have been prepared from aryl iodides using 2 molar equivalents of NiCl2 in DMF, with microwave irradiation.267 Aryl and vinyl triflates can be converted to the corresponding bromide or chloride using a Pd catalyst.268
F, which means that iodides cannot normally be made by this method, the SNAr mechanism is probably not operating.263 However, aryl iodides have been prepared from bromides, by the use of Cu supported on charcoal or Al2O3,264 with an excess of NaI and a Cu catalyst,265 and by treatment with excess KI and a Ni catalyst.266Interestingly, aryl chlorides have been prepared from aryl iodides using 2 molar equivalents of NiCl2 in DMF, with microwave irradiation.267 Aryl and vinyl triflates can be converted to the corresponding bromide or chloride using a Pd catalyst.268
Aryl iodides269 and fluorides can be prepared from arylthallium bis(trifluoroacetates) (see Reaction 12-23), indirectly achieving the ArH → ArI and ArH → ArF conversions. The bis(trifluoroacetates) react with KI to give ArI in high yields.270 Aryllead triacetates [ArPb(OAc)3] can be converted to aryl fluorides by treatment with BF3–etherate.271 Treatment of PhB(OH)2 with NIS gives iodobenzene.272 Arylboronic acids (Reaction 12-28) can be converted to the corresponding aryl bromides by reaction with 1,3-dibromo-5,5-dimethylhydantoin and 5 mol% NaOMe.273 Other aryl halides can be prepared using 1,3-dihalo-5,5-dimethylhydantoins.
OS III, 194, 272, 475; V, 142, 478; VIII, 57; 81, 98.
The reduction of phenols and phenolic esters and ethers is discussed in Reactions 19-38 and 19-35. The reaction ArX → ArH is treated in 11-39, although, depending on reagent and conditions, it can be nucleophilic or free radical substitution, as well as electrophilic.
E. Carbon Nucleophiles274
Some formations of new aryl–carbon bonds formed from aryl substrates have been considered in Reactions 10-57, 10-68, 10-76, and 10-77.
13-8 Cyanation of Aromatic Rings
Cyano-de-halogenation
Cyano-de-metalation
![]()
The reaction between aryl halides and cuprous cyanide is called the Rosenmund–von Braun reaction.275 Reactivity of the aryl halide is in the order I > Br > Cl > F, indicating that the SNAr mechanism does not apply.276 Cyanides (e.g., KCN and NaCN) do not react with aryl halides, even activated ones, but this reaction has been done in ionic liquids using CuCN.277 The reaction has also been done in water using CuCN, a phase-transfer catalyst, and microwave irradiation.278l-Proline has been used to promote the reaction.279
Aryl halides react with metal cyanides, often with another transition metal catalyst, to give aryl nitriles (aryl cyanides). Alkali cyanides convert aryl halides to nitriles280 in dipolar aprotic solvents in the presence of Pd(II) salts281or Cu282 or Ni283 complexes. In the presence of a Pd- catalyst,284 several cyanide-containing compounds react with aryl halides. Several different sources of cyanide may be used with the Pd catalyzed reaction, including: Zn(CN)2,285CuCN,286 sodium cyanoborohydride/catechol,287 potassium ferricyanide,288 and KCN.289 Microwave irradiation has been used to facilitate Pd catalyzed cyanation.290 Aryl triflates may be used in aryl cyanation reactions, as well as aryl halides.291 Benzylthiocyanate reacts with boronic acids to give aryl cyanides in a “cyanide-free” reaction, catalyzed by a Pd complex and mediated by Cu(I).292 Cyanation reactions catalyzed by copper salts are common.293 Aryl bromides react with Ni(CN)2 with microwave irradiation to give ArCN.294 A nickel complex also catalyzes the reaction between aryl triflates and KCN to give aryl nitriles.295 Iridium-catalyzed borylation of arenes also leads to aryl nitriles.296
Alternative procedures are available. One uses excess aq KCN followed by photolysis of the resulting complex ion [ArTl(CN)3−] in the presence of excess KCN.297 Alternatively, arylthallium acetates react with Cu(CN)2 or CuCN to give aryl nitriles.298 Yields from this procedure are variable, ranging from almost nothing to 90 or 100%.
Metal-free methods have been reported. For example, conversion of an aromatic compound to the corresponding iminium salt used POCl3 and DMF, and subsequent reaction with molecular iodine in aq ammonia gave the nitrile.299 An indirect method involves the reaction of an aromatic ring with tert-butyllithium, particularly when there is a directing group (see Reaction 13-17), followed by reaction with PhOCN (phenyl cyanate) to give the aryl nitrile.300 Aromatic ethers (ArOR)301 have been photochemically converted to ArCN.
OS III, 212, 631.
13-9 Coupling of Aryl and Alkyl Organometallic Compounds with Functionalized Aryl Compounds
Aryl-de-halogenation, and so on
![]()
A number of methods involving transition metals have been used to prepare unsymmetrical biaryls302 (see also, Reaction 13-11). The uncatalyzed coupling of aryl halides and metalated aryls (particularly aryllithium reagents) is also known, including cyclization of organolithium reagents to aromatic rings.303 Noncatalyzed coupling reactions of aryllithium reagents and haloarenes can proceed via the well-known aryne route, but a novel addition–elimination pathway is possible when substituents facilitate a chelation-driven nucleophilic substitution pathway.304 Such noncatalyzed coupling reactions often proceed with high regioselectivity and high yield.304 2-Bromopyridine reacts with pyrrolidine, at 130 °C with microwave irradiation, to give 2-(2-pyrrolidino)pyridine.305 Aryl iodides undergo homocoupling to give the biaryl by heating with triethylamine in an ionic liquid.306 Note that alkyl bromides are coupled to pyrrolidine by heating in an ionic liquid.307
Palladium-catalyzed coupling reactions that generate biaryls are increasingly important in synthesis. Aryl halides undergo homocoupling to give the biaryl with a Pd308 or a Ni catalyst.309 Aryl iodides have been coupled to form symmetric biphenyls with a Pd catalyst,310 and homocoupling occurs with aryl triflates under electrolysis conditions with a Pd catalyst.311 A recyclable Pd catalyst for use in ionic liquids has been developed.312 Other alternative solvents include PEG.313
Thiophene derivatives,314 pyrrole,315 azoles,316 quinoline,317 and indolizine318 have been coupled to aryl halides using a Pd catalyst. Fused polycyclic aromatic compounds can also be prepared from halobiaryls.319Trimethylsilylpyridine derivatives are coupled to aryl halides in the presence of a Pd catalyst.320 A related reaction is the Pd catalyzed decarboxylative coupling of arylcarboxylic acids with aryl iodides.321
Palladium catalysts are often used in conjunction with another metal compound or complex. Arylgermanium compounds are coupled with aryl iodides using tetrabutylammonium fluoride and a Pd catalyst.322 Homocoupling of triphenylbismuth is known,323 as well as the coupling of arylbismuth reagents to aryliodonium salts324 and to aryltin compounds325 with Pd compounds or complexes. Aryl triflates were coupled to triphenylbismuth using a Pd catalyst.326 Specialized arylbismuth compounds have been used with a Pd catalyst to convert aryl chlorides to biaryls.327 An aryltin–aryl halide coupling has been done in ionic liquids.328 Aryl halides react with cyclopentadiene and Cp2ZrCl2 and a Pd catalyst to give pentaphenylcyclopentadiene.329 The homocoupling of arylzinc iodides with a Pd catalyst has been reported.330 In a related reaction, arylsulfonyl chlorides also react with ArSnBu3 with Pd and Cu catalysts to give the biaryl.331
Aryl triflates (halides) couple with ArZn(halide) reagents in the presence of a Ni catalyst.332 A homocoupling-type reaction was reported in which PhSnBu3 was treated with 10% CuCl2, 0.5 equiv of iodine, and heated in DMF to give biphenyl.333 Similar coupling was accomplished with aryltellurium compounds.334 A Co(II) catalyzed cross-coupling of arylcopper compounds with aryl halides gives the corresponding biaryl.335 Another homocoupling reaction of pyridyl bromides was reported using NiBr2 under electrolytic conditions. Both alkylmanganese compounds (RMnCl)336 and Ph3In337 react with aryl halides or aryl triflates to give the arene. Aryl halides also react with phenols to form biaryls using a Rh catalyst.338 Diaryliodonium salts react with PhPb(OAc)3 and a Pd catalyst to give the biaryl.339 Coupling of trialkylbismuth compounds with aryl halides leads to the arene in the presence of a Pd catalyst.340 A cross-coupling reaction of benzylindium compounds and aryl halides in the presence of a Pd catalyst has been reported.341
Grignard reagents couple with aryl halides without a Pd catalyst, by the benzyne mechanism,342 but metal-catalyzed reactions are known. Typical catalysts include Fe,343 Ni344 Co,345 or Ti,346 and Pd- catalyzed reactions are important.347 Aryl Grignard reagents react with aryltrimethylammonium triflates in the presence of a Pd catalyst to give the corresponding biaryl.348 Arylmagnesium halides couple with aryl tosylates in the presence of a Pd catalyst to give unsymmetrical biaryls,349 and to halopyridines to give the arylated pyridine.350 Aryl Grignard reagents are coupled to aryliodonium salts, with ZnCl2 and a Pd catalyst, to give the biaryl.351
The coupling reaction of an excess of a Grignard reagent (RMgX) with methoxy aromatic compounds, when the aromatic ring contains multiple alkoxy groups, proceeds with replacement of the OMe group by R.352 Aryl sulfones were coupled with aryl Grignard reagents in the presence of a Ni catalyst.353
Unsymmetrical binaphthyls were synthesized by photochemically stimulated reaction of naphthyl iodides with naphthoxide ions in an SRN1 reaction.354 Methyl chloroacetate coupled with aryl iodides under electrolysis conditions, using a Ni catalyst.355 Unsymmetrical biaryls were prepared from two aryl iodides using a CuI catalyst and microwave irradiation.356
It is possible to couple metalated alkyl compounds to aryl compounds. Alkyl halides357 are coupled to aryl halide using a Ni catalyzed reductive cross coupling.358 Organozinc compounds, available by reaction of an alkyl halide and zinc metal, are coupled to aryl halides using a Pd- catalyst.359 Specialized alkyl indium complexes have been used with a Pd catalyst to give arenes.360 Iron complexes have been used for coupling reactions.361 Triarylbismuth compounds are coupled with aryl bromides in the presence of a Pd catalyst.362 Organolithium reagents are coupled to aryl bromides in the presence of a Ni catalyst,363 as are arylzinc compounds.364 The lithium enolate anion of an ester was coupled to an aryl halide using a Pd catalyst.365 Other ketones can be coupled to aryl triflates, in the presence of a Pd catalyst, and with good enantioselectivity.366
OS VI, 916; VIII, 430, 586; X, 9, 448.
13-10 Arylation and Alkylation of Alkenes
Alkylation or Alkyl-de-hydrogenation, and so on
![]()
Arylation of alkenes367 is an important reaction using an “arylpalladium” reagent, typically generated in situ from an aryl halide or other suitably functionalized aromatic compound and a Pd catalyst.368 The Pd catalyzed aryl–alkene coupling reaction is known as the Heck reaction.369 Mizoroki370 had earlier described the coupling between iodobenzene and styrene to form stilbene in methanol at 120 °C in the presence of potassium acetate and a palladium chloride catalyst leading some to call this the Mizoroki–Heck reaction. The reaction works best with aryl iodides, but conditions are available for aryl bromides and aryl chlorides.371 Aryldiazonium salts (see Reactions 13-25 and 13-26) have also been used.372 Activated aromatic compounds couple readily,373 but unactivated aromatic compounds often require special reaction conditions. The Heck reaction can be done with heterocyclic compounds,374 and heteroaryl halides can be used in the coupling reaction.375 Intramolecular Heck reactions are increasingly important.376 A silane-tethered, intramolecular Heck reaction is known.377 Other nucleophiles can be coupled to aryl halides.378
Phosphine–free catalysts,379 halogen-free reactions,380 and base-free reactions381 have been developed for the Heck reaction. Improvements to the Pd catalyst system are constantly being reported,382 including polymer-supported383 silca-supported,384 and recoverable catalysts.385 Considerable work has been done to study and improve the ligand.386 Efforts have been made to produce a homogeneous catalyst for the Heck reaction.387 The Heck reaction can be done in aq media,388 in perfluorinated solvents,389 in polyethylene glycol,390 in neat tricaprylmethylammonium chloride,391 and in supercritical CO2 (See Sec. 9.D.ii).392 The reaction has been done on solid support,393including Montmorillonite clay,394 glass beads,395 on a reverse-phase silica support,396 and using microwave irradiation.397 A microwave irradiated Heck coupling was done in water using a Pd catalyst.398 A noncatalytic reaction was reported using supercritical water.399 The effects of high pressure have been studied.400 The Heck reaction has also been done in ionic liquids,401 and it is known that the nature of the halide is important in such reactions.402 Ionic liquids have been shown to actually promote the Heck reaction.403
Ethylene is the most reactive alkene, and increasing substitution lowers the reactivity. Coupling generally takes place at the less highly substituted side of the double bond.404 Unlike the diazonium coupling reaction in Reaction 13-26, the Heck reaction is not limited to activated substrates. Both electron-deficient alkenes (e.g., acrylates),405 and electron-rich alkenes406 undergo the Heck reaction. The substrate can be an unactivated alkene,407 and the alkene can contain a variety of functional groups (e.g., esters, ether,408 enol ethers,409 enamides,410 carboxyl, phenolic, or cyano groups).411 The aryl halide or aryl triflate can be coupled to dienes,412 allenes,413 allylic acetates,414 allylic silanes,415 allylic amines,416 vinyl phosphonate esters,417 and with terminal alkynes.418 Aryliodonium salts can be coupled to conjugated alkenes in a Heck-like manner using a Pd catalyst.419 Double-coupling reactions have been reported, generating diaryl aromatic compounds.420 Heck-type reactions have been reported with imines.421
Control of regiochemistry is a serious problem in the coupling to unsymmetrical alkenes. Some regioselectivity can be obtained by the use of alkenes attached to an auxiliary coordinating group,422 or by using special ligands and acrylate or styrene as substrates.423 Neighboring-group effects play a role in the Heck reaction.424 Steric effects are thought to control regioselectivity,425 but electronic influences have also been proposed.426 It has been shown that the presence of steric effects generally improves 1,2-selectivity, and that electronic effects can be used to favor 1,2- or 2,1-selectivity.427 A 1,4-Pd migration between the o- and o′-positions of biaryls has been observed in organopalladium intermediates derived from o-halobiaryls.428 Migration of the double bond is a problem in some cases, and reaction conditions play a significant role in such migrations.429 It has been reported that double-bond isomerization can be suppressed in intramolecular Heck reactions done in supercritical CO2 (see Sec. 9.D.ii).430
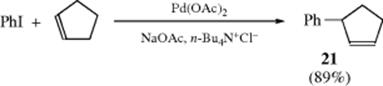
The Pd catalyzed reactions are usually stereospecific,431 yielding products expected from syn addition followed by syn elimination.432 Because the product is formed by an elimination step, with suitable substrates, double-bond migration can occur, resulting in allylic rearrangement (as in the reaction of cyclopentene and iodobenzene to give 21).433 Asymmetric Heck reactions are known,434 including asymmetric intramolecular Heck reactions.435Dihydrofurans react with aryl triflates and a Pd catalyst that includes a chiral ligand, to give the 5-phenyl-3,4-dihydrofuran with good enantioselectivity.436 A similar reaction was reported for an N-carbamoyl dihydropyrrole.437
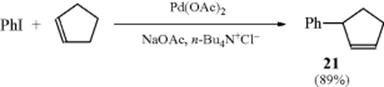
An addition–elimination mechanism (addition of ArPdX followed by elimination of HPdX) operates in most cases.438 In the conventionally accepted reaction mechanism,439 which is shown,440 a four-coordinate arylPd(II) intermediate (a palladacycle)441 is formed by oxidative addition of the aryl halide to a Pd(0) complex prior to olefin addition.442 This description suggests that cleavage of the dimeric precursor complex, reduction
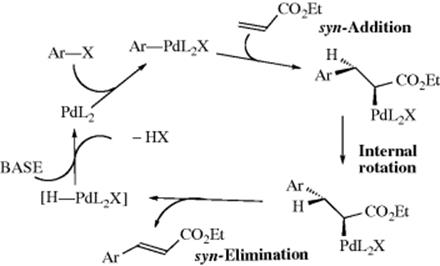
of Pd2+, and ligand dissociation combine to give a viable catalytic species.443 σ-Alkyl Pd(II) intermediates are thought to be involved.444 An analysis of reaction kinetics under dry conditions was reported.435 In this study, the mechanism requires a first-order dependence on olefin concentration, and anomalous kinetics may be observed when the rate-limiting step is not directly on the catalytic cycle.435 The mechanism requires a proton abstraction step, and there are substituent effects for this step.445 A mechanistic study has been reported for the Pd catalyzed decarboxylative reaction of alkenes and arylcarboxylic acids.446 The kinetics and mechanism of the Heck reaction promoted by a C–N palladacycle has been studied.447 The mechanistic implications of asymmetric Heck reactions have been examined.448
There are a number of variations of this reaction, including the use of transition metal catalysts other than Pd. Rhodium-catalyzed Heck reactions are known,449 and there are Co-,450 Ru-,451 and Ni catalyzed variations.452 Iron-mediated arylation of alkenes has been reported,453 and vinylgermanes are coupled to aryl halides with a Pd catalyst.454 Aryl chlorides were coupled to conjugated esters using a RuCl3•3 H2O, in an atmosphere of O2 and CO.455 Aryl halides have been coupled to allenyltin compounds (C=C=C-SnR3).456 Divinylindium chloride [(CH2=CH)2InCl] reacted with an aryl iodide in aq THF with a Pd catalyst to give the styrene derivative.457 Trialkenylindium reagents reacted similarly with aryl halides in the presence of a Pd catalyst.458 Arylzinc chlorides (ArZnCl) were coupled to vinyl chlorides using a Pd catalyst,459 and vinyl zinc compounds were coupled to aryl iodides.460 In the presence of trimethylsilylmagnesium chloride, primary alkyl halides coupled to aryl alkenes to give the substituted alkene (R′–CH=CHAr), using a Co catalyst.461
Potassium vinyltrifluoroborates can be coupled to aryl halides in Heck-like coupling reactions.462 Likewise, the reaction of aryltrifluoroborates with vinyl halides, in the presence of a Pd catalyst, leads to the aryl alkene.463 In a related reaction, alkyltrifluoroborates react with aryl halides in the presence of a Pd and a Rh catalyst.464
Although related to chemistry found in Reaction 10-57, metal-catalyzed alkylation reactions are easily viewed as Heck-like reactions. For that reason, they are discussed here. Alkyl halides are coupled to alkenes to form substituted alkenes using a Co catalyst, promoted by Me3SiCH2MgCl.465 Alkylation requires that the alkyl group lacks a β-hydrogen, and the reaction is successful for the introduction of methyl, benzyl, and neopentyl groups.466However, vinylic groups, even those possessing β-hydrogen atoms, have been successfully introduced (to give 1,3-dienes) by the reaction of the alkene with a vinylic halide in the presence of a trialkylamine and a Pd(0) catalyst.467
OS VI, 815; VII, 361; 81, 42, 54, 63, 263
13-11 Homo-Coupling of Aryl Halides: The Ullmann Reaction
de-halogen-coupling
![]()
The coupling of aryl halides with copper is called the Ullmann reaction.468 The reaction is clearly related to Reaction 13-9, but involves aryl copper intermediates. The reaction is of broad scope and has been used to prepare many symmetrical and unsymmetrical biaryls.469 When a mixture of two different aryl halides is used, there are three possible products, but often only one is obtained. The best leaving group is iodo, and the reaction is most often done on aryl iodides, but bromides, chlorides, and even thiocyanates have been used. New ligands have been developed to promote the reaction, including an air-stable diazaphospholane ligand.470 The Cu catalyst has been immobilized.471Intramolecular reactions are known.472 The coupling reaction can be applied to heterocyclic compounds.473
The effects of other groups on the ring are not easily predicted. The nitro group is strongly activating, but only in the ortho (not meta or para) position.474 Both R and OR groups activate in all positions. Not only do OH, NH2, NHR, and NHCOR inhibit the reaction, as would be expected for aromatic nucleophilic substitution, but so do CO2H (but not CO2R), SO2NH2, and similar groups for which the reaction fails completely. These groups inhibit the coupling reaction by causing side reactions.
The mechanism is not known with certainty. It seems likely that it is basically a two-step process, similar to that of the Wurtz reaction (10-56), which can be represented schematically by
![]()
Organocopper compounds have been trapped by coordination with organic bases.475 In addition, aryl copper compounds (ArCu) have been independently prepared and shown to give biaryls (Ar–Ar′) when treated with aryl iodides (Ar′I).476 A similar reaction has been used for ring closure477: Copper-catalyzed coupling of aryl halides and heterocycles has been reported.478
An important alternative to the Ullmann method is the use of certain Ni complexes.479 Aryl halides (ArX) can also be converted to Ar–Ar480 by treatment with activated Ni metal,481 with Zn and Ni complexes,482 with aq alkaline sodium formate, Pd–C and a phase-transfer catalyst,483 and in an electrochemical process catalyzed by a Ni complex.484
An asymmetric Ullmann reaction has been reported.485
OS III, 339; V, 1120.
13-12 Coupling of Aryl Compounds with Arylboronic Acid Derivatives
Aryl-de-halogenation, and so on
Aryl-de-boronylation, and so on
![]()
Arylboronic acids are coupled to aryl halides using a Pd catalyst to give the arene in what is called Suzuki coupling (or Suzuki–Miyaura coupling).486 Aryl triflates react with arylboronic acids [ArB(OH)2, Reaction 12-28],487 or with organoboranes,488 in the presence of a Pd catalyst.489 Even hindered boronic acids give good yields of the coupled product.490 Homocoupling of arylboronic acids has been reported.491 Some aromatic compounds are so reactive that a catalyst may not be required. Using tetrabutylammonium bromide, phenylboronic acid was coupled to 2-bromofuran without a catalyst.492
Different conditions (including additives and solvent) for the reaction have been reported,493 often focusing on the Pd catalyst494 or the ligand.495 Phosphine-496 and ligand-free conditions497 have been developed. Recyclable catalysts have been developed.498 Catalysts have been developed for deactivated aryl chlorides.499 Suzuki coupling has also been done in ionic liquids,500 in supercritical CO2501 (see Sec. 9.D.ii), and there are solvent-free procedures.502 Several procedures for coupling in aqueous media have been reported.503 The reaction has been done neat on alumina,504 and on alumina with microwave irradiation.505 Several procedures have been reported using microwave irradiation.506 Modifications to the basic procedure include tethering the aryl triflate507 or the boronic acid508 to a polymer, allowing a polymer-supported Suzuki reaction. Polymer-bound Pd complexes have been used.509There is even a Pd free cross-coupling.510
Arylboronic acids have been coupled to vinyl halides511 or vinyl tosylates512 using a Pd catalyst. Halogenated heteroaromatic compounds react, as do aryl carbamates, carbonates and sulfamates,513 and aryl phosphoramides.514Aryl sulfonates have been used.515 Arylboronic acids couple with aryl sulfonate esters.516 Many different heterocycles have been arylated.517 4-Pyridylboronic acids have been used.518 3-Iodopyridine reacted with NaBPh and palladium acetate, with microwave irradiation, to give 3-phenylpyridine.519
A variety of functional groups are compatible with Suzuki coupling, including Ar2P=O,520 CHO,521 C=O of a ketone,522 CO2R,523 cyclopropyl,524 NO2,525 CN, and halogen substituents.526 Vinyl halides react with arylboronic acids to give alkenyl derivatives (vinyl arenes, C=C–Ar),527 in what is clearly a Heck-like reaction. In a variation, vinylboronic acids coupled to aryl halides to give the vinyl-coupling product.528 Vinylboronic acids have been coupled to aryldiazonium salts (see Reaction 13-25) without added base, using a Pd catalyst with an imidazolium ligand.529
Alkylation can accompany arylation if alkyl halides are added, as in the conversion of iodobenzene to 2,6-dibutylbiphenyl.530 Alkyl groups may be coupled to aryls, as in the Pd catalyzed reaction of arylboronic acids with benzylic carbonates.531 Arylboronic acids also couple with alkyl halides using either a palladium(II) acetate532 or a Ni catalyst.533 Conversely, arylboronic acids can be coupled to aliphatic halides.534 Arylboronic acids can be coupled to allylic alcohols as well.535 Benzylic phosphonates have also been used.536 Double Suzuki coupling reactions are known.537 An alkyl–alkyl coupling reaction has been classified as a Suzuki cross-coupling reaction.538 The reaction of an arylboronic acid and 1,2-dibromoethane, with KOH and a Pd catalyst, leads to the styrene derivative.539
Since many biaryls are chiral due to atropisomerism (see Sec. 4.C, category 5), the use of a chiral catalyst, and/or a chiral ligand can lead to enantioselectivity in the Suzuki coupling.540
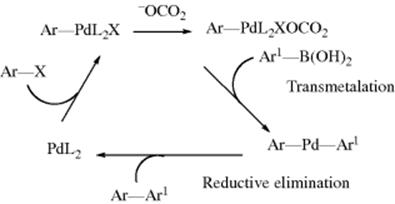
For a mechanistic viewpoint,541 the Suzuki coupling proceeds via oxidative addition of areneboronic acids to give a Pd species, followed by 1,2-arene migration to an electron-deficient Pd atom, eventually leading to very fast reductive elimination to afford biaryls.542 This mechanism is illustrated.543 Several intermediates of the oxidative coupling process have been identified by electrospray ionization mass spectrometry.544 Palladium peroxo complexes have been shown to be key intermediates.545
Other transition metals have been employed in these coupling reactions, sometimes as cocatalysts. Arylboronic acids have been coupled to conjugated alkenes to give the aryl–alkene coupling product using a Pd catalyst,546 a Ru catalyst with copper(II) acetate,547 a Ni catalyst,548 or a Rh catalyst.549 Aryl boronic acids are coupled with aryl ammonium salts to give the biaryl, with a Ni catalyst.550 Allylic acetates have been coupled to arylboronic acids using nickel bis(acetylacetonate) and diisobutylaluminum hydride.551 Arylboronic acids (Reaction 12-28) were shown to react directly with benzene in the presence of Mn(OAc)3.552 Aryl halides couple with ArB(IR′2) species with a Pd catalyst.553 Iron catalysts have been developed for coupling with alkyl halides.554 Arylboronic acids couple with the phenyl group of Ph2TeCl2 with a Pd catalyst.555 Tributyltin aryl compounds were coupled to the aryl group of Ar2I+BF4− with a Ni catalyst.556
Suzuki-type coupling reactions have been reported involving acyl halides. When arylboronic acids were reacted with benzoyl chloride and PdCl2, the product was the diaryl ketone.557 This coupling reaction was also accomplished using a Pd(0) catalyst.558 Cyclopropylboronic acids couple with benzoyl chloride, in the presence of Ag2O and a Pd catalyst, to give the cyclopropyl ketone.559 A Ni catalyst has been used,560 and Ph3P/Ni/C–BuLi has also been used.561 Arylboronic acids have also been coupled to anhydrides,562 and the methoxy group of anisole derivatives has been replaced with phenyl using phenylboronic acid and a Ru catalyst.563
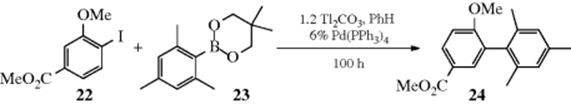
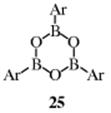
Arylborates (see Reaction 12-28), ArB(OR)2, can be used in place of the boronic acid.564 The coupling reaction of aryl iodide (22) with boronate (23), for example, gave biaryl 24.565 Base-free conditions, using nitrogen ligands, have been reported.566 Aryl and heteroarylboroxines (25) can be coupled to aryl halides using a Pd catalyst.567 Vinylboranes have been coupled to aryl iodides to give the aryl alkene, in the presence of a Pd catalyst.568Organoboranes are coupled to aryl halides with a Pd catalyst.569
In a useful variation, aryltrifluoroborates (ArBF3+ X−) (Reaction 12-28), are coupled to aryl halides with a Pd catalyst to give the biaryl.570 Alkyltrifluoroborates571 (RBF3K, see Reaction 12-28) react with aryl triflates572 aryl halides,573 or aryliodonium salts574 with a Pd catalyst, to give the arene. In a related reaction, vinyltrifluoroborates (C=C–BF3+ X− see Reaction 12-28), are coupled to aryl halides with a Pd catalyst to give the styrene derivative.575Suzuki coupling with trifluoroborates has also been done using microwave irradiation.576 Aryltellurides have been used in this reaction.577 A Pd catalyzed reaction with enamino ketones, mediated by Cu(II) salts, led to coupling to give the aryl derivative.578 Ruthenium catalysts have also been used.579
OS 75, 53, 61
The coupling reactions of alkylboronic acids are covered in Reaction 13-17.
OS CV, 102, 467; OS 81, 89.
13-13 Aryl–Alkyne Coupling Reactions
Alkynyl-de-halogenation, and so on
![]()
When aryl halides react with copper acetylides to give 1-aryl alkynes, the reaction is known as Stephens–Castro coupling.580 Both aliphatic and aromatic substituents can be attached to the alkyne unit, and a variety of aryl iodides have been used. Benzonitrile was shown to react with alkynyl zinc bromides, with a Ni catalyst and after electrolysis to give the diarylalkyne, where the cyano unit was replaced with an alkyne unit.581
![]()
A Pd catalyzed variation is known in which an aryl halide reacts with a terminal alkyne to give 1-aryl alkynes is called Sonogashira coupling.582 Terminal aryl alkynes react with aryl iodides and Pd(0)583 to give the corresponding diaryl alkyne,584 but monoalkynes are easily prepared.585 Aryl iodides are more reactive than aryl fluorides.586 Alkynes can be coupled to heteroaromatic compounds.587 As with all of the metal-catalyzed reactions in this chapter, work has been done to vary reaction conditions, including the catalyst,588 the ligand,589 the solvent, and additives.590 There are copper-591 and ligand-free variations.592 Transition metal free coupling has been reported using a 2,2,6,6-tetramethylpiperidine-N-oxyl radical as an oxidant.593 Variations include Sonogashira coupling in water, without Cu,594 and other reactions are done in aqueous media.595 The coupling has been done in aq polyethylene glycol,596 and in ionic liquids.597 Microwave irradiation is an important tool in this reaction.598 Sonogashira coupling was reported on microbeads,599 with nanoparticulate nickel powder,600 and the aryl iodide was tethered to a polymer for a solid-state reaction.601 Polymer-supported catalysts are known.602 A variation scavenged the triphenylphosphine byproduct by addition of Merrifield resin.603
Coupling of the alkynes to form a diyne (see Reaction 14-16) can be a problem is some cases, although the aryl–alkyne coupling usually predominates.604 There are many variations. Alkyl groups are coupled to alkynes under Sonogashira conditions, including unactivated secondary alkyl halides.605 Coupling propargyl bromide and an aryl iodide, in the presence of an amine, gives the aryl aminomethylalkyne.606 4-Chloroacetophenone reacts with 1-phenylethyne, showing that the carbonyl group is compatible with this reaction.607 Arenediazonium salts can be used for the coupling reaction.608 Similar Pd catalyzed coupling of bromoalkynes with heterocycles also leads to the alkynyl derivative.609 Ynamides are coupled using cooper-free conditions.610
There are variations of the Sonogashira reaction that use other metals as catalysts or cocatalysts. A Pd free reaction is known, using Cu complexes as the catalyst.611 An In catalyzed reaction is known that does not use Cu, Pd, or phosphine ligands.612 Gold(I) has been used to catalyze Sonogashira reactions,613 and there are Ni614 and Fe catalyzed615 reactions. Silver iodide has been used to catalyze the reaction.616 Conversion of 1-lithioalkynes to the corresponding alkynyl zinc reagent allows coupling with aryl iodides when a Pd catalyst is used.617 A 1-lithioalkyne was directly coupled to aryl bromides in the presence of B(OiPr)3 and a Pd catalyst,618 where an alkynylboronic acid was generated in situ. Lithium alkynyltrimethylborates are coupled to aryl chlorides as well.619 Terminal alkynes are coupled to acylpyridinium salts in the presence of a Cu catalyst, giving a product with high enantioselectivity when a chiral ligand is used.620 Coupling with alkynyl tin compounds is known.621 A triphenylstibine [Ph3Sb(OAc)2] was used to transfer a phenyl group to the alkyne carbon of PhC![]() CSiMe3, using Pd and CuI catalysts.622
CSiMe3, using Pd and CuI catalysts.622
A variation of this aryl–alkyne coupling reaction reacted methylthioalkynes (R–C![]() C–SMe) with arylboronic acids and a Pd catalyst to give the aryl alkyne (R–C
C–SMe) with arylboronic acids and a Pd catalyst to give the aryl alkyne (R–C![]() C–Ar).623 The boron trifluoride induced Pd catalyzed cross-coupling reaction of 1-aryltriazenes with areneboronic acids has been reported.624 Aryl halides are coupled to alkynyltrifluoroboronates (R–C
C–Ar).623 The boron trifluoride induced Pd catalyzed cross-coupling reaction of 1-aryltriazenes with areneboronic acids has been reported.624 Aryl halides are coupled to alkynyltrifluoroboronates (R–C![]() C–BF3K, Reaction 12-28) using a Pd catalyst.625 Aryl iodides were also coupled to lithium alkynyl borate complexes, Li[R–C
C–BF3K, Reaction 12-28) using a Pd catalyst.625 Aryl iodides were also coupled to lithium alkynyl borate complexes, Li[R–C![]() C–B(OR′)3], to give the aryl alkyne.626
C–B(OR′)3], to give the aryl alkyne.626
Diaryliodonium salts react with terminal alkynes to give the phenyl alkyne.627 A variation couples the phenyl group of Ph2I+OTf− with an en-yne using a Pd catalyst.628 Aryl sulfonate esters can be coupled to terminal alkynes using a Pd catalyst in polymethylhydrosiloxane.629
OS 11, 2009, 234.
13-14 Arylation at a Carbon Containing an Active Hydrogen
Bis(ethoxycarbonyl)methyl-de-halogenation, and so on
![]()
The arylation of compounds of the form ZCH2Z′ is analogous to Reaction 10-67, where Z is defined as an electron-withdrawing group (ester, cyano, sulfonyl, etc.). Activated aryl halides generally give good results.630 Treatment with an aryl halide in liquid ammonia containing Na or K, for example, leads in the formation of 26 and 27.631 When the solution is irradiated with near-UV light, but Na or K is omitted, the same products are
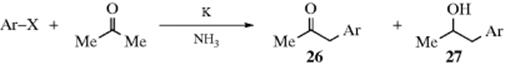
obtained (though in different proportions).632 In either case, other leaving groups can be used instead of halogens (e.g., NR3+, SAr) and the mechanism is the SRN1 mechanism. N-Heterocyclic carbene ligands in the presence of alkoxide bases lead to coupling of ketones and aryl halides, at the α-position of the ketone.633 The reaction can also take place without an added initiator. The reaction of 2-fluoroanisole and KHMDS, and 4 equiv of 2-cyanopropane, leads to substitution of the fluorine atom by CMe2CN.634 β-Keto esters were coupled to aryl fluorides using CsOH and a chiral quaternary ammonium salt, leading to the aryl substitution product with good enantioselectivity.635
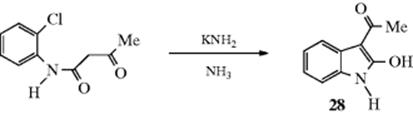
Even unactivated aryl halides can be employed if the reaction is carried out in the presence of a strong base (e.g., NaNH2636 or LDA). Compounds of the form ZCH2Z′, and even simple ketones637 or carboxylic esters have been arylated in this manner. The reaction with unactivated halides proceeds by the benzyne mechanism and represents a method for extending the malonic ester (and similar) syntheses to aromatic compounds. The base performs two functions: It removes a proton from ZCH2Z′ and catalyzes the benzyne mechanism. The reaction has been used for ring closure, as in the formation of indole 28.638
A similar reaction was reported using a Pd catalyst.639 Nitroethane was converted to 2-phenylnitroethane using bromobenzene and a Pd catalyst.640 Palladium catalysts have been developed for the α-arylation of ketones.641 α-Arylation of esters has been accomplished using Pd catalysts.642 Malonate esters are coupled unactivated aryl halides using a Pd catalyst.643 Bis-(sulfones) [CH2(SO2Ar)2], react with aryl halides in the presence of a Pd catalyst.644Iron(II) salts have also been used to initiate this reaction.645 The coupling of active methylene compounds and unactivated aryl halides can also be done with copper halide catalysts103 (the Hurtley reaction).646 Similar coupling was accomplished with CH2(CN)2 and a Ni catalyst.647 A variation of α-arylation reacts α-halocarbonyl compounds with arylboronic acids, in the presence of a Ni catalyst.648 Malonic and β-keto esters can be arylated at the α-carbon in high yields by treatment with aryllead tricarboxylates [ArPb(OAc)3],649 with triphenylbismuth carbonate (Ph2BiCO3),650 and other Bi reagents.651 In a related process, manganese(III) acetate was used to convert a mixture of ArH and ZCH2Z′ to ArCHZZ′.652 Arylzinc reagents have also been used.653
Enolate ions of ketones react with PhI in the dark.654 In this case, it has been suggested655 that initiation takes place by formation of a radical (e.g., 29).

This is a SET mechanism (see Sec. 10.B). The photostimulated reaction has also been used for ring closure.656 In certain instances of the intermolecular reaction, there is evidence that the leaving group exerts an influence on the product ratios, even when it has already departed at the time that product selection takes place.657
The reaction of the enolate anions of ketones and aldehydes, generated in situ by addition of a suitable base, with aryl halides can be accomplished by treatment with a Pd catalyst.658 Formation of an enolate anion of a conjugated ketone (cyclohexenone) via reaction with LDA (see Sec. 8.F, category 7), in the presence of Ph3BiCl2, leads to the α-phenyl conjugated ketone (6-phenylcyclohex-2-enone).659 An ester reacted with TiCl4 and N,N-dimethylanline to give the para-substitution product (Me2N–Ar–2Et).660 Nickel-catalyzed α-arylation of ketone enolate anions is also known.661 The enolate anion of lactams will react with aryl halides in the presence of a Pd catalyst via the 3-aryl lactam.662 When the enolate anion of a ketone is generated in the presence of a Pd catalyst and a chiral phosphine ligand, the α-aryl ketone is formed with good enantioselectivity.663
OS V, 12, 263; VI, 36, 873, 928; VII, 229.
13-15 Conversion of Aryl Substrates to Carboxylic Acids, Their Derivatives, Aldehydes, and Ketones664
Alkoxycarbonyl-de-halogenation, and so on
![]()
Carbonylation of aryl halides665 and aryl triflates666 with CO, an alcohol and a base (which gives an alkoxide), and a Pd catalyst, gives carboxylic esters. Similar carbonylation reactions are possible with alkyl halides. Aryl carboxylic acids were also prepared from aryl iodides by heating in DMF with lithium formate, LiCl, acetic anhydride, and a Pd catalyst.667 Even very sterically hindered alkoxides can be used to produce the corresponding ester.668The use of H2O, RNH2, or an alkali metal or calcium carboxylate669 instead of ROH, gives the carboxylic acid,670 amide,671 or mixed anhydride, respectively.672 Ester formation via carbonylation was done is supercritical CO2 (see Sec. 9.D.ii).673 Microwave promoted carbonylation reactions have been reported.674
Variations including a silica-supported Pd reagent have been used to convert iodobenzene to butyl benzoate, in the presence of CO and butanol.675 2-Chloropyridine was converted to the butyl pyridine 2-carboxylate with this procedure.676 Dicobalt octacarbonyl [Co2(CO)8] may be used as a surrogate reagent for CO.677 Heating an aryl iodide, CO in ethanol and DBU, with a Pd catalyst, gave the ethyl ester of the aryl carboxylic acid.678 A similar result was obtained when an aryl iodide was heated in ethanol with triethylamine, CO, and Pd/C.679 Phenols and aryl halides react with a Pd catalyst, and carbonylation leads to the phenyl ester.680 Arylthallium bis(trifluoroacetates) [ArTl(O2CCF3)2, (see Reaction 12-23], can be carbonylated with CO, an alcohol, and a PdCl2 catalyst to give esters.681 Note that seleno esters (ArCOSeAr) were prepared from aryl iodides, CO, PhSeSnBu3, and a Pd catalyst.682Aminocarbonylation reactions are known.683
Modification of this approach allows the synthesis of ketones, and aryl iodides can be converted to aldehydes.684 Aryl ketones can be prepared from aryltrimethylsilanes (ArSiMe3) and acyl chlorides in the presence of AlCl3.685Aryllithium and Grignard reagents react with iron pentacarbonyl to give aldehydes ArCHO.686 The reaction of CO with aryllithium may occur by electron transfer.687 Aryl iodides are converted to unsymmetrical diaryl ketones on treatment with arylmercury halides and nickel carbonyl: ArI + Ar′HgX + Ni(CO)4 → ArCOAr′.688 Aryl iodides are carbonylated to give the aryl alkyl ketone with CO and R3In.689 Aryl iodides are coupled with aryl acid chlorides in the presence of an In complex to form a diaryl ketone.690 Organomercury compounds undergo a similar reaction.691 The aryllead reagent [PhPb(OAc)2] was converted to benzophenone using NaOMe, CO, and a Pd catalyst.692 Aryl iodides containing an ortho substituent with a β-cyano group that served as the source of a carbonyl group, was converted to a bicyclic ketone with a Pd catalyst at 130 °C in aq DMF.693
Diaryl ketones can also be prepared by coupling aryl iodides with phenylboronic acid (Reaction 12-28), in the presence of CO and a Pd catalyst.694 This reaction has been extended to heteroaromatic systems, with the preparation of phenyl 4-pyridyl ketone from phenylboronic acid and 4-iodopyridine.695 2-Bromopyridine as coupled with phenylboronic acid, CO, and a Pd catalyst to give phenyl 2-pyridyl ketone.696 Carbonylation of an alkyne and an aryl halide, with CO and Pd and Cu catalysts, gave the alkynyl ketone RC![]() C(C=O)Ar.697
C(C=O)Ar.697
13-16 Arylation of Silanes
Silyl and Silyloxy-de-halogenation, Aryl-de-silylation, and so on
![]()
In the presence of transition metal catalysts (e.g., Pd), trialkoxysilanes [HSi(OR)3] react with aryl halides to give the corresponding arylsilane.698 This transformation is an alternative to the Suzuki coupling (Reaction 13-12).699The influence of silicon substituents on the cross-coupling reaction has been studied.700 A similar reaction was reported using a Rh catalyst.701 Similar coupling of aryl halides with trialkylsilanes (HSiR3) in the presence of a Pd,702or Rh catalyst,703 or PtO2704 gives the arylsilane. Cyclic alkoxysilanes are prepared using Pd catalyzed cross-coupling reactions.705 Vinylsilanes are coupled to aryl halides to give the aryl alkene.706 Disilanes have also been employed, using a Pd catalyst.707 Arylsilanes can be coupled to aryl iodides using a Pd catalyst708 and in aqueous media.709 Arylsilanes react with alkyl halides to give the corresponding arene, in the presence of a Pd catalyst.710
Other variations include the conversion of vinyl silanes to styrene derivatives upon treatment with Bu4NF (TBAF), an aryl iodide and a Pd catalyst.711 Arylsilanes were coupled to alkenes to give the styrene derivative using palladium acetate in an oxygen atmosphere,712 or TBAF and an Ir catalyst.713 Aryl iodides can be coupled to 1-methyl-1-vinyl- and 1-methyl-1-(prop-2-enyl)silacyclobutane with desilyation, using a Pd catalyst and TBAF, to give the corresponding styrene derivative.714 Aryl silanes can be coupled to aryl iodides using Ag2O and a Pd catalyst,715 and arylsiloxanes [ArSi(OR)3] are coupled to aryl halides with TBAF and a Pd catalyst.716 1-Trialkylsilylalkynes (R3Si–C![]() C–R′) were coupled to aryl iodides using a Pd catalyst.717
C–R′) were coupled to aryl iodides using a Pd catalyst.717
An alternative approach reacts aryllithium reagents with siloxanes [Si(OR)4], to give the aryl derivative ArSi(OR)3.718 Biaryl derivatives are similarly prepared from aryl halides.719 Arylsiloxanes are similarly coupled at the ortho position of anilide derivatives.720
The reaction of NaBPh4 (sodium tetraphenylborate) and a silyl dichloride (Ph2SiCl2) gives biphenyl.721
13.C.ii. Hydrogen as Leaving Group722
13-17 Alkylation and Arylation
Alkylation or Alkyl-de-hydrogenation, and so on
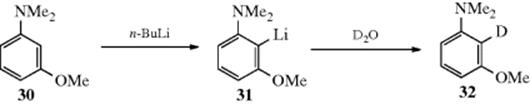
The alkylation of aromatic rings was introduced, in part, in Reaction 10-57. The reaction of an aromatic ring with an organolithium reagent can give H–Li exchange to form an aryllithium. This reaction tends to be slow if there are activating substituents on the aryl halide, or in the absence of diamine additives.723 When heteroatom substituents are present as in 30, however, the reaction is facile and the Li goes into the 2 position (as in 31).724 This regioselectivity can be quite valuable synthetically, and is now known as directed ortho metalation725 (see Reaction 10-57). Subsequent reaction with a suitable electrophilic agent (e.g., D2O) leads to 32 in this case. Aryllithium reagents give arylation. The reaction occurs by an addition–elimination mechanism and the adduct can be isolated.726 Upon heating of the adduct, elimination of LiH occurs and an alkylated product is obtained. With respect to C-2, the first step is the same as that of the SNAr mechanism. The difference is that the unshared pair of electrons on the nitrogen combines with the lithium, so the extra pair of ring electrons has a place to go: It becomes the new unshared pair on the nitrogen.
With TMEDA/n-butyllithium mediated arene lithiation reactions, the viability of directive effects (complex-induced proximate effects) has been questioned,727 although it is not clear if this extends to other systems (particularly when there is a strong coordinating group (e.g., carbamate).728 The 2 position is much more acidic than the 3 position (see Table 8.1), but a negative charge at C-3 is in a more favorable position to be stabilized by the Li+. Lithiation reactions do not necessarily rely on a complex-induced proximity effect.729N,N-Dialkyl aryl-O-sulfamates are substrates for ortho metalation.730 Phenylaziridines also undergo this reaction.731 Formation of the ortho arylmagnesium compound has been accomplished with bases of the form (R2N)2Mg.732 A directed meta metalation has been reported, using alkali metal mediated zincation.733 Note that the substrate-dependent mechanism of the ortho lithiation of aryloxazolines using butyllithium has been studied.734
Benzene, naphthalene, and phenanthrene have been alkylated with alkyllithium reagents, although the usual reaction with these reagents is Reaction 12-22,735 and Grignard reagents have been used to alkylate naphthalene.736 The addition–elimination mechanism apparently applies in these cases too. A protected form of benzaldehyde (protected as the benzyl imine) has been similarly alkylated at the ortho- position with butyllithium.737 The alkylation of heterocyclic nitrogen compounds738 with alkyllithium reagents is called Ziegler alkylation. The reaction of 2-chloropyridine with 3 equiv of butyllithium–Me2NCH2CH2OLi and then iodomethane gave 2-chloro-6-methylpyridine.739Note that H–Li exchange can be faster than Cl–Li exchange. Treatment of 2-chloro-5-phenylpyridine with tert-butyllithium leads to lithiation on the phenyl ring rather than Li–Cl exchange, and subsequent treatment with dimethyl sulfate gave 2-chloro-5-(2-methylphenyl)pyridine.740 The reaction of N-triisopropylsilyl indole with tert-butyllithium and then iodomethane gave the 3-methyl derivative.741 Heteroaromatic compounds can be alkylated. Pyrrole, for example, reacts with an allylic halide and zinc to give primarily the 3-substituted pyrrole.742
Mercuration of aromatic compounds743 can be accomplished with mercuric salts, most often Hg(OAc)2,744 to give ArHgOAc. This is ordinary electrophilic aromatic substitution and takes place by the arenium ion mechanism (Sec. 11.A.i).745 Aromatic compounds can also be converted to arylthallium bis(trifluoroacetates) [ArTl(OOCCF3)2] by treatment with thallium(III) trifluoroacetate746 in trifluoroacetic acid.747 These arylthallium compounds can be converted to phenols, aryl iodides or fluorides (Reaction 12-31), aryl cyanides (Reaction 12-34), aryl nitro compounds,748 or aryl esters (Reaction 12-33). The mechanism of thallation appears to be complex, with electrophilic and electron-transfer mechanisms both taking place.749 Transient metalated aryl complexes can be formed that react with another aromatic compound. Aryl iodides reacted with benzene to form a biaryl in the presence of an Ir catalyst.750Aniline derivatives reacted with TiCl4 to give the para-homo coupling product (R2N–Ar–Ar–NR2).751
Aromatic nitro compounds can be methylated with dimethyloxosulfonium methylid752 or the methylsulfinyl carbanion (obtained by treatment of DMSO with a strong base)753:
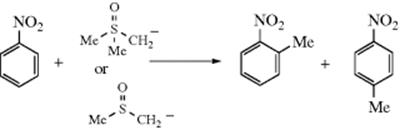
The reactions with the sulfur carbanions are especially useful, since none of these substrates can be methylated by the Friedel–Crafts procedure (Reaction 11-10).
A different kind of alkylation of nitro compounds uses carbanion nucleophiles that have a chlorine at the carbanionic carbon. The following process takes place:754
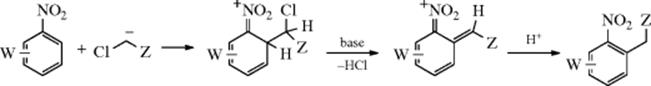
This type of process is called vicarious nucleophilic substitution of hydrogen.755 The Z group is electron withdrawing (e.g., SO2R, SO2OR, SO2NR2, COOR, or CN); it stabilizes the negative charge. The carbanion attacks the activated ring ortho or para to the nitro group.756 Hydride ion (H−) is not normally a leaving group, but in this case the presence of the adjacent Cl allows the hydrogen to be replaced. Hence, Cl is a “vicarious” leaving group. Other leaving groups have been used (e.g., OMe and SPh), but Cl is generally the best. Many W groups in the ortho, meta, or para positions do not interfere. The reaction is also successful for di- and trinitro compounds, for nitronaphthalenes,757 and for many nitro heterocycles, ![]() may also be used.758 When the nucleophile is Br3C− or Cl3C−, the product is ArCHX2, which can easily be hydrolyzed to ArCHO.759 This is therefore an indirect way of formylating an aromatic ring containing one or more NO2 groups, which cannot be done by any of the formylations mentioned in Reaction 11-1–11-18.
may also be used.758 When the nucleophile is Br3C− or Cl3C−, the product is ArCHX2, which can easily be hydrolyzed to ArCHO.759 This is therefore an indirect way of formylating an aromatic ring containing one or more NO2 groups, which cannot be done by any of the formylations mentioned in Reaction 11-1–11-18.
Replacement of an amino group is possible. When aniline derivatives were treated with allyl bromide and tert-butyl nitrite (t-BuONO), the aryl–allyl coupling product was formed (Ar–NH2 → Ar–CH2CH=CH2).760
For the introduction of CH2SR groups into phenols, see Reaction 11-23. See also, Reaction 14-19.
OS II, 517.
13-18 Amination of Nitrogen Heterocycles
Amination or Amino-de-hydrogenation
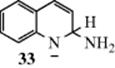
![]()
Pyridine and other heterocyclic nitrogen compounds can be aminated with alkali metal amides in a process called the Chichibabin reaction.761 The attack is always in the 2 position unless both such positions are filled, in which case the 4 position is attacked. Substituted alkali metal amides (e.g., RNH− and R2N−), have also been used. The mechanism is probably similar to that of Reaction 13-17. The existence of intermediate ions (e.g., 33)
(from quinoline) has been demonstrated by NMR spectra.762 A pyridyne type of intermediate was ruled out by several observations including the facts that 3-ethylpyridine gave 2-amino-3-ethylpyridine763 and that certain heterocycles that cannot form an aryne could nevertheless be successfully aminated. Nitro compounds do not give this reaction,764 but they have been aminated (ArH → ArNH2 or ArNHR) via the vicarious substitution principle (see Reaction 13-17), using 4-amino- or 4-alkylamino-1,2,4-triazoles as nucleophiles.765 The vicarious leaving group in this case is the triazole ring. Note, however, that 3-nitropyridine was converted to 6-amino-3-nitropyridine by reaction with KOH, hydroxylamine, and ZnCl2.766
Analogous reactions have been carried out with hydrazide ions (R2NNH−).767 A mixture of NO2 and O3, with excess NaHSO3, converted pyridine to 3-aminopyridine.768 For other methods of aminating aromatic rings, see Reaction 11-6.
There are no Organic Syntheses references, but see OS V, 977, for a related reaction.
13.C.iii. Nitrogen as Leaving Group
The diazonium group can be replaced by a number of groups.769 Some of these are nucleophilic substitutions, with SN1 mechanisms (Sec. 10.A.ii), but others are free radical reactions and are treated in Chapter 14. The solvent in diazonium group reactions is usually water. With other solvents it has been shown that the SN1 mechanism is favored by solvents of low nucleophilicity, while those of high nucleophilicity favor free radical mechanisms.770 The N2+group771 can be replaced by Cl−, Br−, and CN−, by a nucleophilic mechanism (see OS IV, 182), but the Sandmeyer reaction is much more useful (Reaction 14-20). Transition metal catalyzed reactions are known involving aryldiazonium salts, and diazonium variants of the Heck reaction (13-10) and Suzuki coupling (13-12) were mentioned previously. As mentioned in Section 13.B.i, it must be kept in mind that the N2+ group can activate the removal of another group on the ring. In a few cases, nitrogen groups (e.g., nitro or ammonium) can be replaced.
13-19 Diazotization
![]()
When primary aromatic amines are treated with nitrous acid, diazonium salts are formed.772 The reaction also occurs with aliphatic primary amines, but aliphatic diazonium ions are extremely unstable, even in solution (see Sec. 10.G.iii). Aromatic diazonium ions are more stable, because of the resonance interaction between the nitrogen atoms and the ring:
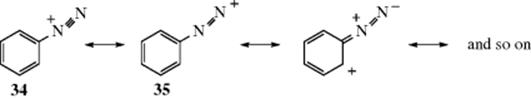
Incidentally, 34 contributes more than 35, as shown by bond-distance measurements.773 In benzenediazonium chloride, the C–N distance is ~1.42 Å, and the N–N distance ~1.08 Å,774 and these values fit more closely to a single and a triple bond than to two double bonds (see Table 1.5). Even aromatic diazonium salts are unstable at temperatures other than about <5 °C. A few are more stable (e.g., the diazonium salt obtained from sulfanilic acid), which is stable up to 10 or 15 °C. Diazonium salts are usually prepared in aqueous solution and used without isolation.775 While it is possible to prepare solid diazonium salts (see Reaction 13-23), many dry diazonium salts are explosive if not handled with great care and extreme caution should be exercised. The stability of aryl diazonium salts can be increased by crown ether complexion.776
For aromatic amines, the reaction is very general. Halogen, nitro, alkyl, aldehyde, sulfonic acid, and so on, groups do not interfere. Since aliphatic amines do not react with nitrous acid below a pH of ~3, it is even possible, by working at a pH ~1, to diazotize an aromatic amine without disturbing an aliphatic amino group in the same molecule.777
![]()
If an aliphatic amino group is α to a COOR, CN, CHO, COR, and so on, and has a hydrogen, treatment with nitrous acid gives not a diazonium salt, but a diazo compound.778 Such diazo compounds can also be prepared, often more conveniently, by treatment of the substrate with isoamyl nitrite (Me2CHCH2CH2ONO) and a small amount of acid.779 Certain heterocyclic amines also give diazo compounds rather than diazonium salts.780
Despite the fact that diazotization takes place in acid solution, the actual reactive species is not the salt of the amine, but is the small amount of free amine present.781 Because aliphatic amines are stronger bases than aromatic ones that at pH values <3 there is not enough free amine present for the former to be diazotized, while the latter still undergo the reaction. In dilute acid, the actual attacking species is N2O3, which acts as a carrier of NO+. Evidence is that the reaction is second order in nitrous acid and, at sufficiently low acidities, the amine does not appear in the rate expression.782 Under these conditions the mechanism is
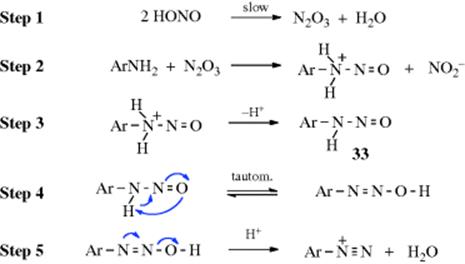
Other evidence exists for this mechanism.783 Other attacking species can be NOCl, H2NO2+, and at high acidities even NO+. Nucleophiles (e.g., Cl−, SCN−, and thiourea) catalyze the reaction by converting the HONO to a better electrophile (e.g., HNO2 + Cl− + H+ → NOCl + H2O).784
N-Aryl ureas are converted to the aryldiazonium nitrate upon treatment with NaNO2 and H2SO4 in dioxane785 or with DMF–NO2 in DMF.786
There are many preparations of diazonium salts listed in Organic Syntheses, but they are always prepared for use in other reactions. They are listed under reactions in which they are used. The preparation of aliphatic diazo compounds can be found in OS III, 392; IV, 424. See also, OS VI, 840.
13-20 Hydroxylation of Aryldiazonium Salts
Hydroxy-de-diazoniation
![]()
This reaction is formally analogous to 13-1, but with a N2+ leaving group rather than a halide. Water is usually present whenever diazonium salts are made, but at these temperatures (0–5°C) the reaction proceeds very slowly. When it is desired to have OH replace the diazonium group, the excess nitrous acid is destroyed and the solution is usually boiled. Some diazonium salts require even more vigorous treatment, for example, boiling with aq H2SO4 or with trifluoroacetic acid containing potassium trifluoroacetate.787 The reaction can be performed on solutions of any diazonium salts, but hydrogen sulfates are preferred to chlorides or nitrates, since in these cases there is competition from the nucleophiles Cl− or NO3−.
A better method, which is faster, avoids side reactions, takes place at room temperature, and gives higher yields consists of adding Cu2O to a dilute solution of the diazonium salt dissolved in a solution containing a large excess of Cu(NO3)2.788 Aryl radicals are intermediates when this method is used. It has been shown that aryl radicals are at least partly involved when ordinary hydroxy-de-diazoniation is carried out in weakly alkaline aqueous solution.789Decomposition of arenediazonium tetrafluoroborates in F3CSO2OH gives aryl triflates directly, in high yields.790
OS I, 404; III, 130, 453, 564; V, 1130.
13-21 Replacement by Sulfur-Containing Groups
Mercapto-de-diazoniation, and so on
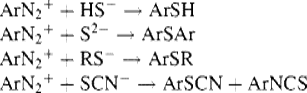
These reactions are convenient methods for incorporating a sulfur-containing group onto an aromatic ring. With Ar′S−, diazosulfides (Ar–N=N–S–Ar′) are intermediates,791 which can in some cases be isolated.792 Thiophenols can be made as shown above, but more often the diazonium ion is treated with EtO–CSS− or S22−, which give the expected products, and these are easily convertible to thiophenols. Aryldiazonium salts are prepared by the reaction of an aniline derivative with an alkyl nitrite (RONO), and when formed in the presence of dimethyl disulfide (MeS–SMe), the product is the thioether (Ar–S–Me).793 Aryl triflates have been converted to the aryl thiol using NaST(P5) and a Pd catalyst, followed by treatment with tetrabutylammonium fluoride794 (see also, Reaction 14-22).
OS II, 580; III, 809 (but see OS V, 1050). Also see, OS II, 238.
13-22 Replacement by Iodine
Iodo-de-diazoniation
![]()
One of the best methods for the introduction of iodine into aromatic rings (see Reaction 13-7) is the reaction of diazonium salts with iodide ions.795 Analogous reactions with chloride, bromide, and fluoride ions give poorer results, and Reactions 14-20 and 13-23 are preferred for the preparation of aryl chlorides, bromides, and fluorides. However, when other diazonium reactions are carried out in the presence of these ions, halides are usually side products. Aniline has also been converted to fluorobenzene by treatment with t-BuONO and SiF4 followed by heating.796 A related reaction between PhN=N–NR2 and iodine gave iodobenzene.797
The actual attacking species is probably not only I− if it is I− at all. The iodide ion is oxidized (by the diazonium ion, nitrous acid, or some other oxidizing agent) to iodine, which in a solution containing iodide ions is converted to I3−; this is the actual attacking species, at least partly. This was shown by isolation of ArN2+ I3− salts, which, on standing, gave ArI.798 From this, it can be inferred that the reason the other halide ions give poor results is not that they are poor nucleophiles but rather they are poor reducing agents (compared with iodide). There is also evidence for a free radical mechanism.799
The hydroxyl group of a phenol can be replaced with iodine. The reaction of phenol with a boronic ester and a Pd catalyst, followed by reaction with NaI and chloramine-T (see Reaction 15-52) converts phenol to iodobenzene.800Arylboronic acids are converted to the aryl fluoride with Selectfluor, in the presence of a Pd catalyst.801
OS II, 351, 355, 604; V, 1120.
13-23 The Schiemann Reaction
Fluoro-de-diazoniation (overall transformation)
![]()
Heating diazonium fluoroborates (the Schiemann or Balz–Schiemann reaction) is by far the best way to introduce fluorine into an aromatic ring.802 In the most common procedure, the tetrafluoroborate salts are prepared by diazotizing as usual with nitrous acid and HCl and then adding a cold aqueous solution of NaBF4, HBF4, or NH4BF4. A precipitate forms, which is dried, and the salt is heated in the dry state. These salts are unusually stable for diazonium salts, and the reaction is usually successful (since diazonium salts are generally unstable, care should be exercised any time a diazonium salt is dried). In general, any aromatic amine that can be diazotized will form a BF4−salt, usually with high yields. The diazonium fluoroborates can be formed directly from primary aromatic amines with tert-butyl nitrite and BF3–etherate.803 The reaction has also been carried out on ArN2+ PF6− ArN2+ SbF6−, and ArN2+ AsF6− salts, in many cases with better yields.804 Aryl chlorides and bromides are commonly prepared by the Sandmeyer reaction (14-20). In an alternative procedure, aryl fluorides are prepared by treatment of aryltriazenes Ar–N=N–NR2 with 70% HF in pyridine.805
The mechanism is of the SN1 type. That aryl cations are intermediates was shown by the following experiments806: Aryl diazonium chlorides are known to arylate other aromatic rings by a free radical mechanism (see Reaction 13-27). In radical arylation it does not matter whether the other ring contains electron-withdrawing or electron-donating groups; in either case a mixture of isomers is obtained, since the attack is not by a charged species. If an aryl radical were an intermediate in the Schiemann reaction and the reaction were run in the presence of other rings, it should not matter what kinds of groups were on these other rings: Mixtures of biaryls should be obtained in all cases. But if an aryl cation is an intermediate in the Schiemann reaction, compounds containing meta-directing groups (i.e., meta-directing for electrophilic substitutions), should be meta arylated and those containing ortho–para directing groups should be ortho and para arylated, since an aryl cation should behave in this respect like any electrophile (see Chapter 11). Experiments have shown807 that such orientation is observed, demonstrating that the Schiemann reaction has a positively charged intermediate. The attacking species, in at least some instances, is not F− but BF4−.808
OS II, 188, 295, 299; V, 133.
13-24 Conversion of Amines to Azo Compounds
N-Arylimino-de-dihydro-bisubstitution
![]()
Aromatic nitroso compounds combine with primary arylamines in glacial acetic acid to give symmetrical or unsymmetrical azo compounds (the Mills reaction).809 A wide variety of substituents may be present in both aryl groups. Unsymmetrical azo compounds have also been prepared by the reaction between aromatic nitro compounds (ArNO2) and N-acyl aromatic amines (Ar′NHAc).810 The use of phase-transfer catalysis increased the yields.
13-25 Methylation, Vinylation, and Arylation of Diazonium Salts
Methyl-de-diazoniation, and so on
![]()
A methyl group can be introduced into an aromatic ring by treatment of diazonium salts with tetramethyltin and a Pd catalyst.811 The reaction has been performed with Me, Cl, Br, and NO2 groups on the ring. A vinylic group can be introduced with CH2=CHSnBu3. When an aryl amine is treated with tert-butyl hyponitrite (t-BuONO) and allyl bromide, the nitrogen is displaced to give the allyl–aryl compound.812
Aryl diazonium salts can be used coupled with alkenes in a Heck-like reaction (Reaction 13-10).813 Other reactive aryl species also couple with aryldiazonium salts in the presence of a Pd catalyst.814 A Suzuki-type coupling (Reaction 13-12) has also been reported using arylboronic acids, aryldiazonium salts, and a Pd catalyst.815
Aryltrifluoroborates (Reaction 12-28) react with aryldiazonium salts in the presence of a Pd catalyst to give the corresponding biaryl.816 See Reaction 13-12. Arylborate esters also react using a Pd catalyst, and the aryl diazonium unit reacts faster than an aryl halide.817
13-26 Arylation of Activated Alkenes by Diazonium Salts: Meerwein Arylation
Arylation or Aryl-de-hydrogenation
![]()
Alkenes activated by an electron-withdrawing group (Z may be C=C, halogen, C=O, Ar, CN, etc.) can be arylated by treatment with a diazonium salt and a cupric chloride818 catalyst. This is called the Meerwein arylation reaction.819 Addition of ArCl to the double bond (to give Z(Cl)C–CHAr) is a side reaction (15-46). In an improved procedure, an arylamine is treated with an alkyl nitrite (generating ArN2+in situ) and a copper(II) halide in the presence of the alkene.820
The mechanism is probably of the free radical type, with Ar√ (36) forming as in Reaction 14-20, and then halogen transfer to give 37 or elimination to give 38.821
The radical 36 can react with cupric chloride by two pathways, one of which leads to addition and the other to substitution. Even when the addition pathway is taken, however, the substitution product may still be formed by subsequent elimination of HCl. Note that radical reactions are presented in Chapter 14, but the coupling of an alkene with an aromatic compound containing a leaving group prompted its placement here. Note the similarity to the Heck reaction in Reaction 13-10.
OS IV, 15.
13-27 Arylation of Aromatic Compounds by Diazonium Salts
Arylation or Aryl-de-hydrogenation
![]()
When the normally acidic solution of a diazonium salt is made alkaline, the aryl portion of the diazonium salt can couple with another aromatic ring. Known as the Gomberg or Gomberg–Bachmann reaction,822 it has been performed on several types of aromatic rings and on quinones. Yields are not high (usually <40%) because of the many side reactions undergone by diazonium salts, although higher yields have been obtained under phase-transfer conditions.823 The conditions of the Meerwein reaction (13-26), treatment of the solution with a copper-ion catalyst, have also been used, as has the addition of sodium nitrite in DMSO (to benzenediazonium tetrafluoroborate).824When the Gomberg–Bachmann reaction is performed intramolecularly as in the formation of 39, either by the alkaline solution or by the copper-ion procedure, it is called the Pschorr reaction825 and yields are usually somewhat higher. Still higher yields have been obtained by carrying out the Pschorr reaction electrochemically.826 The Pschorr reaction has been carried out for Z = CH=CH, CH2CH2, NH, C=O, CH2, and quite a few others. A rapid and convenient way to diazotize the amine substrate uses isopropyl nitrite in the presence of sodium iodide, in which case the ring-closed product is formed in one step.827 Palladium-catalyzed arylation of arenediazonium salts are known.828
Other compounds with nitrogen–nitrogen bonds have been used instead of diazonium salts. Among these are N-nitroso amides [ArN(NO)COR], triazenes,829 and azo compounds. Still another method involves treatment of an aromatic primary amine directly with an alkyl nitrite in an aromatic substrate as solvent.830
In each case, the mechanism involves generation of an aryl radical from a covalent azo compound. In acid solution, diazonium salts are ionic and their reactions are polar. When they cleave, the product is an aryl cation (see Sec. 13.A.i). However, in neutral or basic solution, diazonium ions are converted to covalent compounds, and these cleave to give free radicals (Ar√ and Z√). Note that radical reactions are presented in Chapter 14, but the coupling of an aromatic ring with an aromatic compound containing a leaving group prompted its placement here. Note the similarity to the Suzuki Reaction in 13-12.
![]()
Under Gomberg–Bachmann conditions, the species that cleaves is the anhydride (40).831
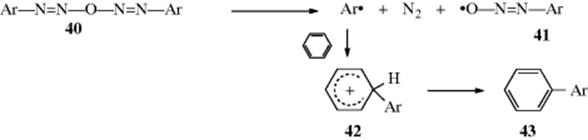
The aryl radical thus formed attacks the substrate to give the aryl cation832 intermediate 42 (see Sec. 14.A.iii), from which the radical 41 abstracts hydrogen to give the product (43). N-Nitroso amides probably rearrange to N-acyloxy compounds (44), which cleave to give aryl radicals.833 There is evidence that the reaction with alkyl nitrites also involves attack by aryl radicals.834
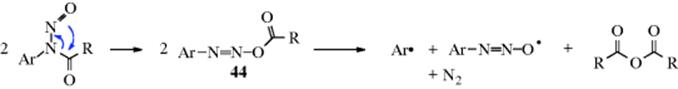
The Pschorr reaction can take place by two different mechanisms, depending on conditions: (1) attack by an aryl radical (as in the Gomberg–Bachmann reaction) or (2) attack by an aryl cation (similar to the SN1 mechanism discussed in Sec. 13.A.ii).835 Under certain conditions the ordinary Gomberg–Bachmann reaction can also involve attack by aryl cations.836
OS I, 113; IV, 718.
13-28 Aryl Dimerization with Diazonium Salts
De-diazonio-coupling; Arylazo-de-diazonio-substitution
![]()
When diazonium salts are treated with cuprous ion (or with Cu and acid, in which case it is called the Gatterman method), two products are possible. If the ring contains electron-withdrawing groups, the main product is the biaryl, but the presence of electron-donating groups leads mainly to the azo compound. This reaction is different from Reaction 13-27 (and from 19-14) in that both aryl groups in the product originate from ArN2+, that is, hydrogen is not a leaving group in this reaction. The mechanism probably involves free radicals.837
OS I, 222; IV, 872. Also see, OS IV, 273.
13-29 Replacement of Nitro
Alkyl-de-nitration, Hydroxy and alkoxy-de-nitration, Halo-de-nitration
![]()
In some cases, the nitrogen group of an aromatic nitro compound can be replaced with an alkyl group. The reaction of 1,4-dinitrobenzene with potassium tert-butoxide in the presence of BEt3, for example, gave 4-ethylnitrobenzene.838
Other nucleophiles can replace a nitrogen-containing group. The reaction of hydroxide with Ar–Y, where Y = nitro,839 azide, NR3+, and so on gives the corresponding phenol. This latter reaction works with alkoxide nucleophiles to give the corresponding aryl ether. The nitro can be replaced with chloro by use of NH4Cl, PCl5, SOCl2, HCl, Cl2, or CCl4. Some of these reagents operate only at high temperatures and the mechanism is not always nucleophilic substitution. Activated aromatic nitro compounds can be converted to fluorides with fluoride ion.840
The reaction of vinyl nitro compounds (C=C–NO2) and aryl iodide to give the styrene compound (C=C–Ar) was reported using BEt3 and exposure to air.841
13.C.iv. Rearrangements
13-30 The von Richter Rearrangement
Hydro-de-nitro-cine-substitution
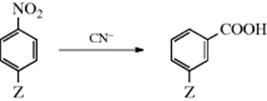
When aromatic nitro compounds are treated with cyanide ion, the nitro group is displaced and a carboxyl group enters with cine substitution (Sec. 13.A.iii), always ortho to the displaced group, never meta or para. The scope of this reaction, called the von Richter rearrangement, is variable.842 As with other nucleophilic aromatic substitutions, the reaction gives best results when electron-withdrawing groups are in ortho and para positions, but yields are low, usually <20% and never >50%.
At one time, it was believed that a nitrile (ArCN) was an intermediate, since cyanide is the reagent and nitriles are hydrolyzable to carboxylic acids under the reaction conditions (16-4). However, a remarkable series of results proved this belief to be in error. Bunnett and Rauhut843 demonstrated that α-naphthyl cyanide is not hydrolyzable to α-naphthoic acid under conditions at which β-nitronaphthalene undergoes the von Richter rearrangement to give α-naphthoic acid. This proved that the nitrile could not be an intermediate. It was subsequently demonstrated that N2 is a major product of the reaction.844 It had previously been assumed that all the nitrogen in the reaction was converted to ammonia, which would be compatible with a nitrile intermediate, since ammonia is a hydrolysis product of nitriles. At the same time it was shown that NO2+ is not a major product. The discovery of nitrogen indicated that a nitrogen–nitrogen bond must be formed during the course of the reaction. Rosenblum proposed a mechanism in accord with all the facts843:
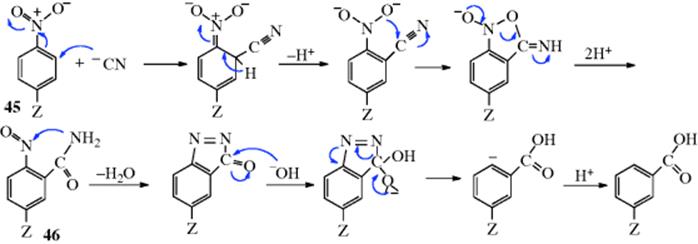
Note that 46 is a stable compound: Hence, it should be possible to prepare it independently and to subject it to the conditions of the von Richter rearrangement. This was done and the correct products are obtained.845 Further evidence is that when 45 (Z = Cl or Br) was treated with cyanide in ![]() , half the oxygen in the product was labeled, showing that one of the oxygen atoms of the carboxyl group came from the nitro group and one from the solvent, as required by this mechanism.846
, half the oxygen in the product was labeled, showing that one of the oxygen atoms of the carboxyl group came from the nitro group and one from the solvent, as required by this mechanism.846
13-31 The Sommelet–Hauser Rearrangement
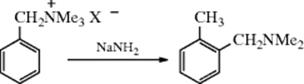
Benzylic quaternary ammonium salts, when treated with alkali metal amides, undergo a rearrangement called the Sommelet–Hauser rearrangement.847 Since the product is a benzylic tertiary amine, it can be further alkylated and the product again subjected to the rearrangement. This process can be continued around the ring until an ortho position is blocked.848
The rearrangement occurs with high yields and can be performed with various groups present in the ring.849 The reaction is most often carried out with three methyl groups on the nitrogen, but other groups can also be used, though if a β-hydrogen is present, Hofmann elimination (Reaction 17-7) often competes. The Stevens rearrangement (Reaction 18-21) is also a competing process.850 When both rearrangements are possible, the Stevens rearrangement is favored at high temperatures and the Sommelet–Hauser at low temperatures.851 The mechanism is:
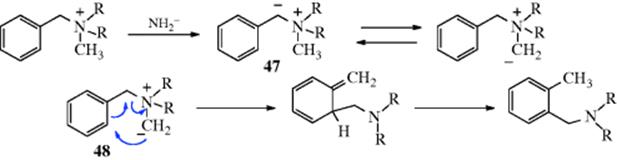
The benzylic hydrogen is most acidic and is the one that first loses a proton to give the ylid (47). However, 48, which is present in a smaller amount, is the species that undergoes the rearrangement, shifting the equilibrium in its favor. This mechanism is an example of a [2,3]-sigmatropic rearrangement (see Reaction 18-35). Another mechanism that might be proposed is one in which a methyl group actually breaks away (in some form) from the nitrogen and then attaches itself to the ring. That this is not so was shown by a product study.852 If the second mechanism were true, 49 should give 50, but the first mechanism predicts the formation of 51, which is what was actually obtained.853
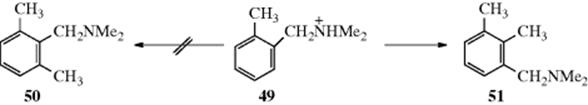
The mechanism as shown can lead only to an ortho product. However, a small amount of para product has been obtained in some cases.854 A mechanism855 in which there is a dissociation of the ArC–N bond (similar to the ion-pair mechanism of the Stevens rearrangement, Reaction 18-21) has been invoked to explain the para products that are observed.
Sulfur ylids containing a benzylic group (analogous to 48) undergo an analogous rearrangement.856
OS IV, 585.
13-32 Rearrangement of Aryl Hydroxylamines
1/C-Hydro-5/N-hydroxy-interchange
![]()
Aryl hydroxylamines treated with acids rearrange to aminophenols.857 Although this reaction (known as the Bamberger rearrangement) is similar in appearance to Reactions 11-28–11-32, the attack on the ring is not electrophilic but nucleophilic. The rearrangement is intermolecular, with the following mechanism:
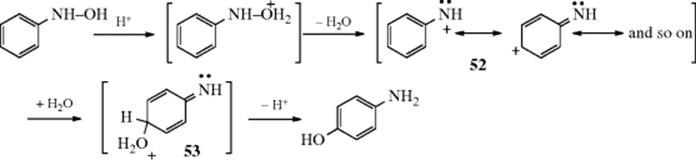
Among the evidence858 for this mechanism are the facts that other products are obtained when the reaction is run in the presence of competing nucleophiles (e.g., p-ethoxyaniline) when ethanol is present, and that when the para position is blocked, compounds similar to 53 are isolated. In the case of 2,6-dimethylphenylhydroxylamine, the intermediate nitrenium ion (52) was trapped, and its lifetime in solution was measured.859 The reaction of 52 with water was found to be diffusion controlled.860
There is a base-mediated rearrangement of aromatic hydroxamic acids to anilines.861
OS IV, 148.
13-33 The Smiles Rearrangement

The Smiles rearrangement actually comprises a group of rearrangements that follow the pattern given above.862 A specific example is the reaction of 54 with hydroxide to give 55.
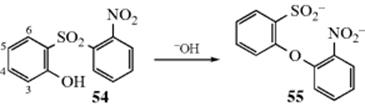
Smiles rearrangements are simply intramolecular nucleophilic substitutions. In the example given, SO2Ar is the leaving group and ArO− is the nucleophile. The nitro group serves to activate its ortho position. Halogens also serve as activating groups.863 The ring at which the substitution takes place is nearly always activated, usually by ortho or para nitro groups. Here X is usually S, SO, SO2,864 O, or CO2, and Y is usually the conjugate base of OH, NH2, NHR, or SH. The reaction has even been carried out with Y = CH2− (phenyllithium was the base here).865
The reaction rate is greatly enhanced by substitution in the 6 position of the attacking ring, for steric reasons. For example, a methyl, chloro, or bromo group in the 6 position of 54 caused the rate to be ~105 times faster than when the same groups were in the 4 position,866 although electrical effects should be similar at these positions. The enhanced rate comes about because the most favorable conformation of the molecule can adapt to suit the bulk of the 6-substituent is also the conformation required for the rearrangement. Thus, less entropy of activation is required.
Although the Smiles rearrangement is usually carried out on compounds containing two rings, this need not be the case, as in the formation of 56.867
In this case the, sulfenic acid (56) is unstable868 and the actual products isolated were the corresponding sulfinic acid (RSO2H) and disulfide (R2S2).
In the Smiles rearrangement, the nucleophile Y is most often the conjugate base of SH, SO2NHR, SO2NH2, NH2, NHR, OH, and OR. There are few examples where Y is a carbanion, and the most common example is probably the Truce–Smiles rearrangement, where L–YH is an o-tolyl group.869 The prototypical Truce–Smiles rearrangement requires use of a strong base to form the benzylic carbanion that undergoes the rearrangement. When sulfone (57) was treated with butyllithium, for example, deprotonation led to the benzylic lithium compound (58). Truce–Smiles rearrangement led to 59, and hydrolysis gave the sulfinic acid (60).870 Truce–Smiles rearrangements with stabilized benzylic carbanions are known,870 and rearrangements of carbanions in general fall under this category.871 Relatively few examples have been reported, however.872 Truce–Smiles rearrangements of sulfones that proceed through a six-membered transition state have been reported.873 In another example, displacement of an activated aryl fluoride with o-hydroxyacetophenone gave a product that was C-arylated adjacent to the ketone.874
Notes
1. See Zoltewicz, J.A. Top. Curr. Chem. 1975, 59, 33.
2. See Fairlamb, I.J.S. Tetrahedron 2005, 61, 9661.
3. Acevedo, O.; Jorgensen, W.L. Org. Lett. 2004, 6, 2881.
4. See Miller, J. Aromatic Nucleophilic Substitution, Elsevier, NY, 1968. For reviews, see Bernasconi, C.F. Chimia 1980, 34, 1; Acc. Chem. Res. 1978, 11, 147; Bunnett, J.F. J. Chem. Educ. 1974, 51, 312; Ross, S.D. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 13, Elsevier, NY, 1972, pp. 407–431; Buck, P. Angew. Chem, Int. Ed. 1969, 8, 120; Buncel, E.; Norris, A.R.; Russell, K.E. Q. Rev. Chem. Soc. 1968, 22, 123; Bunnett, J.F. Tetrahedron 1993, 49, 4477; Zoltewicz, J.A. Top. Curr. Chem. 1975, 59, 33.
5. See Barrett, I.C.; Kerr, M.A. Tetrahedron Lett. 1999, 40, 2439.
6. Also see Wu, Z.; Glaser, R. J. Am. Chem. Soc. 2004, 126, 10632; Terrier, F.; Mokhtari, M.; Goumont, T.; Hallé, J.-C.; Buncel, E. Org. Biomol. Chem. 2003, 1, 1757.
7. Meisenheimer, J. Liebigs Ann. Chem. 1902, 323, 205; Jackson, C.L.; see Jackson, C.L.; Gazzolo, F.H. Am. Chem. J. 1900, 23, 376; Jackson, C.L.; Earle, R.B. Am. Chem. J., 1903, 29, 89.
8. For heteroatom nucleophiles see Gallardo, I.; Guirado, G.; Marquet, J. J. Org. Chem. 2002, 67, 2548.
9. See Buncel, E.; Crampton, M.R.; Strauss, M.J.; Terrier, F. Electron Deficient Aromatic- and Heteroaromatic-Base Interactions, Elsevier, NY, 1984; Illuminati, G.; Stegel, F. Adv. Heterocycl. Chem. 1983, 34, 305; Terrier, F. Chem. Rev. 1982, 82, 77; Strauss, M.J. Acc. Chem. Res. 1974, 7, 181; Hall, T.N.; Poranski, Jr., C.F. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, pt. 2, Wiley, NY, 1970, pp. 329–384; Foster, R.; Fyfe, C.A. Rev. Pure Appl. Chem. 1966, 16, 61.
10. Crampton, M.R.; Gold, V. J. Chem. Soc. B 1966, 893. See Buncel, E.; Crampton, M.R.; Strauss, M.J.; Terrier, F. Electron Deficient Aromatic- and Heteroaromatic-Base Interactions, Elsevier, NY, 1984, pp. 15–133.
11. Destro, R.; Gramaccioli, C.M.; Simonetta, M. Acta Crystallogr. 1968, 24, 1369; Ueda, H.; Sakabe, M.; Tanaka, J.; Furusaki, A. Bull. Chem. Soc. Jpn. 1968, 41, 2866; Messmer, G.G.; Palenik, G.J. Chem. Commun. 1969, 470.
12. Grossi, L. Tetrahedron Lett. 1992, 33, 5645.
13. Bunnett, J.F.; Garbisch, Jr., E.W.; Pruitt, K.M. J. Am. Chem. Soc. 1957, 79, 385. See Gandler, J.R.; Setiarahardjo, I.U.; Tufon, C.; Chen, C. J. Org. Chem. 1992, 57, 4169.
14. See Fernndez, I.; Frenking, G.; Uggerud, E. J. Org. Chem. 2010, 75, 2971.
15. Chiacchiera, S.M.; Singh, J.O.; Anunziata, J.D.; Silber, J.J. J. Chem. Soc. Perkin Trans. 2 1987, 987.
16. Bernasconi, C.F.; de Rossi, R.H.; Schmid, P. J. Am. Chem. Soc. 1977, 99, 4090.
17. Bunnett, J.F.; Sekiguchi, S.; Smith, L.A. J. Am. Chem. Soc. 1981, 103, 4865.
18. See Nudelman, N.S. J. Phys. Org. Chem. 1989, 2, 1. See also, Nudelman, N.S.; Montserrat, J.M. J. Chem. Soc. Perkin Trans. 2 1990, 1073.
19. Jain, A.K.; Gupta, V.K.; Kumar, A. J. Chem. Soc. Perkin Trans. 2 1990, 11.
20. Ayrey, G.; Wylie, W.A. J. Chem. Soc. B 1970, 738.
21. Bacaloglu, R.; Blaskó, A.; Bunton, C.A.; Dorwin, E.; Ortega, F.; Zucco, C. J. Am. Chem. Soc. 1991, 113, 238; Crampton, M.R.; Davis, A.B.; Greenhalgh, C.; Stevens, J.A. J. Chem. Soc. Perkin Trans. 2 1989, 675.
22. See Muscio, Jr., O.J.; Rutherford, D.R. J. Org. Chem. 1987, 52, 5194.
23. Quiñonero, D.; Garau, C.; Rotger, C.; Frontera, A.; Ballester, P.; Costa, A.; Deyà. P.M. Angew. Chem. Int. Ed. 2002, 41, 3389.
24. Himeshima, Y.; Kobayashi, H.; Sonoda, T. J. Am. Chem. Soc. 1985, 107, 5286.
25. See Glaser, R.; Horan, C.J.; Nelson, E.D.; Hall, M.K. J. Org. Chem. 1992, 57, 215.
26. Aryl iodonium salts Ar2I+ also undergo substitutions by this mechanism (and by a free radical mechanism).
27. Also see Lorand, J.P. Tetrahedron Lett. 1989, 30, 7337.
28. See Ambroz, H.B.; Kemp, T.J. Chem. Soc. Rev. 1979, 8, 353.
29. Stang, P.J.; Rappoport, Z.; Hanack, M.; Subramanian, L.R. Vinyl Cations, Academic Press, NY, 1979. See Hanack, M. Pure Appl. Chem. 1984, 56, 1819; Rappoport, Z. Reactiv. Intermed. (Plenum) 1983, 3, 427; Ambroz, H.B.; Kemp, T.J. Chem. Soc. Rev. 1979, 8, 353. See also, Charton, M. Mol. Struct. Energ. 1987, 4, 271; Glaser, R.; Horan, C.J.; Lewis, M.; Zollinger, H. J. Org. Chem. 1999, 64, 902.
30. See Zollinger, H. Angew. Chem, Int. Ed. 1978, 17, 141; Swain, C.G.; Sheats, J.E.; Harbison, K.G. J. Am. Chem. Soc. 1975, 97, 783, 796; Burri, P.; Wahl, Jr., G.H.; Zollinger, H. Helv. Chim. Acta 1974, 57, 2099; Richey, Jr., H.G.; Richey, J.M. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, pp. 922–931; Miller, J. Aromatic Nucleophilic Substitution, Elsevier, NY, 1968, pp. 29–40.
31. Lewis, E.S.; Miller, E.B. J. Am. Chem. Soc. 1953, 75, 429.
32. Swain, C.G.; Sheats, J.E.; Gorenstein, D.G.; Harbison, K.G. J. Am. Chem. Soc. 1975, 97, 791.
33. See Apeloig, Y.; Arad, D. J. Am. Chem. Soc. 1985, 107, 5285.
34. See Williams, D.L.H.; Buncel, E. Isot. Org. Chem. Vol. 5, Elsevier, Amsterdam, The Netherlands, 1980, pp. 147, 212; Zollinger, H. Pure Appl. Chem. 1983, 55, 401.
35. Lewis, E.S.; Kotcher, P.G. Tetrahedron 1969, 25, 4873; Lewis, E.S.; Holliday, R.E. J. Am. Chem. Soc. 1969, 91, 426; Tröndlin, F.; Medina, R.; Rüchardt, C. Chem. Ber. 1979, 112, 1835.
36. Bergstrom, R.G.; Landell, R.G.M.; Wahl, Jr., G.H.; Zollinger, H. J. Am. Chem. Soc. 1976, 98, 3301.
37. Szele, I.; Zollinger, H. Helv. Chim. Acta 1981, 64, 2728.
38. Ravenscroft, M.D.; Skrabal, P.; Weiss, B.; Zollinger, H. Helv. Chim. Acta 1988, 71, 515.
39. See Hoffmann, R.W. Dehydrobenzene and Cycloalkynes, Academic Press, NY, 1967; Gilchrist, T.L. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt. 1, Wiley, NY, 1983, pp. 383–419; Bryce, M.R.; Vernon, J.M. Adv. Heterocycl. Chem. 1981, 28, 183; Levin, R.H. React. Intermed. (Wiley) 1985, 3, 1; Fields, E.K. in McManus, S.P. Organic Reactive Intermediates, Academic Press, NY, 1973, pp. 449–508.
40. Roberts, J.D.; Semenow, D.A.; Simmons, H.E.; Carlsmith, L.A. J. Am. Chem. Soc. 1965, 78, 601.
41. See Hess, Jr., B.A. Eur. J. Org. Chem. 2001, 2185.
42. Wentrup, C. Austr. J. Chem. 2010, 63, 979.
43. See Kitamura, T.; Meng, Z.; Fujiwara, Y. Tetrahedron Lett. 2000, 41, 6611; Kawabata, H.; Nishino, T.; Nishiyama, Y.; Sonoda, N. Tetrahedron Lett. 2002, 43, 4911. Microwave spectroscopy can be used to detect benzynes: see Godfrey, P.D. Austr. J. Chem. 2010, 63, 1061.
44. See Suwinski, J.; Swierczek, K. Tetrahedron 2001, 57, 1639.
45. See Gilman, H.; Avakian, S. J. Am. Chem. Soc. 1945, 67, 349. For a table of many such examples, see Bunnett, J.F.; Zahler, R.E. Chem. Rev. 1951, 49, 273, p. 385.
46. Riggs, J.C.; Ramirez, A.; Cremeens, M.E.; Bashore, C.G.; Candler, J.; Wirtz, M.C.; Coe, J.C.; Collum, D.B. J. Am. Chem. Soc. 2008,130, 3406.
47. Bunnett, J.F.; Kearley, Jr., F.J. J. Org. Chem. 1971, 36, 184.
48. See Kalendra, D.M.; Sickles, B.R. J. Org. Chem. 2003, 68, 1594.
49. See Pellissier, H.; Santelli, M. Tetrahedron 2003, 59, 701.
50. See Gaviña, F.; Luis, S.V.; Costero, A.M.; Gil, P. Tetrahedron 1986, 42, 155.
51. Chapman, O.L.; Mattes, K.; McIntosh, C.L.; Pacansky, J.; Calder, G.V.; Orr, G. J. Am. Chem. Soc. 1973, 95, 6134. For the IR spectrum of pyridyne trapped in a matrix, see Nam, H.; Leroi, G.E. J. Am. Chem. Soc. 1988, 110,4096. See Brown, R.D.; Godfrey, P.D.; Rodler, M. J. Am. Chem. Soc. 1986, 108, 1296.
52. DeProft, F.; Schleyer, P.v.R.; van Lenthe, J.H.; Stahl, F.; Geerlings, P. Chem. Eur. J. 2002, 8, 3402.
53. Nash, J.J.; Nizzi, K.E.; Adeuya, A.; Yurkovich, M.J.; Cramer, C.J.; Kenttämaa, H.I. J. Am. Chem. Soc. 2005, 127, 5760.
54. For reviews of hetarynes, see van der Plas, H.C.; Roeterdink, F. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 1, Wiley, NY, 1983, pp. 421–511; Reinecke, M.G. Tetrahedron 1982, 38, 427; den Hertog, H.J.; van der Plas, H.C. Adv. Heterocycl. Chem. 1971, 40, 121; Kauffmann, T.; Wirthwein, R. Angew. Chem, Int. Ed. 1971, 10, 20.
55. Hamura, T.; Ibusuki, Y.; Sato, K.; Matsumoto, T.; Osamura, Y.; Suzuki, K. Org. Lett. 2003, 5, 3551.
56. Kim, J.K.; Bunnett, J.F. J. Am. Chem. Soc. 1970, 92, 7463, 7464.
57. See Rossi, R.A.; de Rossi, R.H. Aromatic Substitution by the SRN1 Mechanism, American Chemical Society, Washington, 1983; Savéant, J. Adv. Phys. Org. Chem. 1990, 26, 1; Norris, R.K. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 1, Wiley, NY, 1983, pp. 681–701; Chanon, M.; Tobe, M.L. Angew. Chem, Int. Ed. 1982, 21, 1; Rossi, R.A. Acc. Chem. Res. 1982, 15, 164. Also see Rossi, R.A.; Pierini, A.B.; Palacios, S.M. Adv. Free Radical Chem. (Greenwich, Conn.) 1990, 1, 193; Costentin, C.; Hapiot, P.; Médebielle, M.; Savéant, J.-M. J. Am. Chem. Soc. 1999, 121, 4451.
58. The symbol T is used for electron transfer.
59. See Amatore, C.; Pinson, J.; Savéant, J.; Thiébault, A. J. Am. Chem. Soc. 1981, 103, 6930.
60. Bunnett, J.F. Acc. Chem. Res. 1978, 11, 413.
61. Savéant, J.-M. Tetrahedron 1994, 50, 10117.
62. See Cornelisse, J.; de Gunst, G.P.; Havinga, E. Adv. Phys. Org. Chem. 1975, 11, 225; Cornelisse, J. Pure Appl. Chem. 1975, 41, 433; Pietra, F. Q. Rev. Chem. Soc. 1969, 23, 504, p. 519.
63. See Savéant, J. Acc. Chem. Res. 1980, 13, 323. See also, Alam, N.; Amatore, C.; Combellas, C.; Thiébault, A.; Verpeaux, J.N. J. Org. Chem. 1990, 55, 6347.
64. Swartz, J.E.; Bunnett, J.F. J. Org. Chem. 1979, 44, 340, and references cited therein.
65. Galli, C.; Gentili, P.; Guarnieri, A. Gazz. Chim. Ital., 1995, 125, 409.
66. Borosky, G.L.; Pierini, A.B.; Rossi, R.A. J. Org. Chem. 1992, 57, 247.
67. Manzo, P.G.; Palacios, S.M.; Alonso, R.A. Tetrahedron Lett. 1994, 35, 677.
68. Galli, C.; Gentili, P. J. Chem. Soc. Perkin Trans. 2 1993, 1135.
69. Marquet, J.; Jiang, Z.; Gallardo, I.; Batlle, A.; Cayón, E. Tetrahedron Lett. 1993, 34, 2801. Also see, Keegstra, M.A. Tetrahedron 1992, 48, 2681.
70. Rossi, R.A.; Palacios, S.M. Tetrahedron 1993, 49, 4485.
71. Marquet, J.; Casado, F.; Cervera, M.; Espín, M.; Gallardo, I.; Mir, M.; Niat, M. Pure Appl. Chem. 1995, 67, 703.
72. With meta substituents, electron-withdrawing groups also increase the rate: See Nurgatin, V.V.; Sharnin, G.P.; Ginzburg, B.M. J. Org. Chem, USSR 1983, 19, 343.
73. See Miller, J. Aromatic Nucleophilic Substitution, Elsevier, NY, 1968, pp. 61–136.
74. For reviews of reactivity of nitrogen-containing heterocycles, see Illuminati, G. Adv. Heterocycl. Chem. 1964, 3, 285; Shepherd, R.G.; Fedrick, J.L. Adv. Heterocycl. Chem. 1965, 4, 145.
75. See Albini, A.; Pietra, S. Heterocyclic N-Oxides, CRC Press, Boca Raton, FL, 1991, pp. 142–180; Katritzky, A.R.; Lagowski, J.M. Chemistry of the Heterocyclic N-Oxides, Academic Press, NY, 1971, pp. 258–319, 550–553.
76. Bunnett, J.F.; Zahler, R.E. Chem. Rev. 1951, 49, 273, pp. 308.
77. Miller, J.; Parker, A.J. Aust. J. Chem. 1958, 11, 302.
78. Berliner, E.; Monack, L.C. J. Am. Chem. Soc. 1952, 74, 1574.
79. See de Boer, T.J.; Dirkx, I.P. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, pt. 1, Wiley, NY, 1970, pp. 487–612.
80. Fluorine significantly activates ortho and meta positions, and slightly deactivates para positions (see Table 13.1): See Chambers, R.D.; Seabury, N.J.; Williams, D.L.H.; Hughes, N. J. Chem. Soc. Perkin Trans. 1 1988, 255.
81. See Yakobson, G.G.; Vlasov, V.M. Synthesis 1976, 652; Kobrina, L.S. Fluorine Chem. Rev. 1974, 7, 1.
82. See Balas, L.; Jhurry, D.; Latxague, L.; Grelier, S.; Morel, Y.; Hamdani, M.; Ardoin, N.; Astruc, D. Bull. Soc. Chim. Fr. 1990, 401.
83. See Bartoli, G.; Todesco, P.E. Acc. Chem. Res. 1977, 10, 125; a list of σ− values in Table 9.4.
84. From Roberts, J.D.; Vaughan, C.W.; Carlsmith, L.A.; Semenow, D.A. J. Am. Chem. Soc. 1956, 78, 611. See Hoffmann, R.W. Dehydrobenzene and Cycloalkynes, Academic Press, NY, 1973, pp. 134–150.
85. Wotiz, J.H.; Huba, F. J. Org. Chem. 1959, 24, 595. See also, Biehl, E.R.; Razzuk, A.; Jovanovic, M.V.; Khanapure, S.P. J. Org. Chem. 1986, 51, 5157.
86. See Miller, J. Aromatic Nucleophilic Substitution, Elsevier, NY, 1968, pp. 137–179.
87. See Furukawa, N.; Ogawa, S.; Kawai, T.; Oae, S. J. Chem. Soc. Perkin Trans. 1 1984, 1839.
88. See Beck, J.R. Tetrahedron 1978, 34, 2057. See also, Effenberger, F.; Koch, M.; Streicher, W. Chem. Ber. 1991, 24, 163.
89. Loudon, J.D.; Shulman, N. J. Chem. Soc. 1941, 772; Suhr, H. Chem. Ber. 1963, 97, 3268.
90. Kobrina, L.S.; Yakobson, G.G. J. Gen. Chem. USSR 1963, 33, 3238.
91. Reinheimer, J.D.; Taylor, R.C.; Rohrbaugh, P.E. J. Am. Chem. Soc. 1961, 83, 835; Ross, S.D. J. Am. Chem. Soc. 1959, 81, 2113; Litvinenko, L.M.; Shpan'ko, L.V.; Korostylev, A.P. Doklad. Chem. 1982, 266, 309.
92. See Miller, J. Aromatic Nucleophilic Substitution, Elsevier, NY, 1968, pp. 180–233.
93. From Bunnett, J.F.; Zahler, R.E. Chem. Rev. 1951, 49, 273, p. 340; Sauer, J.; Huisgen, R. Angew. Chem. 1960, 72, 294, p. 311; Bunnett, J.F. Annu. Rev. Phys. Chem. 1963, 14, 271.
94. See Amatore, C.; Combellas, C.; Robveille, S.; Savéant, J.; Thiébault, A. J. Am. Chem. Soc. 1986, 108, 4754, and references cited therein.
95. Seeliger, F.; Bła![]() ej, S.; Bernhardt, S.; M
ej, S.; Bernhardt, S.; M![]() kosza, M.; Mayr, H. Chemistry: European J. 2008, 14, 6108.
kosza, M.; Mayr, H. Chemistry: European J. 2008, 14, 6108.
96. See Fyfe, C.A. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pp. 83–124.
97. This benzyne mechanism is also supported by ![]() labeling experiments: Bottini, A.T.; Roberts, J.D. J. Am. Chem. Soc. 1957, 79, 1458; Dalman, G.W.; Neumann, F.W. J. Am. Chem. Soc. 1968, 90, 1601.
labeling experiments: Bottini, A.T.; Roberts, J.D. J. Am. Chem. Soc. 1957, 79, 1458; Dalman, G.W.; Neumann, F.W. J. Am. Chem. Soc. 1968, 90, 1601.
98. Anderson, K.W.; Ikawa, T.; Tundel, R.E.; Buchwald, S.L. J. Am. Chem. Soc. 2006, 128, 10694. Also see Schulz, T.; Torborg, C.; Schäffner, B.; Huang, J.; Zapf, A.; Kadyrov, R.; Börner, A.; Beller, M. Angew. Chem. Int. Ed.2009, 48, 918; Sergeev, A.G.; Schulz, T.; Torborg, C.; Spannenberg, A.; Neumann, H.; Beller, M. Angew. Chem. Int. Ed. 2009, 48, 7595.
99. Hashemi, M.M.; Akhbari, M. Synth. Commun. 2004, 34, 2783.
100. Maurer, S.; Liu, W.; Zhang, X.; Jiang, Y.; Ma, D. Synlett 2010, 976. See also Jing, L.; Wei, J.; Zhou, L.; Huang, Z.; Li, Z.; Zhou, X. Chem. Commun. 2010, 4767.
101. Cantrell, Jr., W.R.; Bauta, W.E.; Engles, T. Tetrahedron Lett. 2006, 47, 4249.
102. Kormos, C.M.; Leadbeater, N.E. Tetrahedron 2006, 62, 4728.
103. See Seeboth, H. Angew. Chem, Int. Ed. 1967, 6, 307; Gilbert, E.E. Sulfonation and Related Reactions; Wiley, NY, 1965, pp. 166–169.
104. Hawthorne, M.F. J. Org. Chem. 1957, 22, 1001. For other procedures, see Lewis, N.J.; Gabhe, S.Y. Aust. J. Chem. 1978, 31, 2091; Hoffmann, R.W.; Ditrich, K. Synthesis 1983, 107.
105. Pickles, G.M.; Thorpe, F.G. J. Organomet. Chem. 1974, 76, C23.
106. Simon, J.; Salzbrunn, S.; Prakash, G.K.S.; Petasis, N.A.; Olah, G.A. J. Org. Chem. 2001, 66, 633. Also see Xu, J.; Wang, X.; Shao, C.; Su, D.; Cheng, G.; Hu, Y. Org. Lett. 2010, 12, 1964.
107. Maleczka, Jr., R.E.; Shi, F.; Holmes, D.; Smith III, M.R. J. Am. Chem. Soc. 2003, 125, 7792.
108. Parker, K.A.; Koziski, K.A. J. Org. Chem. 1987, 52, 674. See Taddei, M.; Ricci, A. Synthesis 1986, 633; Einhorn, J.; Luche, J.; Demerseman, P. J. Chem. Soc. Chem. Commun. 1988, 1350.
109. Taylor, E.C.; Altland, H.W.; Danforth, R.H.; McGillivray, G.; McKillop, A. J. Am. Chem. Soc. 1970, 92, 3520.
110. Taylor, E.C.; Altland, H.W.; McKillop, A. J. Org. Chem. 1975, 40, 2351.
111. Buzbee, L.R. J. Org. Chem. 1966, 31, 3289; Oae, S.; Furukawa, N.; Kise, M.; Kawanishi, M. Bull. Chem. Soc. Jpn. 1966, 39, 1212.
112. See Gujadhur, R.; Venkataraman, D. Synth. Commun. 2001, 31, 2865.
113. Testaferri, L.; Tiecco, M.; Tingoli, M.; Chianelli, D.; Montanucci, M. Tetrahedron 1983, 39, 193.
114. Rebeiro, G.L.; Khadilkar, B.M. Synth. Commun. 2003, 33, 1405.
115. Chauhan, S.M.S.; Jain, N.; Kumar, A.; Srinivas, K.A. Synth. Commun. 2003, 33, 3607.
116. Kizner, T.A.; Shteingarts, V.D. J. Org. Chem, USSR 1984, 20, 991.
117. Artamanova, N.N.; Seregina, V.F.; Shner, V.F.; Salov, B.V.; Kokhlova, V.M.; Zhdamarova, V.N. J. Org. Chem, USSR 1989, 25, 554.
118. See Agejas, J.; Bueno, A.B. Tetrahedron Lett. 2006, 47, 5661.
119. Chaouchi, M.; Loupy, A.; Marque, S.; Petit, A. Eur. J. Org. Chem. 2002, 1278.
120. Chen, C.-y.; Dormer, P.G. J. Org. Chem. 2005, 70, 6964.
121. Luo, Y.; Wu, J.X.; Ren, R.X. Synlett 2003, 1734.
122. Oae, S.; Kiritani, R. Bull. Chem. Soc. Jpn. 1964, 37, 770; 1966, 39, 611.
123. See Hosseinzadeh, R.; Tajbakhsh, M.; Mohadjerani, M.; Alikarami, M. Synlett 2005, 1101.
124. Naidu, A.B.; Raghunath, O.R.; Prasad, D.J.C.; Sekar, G. Tetrahedron Lett. 2008, 49, 1057; Naidu, A.B.; Sekar, G. Tetrahedron Lett. 2008, 49, 3147. See Moroz, A.A.; Shvartsberg, M.S. Russ. Chem. Rev. 1974, 43, 679; Kunz, K.; Scholz, U.; Ganzer, D. Synlett 2003, 2428.
125. Weingarten, H. J. Org. Chem. 1964, 29, 977, 3624. See Cai, Q.; Zou, B.; Ma, D. Angew. Chem. Int. Ed. 2006, 45, 1276.
126. Chang, J.W.W.; Chee, S.; Mak, S.; Buranaprasertsuk, P.; Chavasiri, W.; Chan, P.W.H. Tetrahedron Lett. 2008, 49, 2018.
127. Kawaki, T.; Hashimoto, H. Bull. Chem. Soc. Jpn. 1972, 45, 1499.
128. Whitesides, G.M.; Sadowski, J.S.; Lilburn, J. J. Am. Chem. Soc. 1974, 96, 2829.
129. Lipshutz, B.H.; Unger, J.B.; Taft, B.R. Org. Lett. 2007, 9, 1089; Maiti, D.; Buchwald, S.L. J. Org. Chem. 2010, 75, 1791; Maiti, D.; Buchwald, S.L. J. Am. Chem. Soc. 2009, 131, 17423; Tlili, A.; Monnier, F.; Taillefer, M. Chemistry: Eur. J. 2010, 16, 12299; Zhao, D.; Wu, N.; Zhang, S.; Xi, P.; Su, X.; Lan, J.; You, J. Angew. Chem. Int. Ed. 2009, 48, 8729.
130. Parrish, C.A.; Buchwald, S.L. J. Org. Chem. 2001, 66, 2498; Torraca, K.E.; Huang, X.; Parrish, C.A.; Buchwald, S.L. J. Am. Chem. Soc. 2001, 123, 10770.
131. Burgos, C.H.; Barder, T.E.; Huang, X.; Buchwald, S.L. Angew. Chem. Int. Ed. 2006, 45, 4321.
132. Kuwabe, S.-i.; Torraca, K.E.; Buchwald, S.L. J. Am. Chem. Soc. 2001, 123, 12202.
133. Manolikakes, G.; Dastbaravardeh, N.; Knochel, P. Synlett 2007, 2077.
134. Xu, L.-W.; Xia, C.-G.; Li, J.-W.; Hu, X.-X. Synlett 2003, 2071.
135. Bistri, O.; Correa, A.; Bolm, C. Angew. Chem. Int. Ed. 2008, 47, 586.
136. See Desai, L.V.; Stowers, K.J.; Sanford, M.S. J. Am. Chem. Soc. 2008, 130, 13285.
137. Jönsson, L.; Wistrand, L. J. Org. Chem. 1984, 49, 3340.
138. First proposed by Alder, R.W. J. Chem. Soc. Chem. Commun. 1980, 1184.
139. See Peach, M.E. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, pp. 735–744.
140. Bieniarz, C.; Cornwell, M.J. Tetrahedron Lett. 1993, 34, 939.
141. See Palomo, C.; Oiarbide, M.; López, R.; Gómez-Bengoa, E. Tetrahedron Lett. 2000, 41, 1283.
142. Testaferri, L.; Tiecco, M.; Tingoli, M.; Chianelli, D.; Montanucci, M. Synthesis 1983, 751. See Tiecco, M.; Testaferri, L.; Tingoli, M.; Chianelli, D.; Montanucci, M. J. Org. Chem. 1983, 48, 4289.
143. Bradshaw, J.S.; South, J.A.; Hales, R.H. J. Org. Chem. 1972, 37, 2381.
144. Caruso, A.J.; Colley, A.M.; Bryant, G.L. J. Org. Chem. 1991, 56, 862; Shaw, J.E. J. Org. Chem. 1991, 56, 3728.
145. Cogolli, P.; Maiolo, F.; Testaferri, L.; Tingoli, M.; Tiecco, M. J. Org. Chem. 1979, 44, 2642. See also, Testaferri, L.; Tingoli, M.; Tiecco, M. Tetrahedron Lett. 1980, 21, 3099; Suzuki, H.; Abe, H.; Osuka, A. Chem. Lett. 1980, 1363.
146. Lee, S.B.; Hong, J.-I. Tetrahedron Lett. 1995, 36, 8439.
147. Fernández-Rodríguez, M.A.; Shen, Q.; Hartwig, J.F. J. Am. Chem. Soc. 2006, 128, 2180.
148. Sperotto, E.; van Klink, G.P.M.; de Vries, J.G.; van Koten, G. J. Org. Chem. 2008, 73, 5625; Zhu, D.; Xu, L.; Wu, F.; Wan, B. Tetrahedron Lett. 2006, 47, 5781; Feng, Y.-S.; Li, Y.-Y.; Tang, L.; Wu, W.; Xu, H.-J. Tetrahedron Lett. 2010, 51, 2489; Feng, Y.; Wang, H.; Sun, F.; Li,Y.; Fu, X.; Jin, K. Tetrahedron 2009, 65, 9737.
149. Rout, L.; Saha, P.; Jammi, S.; Punniyamurthy, T. Eur. J. Org. Chem. 2008, 640.
150. Gómez-Benítez, V.; Baldovino-Pantaleón, O.; Herrera-Álvarez, C.; Toscano, R.A.; Morales-Morales, D. Tetrahedron Lett. 2006, 47, 5059; Yoon, H.-J.; Choi, J.-W.; Kang, H.; Kang, T.; Lee, S.-M.; Jun, B.-H.; Lee, Y.-S. Synlett 2010, 2518.
151. Reddy, V.P.; Swapna, K.; Kumar, A.V.; Rama Rao, K. J. Org. Chem. 2009, 74, 3189.
152. Buranaprasertsuk, P.; Chang, J.W.W.; Chavasiri, W.; Chan, P.W.H. Tetrahedron Lett. 2008, 49, 2023.
153. Tankguchi, N. J. Org. Chem. 204, 69, 6904.
154. Bunnett, J.F.; Creary, X. J. Org. Chem. 1974, 39, 3173, 3611.
155. Meyer, G.; Troupel, M. J. Organomet. Chem. 1988, 354, 249.
156. Wang, L.; Chen, Z.-C. Synth. Commun. 2001, 31, 1227.
157. Herradua, P.S.; Pendola, K.A.; Guy, R.K. Org. Lett. 2000, 2, 2019.
158. Savarin, C.; Srogl, J.; Liebeskind, L.S. Org. Lett. 2002, 4, 4309.
159. Suzuki, H.; Abe, H. Tetrahedron Lett. 1995, 36, 6239.
160. Maitro, G.; Vogel, S.; Prestat, G.; Madec, D.; Poli, G. Org. Lett. 2006, 8, 5951.
161. Baskin, J.M.; Wang, Z. Org. Lett. 2002, 4, 4423.
162. Kar, A.; Sayyed, I.A.; Lo, W.F.; Kaiser, H.M.; Beller, M.; Tse, M.K. Org. Lett. 2007, 9, 3405.
163. Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Parisi, L.M.; Bernini, R. J. Org. Chem. 2004, 69, 5608.
164. Taniguchi, N.; Onami, T. J. Org. Chem.2004, 69, 915; Taniguchi, N.; Onami, T. Synlett 2003, 829.
165. Beletskaya, I.P.; Sigeev, A.S.; Peregudov, A.S.; Petrovlskii, P.V. Tetrahedron Lett. 2003, 44, 7039.
166. Li, Y.; Wang, H.; Li, X.; Chen, T.; Zhao, D. Tetrahedron 2010, 66, 8583.
167. Clark, J.H.; Jones, C.W.; Duke, C.V.A.; Miller, J.M. J. Chem. Soc. Chem. Commun. 1989, 81. See also, Yadav, J.S.; Reddy, B.V.S.; Shubashree, S.; Sadashiv, K. Tetrahedron Lett. 2004, 45, 2951.
168. Xu, Z.-B.; Lu, Y.; Guo, Z.-R. Synlett 2003, 564. See Li, W.; Yun, L.; Wang, H. Synth. Commun. 2002, 32, 2657.
169. See Yang, T.; Cho, B.P. Tetrahedron Lett. 2003, 44, 7549.
170. Kazankov, M.V.; Ginodman, L.G. J. Org. Chem, USSR 1975, 11, 451.
171. Sekiguchi, S.; Horie, T.; Suzuki, T. J. Chem. Soc. Chem. Commun. 1988, 698.
172. Xu, G.; Wang, Y.-G. Org. Lett. 2004, 6, 985.
173. Hertas, I.; Gallardo, I.; Marquet, J. Tetrahedron Lett. 2000, 41, 279.
174. Yadav, J.S.; Reddy, B.V.S.; Basak, A.K.; Narsaiah, A.V. Tetrahedron Lett. 2003, 44, 2217.
175. Smith, C.J.; Tsang, M.W.S.; Holmes, A.B.; Danheiser, R.L.; Tester, J.W. Org. Biomol. Chem. 2005, 3, 3767.
176. Hamper, B.C.; Tesfu, E. Synlett 2007, 2257.
177. See Heaney, H. Chem Rev. 1962, 62, 81, see p. 83.
178. Tripathy, S.; Le Blanc, R.; Durst, T. Org. Lett. 1999, 1, 1973. See Kanth, J.V.B.; Periasamy, M. J. Org. Chem. 1993, 58, 3156.
179. Shi, L.; Wang, M.; Fan, C.-A.; Zhang, F.-M.; Tu, Y.-Q. Org. Lett. 2003, 5, 3515.
180. Neunhoeffer, O.; Heitmann, P. Chem. Ber. 1961, 94, 2511.
181. Smith III, W.J.; Sawyer, J.S. Tetrahedron Lett. 1996, 37, 299.
182. Magdolen, P.; Meciarová, M.; Toma, S. Tetrahedron 2001, 57, 4781.
183. Kidwai, M.; Sapra, P.; Dave, B. Synth. Commun. 2000, 30, 4479.
184. Thomas, S.; Roberts, S.; Pasumansky, .L; Gamsey, S.; Singaram, B. Org. Lett. 2003, 5, 3867.
185. Sanger, F The Biochem. J. 1945, 39, 507.
186. Ali, M.H.; Buchwald, S.L. J. Org. Chem. 2001, 66, 2560; Kuwano, R.; Utsunomiya, M.; Hartwig, J.F. J. Org. Chem. 2002, 67, 6479; Reddy, Ch.V.; Kingston, J.V.; Verkade, J.G. J. Org. Chem. 2008, 73, 3047; Shen, Q.; Hartwig, J.F. Org. Lett. 2008, 10, 4109.
187. Harris, M.C.; Huang, X.; Buchwald, S.L. Org. Lett. 2003, 4, 2885. See Coldham, I.; Leonori, D. Org. Lett. 2008, 10, 3923; Shen, Q.; Hartwig, J.F. J. Am. Chem. Soc. 2006, 128, 10028. For a discussion about the influence of water on this reaction, see Dallas, A.S.; Gothelf, K.V. J. Org. Chem. 2005, 70, 3321.
188. See Anderson, K.W.; Tundel, R.E.; Ikawa, T.; Altman, R.A.; Buchwald, S.L. Angew. Chem. Int. Ed. 2006, 45, 6523.
189. Gajare, A.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. J. Org. Chem. 2004, 69, 6504; Huang, X.; Anderson, K.W.; Zim, D.; Jiang, L.; Klapars, A.; Buchwald, S.L. J. Am. Chem. Soc. 2004, 125, 6653; Singer, R.A.; Tom, N.J.; Frost, H.N.; Simon, W.M. Tetrahedron Lett. 2004, 45, 4715; Smith, C.J.; Early, T.R.; Holmes, A.B.; Shute, R.E. Chem. Commun. 2004, 1976.
190. See Singh, U.K.; Strieter, E.R.; Blackmond, D.G.; Buchwald, S.L. J. Am. Chem. Soc. 2002, 124, 14104. For a study of rate enhancement by the added base, see Meyers, C.; Maes, B.U.W.; Loones, K.T.J.; Bal, G.; Lemière, G.L.F.; Dommisse, R.A. J. Org. Chem. 2004, 69, 6010.
191. See Strieter, E.R.; Buchwald, S.L. Angew. Chem. Int. Ed. 2006, 45, 925; Fors, B.P.; Davis, N.R.; Buchwald, S.L. J. Am. Chem. Soc. 2009, 131, 5766.
192. Guinó, M.; Hii, K.K. Tetrahedron Lett. 2005, 46, 7363. See Inasaki, T.; Ueno, M.; Miyamoto, S.; Kobayashi, S. Synlett 2007, 3209.
193. Parrish, C.A.; Buchwald, S.L. J. Org. Chem. 2001, 66, 3820.
194. Weigand, K.; Pelka, S. Org. Lett. 2002, 4, 4689.
195. See Grasa, G.A.; Viciu, M.S.; Huang, J.; Nolan, S.P. J. Org. Chem. 2001, 66, 7729.
196. Jensen, T.A.; Liang, X.; Tanner, D.; Skjaerbaek, N. J. Org. Chem. 2004, 69, 4936.; Loones, K.T.J.; Maes, B.U.W.; Rombouts, G.; Hostyn, S.; Diels, G. Tetrahedron 2005, 61, 10338.
197. Xu, C.; Gong, J.-F.; Wu, Y.-J. Tetrahedron Lett. 2007, 48, 1619.
198. So, C.M.; Zhou, Z.; Lau, C.P.; Kwong, F.Y. Angew. Chem. Int. Ed. 2008, 47, 6402.
199. Tagashira, J.; Imao, D.; Yamamoto, T.; Ohta, T.; Furukawa, I.; Ito, Y. Tetrahedron Asymmetry 2005, 16, 230.
200. Huang, X.; Buchwald, S.L. Org. Lett. 2001, 3, 3417.
201. Kang, S.-K.; Lee, H.-W.; Choi, W.-K.; Hong, R.-K.; Kim, J.-S. Synth. Commun. 1996, 26, 4219. See Carroll, M.A.; Wood, R.A. Tetrahedron 2007, 63, 11349.
202. Kang, S.-K.; Lee, S.-H.; Lee, D. Synlett 2000, 1022.
203. Ogawa, K.; Radke, K.R.; Rothstein, S.D.; Rasmussen, S.C. J. Org. Chem. 2001, 66, 9067.
204. Junckers, T.H.M.; Maes, B.U.W.; Lemière, G.L.F.; Dommisse, R. Tetrahedron 2001, 57, 7027; Basu, B.; Jha, S.; Mridha, N.K.; Bhuiyan, Md.M.H. Tetrahedron Lett. 2002, 43, 7967.
205. Choudary, B.M.; Sridhar, C.; Kantam, M.L.; Venkanna, G.T.; Sreedhar, B. J. Am. Chem. Soc. 2005, 127, 9948; Shafir, A.; Buchwald, S.L. J. Am. Chem. Soc. 2006, 128, 8742; Wolf, C.; Liu, S.; Mei, X.; August, A.T.; Casimir, M.D. J. Org. Chem. 2006, 71, 3270; Jiang, D.; Fu, H.; Jiang, Y.; Zhao, Y. J. Org. Chem. 2007, 72, 672; Wang, H.; Li, Y.; Sun, F.; Feng, Y.; Jin, K.; Wang, X. J. Org. Chem. 2008, 73, 8639; Cai, Q.; Zhu, W.; Zhang, H.; Zhang, Y.; Ma, D. Synthesis 2005, 496. For a ligand-free reaction, see Yong, F.F.; Teo, Y.-C. Synlett 2010, 3068.
206. Shafir, A.; Lichtor, P.A.; Buchwald, S.L. J. Am. Chem. Soc. 2007, 129, 3490. For an amination using aq ammonia, see Meng, F.; Zhu, X.; Li, Y.; Xie, J.; Wang, B.; Yao, J.; Wan, Y. Eur. J. Org. Chem. 2010, 6149.
207. Lu, Z.; Twieg, R.J. Tetrahedron Lett. 2005, 46, 2997.
208. Kim, J.; Chang, S. Chem. Commun. 2008, 3052.
209. See Renger, B. Synthesis 1985, 856.
210. Wolfe, J.P.; Buchwald, S.L. J. Am. Chem. Soc. 1997, 119, 6054; Lipshutz, B.H.; Ueda, H. Angew. Chem. Int. Ed. 2000, 39, 4492.; Brenner, E.; Schneider, R.; Fort, Y. Tetrahedron 2002, 58, 6913; Chen, C.; Yang, L.-M. J. Org. Chem. 2007, 72, 6324.
211. Manolikakes, G.; Gavryushin, A.; Knochel, P. J. Org. Chem. 2008, 73, 1429; Gao, C.-Y.; Cao, X.; Yang, L.-M. Org. Biomol. Chem. 2009, 7, 3922.
212. Tasler, S.; Lipshutz, B.H. J. Org. Chem. 2003, 68, 1190.
213. Omar-Amrani, R.; Thomas, A.; Brenner, E.; Schneider, R.; Fort, Y. Org. Lett. 2003, 5, 2311.
214. Fedorov, A.Yu.; Finet, J.-P. J. Chem. Soc. Perkin Trans. 1 2000, 3775.
215. Berman, A.M.; Johnson, J.S. J. Am. Chem. Soc. 2004, 126, 5680.
216. Berman, A.M.; Johnson, J.S. Synlett 2005, 1799.
217. Lee, D.-Y.; Hartwig, J.F. Org. Lett. 2005, 7, 1169.
218. Guo, D.; Huang, H.; Xu, J.; Jiang, H.; Liu, H. Org. Lett. 2008, 10, 4513; Correa, A.; Bolm, C. Angew. Chem. Int. Ed. 2007, 46, 8862; Correa, A.; Carril, M.; Bolm, C. Chemistry: European J. 2008, 14, 10919.
219. Appukkuttan, P.; Axelsson, L.; Van der Eycken, E.; Larhed, M. Tetrahedron Lett. 2008, 49, 5625.
220. See Jordan-Hore, J.A.; Johansson, C.C.C.; Gulias, M.; Beck, E.M.; Gaunt, M.J. J. Am. Chem. Soc. 2008, 130, 16184; Kuwahara, A.; Nakano, K.; Nozaki, K. J. Org. Chem. 2005, 70, 413.
221. See Bunnett, J.F.; Hrutfiord, B.F. J. Am. Chem. Soc. 1961, 83, 1691. Also see Hoffmann, R.W. Dehydrobenzene and Cycloalkynes, Academic Press, NY, 1973, pp. 150–164.
222. Chiang, G.C.H.; Olsson, T. Org. Lett. 2004, 6, 3079; Lan, J.-B.; Zhang, G.-L.; Yu, X.-Q.; You, J.-S.; Chen, L.; Yan, M.; Xie, R.-G. Synlett 2004, 1095.
223. Jiang, Z.; Wu, Z.; Wang, L.; Wu, D.; Zhou, X. Can. J. Chem. 2010, 88, 964.
224. Chernick, E.T.; Ahrens, M.J.; Scheidt, K.A.; Wasielewski, M.R. J. Org. Chem. 2005, 70, 1486.
225. Quach, T.D.; Batey, R.A. Org. Lett. 2003, 5, 4397.
226. See Finet, J. Chem. Rev. 1989, 89, 1487.
227. See Barton, D.H.R.; Yadav-Bhatnagar, N.; Finet, J.; Khamsi, J. Tetrahedron Lett. 1987, 28, 3111.
228. See Bethell, D.; Jenkins, I.L.; Quan, P.M. J. Chem. Soc. Perkin Trans. 1 1985, 1789; Paine, A.J. J. Am. Chem. Soc. 1987, 109, 1496.
229. Gauthier, S.; Fréchet, J.M.J. Synthesis 1987, 383.
230. See van der Plas, H.C. Tetrahedron 1985, 41, 237; Acc. Chem. Res. 1978, 11, 462.
231. See Chupakhin, O.N.; Postovskii, I.Ya. Russ. Chem. Rev. 1976, 45, 454, p. 456.
232. Kabalka, G.W.; Li, G. Tetrahedron Lett. 1997, 38, 5777.
233. Sapountzis, I.; Knochel, P. J. Am.Chem. Soc. 2002, 124, 9390.
234. Kitagawa, O.; Takahashi, M.; Yoshikawa, M.; Taguchi, T. J. Am. Chem. Soc. 2005, 127, 3676; Fors, B.P.; Dooleweerdt, K.; Zeng, Q.; Buchwald, S.L. Tetrahedron 2009, 65, 6576. For an intramolecular reaction, see Yang, B.H.; Buchwald, S.L. Org. Lett. 1999, 1, 35.
235. Hosseinzadeh, R.; Tajbakhsh, M.; Mohadjerani, M.; Mehdinejad, H. Synlett 2004, 1517.
236. Browning, R.G.; Badaringarayana, V.; Mahmud, H.; Lovely, C.J. Tetrahedron 2004, 60, 359; Deng, W.; Wang, Y.-F.; Zou, Y.; Liu, L.; Guo, Q.-X. Tetrahedron Lett. 2004, 45, 2311. Also see Klapars, A.; Huang, X.; Buchwald, S.L. J. Am. Chem. Soc. 2002, 124, 7421; Ferraccioli, R.; Carenzi, D.; Rombolà, O.; Catellani, M. Org. Lett. 2004, 6, 4759.
237. Also see Klapars, A.; Parris, S.; Anderson, K.W.; Buchwald, S.L. J. Am. Chem. Soc. 2004, 126, 3529.
238. Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Zappia, G. Org. Lett. 2001, 3, 2539.
239. Lam, P.Y.S.; Deudon, S.; Hauptman, E.; Clark, C.G. Tetrahedron Lett. 2001, 42, 2427.
240. Wolter, M.; Klapars, A.; Buchwald, S.L. Org. Lett. 2001, 3, 3803.
241. Padwa, A.; Crawford, K.R.; Rashatasakhon, P.; Rose, M. J. Org. Chem. 2003, 68, 2609.
242. Kang, S.-K.; Kim, D.-H.; Park, J.-N. Synlett 2002, 427.
243. Nandakumar, M.V. Tetrahedron Lett. 2004, 45, 1989.
244. Gelpke, A.E.S.; Kooijman, H.; Spek, A.L.; Hiemstra, H. Chem. Eur. J. 1999, 5, 2472; Ding, K.; Wang, Y.; Yun, H.; Liu, J.; Wu, Y.; Terada, M.; Okubo, Y.; Mikami, K. Chem. Eur. J. 1999, 5, 1734; Vyskocil, S.; Smrcina, M.; Hanus, V.; Polasek, M.; Kocovsky, P. J. Org. Chem. 1998, 63, 7738; Martorell, G.; Garcias, X.; Janura, M.; Saá, J.M. J. Org. Chem. 1998, 63, 3463; Lipshutz, B.H.; Buzard, D.H.; Yun, C.S. Tetrahedron Lett. 1999, 40, 201.
245. Ager, D.J.; Laneman, S. Chem. Commun. 1997, 2359.
246. Tunney, B.H.; Stille, J.K. J. Org. Chem. 1987, 52, 748.
247. Van Allen, D.; Venkataraman, D. J. Org. Chem. 2003, 68, 4590.
248. Stadler, A.; Kappe, C.O. Org. Lett. 2002, 4, 3541.
249. Kwong, F.Y.; Lai, C.W.; Chan, K.S. Tetrahedron Lett. 2002, 43, 3537.
250. Marcoux, D.; Charette, A.B. J. Org. Chem. 2008, 73, 590.
251. Rieche, A.; Seeboth, H. Liebigs Ann. Chem. 1960, 638, 66.
252. Rieche, A.; Seeboth, H. Liebigs Ann. Chem. 1960, 638, 43, 57.
253. Kozlov, V.V.; Veselovskaia, I.K. J. Gen. Chem. USSR 1958, 28, 3359.
254. Rieche, A.; Seeboth, H. Liebigs Ann. Chem. 1960, 638, 76.
255. Rossi, R.A.; Bunnett, J.F. J. Org. Chem. 1972, 37, 3570.
256. See Scherrer, R.A.; Beatty, H.R. J. Org. Chem. 1972, 37, 1681.
257. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 671–672.
258. Sauer, J.; Huisgen, R. Angew. Chem. 1960, 72, 294, p. 297.
259. Schaefer, J.P.; Higgins, J. J. Org. Chem. 1967, 32, 1607.
260. Bay, E.; Bak, D.A.; Timony, P.E.; Leone-Bay, A. J. Org. Chem. 1990, 55, 3415.
261. Kimura, Y.; Suzuki, H. Tetrahedron Lett. 1989, 30, 1271. See Dolby-Glover, L. Chem. Ind. (London) 1986, 518.
262. Uchibori, Y.; Umeno, M.; Seto, H.; Qian, Z.; Yoshioka, H. Synlett 1992, 345.
263. Bacon, R.G.R.; Hill, H.A.O. J. Chem. Soc. 1964, 1097, 1108. See also, Clark, J.H.; Jones, C.W.; Duke, C.V.A.; Miller, J.M. J. Chem. Res. (S) 1989, 238.
264. Clark, J.H.; Jones, C.W. J. Chem. Soc. Chem. Commun. 1987, 1409.
265. Klapars, A.; Buchwald, S.L. J. Am. Chem. Soc. 2002, 124, 14844.
266. Yang, S.H.; Li, C.S.; Cheng, C.H. J. Org. Chem. 1987, 52, 691.
267. Arvela, R.K.; Leadbeater, N.E. Synlett 2003, 1145.
268. Shen, X.; Hyde, A.M.; Buchwald, S.L. J. Am. Chem. Soc. 2010, 132, 14076.
269. See Merkushev, E.B. Synthesis 1988, 923; Russ. Chem. Rev. 1984, 53, 343.
270. Taylor, E.C.; Kienzle, F.; McKillop, A. Org. Synth. VI, 826; Taylor, E.C.; Katz, A.H.; Alvarado, S.I.; McKillop, A. J. Organomet. Chem. 1985, 285, C9. See Usyatinskii, A.Ya.; Bregadze, V.I. Russ. Chem. Rev. 1988, 57, 1054; Taylor, E.C.; Altland, H.W.; McKillop, A. J. Org. Chem. 1975, 40, 2351.
271. De Meio, G.V.; Pinhey, J.T. J. Chem. Soc. Chem. Commun. 1990, 1065.
272. Thiebes, C.; Prakash, G.K.S.; Petasis, N.A.; Olah, G.A. Synlett 1998, 141.
273. Szumigala, Jr., R.H.; Devine, P.N.; Gauthier, Jr., D.R.; Volante, R.P. J. Org. Chem. 2004, 69, 566.
274. See Artamkina, G.A.; Kovalenko, S.V.; Beletskaya, I.P.; Reutov, O.A. Russ. Chem. Rev. 1990, 59, 750.
275. See Ellis, G.P.; Romney-Alexander, T.M. Chem. Rev. 1987, 87, 779.
276. See Connor, J.A.; Leeming, S.W.; Price, R. J. Chem. Soc. Perkin Trans. 1 1990, 1127.
277. Wu, J.X.; Beck, B.; Ren, R.X. Tetrahedron Lett. 2002, 43, 387.
278. Arvela, R.K.; Leadbeater, N.W.; Torenius, H.M.; Tye, H. Org. Biomol. Chem. 2003, 1, 1119.
279. Wang, D.; Kuang, L.; Li, Z.; Ding, K. Synlett 2008, 69.
280. For a list of reagents that convert aryl halides to cyanides, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1705–1709.
281. Takagi, K.; Sasaki, K.; Sakakibara, Y. Bull. Chem. Soc. Jpn. 1991, 64, 1118.
282. Connor, J.A.; Gibson, D.; Price, R. J. Chem. Soc. Perkin Trans. 1 1987, 619.
283. Sakakibara, Y.; Okuda, F.; Shimobayashi, A.; Kirino, K.; Sakai, M.; Uchino, N.; Takagi, K. Bull. Chem. Soc. Jpn. 1988, 61, 1985.
284. See Stazi, F.; Palmisano, G.; Turconi, M.; Santagostino, M. Tetrahedron Lett. 2005, 46, 1815; Zhu, Y.-Z.; Cai, C. Eur. J. Org. Chem. 2007, 2401.
285. Marcantonio, K.M.; Frey, L.F.; Liu, Y.; Chen, Y.; Strine, J.; Phenix, B.; Wallace, D.J.; Chen, C.-y. Org. Lett. 2004, 6, 3723. See Erker, T.; Nemec, S. Synthesis 2004, 23.
286. Sakamoto, T.; Ohsawa, K. J. Chem. Soc. Perkin Trans. 1 1999, 2323.
287. Jiang, B.; Kan, Y.; Zhang, A. Tetrahedron 2001, 57, 1581.
288. Mariampillai, B.; Alliot, J.; Li, M.; Lautens, M. J. Am. Chem. Soc. 2007, 129, 15372; Weissman, S.A.; Zewge, D.; Chen, C. J. Org. Chem. 2005, 70, 1508; Schareina, T.; Zapf, A.; Mägerlein, W.; Müller, N.; Beller, M. Tetrahedron Lett. 2007, 48, 1087; Velmathi, S.; Leadbeater, N.E. Tetrahedron Lett. 2008, 49, 4693.
289. Yang, C.; Williams, J.M. Org. Lett. 2004, 6, 2837.
290. Chobanian, H.R.; Fors, B.P.; Lin, L.S. Tetrahedron Lett. 2006, 47, 3303.
291. Zhu, Y.-Z.; Cai, C. Synth. Commun. 2008, 38, 2753; Yeung, P.Y.; So, C.M.; Lau. C.-P.; Kwong, F.Y. Angew. Chem. Int. Ed. 2010, 49, 8918.
292. Zhang, Z.; Liebeskind, L.S. Org. Lett. 2006, 8, 4331.
293. Cristau, H.-J.; Ouali, A.; Spindler, J.-F.; Taillefer, M. Chemistry: European J. 2005, 11, 2483.
294. Arvela, R.K.; Leadbeater, N.E. J. Org. Chem. 2003, 68, 9122.
295. Chambers, M.R.I.; Widdowson, D.A. J. Chem. Soc. Perkin Trans. 1 1989, 1365; Takagi, K.; Sakakibara, Y. Chem. Lett. 1989, 1957.
296. Liskey, C.W.; Liao, X.; Hartwig, J.F. J. Am. Chem. Soc. 2010, 132, 11389.
297. Taylor, E.C.; Altland, H.W.; McKillop, A. J. Org. Chem. 1975, 40, 2351.
298. Uemura, S.; Ikeda, Y.; Ichikawa, K. Tetrahedron 1972, 28, 3025.
299. Ushijima, S.; Togo, H. Synlett 2010, 1067; Ushijima, S.; Togo, H. Synlett 2010, 1562.
300. Sato, N. Tetrahedron Lett. 2002, 43, 6403.
301. Letsinger, R.L.; Colb, A.L. J. Am. Chem. Soc. 1972, 94, 3665.
302. Alberico, D.; Scott, M.E.; Lautens, M. Chem. Rev. 2007, 107, 174.
303. See Clayden, J.; Kenworthy, M.N. Synthesis 2004, 1721.
304. See Becht, J.-M.; Gissot, A.; Wagner, A.; Mioskowski, C. Chem. Eur. J. 2003, 9, 3209.
305. Narayan, S.; Seelhammer, T.; Gawley, R.E. Tetrahedron Lett. 2004, 45, 757.
306. Park, S.B.; Alper, H. Tetrahedron Lett. 2004, 45, 5515.
307. Jorapur, Y.R.; Lee, C.-H.; Chi, D.Y. Org. Lett. 2005, 7, 1231.
308. Silveira, P.B.; Lando, V.R.; Dupont, J.; Monteiro, A.L. Tetrahedron Lett. 2002, 43, 2327; Kuroboshi, M.; Waki, Y.; Tanaka, H. Synlett 2002, 637. See also, Venkatraman, S.; Li, C.-J. Org. Lett. 1999, 1, 1133.
309. Leadbeater, N.E.; Resouly, S.M. Tetrahedron Lett. 1999, 40, 4243.
310. Penalva, V.; Hassan, J.; Lavenot, L.; Gozzi, C.; Lemaire, M. Tetrahedron Lett. 1998, 39, 2559.
311. de Franca, K.W.R.; Navarro, M.; Léonel, É; Durandetti, M.; Nédélec, J.-Y. J. Org. Chem. 2002, 67, 1838.
312. Wang, R.; Twamley, B.; Shreeve, J.M. J. Org. Chem. 2006, 71, 426.
313. Wang, L.; Zhang, Y.; Liu, L.; Wang, Y. J. Org. Chem. 2006, 71, 1284.
314. Glover, B.; Harvey, K.A.; Liu, B.; Sharp, M.J.; Tymoschenko, M.F. Org. Lett. 2003, 5, 301.
315. See Rieth, R.D.; Mankand, N.P.; Calimano, E.; Sadighi, J.P. Org. Lett. 2004, 6, 3981.
316. Sezen, B.; Sames, D. Org. Lett. 2003, 5, 3607.
317. Quintin, J.; Franck, X.; Hocquemiller, R.; Figadère, B. Tetrahedron Lett. 2002, 43, 3547.
318. Park, C.-H.; Ryabova, V.; Seregin, I. V.; Sromek, A. W.; Gevorgyan, V. Org. Lett. 2004, 6, 1159.
319. Liu, Z.; Zhang, X.; Larock, R.C. J. Am. Chem. Soc. 2005, 127, 15716.
320. Napier, S.; Marcuccio, S.M.; Tye, H.; Whittaker, M. Tetrahedron Lett. 2008, 49, 6314. See also, Denmark, S.E.; Smith, R.C.; Chang, W.-T.T.; Muhuhi, J.M. J. Am. Chem. Soc. 2009, 131, 3104.
321. Wang, Z.; Ding, Q.; He, X.; Wu, J. Tetrahedron 2009, 65, 4635.
322. Nakamura, T.; Kinoshita, H.; Shinokubo, H.; Oshima, K. Org. Lett. 2002, 4, 3165.
323. Ohe, T.; Tanaka, T.; Kuroda, M.; Cho, C.S.; Ohe, K.; Uemura, S. Bull. Chem. Soc. Jpn. 1999, 72, 1851.
324. Kang, S.-K.; Ryu, H.-C.; Kim, J.-W. Synth. Commun. 2001, 31, 1021.
325. Wang, J.; Scott, A.I. Tetrahedron Lett. 1996, 37, 3247; Kim, Y.M.; Yu, S. J. Am. Chem. Soc. 2003, 125, 1696.
326. Rao, M.L.N.; Yamazaki, O.; Shimada, S.; Tanaka, T.; Suzuki, Y.; Tanaka, M. Org. Lett. 2001, 3, 4103.
327. Yamazaki, O.; Tanaka, T.; Shimada, S.; Suzuki, Y.; Tanaka, M. Synlett 2004, 1921.
328. Grasa, G.A.; Nolan, S.P. Org. Lett. 2001, 3, 119.
329. Dyker, G.; Heiermann, J.; Miura, M.; Inoh, J.-I.; Pivsa-Ast, S.; Satoh, T.; Nomura, M. Chem. Eur. J. 2000, 6, 3426.
330. With NCS, Hossain, K.M.; Kameyama, T.; Shibata, T.; Takagi, K. Bull. Chem. Soc. Jpn. 2001, 74, 2415. See also, Venkatraman, S.; Li, C.-J. Tetrahedron Lett. 2000, 41, 4831.
331. Dubbaka, S.R.; Vogel, P. J. Am. Chem. Soc. 2003, 125, 15292.
332. Chen, C. Synlett 2000, 1491. See Walla, P.; Kappe, C.O. Chem. Commun. 2004, 564.
333. Kang, S.-K.; Baik, T.-G.; Jiao, X.H.; Lee, Y.-T. Tetrahedron Lett. 1999, 40, 2383.
334. Kang, S.-K.; Lee, S.-W.; Kim, M.-S.; Kwon, H.S. Synth. Commun. 2001, 31, 1721.
335. Korn, T.J.; Knochel, P. Angew. Chem. Int. Ed. 2005, 44, 2947.
336. Cahiez, G.; Luart, D.; Lecomte, F. Org. Lett. 2004, 6, 4395.
337. Pérez, I.; Sestelo, J.P.; Sarandeses, L.A. J. Am. Chem. Soc. 2001, 123, 4155.
338. Bedford, R.B.; Limmert, M.E. J. Org. Chem. 2003, 68, 8669.
339. Kang, S.-K.; Choi, S.-C.; Baik, T.-G. Synth. Commun. 1999, 29, 2493.
340. Gagnon, A.; Duplessis, M.; Alsabeh, P.; Barabé, F. J. Org. Chem. 2008, 73, 3604.
341. Chupak, L.S.; Wolkowski, J.P.; Chantigny, Y.A. J. Org. Chem. 2009, 74, 1388.
342. Du, C.F.; Hart, H.; Ng, K.D. J. Org. Chem. 1986, 51, 3162.
343. Cahiez, G.; Chaboche, C.; Mahuteau-Betzer, F.; Ahr, M. Org. Lett. 2005, 7, 1943.
344. Yoshikai, N.; Mashima, H.; Nakamura, E. J. Am. Chem. Soc. 2005, 127, 17978.
345. Korn, T.J.; Cahiez, G.; Knochel, P. Synlett 2003, 1892.
346. Inoue, A.; Kitagawa, K.; Shinokubo, H.; Oshima, K. Tetrahedron 2000, 56, 9601.
347. Manabe, K.; Ishikawa, S. Synthesis 2008, 2645.
348. Reeves, J.T.; Fandrick, D.R.; Tan, Z.; Song, J.J.; Lee, H.; Yee, N.K.; Senanayake, C.H. Org. Lett. 2010, 12, 4388.
349. Roy, A.H.; Hartwig, J.F. J. Am. Chem. Soc. 2003, 125, 8704.
350. Bonnet, V.; Mongin, F.; Trècourt, F.; Quèguiner, G.; Knochel, P. Tetrahedron Lett. 2001, 42, 5717.
351. Wang, L.; Chen, Z.-C. Synth. Commun. 2000, 30, 3607.
352. Kojima, T.; Ohishi, T.; Yamamoto, I.; Matsuoka, T.; Kotsuki, H. Tetrahedron Lett. 2001, 42, 1709.
353. Clayden, J.; Cooney, J.J.A.; Julia, M. J. Chem. Soc. Perkin Trans. 1 1995, 7.
354. Beugelmans, R.; Bois-Choussy, M.; Tang, Q. Tetrahedron Lett. 1988, 29, 1705. See Pierini, A.B.; Baumgartner, M.T.; Rossi, R.A. Tetrahedron Lett. 1988, 29, 3429.
355. Durandetti, M.; Nédélec, J.-Y.; Périchon, J. J. Org. Chem. 1996, 61, 1748.
356. He, H.; Wu, Y.-J. Tetrahedron Lett. 2003, 44, 3445.
357. For a review of coupling reactions using various metals, see Terao, J.; Kambe, N. Acc. Chem. Res. 2008, 41,1545.
358. Everson, D.A.; Shrestha, R.; Weix, D.J. J. Am. Chem. Soc. 2010, 132, 920.
359. Hama, T.; Culkin, D.A.; Hartwig, J.F. J. Am. Chem. Soc. 2006, 128, 4976.
360. Shenglof, M.; Gelman, D.; Heymer, B.; Schumann, H.; Molander, G.A.; Blum, J. Synthesis 2003, 302.
361. Sherry, B.D.; Fürstner, A. Acc. Chem. Res. 2008, 41,1500.
362. Rao, M.L.N.; Banerjee, D.; Jadhav, D.N. Tetrahedron Lett. 2007, 48, 2707.
363. Jhaveri, S.B.; Carter, K.R. Chemistry: European J. 2008, 14, 685.
364. Wang, L.; Wang, Z.-X. Org. Lett. 2007, 9, 4335.
365. Moradi, W.A.; Buchwald, S.L. J. Am. Chem. Soc. 2001, 123, 7996.
366. Liao, X.; Weng, Z.; Hartwig, J.F. J. Am. Chem. Soc. 2008, 130, 195.
367. See Heck, R.F. Palladium Reagents in Organic Syntheses, Academic Press, NY, 1985, pp. 179–321; Ryabov, A.D. Synthesis 1985, 233; Heck, R.F. Org. React. 1982, 27, 345; Moritani, I.; Fujiwara, Y. Synthesis 1973, 524. See Cabri, W.; Candiani, I. Accts. Chem. Res. 1995, 28, 2.
368. See Heck, R.F. Acc. Chem. Res. 1979, 12, 146; Kozhevnikov, I.V. Russ. Chem. Rev. 1983, 52, 138. See also, Spencer, A. J. Organomet. Chem. 1983, 258, 101; Andersson, C.; Karabelas, K.; Hallberg, A.; Andersson, C. J. Org. Chem. 1985, 50, 3891; Larock, R.C.; Johnson, P.L. J. Chem. Soc. Chem. Commun. 1989, 1368.
369. See Alonso, F.; Beletskaya, I.P.; Yus, M. Tetrahedron 2005, 61, 11771.
370. Mizoroki, T.; Mori, K.; Ozaki, A. Bull. Chem. Soc. Jpn 1971, 4, 581.
371. See Littke, A.F.; Fu, G.C. Angew. Chem. Int. Ed. 2002, 41, 4176.
372. Roglans, A.; Pla-Quintana, A.; Moreno-Mañas, M. Chem. Rev. 2006, 106, 4622;
373. Myers, A.G.; Tanaka, D.; Mannion, M.R. J. Am. Chem. Soc. 2002, 124, 11250.
374. Pyridines: Draper, T.L.; Bailey, T.R. Synlett 1995, 157.
375. See Park, S.B.; Alper, H. Org. Lett. 2003, 5, 3209. See also, Zeni, G.; Larock, R.C. Chem. Rev. 2004, 104, 2285.
376. See Firmansjah, L.; Fu, G.C. J. Am. Chem. Soc. 2007, 129, 11340. Also see, Echavarren, A.M.; Gómez-Lor, B.; González, J.J.; de Frutos, Ó. Synlett 2003, 585.
377. Mayasundari, A.; Young, D.G.J. Tetrahedron Lett. 2001, 42, 203.
378. For a review, see Prim, D.; Campagne, J.-M.; Joseph, D.; Andrioletti, B. Tetrahedron 2002, 58, 2041.
379. Consorti, C.S.; Zanini, M.L.; Leal, S.; Ebeling, G.; Dupont, J. Org. Lett. 2003, 5, 983; Senra, J.D.; Malta, L.F.B.; de Souza, A.L.F.; Medeiros, M.E.; Aguiar, L.C.S.; Antunes, O.A.C. Tetrahedron Lett. 2007, 48, 8153.
380. Hirabayashi, K.; Nishihara, Y.; Mori, A.; Hiyama, T. Tetrahedron Lett. 1998, 39, 7893.
381. Ruan, J.; Li, X.; Saidi, O.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 2424; Martinez, R.; Voica, F.; Genet, J.-P.; Darses, S. Org. Lett. 2007, 9, 3213.
382. Selvakumar, K.; Zapf, A.; Beller, M. Org. Lett. 2002, 4, 3031; Feuerstein, M.; Doucet, H.; Santelli, M. Tetrahedron Lett. 2002, 43, 2191. For the use of palladium nanoparticles, see Calò, V.; Nacci, A.; Monopoli, A.; Laera, S.; Cioffi, N. J. Org. Chem. 2003, 68, 2929. See Lindh, J.; Enquist, P.-A.; Pilotti, Å.; Nilsson, P.; Larhed, M. J. Org. Chem. 2007, 72, 7957. See also Seki, M. Synthesis 2006, 2975.
383. Kim, J.-H.; Kim, J.W.; Shokouhimehr, M.; Lee, Y.-S. J. Org. Chem. 2005, 70, 6714.
384. Polshettiwar, V.; Molnár, Á. Tetrahedron 2007, 63, 6949.
385. Karimi, B.; Enders, D. Org. Lett. 2006, 8, 1237; Ambulgekar, G.V.; Bhanage, B.M.; Samant, S.D. Tetrahedron Lett. 2005, 46, 2483; Papp, A.; Galbács, G.; Molnár, Á. Tetrahedron Lett. 2005, 46, 7725; Li, H.; Wang, L.; Li, P. Synthesis 2007, 1635.
386. See Qadir, M.; Möchel, T.; Hii, K.K. Tetrahedron 2000, 56, 7975. Also see Tani, M.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2004, 69, 1221; Yang, D.; Chen, Y.-C.; Zhu, N.-Y. Org. Lett. 2004, 6, 1577; Eberhard, M.R. Org. Lett. 2004, 6, 2125; Liu, J.; Li, S.; Xie, H.; Zhang, S.; Lin, Y.; Xu, J.; Cao, J. Synlett 2005, 1885; Schultz, T.; Pfaltz, A. Synthesis 2005, 1005; Iranpoor, N.; Firouzabadi, H.; Tarassoli, A.; Fereidoonnezhad, M. Tetrahedron 2010, 66, 2415.
387. Nair, D.; Scarpello, J.T.; White, L.S.; dos Santes, L.M.F.; Vankelecom, I.F.J.; Livingston, A.G. Tetrahedron Lett. 2001, 42, 8219.
388. See Botella, L.; Nájera, C. J. Org. Chem. 2005, 70, 4360; Zhang, Z.; Zha, Z.; Gan, C.; Pan, C.; Zhou, Y.; Wang, Z.; Zhou, M.-M. J. Org. Chem. 2006, 71, 4339; Bhattacharya, S.; Srivastava, A.; Sengupta, S. Tetrahedron Lett.2005, 46, 3557.
389. Moineau, J.; Pozzi, G.; Quici, S.; Sinou, D. Tetrahedron Lett. 1999, 40, 7683.
390. Chandrasekhar, S.; Narsihmulu, Ch.; Sultana, S.S.; Reddy, N.R. Org. Lett. 2002, 4, 4399.
391. Perosa, A.; Tundo, P.; Selva, M.; Zinovyev, S.; Testa, A. Org. Biomol. Chem. 2004, 2, 2249.
392. Early, T.R.; Gordon, R.S.; Carroll, M.A.; Holmes, A.B.; Shute, R.E.; McConvey, I.F. Chem. Commun. 2001, 1966. See Kayaki, Y.; Noguchi, Y.; Ikariya, T. Chem. Commun. 2000, 2240.
393. Franzén, R. Can. J. Chem. 2000, 78, 457.
394. Ramchandani, R.K.; Uphade, B.S.; Vinod, M.P.; Wakharkar, R.D.; Choudhary, V.R.; Sudalai, A. Chem. Commun. 1997, 2071.
395. Tonks, L.; Anson, M.S.; Hellgardt, K.; Mirza, A.R.; Thompson, D.F.; Williams, J.M.J. Tetrahedron Lett. 1997, 38, 4319.
396. Anson, M.S.; Mirza, A.R.; Tonks, L.; Williams, J.M.J. Tetrahedron Lett. 1999, 40, 7147.
397. Arvela, R.K.; Leadbeater, N.E. J. Org. Chem. 2005, 70, 1786; Leadbeater, N.E.; Williams, V.A.; Barnard, T.M.; Collins, Jr., M.J. Synlett 2006, 2953; Declerck, V.; Martinez, J.; Lamaty, F. Synlett 2006, 3029; Zhu, M.; Song, Y.; Cao, Y. Synthesis 2007, 853; Du, L.-H.; Wang, Y.-G. Synth. Commun. 2007, 37, 217. See Nilsson, P.; Gold, H.; Larhed, M.; Hallberg, A. Synthesis 2002, 1611.
398. Wang, J.-X.; Liu, Z.; Hu, Y.; Wei, B.; Bai, L. Synth. Commun. 2002, 32, 1607.
399. Zhang, R.; Sato, O.; Zhao, F.; Sato, M.; Ikushima, Y. Chem. Eur. J. 2004, 10, 1501.
400. Buback, M.; Perkovic, T.; Redlich, S.; de Meijere, A. Eur. J. Org. Chem. 2003, 2375.
401. Hagiwara, H.; Sugawara, Y.; Isobe, K.; Hoshi, T.; Suzuki, T. Org. Lett. 2004, 6, 2325; Handy, S.T. Synlett 2006, 3176; Jung, J.-Y.; Taher, A.; Kim, H.-J.; Ahn, W.-S.; Jin, M.-J. Synlett 2009, 39; Zhou, L.; Wang, L. Synthesis2006, 2653; Iranpoor, N.; Firouzabadi, H.; Azadi, R. Eur. J. Org. Chem. 2007, 2197; Cárdenas, J.C.; Fadini, L.; Sierra, C.A. Tetrahedron Lett. 2010, 51, 6867.
402. Handy, S.T.; Okello, M. Tetrahedron Lett. 2003, 44, 8395.
403. Mo, J.; Xu, L.; Xiao, J. J. Am. Chem. Soc. 2005, 127, 751.
404. Heck, R.F. J. Am. Chem. Soc. 1969, 91, 6707; 1971, 93, 6896.
405. Xu, Y.-H.; Lu, J.; Loh, T.-P. J. Am. Chem. Soc. 2009, 131, 1372.
406. Mo, J.; Xiao, J. Angew. Chem. Int. Ed. 2006, 45, 4152.
407. Yokota, T.; Tani, M.; Sakaguchi, S.; Ishii, Y. J. Am. Chem. Soc. 2003, 125, 1476.
408. Larhed, M.; Hallberg, A. J. Org. Chem. 1996, 61, 9582.
409. Battace, A.; Zair, T.; Doucet, H.; Santelli, M. Tetrahedron Lett. 2006, 47, 459; Andappan, M.M.S.; Nilsson, P.; von Schenck, H.; Larhed, M. J. Org. Chem. 2004, 69, 5212. For a review pertaining to enol ethers, see Daves, Jr., G.D. Adv. Met. Org. Chem. 1991, 2, 59.
410. Liu, Z.; Xu, D.; Tang, W.; Xu, L.; Mo, J.; Xiao, J. Tetrahedron Lett. 2008, 49, 2756.
411. See Daves, Jr., G.D.; Hallberg, A. Chem. Rev. 1989, 89, 1433.
412. Jeffery, T. Tetrahedron Lett. 1992, 33, 1989.
413. Chang, H.-M.; Cheng, C.-H. J. Org. Chem. 2000, 65, 1767.
414. Mariampillai, B.; Herse, C.; Lautens, M. Org. Lett. 2005, 7, 4745.
415. Jeffery, T. Tetrahedron Lett. 2000, 41, 8445.
416. Olofsson, K.; Larhed, M.; Hallberg, A. J. Org. Chem. 2000, 65, 7235; Wu, J.; Marcoux, J.-F.; Davies, I.W.; Reider, P.J. Tetrahedron Lett. 2001, 42, 159.
417. Kabalka, G.W.; Guchhait, S.K.; Naravane, A. Tetrahedron Lett. 2004, 45, 4685.
418. Kundu, N.G.; Pal, M.; Mahanty, J.S.; Dasgupta, S.K. J. Chem. Soc. Chem. Commun. 1992, 41. See also, Heck, R.F. Palladium Reagents in Organic Syntheses, Academic Press, NY, 1985, pp. 299–306.
419. Xia, M.; Chen, Z.C. Synth. Commun. 2000, 30, 1281.
420. Handy, S.T.; Wilson, T.; Muth, A. J. Org. Chem. 2007, 72, 8496.
421. Li, Z.; Fu, Y.; Zhang, S.-L.; Guo, Q.-X.; Liu, L. Chemistry: Asian J. 2010, 5, 1475.
422. Nilsson, P.; Larhed, M.; Hallberg, A. J. Am. Chem. Soc. 2001, 123, 8217 and earlier references therein.
423. Ludwig, M.; Strömberg, S.; Svensson, M.; Åkermark, B. Organometallics 1999, 18, 970.
424. Oestreich, M. Eur. J. Org. Chem. 2005, 783.
425. Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry, 2nd ed., University Science Books, Mill Valley, CA, 1987; Cornils, B.; Herrmann, A.W., Eds., Applied Homogeneous Catalysis with Organometallic Compounds, Wiley, NY, 1996; Vol. 2; Heck, R.F. Acc. Chem. Res. 1979, 12, 146.
426. Cabri, W.; Candiani, I. Acc. Chem. Res. 1995, 28, 2.
427. von Schenck, H.; Akermark, B.; Svensson, M. J. Am. Chem. Soc. 2003, 125, 3503.
428. Campo, M.A.; Zhang, H.; Yao, T.; Ibdah, A.; McCulla, R.D.; Huang, Q.; Zhao, J.; Jenks, W.S.; Larock, R.C. J. Am. Chem. Soc. 2007, 129, 6298.
429. Fall, Y.; Berthiol, F.; Doucet, H.; Santelli, M. Synthesis 2007, 1683.
430. Shezad, N.; Clifford, A.A.; Rayner, C.M. Tetrahedron Lett. 2001, 42, 323.
431. Su, Y.; Jiao, N. Org. Lett. 2009, 11, 2980.
432. Heck, R.F. J. Am. Chem. Soc. 1969, 91, 6707; See Masllorens, J.; Moreno-Mañas, M.; Pla-Quintana, A.; Plexats, R.; Roglans, A. Synthesis 2002, 1903. See Tan, Z.; Negishi, E. Angew. Chem. Int. Ed. 2006, 45, 762.
433. Larock, R.C.; Baker, B.E. Tetrahedron Lett. 1988, 29, 905. Also see, Larock, R.C.; Gong, W.H.; Baker, B.E. Tetrahedron Lett. 1989, 30, 2603.
434. Wu, W.-Q.; Peng, Q.; Dong, D.-X.; Hou, X.-L.; Wu, Y.-D. J. Am. Chem. Soc. 2008, 130, 9717.
435. Lapierre, A.J.B.; Geib, S.J.; Curran, D.P. J. Am. Chem. Soc. 2007, 129, 494. See Dounay, A.B.; Overman, L.E. Chem. Rev. 2002, 102, 2945.
436. Gilbertson, S.R.; Xie, D.; Fu, Z. J. Org. Chem. 2001, 66, 7240; Gilbertson, S.R.; Fu, Z. Org. Lett. 2001, 3, 161; Hennessy, A.J.; Connolly, D.J.; Malone, Y.M.; Buiry, P.J. Tetrahedron Lett. 2000, 41, 7757.
437. Servino, E.A.; Correia, C.R.D. Org. Lett. 2000, 2, 3039.
438. Heck, R.F.; Nolley, Jr., J.P. J. Org. Chem. 1972, 3720; Henriksen, S.T.; Norrby, P.-O.; Kaukoranta, P.; Andersson, P.G. J. Am. Chem. Soc. 2008, 130, 10414.
439. Heck, R.F. Comprehensive Organic Synthesis Vol. 4, Trost, B.M.; Fleming, I., Eds., Pergamon, Oxford, NY, 1991, p. 833; de Meijere, A.; Meyer, F.E. Angew. Chem. Int. Ed. 1994, 33, 2379; Cabri, W.; Candiani, I. Acc. Chem. Res. 1995, 28, 2; Crisp, G.T. Chem. Soc. Rev. 1998, 27, 427.
440. See Diederich, F.; Stang, P.J., Eds. Metal-Catalyzed Cross-Coupling Reactions, Wiley–VCH, Weinheim, 1998; Heck, R.F. Palladium Reagents in Organic Syntheses, Academic Press, NY, 1985.
441. Masselot, D.; Charmant, J.P.H.; Gallagher, T. J. Am. Chem. Soc. 2006, 128, 694.
442. See Amatore, C.; Godin, B.; Jutand, A.; Lemaître, F. Chemistry: European J. 2007, 13, 2002.
443. Rosner, T.; Pfaltz, A.; Blackmond, D. G. J. Am. Chem. Soc. 2001, 123, 4621.
444. For related work, see Kalyani, D.; Sanford, M.S. J. Am. Chem. Soc. 2008,130, 2150.
445. García-Cuadrado, D.; de Mendoza, P,; Braga, A.C.; Maseras, F.; Echavarren, A.M. J. Am. Chem. Soc. 2007, 129, 6880.
446. Tanaka, D.; Romeril, S.P.; Myers, A.G. J. Am. Chem. Soc. 2005, 127, 10323.
447. Consorti, C.S.; Flores, F.R.; Dupont, J. J. Am. Chem. Soc. 2005, 127, 12054.
448. Hii, K.K.; Claridge, T.D.W.; Brown, J.M.; Smith, A.; Deeth, R.J. Helv. Chim. Acta 2001, 84, 3043.
449. Kurahashi, T.; Shinokubo, H.; Osuka, A. Angew. Chem. Int. Ed. 2006, 45, 6336.
450. Zhou, P.; Li, Y.; Sun, P.; Zhou, J.; Bao, J. Chem. Commun. 2007, 1418; Amatore, M.; Gosmini, C.; Périchon, J. Eur. J. Org. Chem. 2005, 989.
451. Matsuura, Y.; Tamura, M.; Kochi, T.; Sato, M.; Chatani, N.; Kakiuchi, F. J. Am. Chem. Soc. 2007, 129, 9858.
452. Inamoto, K.; Kuroda, J.; Danjo, T.; Sakamoto, T. Synlett 2005, 1624; Denmark, S.E.; Butler, C.R. Chem. Commun. 2009, 20.
453. Wen, J.; Zhang, J.; Chen, S.-Y.; Li, J.; Yu, X.-Q. Angew. Chem. Int. Ed. 2008, 47, 8897; Liu, W.; Cao, H.; Lei, A. Angew. Chem. Int. Ed. 2010, 49, 2004.
454. Torres, N.M.; Lavis, J.M.; Maleczka, Jr., R.E. Tetrahedron Lett. 2009, 50, 4407.
455. Weissman, H.; Song, X.; Milstein, D. J. Am. Chem. Soc. 2001, 123, 337.
456. Huang, C.-W.; Shanmugasundaram, M.; Chang, H.-M.; Cheng, C.-H. Tetrahedron 2003, 59, 3635.
457. Takami, K.; Yorimitsu, H.; Shinokubo, H.; Matsubara, S.; Oshima, K. Org. Lett. 2001, 3, 1997.
458. Lehmann, U.; Awasthi, S.; Minehan, T. Org. Lett. 2003, 5, 2405.
459. Dai, C.; Fu, G.C. J. Am. Chem. Soc. 2001, 123, 2719.
460. Jalil, A.A.; Kurono, N.; Tokuda, M. Synlett 2001, 1944.
461. Ikeda, Y.; Makamura, T.; Yorimitsu, H.; Oshima, K. J. Am. Chem. Soc. 2002, 124, 6514.
462. Molander, G.A.; Brown, A.R. J. Org. Chem. 2006, 71, 9681. Also see Alacid, E.; Njera, C. J. Org. Chem. 2009, 74, 2321.
463. Molander, G.A.; Fumagalli, T. J. Org. Chem. 2006, 71, 5743.
464. Molander, G.A.; Jean-Gérard, L. J. Org. Chem. 2007, 72, 8422.
465. Affo, W.; Ohmiya, H.; Fujioka T.; Ikeda, Y.; Nakamura, T.; Yorimitsu, H.; Oshima, K.; Imamura, Y.; Mizuta, T.; Miyoshi, K. J. Am. Chem. Soc. 2006, 128, 8068.
466. Heck, R.F. J. Organomet. Chem. 1972, 37, 389; Heck, R.F.; Nolley, Jr., J.P. J. Org. Chem. 1972, 3720.
467. Kim, J.I.; Patel, B.A.; Heck, R.F. J. Org. Chem. 1981, 46, 1067; Heck, R.F. Pure Appl. Chem. 1981, 53, 2323. See also, Jeffery, T. J. Chem. Soc. Chem. Commun. 1991, 324; Larock, R.C.; Gong, W.H. J. Org. Chem. 1989, 54, 2047. Also see Varma, R.S.; Naicker, K.P.; Liesen, P.J. Tetrahedron Lett. 1999, 40, 2075.
468. See Fanta, P.E. Synthesis 1974, 9; Goshaev, M.; Otroshchenko, O.S.; Sadykov, A.S. Russ. Chem. Rev. 1972, 41, 1046.
469. See Bringmann, G.; Walter, R.; Weirich, R. Angew. Chem, Int. Ed. 1990, 29, 977. Also see, Meyers, A.I.; Price, A. J. Org. Chem. 1998, 63, 412.
470. Yang, M.; Liu, F. J. Org. Chem. 2007, 72, 8969.
471. Wu, Q.; Wang, L. Synthesis 2008, 2007.
472. See for example, Karimipour, M.; Semones, A.M.; Asleson, G.L.; Heldrich, F.J. Synlett, 1990, 525.
473. D'Angelo, N.D.; Peterson, J.J.; Booker, S.K.; Fellows, I.; Dominguez, C.; Hungate, R.; Reider, P.J.; Kim, T.-S. Tetrahedron Lett. 2006, 47, 5045.
474. Forrest, J. J. Chem. Soc. 1960, 592.
475. Lewin, A.H.; Cohen, T. Tetrahedron Lett. 1965, 4531.
476. See Mack, A.G.; Suschitzky, H.; Wakefield, B.J. J. Chem. Soc. Perkin Trans. 1 1980, 1682.
477. Salfeld, J.C.; Baume, E. Tetrahedron Lett. 1966, 3365; Lothrop, W.C. J. Am. Chem. Soc. 1941, 63, 1187.
478. Do, H.-Q.; Daugulis, O. J. Am. Chem. Soc. 2007, 129, 12404.
479. See Lourak, M.; Vanderesse, R.; Fort, Y.; Caubere, P. J. Org. Chem. 1989, 54, 4840, 4844; Iyoda, M.; Otsuka, H.; Sato, K.; Nisato, N.; Oda, M. Bull. Chem. Soc. Jpn. 1990, 63, 80. For a review of the mechanism, see Amatore, C.; Jutand, A. Acta Chem. Scand. 1990, 44, 755.
480. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 82–84.
481. Matsumoto, H.; Inaba, S.; Rieke, R.D. J. Org. Chem. 1983, 48, 840; Chao, C.S.; Cheng, C.H.; Chang, C.T. J. Org. Chem. 1983, 48, 4904.
482. Takagi, K.; Hayama, N.; Sasaki, K. Bull. Chem. Soc. Jpn. 1984, 57, 1887.
483. Bamfield, P.; Quan, P.M. Synthesis 1978, 537.
484. Meyer, G.; Rollin, Y.; Perichon, J. J. Organomet. Chem. 1987, 333, 263.
485. Nelson, T.D.; Meyers, A.I. J. Org. Chem. 1994, 59, 2655; Nelson, T.D.; Meyers, A.I. Tetrahedron Lett. 1994, 35, 3259.
486. Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457; Alonso, F.; Beletskaya, I.P.; Yus, M. Tetrahedron 2008, 64, 3047; Doucet, H. Eur. J. Org. Chem. 2008, 2013; Molander, G.A.; Yun, C.-S. Tetrahedron 2002, 58, 1465. See Kotha, S.; Lahiri, K.; Kasshinath, D. Tetrahedron 2002, 58, 9633.
487. Miyaura, N.; Yanagi, T.; Suzuki, A. Synth. Commun. 1981, 11, 513; Badone, D.; Baroni, M.; Cardomone, R.; Ielmini, A.; Guzzi, U. J. Org. Chem. 1997, 62, 7170. See Torrell, E.; Brookes, P. Synthesis 2003, 469.
488. Fürstner, A.; Seidel, G. Synlett, 1998, 161.
489. For a review, see Bellina, F.; Carpita, A.; Rossi, R. Synthesis 2004, 2419.
490. Watanabe, T.; Miyaura, N.; Suzuki, A. Synlett 1992, 207.
491. Lei, A.; Zhang, X. Tetrahedron Lett. 2002, 43, 2525; Parrish, J.P.; Jung, Y.C.; Floyd, R.J.; Jung, K.W. Tetrahedron Lett. 2002, 43, 7899.
492. Bussolari, J.C.; Rehborn, D.C. Org. Lett. 1999, 1, 965.
493. Fairlamb, I.J.S.; Kapdi, A.R.; Lee, A.F. Org. Lett. 2004, 6, 4435; Arentsen, K.; Caddick, S.; Cloke, G.N.; Herring, A.P.; Hitchcock, P.B. Tetrahedron Lett. 2004, 45, 3511; Artok, L.; Bulat, H. Tetrahedron Lett. 2004, 45, 3881, Arcadi, A.; Cerichelli, G.; Chiarini, M.; Correa, M.; Zorzan, D. Eur. J. Org. Chem. 2003, 4080.
494. See Schweizer, S.; Becht, J.-M.; Le Drian, C. Org. Lett. 2007, 9, 3777; Burns, M.J.; Fairlamb, I.J.S.; Kapdi, A.R.; Sehnal, P.; Taylor, R.J.K. Org. Lett. 2007, 9, 5397; Guo, M; Jian, F.; He, R. Tetrahedron Lett. 2006, 47, 2033; Li, J.-H.; Zhu, Q.-M.; Xie, Y.-X. Tetrahedron 2006, 62, 10888; Kantam, M.L.; Subhas, M.S.; Roy, S.; Roy, M. Synlett 2006, 633; Alonso, D.A.; Cívicos, J.F.; Nájera, C. Synlett 2009, 3011; You, E.; Li, P.; Wang, L. Synthesis 2006, 1465; Felpin, F.-X.; Ayad, T.; Mitra, S. Eur. J. Org. Chem. 2006, 2679; Lee, D.-H.; Jung, J.-Y.; Lee, I.-M.; Jin, M.-J. Eur. J. Org. Chem. 2008, 356; Subhas, M.S.; Racharlawar, S.S.; Sridhar, B.; Kennady, P.K.; Likhar, P.R.; Kantam, M.L.; Bhargava, S.K. Org. Biomol. Chem. 2010, 8, 3001; Bhayana, B.; Fors, B.P.; Buchwald, S.L. Org. Lett. 2009, 11, 3954; Nishikata, T.; Abela, A.R.; Huang, S.; Lipshutz, B.H. J. Am. Chem. Soc. 2010, 132, 4978; Guo, M.; Zhang, Q. Tetrahedron Lett. 2009, 50, 1965. For a discussion of catalysts in this reaction, see Barder, T.E.; Walker, S.D.; Martinelli, J.R.; Buchwald, S.L. J. Am. Chem. Soc. 2005, 127, 4685. For a precatalyst that is useful with unstable 2-heteroaryl boronic acids, see Kinzel, T.; Zhang, Y.; Buchwald, S.L. J. Am. Chem. Soc. 2010, 132, 14073.
495. So, C.M.; Yeung, C.C.; Lau, C.P.; Kwong, F.Y.J. Org. Chem. 2008, 73, 7803; Lipshutz, B.H.; Petersen, T.B.; Abela, A.R. Org. Lett. 2008, 10, 1333; Dai, W.-M.; Zhang, Y. Tetrahedron Lett. 2005, 46, 1377; Villemin, D.; Jullien, A.; Bar, N. Tetrahedron Lett. 2007, 48, 4191; Lai, Y.-C.; Chen, H.-Y.; Hung, W.-C.; Lin, C.-C.; Hong, F.-E. Tetrahedron 2005, 61, 9484; Kuriyama, M.; Shimazawa, R.; Shirai, R. Tetrahedron 2007, 63, 9393; Mai, W.; Gao, L. Synlett 2006, 2553; Ghosh, R.; Adarsh, N.N.; Sarkar, A. J. Org. Chem. 2010, 75, 5320.
496. Mino, T.; Shirae, Y.; Sakamoto, M.; Fujita, T. J. Org. Chem. 2005, 70, 2191; Liu, L.; Zhang, Y.; Wang, Y. J. Org. Chem. 2005, 70, 6122; Yamamoto, Y.; Suzuki, R.; Hattori, K.; Nishiyama, H. Synlett 2006, 1027; Mino, T.; Kajiwara, K.; Shirae, Y.; Sakamoto, M.; Fujita, T. Synlett 2008, 2711; Zhang, G. Synthesis 2005, 537; Cui, X.; Qin, T.; Wang, J.R.; Liu, L.; Guo, Q.-X. Synthesis 2007, 393.
497. Liu, L.; Zhang, Y.; Xin, B. J. Org. Chem. 2006, 71, 3994; Korolev, D.N.; Bumagin, N.A. Tetrahedron Lett. 2006, 47, 4225; Li, J.-H.; Li, J.-L.; Xie, Y.-X. Synthesis 2007, 984; Saha, D.; Chattopadhyay, K.; Ranu, B.C. Tetrahedron Lett. 2009, 50, 1003; da Conceição Silva, A.; Senra, J.D.; Aguiar, L.C.S.; Simas, A.B.C.; de Souza, A.L.F.; Malta, L.F.B.; Antunes, O.A.C. Tetrahedron Lett. 2010, 51, 3883; Zhou, W.-J.; Wang, K.-H.; Wang, J.-X.; Gao, Z.-R. Tetrahedron 2010, 66, 7633.
498. Li, J.-H.; Liu, W.-J.; Xie, Y.-X. J. Org. Chem. 2005, 70, 5409; Felpin, F.-X. J. Org. Chem. 2005, 70, 8575; Masllorens, J.; González, I.; Roglans, A. Eur. J. Org. Chem. 2007, 158.
499. Zapf, A.; Ehrentraut, A.; Beller, M. Angew. Chem. Int. Ed. 2000, 39, 4153.
500. See Calò, V.; Nacci, A.; Monopoli, A.; Montingelli, F. J. Org. Chem. 2005, 70, 6040; Yang, C.-H.; Tai, C.C.; Huang, Y.-T.; Sun, I.-W. Tetrahedron 2005, 61, 4857; Hagiwara, H.; Ko, K.H.; Hoshi, T.; Suzuki, T. Chem. Commun. 2007, 2838; Xin, B.; Zhang, Y.; Liu, L.; Wang, Y. Synlett 2005, 3083.
501. Early, T.R.; Gordon, R.S.; Carroll, M.A.; Holmes, A.B.; Shute, R.E.; McConvey, I.F. Chem. Commun. 2001, 1966.
502. Li, J.-H.; Deng, C.-L.; Xie, Y.-X. Synth. Commun. 2007, 37, 2433.
503. For a review, see Franzén, R.; Xu, Y. Can. J. Chem. 2005, 83, 266. See Jiang, N.; Ragauskas, A.J. Tetrahedron Lett. 2006, 47, 197; Arvela, R.K.; Leadbeater, N.E.; Mack, T.L.; Kormos, C.M. Tetrahedron Lett. 2006, 47, 217; Hattori, H.; Fujita, K.; Muraki,T.; Sakaba, A. Tetrahedron Lett. 2007, 48, 6817; Del Zotto, A.; Amoroso, F.; Baratta, W.; Rigo, P. Eur. J. Org. Chem. 2009, 110; Xu, K.; Hao, X.-Q.; Gong, J.-F.; Song, M.-P.; Wu, Y.-J. Austr. J. Chem. 2010, 63, 315.
504. Kabalka, G.W.; Pagni, R.M.; Hair, C.M. Org. Lett. 1999, 1, 1423. See Basu, B.; Das, P.; Bhuiyan, Md.M.H.; Jha, S. Tetrahedron Lett. 2003, 44, 3817.
505. Villemin, D.; Caillot, F. Tetrahdron Lett. 2001, 42, 639.
506. Chanthavong, F.; Leadbeater, N.E. Tetrahedron Lett. 2006, 47, 1909; Arvela, R.K.; Leadbeater, N.E.; Sangi, M.S.; Williams, V.A.; Granados, P.; Singer, R.D. J. Org. Chem. 2005, 70, 161; Miao, G.; Ye, P.; Yu, L.; Baldino, C.M. J. Org. Chem. 2005, 70, 2332; Arvela, R.K.; Leadbeater, N.E. Org. Lett. 2005, 7, 2101; Baxendale, I.R.; Griffiths-Jones, C.M.; Ley, S.V.; Tranmer, G.K. Chemistry: European J. 2006, 12, 4407; Yan, J.; Zhu, M.; Zhou, Z. Eur. J. Org. Chem. 2006, 2060.
507. Blettner, C.G.; König, W.A.; Stenzel, W.; Schotten, T. J. Org. Chem. 1999, 64, 3885. For other reactions on solid support, see Franzén, R. Can. J. Chem. 2000, 78, 957.
508. Hebel, A.; Haag, R. J. Org. Chem. 2002, 67, 9452.
509. Kantam, M.L.; Roy, M.; Roy, S.; Sreedhar, B.; Madhavendra, S.S.; Choudary, B.M.; De, R.L. Tetrahedron 2007, 63, 8002; Schweizer, S.; Becht, J.-M.; Le Drian, C. Tetrahedron 2010, 66, 765; Islam, M.; Mondal, P.; Tuhina, K.; Hossain, D.; Roy, A.S. Chem. Lett. 2010, 39, 1200.
510. Guo, Y.; Young, D.J.; Hor, T.S.A. Tetrahedron Lett. 2008, 49, 5620.
511. Poondra, R.R.; Fischer, P.M.; Turner, N.J. J. Org. Chem. 2004,69, 6920.
512. Wu, J.; Zhu, Q.; Wang, L.; Fathi, R.; Yang, Z. J. Org. Chem. 2003, 68, 670.
513. Quasdorf, K.W.; Riener, M.; Petrova, K.V.; Garg, N.K. J. Am. Chem. Soc. 2009, 131, 17748. See Antoft-Finch, A.; Blackburn, T.; Snieckus, V. J. Am. Chem. Soc. 2009, 131, 17750.
514. Zhao, Y.-L.; Li, Y.; Li, Y.; Gao, L.-X.; Han, F.-S. Chemistry: Eur. J. 2010, 16, 4991.
515. Zhang, W.; Chen, C.H.-T.; Lu, Y.; Nagashima, T. Org. Lett. 2004, 6, 1473; Riggleman, S.; DeShong, P. J. Org. Chem. 2003, 68, 8106.
516. So, C.M.; Lau, C.P.; Chan, A.S.C.; Kwong, F.Y. J. Org. Chem. 2008, 73, 7731; So, C.M.; Lau, C.P.; Kwong, F.Y. Angew. Chem. Int. Ed. 2008, 47, 8059.
517. Lane, B.S.; Sames, D. Org. Lett. 2004, 6, 2897; Denmark, S.E.; Baird, J.D. Org. Lett. 2004, 6, 3649; Prieto, M.; Zurita, E.; Rosa, E.; Muñoz, L.; Lloyd-Williams, P.; Giralt, E. J. Org. Chem. 2004, 69, 6812; Dvorak, C.A.; Rudolph, D.A.; Ma, S.; Carruthers, N.I. J. Org. Chem. 2005, 70, 4186; Kudo, N.; Perseghini, M.; Fu, G.C. Angew. Chem. Int. Ed. 2006, 45, 1282; Yang, S.-D.; Sun, C.-L.; Fang, Z.; Li, B.-J.; Li, Y.-Z.; Shi, Y.-J. Angew. Chem. Int. Ed. 2008, 47, 1473.
518. Morris, G.A.; Nguyen, S.T. Tetrahedron Lett. 2000, 42, 2093.
519. Villemin, D.; Gómez-Escalonilla, M.J.; Saint-Clair, J.-F. Tetrahedron Lett. 2001, 42, 635.
520. Baillie, C.; Chen, W.; Xiao, J. Tetrahedron Lett. 2001, 42, 9085.
521. Phan, N.T.S.; Brown, D.H.; Stgring, P. Tetrahedron Lett. 2004, 45, 7915.
522. Baillie, C.; Zhang, L.; Xiao, J. J. Org. Chem. 2004, 69, 7779.
523. Mutele, I.; Suna, E. Tetrahedron Lett. 2004, 45, 3909.
524. Ma, H.-r.; Wang, X.-L.; Deng, M.-z. Synth. Commun. 1999, 29, 2477.
525. Tao, B.; Boykin, D.W. J. Org. Chem. 2004, 69, 4330; Li, J.-H.; Liu, W.-J. Org. Lett. 2004, 6, 2809.
526. Colacot, T.J.; Shea, H.A. Org. Lett. 2004, 6, 3731; DeVasher, R.B.; Moore, L.R.; Shaughnessy, K.H. J. Org. Chem. 2004, 69, 7919.
527. Shen, W. Synlett 2000, 737.
528. Peyroux, E.; Berthiol, F.; Doucet, H.; Santelli, M. Eur. J. Org. Chem. 2004, 1075.
529. Andrus, M.B.; Song, C.; Zhang, J. Org. Lett. 2002, 4, 2079.
530. Catellani, M.; Motti, E.; Minari, M. Chem. Commun. 2000, 157. See Yin, J.; Buchwald, S.L. J. Am. Chem. Soc. 2000, 122, 112051.
531. Kuwano, R.; Yokogi, M. Org. Lett. 2005, 7, 945.
532. Kirchhoff, J.H.; Netherton, M.R.; Hills, I.D.; Fu, G.C. J. Am. Chem. Soc. 2002, 124, 13662.
533. Zhou, J.; Fu, G.C. J. Am. Chem. Soc. 2004, 126, 1340.
534. Bandgar, B.P.; Bettigeri, S.V.; Phopase, J. Tetrahedron Lett. 2004, 45, 6959.
535. Tsukamoto, H.; Sato, M.; Kondo, Y. Chem. Commun. 2004, 1200; Kayaki, Y.; Koda, T.; Ikariya, T. Eur. J. Org. Chem. 2004, 4989.
536. McLaughlin, M. Org. Lett. 2005, 7, 4875.
537. Habashneh, A.Y.; Dakhil, O.O.; Zein, A.; Georghiou, P.E. Synth. Commun. 2009, 39, 4221.
538. Lu, Z.; Fu, G.C. Angew. Chem. Int. Ed. 2010, 49, 6676.
539. Lando, V.R.; Moneitro, A.L. Org. Lett. 2003, 5, 2891.
540. Navarro, O.; Kelly, III, R.A.; Nolan, S.P. J. Am. Chem. Soc. 2003, 125, 16194; Baudoin, O. Eur. J. Org. Chem. 2005, 4223.
541. See Esponet, P.; Echavarren, A.M. Angew. Chem. Int. Ed. 2004, 43, 4704.
542. Moreno-Mañas, M.; Pérez, M.; Pleixats, R. J. Org. Chem. 1996, 61, 2346.
543. See Metal–Catalyzed Cross–Coupling Reactions, Diederich, F.; Stang, P.J. Wiley–VCH, Weinheim, 1998, p. 212.
544. Aramendia, M.A.; Lafont, F.; Moreno-Mañas, M.; Pleixats, R.; Roglans, A. J. Org. Chem. 1999, 64, 3592.
545. Adamo, C.; Amatore, C.; Ciofini, I.; Jutand, A.; Hakim Lakmini, H. J. Am. Chem. Soc. 2006, 128, 6829.
546. Jung, Y.C.; Mishra, R.K.; Yoon, C.H.; Jung, K.W. Org. Lett. 2003, 5, 2231.
547. Farrington, E.J.; Brown, J.M.; Barnard, C.F.J.; Rowsell, E. Angew. Chem. Int. Ed. 2002, 41, 169.
548. Percec, V.; Golding, G.M.; Smidrkal, J.; Weichold, O. J. Org. Chem. 2004, 69, 3447. See Quasdorf, K.W.; Tian, X.; Garg, N.K. J. Am. Chem. Soc. 2008, 130, 14422; Chen, C.; Yang, L.-M. Tetrahedron Lett. 2007, 48, 2427; González-Bobes, F.; Fu, G.C. J. Am. Chem. Soc. 2006, 128, 5360.
549. Lautens, M.; Roy, A.; Fukuoka, K.; Fagnou, K.; Martín-Matute, B. J. Am. Chem. Soc. 2001, 123, 5358.
550. Blakey, S.B.; MacMillan, D.W.C. J. Am. Chem. Soc. 2003, 125, 6046.
551. Chung, K.-G.; Miyake, Y.; Uemura, S. J. Chem. Soc. Perkin Trans. 1 2000, 15.
552. Demir, A.S.; Reis, Ö; Emrullahoglu, M. J. Org. Chem. 2003, 68, 578.
553. Bumagin, N.A.; Tsarev, D.A. Tetrahedron Lett. 1998, 39, 8155.
554. Hatakeyama, T.; Hashimoto, T.; Kondo, Y.; Fujiwara, Y.; Seike, H.; Takaya, H.; Tamada, Y.; Ono, T.; Nakamura, M. J. Am. Chem. Soc. 2010, 132, 10674. However, see Bedford, R.B.; Nakamura, M.; Gower, N.J.; Haddow, M.F.; Hall, M.A.; Huwe, M.; Hashimoto, T.; Okopie, R.A. Tetrahedron Lett. 2009, 50, 6110.
555. Kang, S.-K.; Hong, Y.-T.; Kim, D.-H.; Lee, S.-H. J. Chem. Res. (S) 2001, 283.
556. Kang, S.-K.; Ryu, H.-C.; Lee, S.-W. J. Chem. Soc. Perkin Trans. 1 1999, 2661.
557. Bumagin, N.A.; Korolev, D.N. Tetrahedron Lett. 1999, 40, 3057.
558. Haddach, M.; McCarthy, J.R. Tetrahedron Lett. 1999, 40, 3109.
559. Chen, H.; Deng, M.-Z. Org. Lett. 2000, 2, 1649.
560. Leadbeater, N.E.; Resouly, S.M. Tetrahedron 1999, 55, 11889.
561. Lipshutz, B.H.; Sclafani, J.A.; Blomgren, P.A. Tetrahedron 2000, 56, 2139.
562. Gooßen, L.J.; Ghosh, K. Angew. Chem. Int. Ed. 2001, 40, 3458.
563. Kakiuchi, F.; Usai, M.; Ueno, S.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2004, 126, 2706.
564. Lu, G.; Franzén, R.; Zhang, Q.; Xu, Y. Tetrahedron Lett. 2005, 46, 4255.
565. Chameil, H.; Signorella, S.; Le Drian, C. Tetrahedron 2000, 56, 9655. Also see Kakiuchi, F.; Matsuura, Y.; Kan, S.; Chatani, N. J. Am. Chem. Soc. 2005, 127, 5936.
566. Yoo, K.S.; Yoon, C.H.; Jung, K.W. J. Am. Chem. Soc. 2006, 128, 16384.
567. Cioffi, C.L.; Spencer, W.T.; Richards, J.J.; Herr, R.J. J. Org.Chem. 2004, 69, 2210.
568. Nishihara, Y.; Miyasaka, M.; Okamoto, M.; Takahashi, H.; Inoue, E.; Tanemura, K.; Takagi, K. J. Am. Chem. Soc. 2007, 129, 12634.
569. Iglesias, B.; Alvarez, R.; de Lera, A.R. Tetrahedron 2001, 57, 3125.
570. Molander, G.A.; Canturk, B.; Kennedy, L.E. J. Org. Chem. 2009, 74, 973; Molander, G.A.; Jean-Grard, L. J. Org. Chem. 2009, 74, 1297; Barder, T.E.; Buchwald, S.L. Og. Lett. 2004, 6, 2649. See Ito, T.; Iwai, T.; Mizuno, T.; Ishino, Y. Synlett 2003, 1435.
571. Darses, S.; Genet, J.-P. Chem. Rev. 2008, 108, 288.
572. Molander, G.A.; Yun, C.-S.; Ribagorda, M.; Biolatto, B. J. Org. Chem. 2003, 68, 5534.
573. Dreher, S.D.; Dormer, P.G.; Sandrock, D.L.; Molander, G.A. J. Am. Chem. Soc. 2008, 130, 9257; Dreher, S.D.; Lim, S.-E.; Sandrock, D.L.; Molander, G.A. J. Org. Chem. 2009, 74, 3626.
574. Xia, M.; Chen, Z.-C. Synth. Commun. 1999, 29, 2457.
575. Molander, G. A.; Bernardi, C.R. J. Org. Chem. 2002, 67, 8424.
576. Kabalka, G.W.; Al-Masum, M. Tetrahedron Lett. 2005, 46, 632; Kabalka, G.W.; Zhou, L.-L.; Naravane, A. Tetrahedron Lett. 2006, 47, 6887; Harker, R.L.; Crouch, R.D. Synthesis 2007, 25; Kabalka, G.W.; Naravane, A.; Zhao, L.L. Tetrahedron Lett. 2007, 48, 7091; Kabalka, G.W.; Al-Masum, M.; Mereddy, A.R.; Dadush, E. Tetrahedron Lett. 2006, 47, 1133.
577. Cella, R.; Cunha, R.L.O.R.; Reis, A.E.S.; Pimenta, D.C.; Klitzke, C.F.; Stefani, H.A. J. Org. Chem. 2006, 71, 244.
578. Ge, H.; Niphakis, M.J.; Georg, G.I. J. Am. Chem. Soc. 2008, 130, 3708.
579. Wu, J.; Zhang, L.; Gao, K. Eur. J. Org. Chem. 2006, 5260.
580. Castro, C.E.; Stephens, R.D. J. Org. Chem. 1963, 28, 2163; Stephens, R.D.; Castro, C.E. J. Org. Chem. 1963, 28, 3313. See Sladkov, A.M.; Gol'ding, I.R. Russ. Chem. Rev. 1979, 48, 868; Bumagin, N.A.; Kalinovskii, I.O.; Ponomarov, A.B.; Beletskaya, I.P. Doklad. Chem. 1982, 265, 262.
581. Penney, J.M.; Miller, J.A. Tetrahedron Lett. 2004, 45, 4989.
582. Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 4467; Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874; Rossi, R.; Carpita, A.; Bellina, F. Org. Prep. Proceed. Int. 1995, 27, 127; Sonogashira, K. in Diederich, F.; Stang, P.J. Metal–Catalyzed Cross–Coupling Reactions, Wiley–VCH, NY, 1998, Chapter 5.
583. See Novák, Z.; Szabó, A.; Répási, J.; Kotschy, A. J. Org. Chem. 2003, 68, 3327.
584. Böhm, V.P.W.; Herrmann, W.A. Eur. J. Org. Chem. 2000, 3679. See Mori, A.; Shimada, T.; Kondo, T.; Sekiguchi, A. Synlett 2001, 649.
585. Liang, B.; Dai, M.; Chen, J.; Yang, Z. J. Org. Chem. 2005, 70, 391.
586. See Mio, M.J.; Kopel, L.C.; Braun, J.B.; Gadzikwa, T.L.; Hull, K.; Brisbois, R.G.; Markworth, C.J.; Grieco, P.A. Org. Lett. 2002, 4, 3199.
587. Wolf, C.; Lerebours, R. Org. Biomol. Chem. 2004, 2, 2161; Chen, Y.; Markina, N.A.; Larock, R.C. Tetrahedron 2009, 65, 8908; R'kyek, O.; Halland, N.; Lindenschmidt, A.; Alonso, J.; Lindemann, P.; Urmann, M.; Nazaré, M. Chemistry: Eur. J. 2010, 16, 9986.
588. Yi, C.; Hua, R. J. Org. Chem. 2006, 71, 2535; Tang, B.X.; Wang, F.; Li, J.-H.; Xie, Y.-X.; Zhang, M.-B. J. Org. Chem. 2007, 72, 6294; Li, P.; Wang, L.; Li, H. Tetrahedron 2005, 61, 8633; Plenio, H. Angew. Chem. Int. Ed.2008, 47, 6954.
589. Mori, S.; Yanase, T.; Aoyagi, S.; Monguchi, Y.; Maegawa, T.; Sajiki, H. Chemistry: European J. 2008, 14, 6994.
590. See Sakai, N.; Annaka, K.; Konakahara, T. Org. Lett. 2004, 6, 1527; Djakovitch, L.; Rollet, P. Tetrahedron Lett. 2004, 45, 1367; Hierso, J.-C.; Fihri, A.; Amardeil, R.; Meunier, P.; Doucet, H.; Santelli, M.; Ivanov, V.V. Org. Lett. 2004, 6, 3473.
591. Yi, C.; Hua, R. J. Org. Chem. 2006, 71, 2535; Cwik, A.; Hell, Z.; Figueras, F. Tetrahedron Lett. 2006, 47, 3023; Komáromi, A.; Tolnai, G.L.; Novák, Z. Tetrahedron Lett. 2008, 49, 7294; Teratani, T.; Ohtaka, A.; Kawashima, T.; Shimomura, O.; Nomura, R. Synlett 2010, 2271; Li, J.-H.; Zhang, X.-D.; Xie, Y.-X. Synthesis 2005, 804; Li, J.-H.; Zhang, X.-D.; Xie, Y.-X. Eur. J. Org. Chem. 2005, 4256; Ren, T.; Zhang, Y.; Zhu, W.; Zhou, J. Synth. Commun.2007, 37, 3279; Bakherad, M.; Keivanloo, A.; Bahramian, B.; Hashemi, M. Tetrahedron Lett. 2009, 50, 1557. For a variation using Au-nanoparticles, see de Souza, R.O.M.A.; Bittar, M.S.; Mendes, L.V.P.; da Silva, C.M.F.; da Silva, V.T.; Antunes, O.A.C. Synlett 2008, 1777.
592. Gholap, A.R.; Venkatesan, K.; Pasricha, R.; Daniel, T.; Lahoti, R.J.; Srinivasan, K.V. J. Org. Chem. 2005, 70, 4869; Pan, C.; Luo, F.; Wang, W.; Ye, Z.; Cheng, J. Tetrahedron Lett. 2009, 50, 5044.
593. Maji, M.S.; Murarka, S.; Studer, A. Org. Lett. 2010, 12, 3878.
594. Lipshutz, B.L.; Chung, D.W.; Rich, B. Org. Lett. 2008, 10, 3793; Guan, J.T.; Weng, T.Q.; Yu, G.-A.; Liu, S.H. Tetrahedron Lett. 2007, 48, 7129.
595. Özdemir, I.; Gürbüz, N.; Gök, Y.; Çetinkaya, E.; Çetinkaya, B. Synlett 2005, 2394; Chen, G.; Zhu, X.; Cai, J.; Wan, Y. Synth. Commun. 2007, 37, 1355.
596. Leadbeater, N.E.; Marco, M.; Tominack, B.J. Org. Lett. 2003, 5, 3919.
597. See Fukuyama, T.; Shinmen, M.; Nishitani, S.; Sato, M.; Ryu, I. Org. Lett. 2002, 4, 1691; Park, S.B.; Alper, H. Chem. Commun. 2004, 1306; de Lima, P.G.; Antunes, O.A.C. Tetrahedron Lett. 2008, 49, 2506.
598. Appukkuttan, P.; Dehaen, W.; van der Eyken, E. Eur. J. Org. Chem. 2003, 4713. See also, Kabalka, G.W.; Wang, L.; Namboodiri, V.; Pagni, R.M. Tetrahedron Lett. 2000, 41, 515.
599. Liao, Y.; Fathi, R.; Reitman, M.; Zhang, Y.; Yang, Z. Tetrahedron Lett. 2001, 42, 1815; Gonthier, E.; Breinbauer, R. Synlett 2003, 1049.
600. Wang, M.; Li, P.; Wang, L. Synth. Commun. 2004, 34, 2803.
601. Erdélyi, M.; Gogoll, A. J. Org. Chem. 2003, 68, 6431.
602. Bakherad, M.; Amin, A.H.; Keivanloo, A.; Bahramian, B.; Raeissi, M. Tetrahedron Lett. 2010, 51, 5653.
603. Lipshutz, B.H.; Blomgren, P.A. Org. Lett. 2001, 3, 1869.
604. See Chow, H.-F.; Wan, C.-W.; Low, K.-H.; Yeung, Y.-Y. J. Org. Chem. 2001, 66, 1910.
605. Altenhoff, G.; Würtz, S.; Glorius, F. Tetrahedron Lett. 2006, 47, 2925.
606. Olivi, N.; Spruyt, P.; Peyrat, J.-F.; Alami, M.; Brion, J.-D. Tetrahedron Lett. 2004, 45, 2607.
607. Feuerstein, M.; Doucet, H.; Santelli, M. Tetrahedron Lett. 2004, 45, 8443.
608. Fabrizi, G.; Goggiamani, AA.; Sferrazza, A.; Cacchi, S. Angew. Chem. Int. Ed. 2010, 49, 4067.
609. Seregin, I.V.; Ryabova, V.; Gevorgyan, V. J. Am. Chem. Soc. 2007, 129, 7742.
610. Wakamatsu, H.; Takeshita, M. Synlett 2010, 2322.
611. Monnier, F.; Turtaut, F.; Duroure, L.; Taillefer, M. Org. Lett. 2008, 10, 3203. For a mcirowave induced variation of this reacation, see Colacino, E.; Daïch, L.; Martinez, J.; Lamaty, F. Synlett 2007, 1279.
612. Borah, H.N.; Prajapati, D.; Boruah, R.C. Synlett 2005, 2823.
613. Li, P.; Wang, L.; Wang, M.; You, F. Eur. J. Org. Chem. 2008, 5946. However, see Lauterbach, T.; Livendahl, M.; Roselln, A.; Espinet, P.; Echavarren, A.M. Org. Lett. 2010, 12, 3006, and Panda, B.; Sarkar, T.K. Tetrahedron Lett. 2010, 51, 301. See also, Beaumont, S.K.; Kyriakou, G.; Lambert, R.M. J. Am. Chem. Soc. 2010, 132, 12246.
614. Bakherad, M.; Keivanloo, A.; Mihanparast, S. Synth. Commun. 2010, 40, 179.
615. Sawant, D.N.; Tambade, P.J.; Wagh, Y.S.; Bhanage, B.M. Tetrahedron Lett. 2010, 51, 2758.
616. Li, P.; Wang, L. Synlett 2006, 2261.
617. Anastasia, L.; Negishi, E. Org. Lett. 2001, 3, 3111.
618. Castanet, A.-S.; Colobert, F.; Schlama, T. Org. Lett. 2000, 2, 3559.
619. Torres, G.H.; Choppin, S.; Colobert, F. Eur. J. Org. Chem. 2006, 1450.
620. Sun, Z.; Yu, S.; Ding, Z.; Ma, D. J. Am. Chem. Soc. 2007, 129, 9300.
621. See Jeganmohan, M.; Cheng, C.-H. Org. Lett. 2004, 6, 2821.
622. Kang, S.-K.; Ryu, H.-C.; Hong, Y-T. J. Chem. Soc. Perkin Trans. 1 2001, 736.
623. Savarin, C.; Srogl, J.; Liebeskind, L.S. Org. Lett. 2001, 3, 91.
624. Saeki, T.; Son, E.-C.; Tamao, K. Org. Lett. 2004, 6, 617.
625. Molander, G.A.; Katona, B.W.; Machrouhi, F. J. Org. Chem. 2002, 67, 8416.
626. Oh, C.H.; Jung, S.H. Tetrahedron Lett. 2000, 41, 8513.
627. Kang, S.-K.; Yoon, S.-K.; Kim, Y.-M. Org. Lett. 2001, 3, 2697.
628. Radhakrishnan, U.; Stang, P.J. Org. Lett. 2001, 3, 859.
629. Gallagher, W.P.; Maleczka, Jr., R.E. J. Org. Chem. 2003, 68, 6775.
630. There is evidence for both the SNAr mechanism (see Leffek, K.T.; Matinopoulos-Scordou, A.E. Can. J. Chem. 1977, 55, 2656, 2664) and the SRN1 mechanism (see Zhang, X.; Yang, D.; Liu, Y.; Chen, W.; Cheng, J. Res. Chem. Intermed. 1989, 11, 281).
631. Rossi, R.A.; Bunnett, J.F. J. Org. Chem. 1973, 38, 3020; Bunnett, J.F.; Gloor, B.F. J. Org. Chem. 1973, 38, 4156; 1974, 39, 382.
632. Rajan, S.; Muralimohan, K. Tetrahedron Lett. 1978, 483; Rossi, R.A.; Alonso, R.A. J. Org. Chem. 1980, 45, 1239; Beugelmans, R. Bull. Soc. Chim. Belg. 1984, 93, 547.
633. Matsubara, K.; Ueno, K.; Koga, Y.; Hara, K. J. Org. Chem. 2007, 72, 5069.
634. Caron, S.; Vasquez, E.; Wojcik, J.M. J. Am. Chem. Soc. 2000, 122, 712.
635. Bella, M.; Kobbelgaard, S.; Jørgensen, K.A. J. Am. Chem. Soc. 2005, 127, 3670.
636. Leake, W.W.; Levine, R. J. Am. Chem. Soc. 1959, 81, 1169, 1627.
637. See Caubere, P.; Guillaumet, G. Bull. Soc. Chim. Fr. 1972, 4643, 4649.
638. Bunnett, J.F.; Kato, T.; Flynn, R.; Skorcz, J.A. J. Org. Chem. 1963, 28, 1. See Biehl, E.R.; Khanapure, S.P. Acc. Chem. Res. 1989, 22, 275; Hoffmann, R.W. Dehydrobenzene and Cycloalkynes, Academic Press, NY, 1967, pp. 150–164. See also, Kessar, S.V. Acc. Chem. Res. 1978, 11, 283.
639. You, J.; Verkade, J.G. Angew. Chem. Int. Ed. 2003, 42, 5051.
640. Vogl, E.M.; Buchwald, S.L. J. Org. Chem. 2002, 67, 106.
641. Grasa, G.A.; Colacot, T.J. Org. Lett. 2007, 9, 5489.
642. Hama, T.; Hartwig, J.F. Org. Lett. 2008, 10, 1545, 1549; Biscoe, M.R.; Buchwald, S.L. Org. Lett. 2009, 11, 1773.
643. Aramendía, M.A.; Borau, V.; Jiménez, C.; Marinas, J.M.; Ruiz, J.R.; Urbano, F.J. Tetrahedron Lett. 2002, 43, 2847.
644. Kashin, A.N.; Mitin, A.V.; Beletskaya, I.P.; Wife, R. Tetrahedron Lett. 2002, 43, 2539.
645. Galli, C.; Bunnett, J.F. J. Org. Chem. 1984, 49, 3041.
646. See Osuka, A.; Kobayashi, T.; Suzuki, H. Synthesis 1983, 67; Hennessy, E.J.; Buchwald, S.L. Org. Lett. 2002, 4, 269.
647. Cristau, H.J.; Vogel, R.; Taillefer, M.; Gadras, A. Tetrahedron Lett. 2000, 41, 8457.
648. Liu, C.; He, C.; Shi, W.; Chen, M.; Lei, A. Org. Lett. 2007, 9, 5601.
649. Elliott, G.I.; Konopelski, J.P.; Olmstead, M.M. Org. Lett. 1999, 1, 1867 and refs. 3–7 therein.
650. See Elliott, G.I.; Konopelski, J.P. Tetrahedron 2001, 57, 5683.
651. Barton, D.H.R.; Blazejewski, J.; Charpiot, B.; Finet, J.; Motherwell, W.B.; Papoula, M.T.B.; Stanforth, S.P. J. Chem. Soc, Perkin Trans. 1 1985, 2667; O'Donnell, M.J.; Bennett, W.D.; Jacobsen, W.N.; Ma, Y. Tetrahedron Lett.1989, 30, 3913.
652. Citterio, A.; Santi, R.; Fiorani, T.; Strologo, S. J. Org. Chem. 1989, 54, 2703; Citterio, A.; Fancelli, D.; Finzi, C.; Pesce, L.; Santi, R. J. Org. Chem. 1989, 54, 2713.
653. Lundin, P.M.; Esquivias, J.; Fu, G.C. Angew. Chem. Int. Ed. 2009, 48, 154.
654. Scamehorn, R.G.; Hardacre, J.M.; Lukanich, J.M.; Sharpe, L.R. J. Org. Chem. 1984, 49, 4881.
655. Aoki, S.; Fujimura, T.; Nakamura, E.; Kuwjima, I. J. Am. Chem. Soc. 1988, 110, 3296.
656. See Semmelhack, M.F.; Bargar, T. J. Am. Chem. Soc. 1980, 102, 7765; Bard, R.R.; Bunnett, J.F. J. Org. Chem. 1980, 45, 1546.
657. Bard, R.R.; Bunnett, J.F.; Creary, X.; Tremelling, M.J. J. Am. Chem. Soc. 1980, 102, 2852; Tremelling, M.J.; Bunnett, J.F. J. Am. Chem. Soc. 1980, 102, 7375.
658. See Culkin, D.A.; Hartwig, J.F. Acc. Chem. Res. 2003, 36, 234.
659. Arnauld, T.; Barton, D.H.R.; Normant, J.-F.; Doris, E. J. Org. Chem. 1999, 64, 6915
660. Periasamy, M.; KishoreBabu N.; Jayakumar, K.N. Tetrahedron Lett. 2003, 44, 8939.
661. Chen, G.; Kwong, F.Y.; Chan, H.O.; Yu, W.-Y.; Chan, A.S.C. Chem. Commun. 2006, 1413.
662. Cossy, J.; de Filippis, A.; Pardo, D.G. Org. Lett. 2003, 5, 3037.
663. Hamada, T.; Chieffi, A.; Åhman, J.; Buchwald, S.L. J. Am. Chem. Soc. 2002, 124, 1261.
664. See Weil, T.A.; Cassar, L.; Foà, M. in Wender, I.; Pino, P. Organic Synthesis Via Metal Carbonyls, Vol. 2, Wiley, NY, 1977, pp. 517–543.
665. Liu, J.; Liang, B.; Shu, D.; Hu, Y.; Yang, Z.; Lei, A. Tetrahedron 2008, 64, 9581; Berger, P.; Bessmernykh, A.; Caille, J.-C.; Mignonac, S. Synthesis 2006, 3106.
666. Garrido, F.; Raeppel, S.; Mann, A.; Lautens, M. Tetrahedron Lett. 2001, 42, 265.
667. Cacchi, S.; Babrizi, G.; Goggiamani, A. Org. Lett. 2003, 5, 4269.
668. Antebi, S.; Arya, P.; Manzer, L.E.; Alper, H. J. Org. Chem. 2002, 67, 6623. For an interesting variation that generated a lactone ring, see Cho, C.S.; Baek, D.Y.; Shim, S.C. J. Heterocyclic Chem. 1999, 36, 289.
669. Pri-Bar, I.; Alper, H. J. Org. Chem. 1989, 54, 36.
670. See Bumagin, N.A.; Nikitin, K.V.; Beletskaya, I.P. Doklad. Chem. 1990, 312, 149.
671. Wan, Y.; Alterman, M.; Larhed, M.; Hallberg, A. J. Org. Chem. 2002, 67, 6232.
672. See Heck, R.F. Palladium Reagents in Organic Synthesis, Academic Press, NY, 1985, pp. 348–358.
673. Albaneze-Walker, J.; Bazaral, C.; Leavey, T.; Dormer, P.G.; Murry, J.A. Org. Lett. 2004, 6, 2097.
674. Kormos, C.M.; Leadbeater, N.E. Synlett 2006, 1663.
675. Cai, M.-Z.; Song, C.-S.; Huang, X. J. Chem. Soc. Perkin Trans. I, 1997, 2273.
676. Beller, M.; Mägerlein, W.; Indolese, A.F.; Fischer, C. Synthesis 2001, 1098.
677. Brunet, J.; Sidot, C.; Caubere, P. J. Org. Chem. 1983, 48, 1166. See also, Kudo, K.; Shibata, T.; Kashimura, T.; Mori, S.; Sugita, N. Chem. Lett. 1987, 577.
678. Ramesh, C.; Kubota, Y.; Miwa, M.; Sugi, Y. Synthesis 2002, 2171.
679. Ramesh, C.; Nakamura, R.; Kubota, Y.; Miwa, M.; Sugi, Y. Synthesis 2003, 501.
680. Watson, D.A.; Fan, X.; Buchwald, S.L. J. Org. Chem. 2008, 73, 7096.
681. Larock, R.C.; Fellows, C.A. J. Am. Chem. Soc. 1982, 104, 1900.
682. Nishiyama, Y.; Tokunaga, K.; Kawamatsu, H.; Sonoda, N. Tetrahedron Lett. 2002, 43, 1507.
683. Wu, X.; Larhed, M. Org. Lett. 2005, 7, 3327; Ju, J.; Jeong, M.; Moon, J.; Jung, H.M.; Lee, S. Org. Lett. 2007, 9, 4615; Csajági, C.; Borcsek, B.; Niesz, K.; Kovács, I.; Székelyhidi, Z.; Bajkó, Z.; Ürge, L.; Darvas, F. Org. Lett.2008, 10, 1589; Letavic, M.A.; Ly, K.S. Tetrahedron Lett. 2007, 48, 2339; Tambade, P.J.; Patil, Y.P.; Bhanushali, M.J.; Bhanage, B.M. Synthesis 2008, 2347.
684. Ryu, I.; Kusano, K.; Masumi, N.; Yamazaki, H.; Ogawa, A.; Sonoda, N. Tetrahedron Lett. 1990, 31, 6887.
685. Dey, K.; Eaborn, C.; Walton, D.R.M. Organomet. Chem. Synth. 1971, 1, 151.
686. Yamashita, M.; Miyoshi, K.; Nakazono, Y.; Suemitsu, R. Bull. Chem. Soc. Jpn. 1982, 55, 1663.
687. Nudelman, N.S.; Doctorovich, F. Tetrahedron 1994, 50, 4651.
688. Ryu, I.; Ryang, M.; Rhee, I.; Omura, H.; Murai, S.; Sonoda, N. Synth. Commun. 1984, 14, 1175 and cited references. See Hatanaka, Y.; Hiyama, T. Chem. Lett. 1989, 2049.
689. Lee, S.W.; Kee, K.; Seomoon, D.; Kim, S.; Kim, H.; Kim, H.; Shim, E.; Lee, M.; Lee, S.; Kim, S.; Lee, P.H. J. Org. Chem. 2004, 69, 4852.
690. Papoian, V.; Minehan, T. J. Org. Chem. 2008, 73, 7376.
691. Baird, Jr., W.C.; Hartgerink, R.L.; Surridge, J.H. J. Org. Chem. 1985, 50, 4601.
692. Kang, S.-K.; Ryu, H.-C.; Choi, S.-C. Synth. Commun. 2001, 31, 1035.
693. Pletnev, A.A.; Larock, R.C. J. Org. Chem. 2002, 67, 9428.
694. Ishiyama, T.; Kizaki, H.; Miyaura, N.; Suzuki, A. Tetrahedron Lett. 1993, 34, 7595.
695. Couve-Bonnaire, S.; Caprentier, J.-F.; Mortreux, A.; Castanet, Y. Tetrahedron Lett. 2001, 42, 3689.
696. Maerten, E.; Hassouna, F.; Couve-Bonnaire, S.; Mortreux, A.; Carpentiere, J.-F.; Castanet, Y. Synlett 2003, 1874.
697. Ahmed, M.S.M.; Mori, A. Org. Lett. 2003, 5, 3057. For a Pd-catalyzed reaction, see Liang, B.; Huang, M.; You, Z.; Xiong, Z.; Lu, K.; Fathi, R.; Chen, J.; Yang, Z. J. Org. Chem. 2005, 70, 6097.
698. Yamanoi, Y.; Nishihara, H. J. Org. Chem. 2008, 73, 6671; Gordillo, Á.; de Jesús, E.; López-Mardomingo, C. Org. Lett. 2006, 8, 3517; Murata, M.; Yamasaki, H.; Ueta, T.; Nagata, M.; Ishikura, M.; Watanabe, S.; Masuda, Y. Tetrahedron 2007, 63, 4087; Murata, M.; Yoshida, S.; Nirei, S.; Watanabe, S.; Masuda, Y. Synlett 2006, 118; Ye, Z.; Chen, F.; Luo, F.; Wang, W.; Lin, B.; Jia, X.; Cheng, J. Synlett 2009, 2198. See also, Denmark, S.E.; Kallemeyn, J.M. J. Am. Chem. Soc. 2006, 128, 15958.
699. Seganish, W.M.; DeShong, P. Org. Lett. 2004, 6, 4379.
700. Denmark, S.E.; Neuville, L.; Christy, M.E.L.; Tymonko, S.A. J. Org. Chem. 2006, 71, 8500.
701. Murata, M.; Ishikura, M.; Nagata, M.; Watanabe, S.; Masuda, Y. Org. Lett. 2002, 4, 1843.
702. Yamanoi, Y. J. Org. Chem. 2005, 70, 9607.
703. Omachi, H.; Itami, K. Chem. Lett. 2009, 38, 186.
704. Hamze, A.; Provot, O.; Alami, M.; Brion, J.-D. Org. Lett. 2006, 8, 931.
705. Nakao, Y.; Imanaka, H.; Sahoo, A.K.; Yada, A.; Hiyama, T. J. Am. Chem. Soc. 2005, 127, 6952.
706. Denmark, S.E.; Tymonko, S.A. J. Am. Chem. Soc. 2005, 127, 8004; Denmark, S.E.; Butler, C.R. J. Am. Chem. Soc. 2008, 130, 3690.
707. McNeill, E.; Barder, T.E.; Buchwald, S.L. Org. Lett. 2007, 9, 3785.
708. Denmark, S.E.; Wu, Z. Org. Lett. 1999, 1, 1495; Lee, H.M.; Nolan, S.P. Org. Lett. 2000, 2, 2053.
709. Denmark, S.E.; Ober, M.H. Org. Lett. 2003, 5, 1357.
710. Lee, J.-y.; Fu, G.C. J. Am. Chem. Soc. 2003, 125, 5616.
711. Hanamoto, T.; Kobayashi, T. J. Org. Chem. 2003, 68, 6354. See also, Taguchi, H.; Ghoroku, K.; Tadaki, M.; Tsubouchi, A.; Takeda, T. J. Org. Chem. 2002, 67, 8450.
712. Parrish, J.P.; Jung, Y.C.; Shin, S.I.; Jung, K.W. J. Org. Chem. 2002, 67, 7127.
713. Koike, T.; Du, X.; Sanada, T.; Danda, Y.; Mori, A. Angew. Chem. Int. Ed. 2003, 42, 89.
714. Denmark, S.E.; Wang, Z. Synthesis 2000, 999.
715. Hirabayashi, K.; Kawashima, J.; Nishihara, Y.; Mori, A.; Hiyama, T. Org. Lett. 1999, 1, 299.
716. Mowery, M.E.; DeShong, P. Org. Lett. 1999, 1, 2137.
717. Kabalka, G.W.; Wang, L.; Pagni, R.M. Tetrahedron 2001, 57, 8017; Denmark, S.E.; Tymonko, S.A. J. Org. Chem. 2003, 68, 9151.
718. Manoso, A.S.; Ahn, C.; Soheili, A.; Handy, C.J.; Correia, R.; Seganish, W.M.; DeShong, P. J. Org. Chem. 2004, 69, 8305.
719. Mori, A.; Suguro, M. Synlett 2001, 845; Murata, M.; Shimazaki, R.; Watanabe, S.; Masuda, Y. Synthesis 2001, 2231.
720. Yang, S.; Li, B.; Wan, X.; Shi, Z. J. Am. Chem. Soc. 2007, 129, 6066.
721. Sakurai, H.; Morimoto, C.; Hirao, T. Chem. Lett. 2001, 1084. See also, Powell, D.A.; Fu, G.C. J. Am. Chem. Soc. 2004, 126, 7788.
722. See Chupakhin, O.N.; Postovskii, I.Ya. Russ. Chem. Rev. 1976, 45, 454. See Chupakhin, O.N.; Charushin, V.N.; van der Plas, H.C. Tetrahedron 1988, 44, 1.
723. See Becht, J.-M.; Gissot, A.; Wagner, A.; Misokowski, C. Tetrahedron Lett. 2004, 45, 9331.
724. Slocum, D.W.; Jennings, C.A. J. Org. Chem. 1976, 41, 3653. However, the regioselectivity can depend on reaction conditions: See Meyers, A.I.; Avila, W.B. Tetrahedron Lett. 1980, 3335.
725. See Snieckus, V. Chem. Rev. 1990, 90, 879; Gschwend, H.W.; Rodriguez, H.R. Org. React. 1979, 26, 1; Green, L.; Chauder, B.; Snieckus, V. J. Heterocylic Chem. 1999, 36, 1453. Also see, Green, L.; Chauder, B.; Snieckus, V. J. Heterocyclic Chem. 1999, 36, 1453; Slocum, D.W.; Dietzel, P. Tetrahedron Lett. 1999, 40, 1823.
726. Armstrong, D.R.; Mulvey, R.E.; Barr, D.; Snaith, R.; Reed, D. J. Organomet. Chem. 1988, 350, 191.
727. Chadwick, S.T.; Rennels, R.A.; Rutherford, J.L.; Collum, D.B. J. Am. Chem. Soc. 2000, 122, 8640.
728. Hay, D. R.; Song, Z.; Smith, S.G.; Beak, P. J. Am. Chem. Soc. 1988, 110, 8145.
729. Chadwick, S.T.; Rennels, R.A.; Rutherford, J.L.; Collum, D.B. J. Am. Chem. Soc. 2000, 122, 8640; Collum, D.B. Acc. Chem. Res. 1992, 25, 448. For a discusison of mechanistic possibilities, see Nguyen, T.-H.; Chau, N.T.T.; Castanet, A.-S.; Nguyen, K.P.P.; Mortier, J. Org. Lett. 2005, 7, 2445.
730. Macklin, T.K.; Snieckus, V. Org. Lett. 2005, 7, 2519.
731. Capriati, V.; Florio, S.; Luisi, R.; Musio, B. Org. Lett. 2005, 7, 3749.
732. Eaton, P.E.; Lee, C.; Xiong, Y. J. Am. Chem. Soc. 1989, 111, 8016. Also see Kronenburg, C.M.P.; Rijnberg, E.; Jastrzebski, J.T.B.H.; Kooijman, H.; Lutz, M.; Spek, A.L.; Gossage, R.A.; van Koten, G. Chemistry: European J.2005, 11, 253.
733. Armstrong, D.R.; Clegg, W.; Dale, S.H.; Hevia, E.; Hogg, L.M.; Honeyman, G.W.; Mulvey, R.E. Angew. Chem. Int. Ed. 2006, 45, 3775.
734. Chadwick, S.T.; Ramirez, A.; Gupta, L.; Collum, D.B. J. Am. Chem. Soc. 2007, 129, 2259.
735. Eppley, R.L.; Dixon, J.A. J. Am. Chem. Soc. 1968, 90, 1606.
736. Bryce-Smith, D.; Wakefield, B.J. Tetrahedron Lett. 1964, 3295.
737. Flippin, L.A.; Carter, D.S.; Dubree, N.J.P. Tetrahedron Lett. 1993, 34, 3255.
738. See Vorbrüggen, H.; Maas, M. Heterocycles, 1988, 27, 2659. Also see Comins, D.L.; O'Connor, S. Adv. Heterocycl. Chem. 1988, 44, 199.
739. Choppin, S.; Gros, P.; Fort, Y. Org. Lett. 2000, 2, 803.
740. Fort, Y.; Rodriguez, A.L. J. Org. Chem. 2003, 68, 4918.
741. Matsuzono, M.; Fukuda, T.; Iwao, M. Tetrahedron Lett. 2001, 42, 7621.
742. Yadav, J.S.; Reddy, B.V.S.; Reddy, P.M.; Srinivas, Ch. Tetrahedron Lett. 2002, 43, 5185.
743. See Larock, R.C. Organomercury Compounds in Organic Synthesis, Springer, NY, 1985, pp. 60–97; Wardell, J.L. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11, VCH, NY, 1988, pp. 308–318.
744. See Butler, R.N. in Pizey, J.S. Synthetic Reagents, Vol. 4, Wiley, NY, 1981, pp. 1–145.
745. See Taylor, R. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 13, Elsevier, NY, 1972, pp. 186–194. An alternative mechanism, involving radial cations, has been reported: Courtneidge, J.L.; Davies, A.G.; McGuchan, D.C.; Yazdi, S.N. J. Organomet. Chem. 1988, 341, 63.
746. See Uemura, S. in Pizey, J.S. Synthetic Reagents, Vol. 5, Wiley, NY, 1983, pp. 165–241.
747. Taylor, E.C.; Kienzle, F.; McKillop, A. Org. Synth. VI, 826; Taylor, E.C.; Katz, A.H.; Alvarado, S.I.; McKillop, A. J. Organomet. Chem. 1985, 285, C9. See Usyatinskii, A.Ya.; Bregadze, V.I. Russ. Chem. Rev. 1988, 57, 1054.
748. Uemura, S.; Toshimitsu, A.; Okano, M. Bull. Chem. Soc. Jpn. 1976, 49, 2582.
749. Lau, W.; Kochi, J.K. J. Am. Chem. Soc. 1984, 106, 7100; 1986, 108, 6720.
750. Fujita, K.-i.; Nonogawa, M.; Yamaguchi, R. Chem. Commun. 2004, 1926.
751. Periasamy, M.; Jayakumar, K.N.; Bharathi, P. J. Org. Chem. 2000, 65, 3548.
752. Traynelis, V.J.; McSweeney, J.V. J. Org. Chem. 1966, 31, 243.
753. Russell, G.A.; Weiner, S.A. J. Org. Chem. 1966, 31, 248.
754. See Stahly, G.P.; Stahly, B.C.; Maloney, J.R. J. Org. Chem. 1988, 53, 690.
755. See Makosza, M. Synthesis 1991, 103; Russ. Chem. Rev. 1989, 58, 747; Makosza, M.; Winiarski, J. Acc. Chem. Res. 1987, 20, 282.
756. For a discussion of the mechanism of vicarious nucleophilic aromatic substitution, see Makosza, M.; Lemek, T.; Kwast, A.; Terrier, F. J. Org. Chem. 2002, 67, 394.
757. Makosza, M.; Danikiewicz, W.; Wojciechowski, K. Liebigs Ann. Chem. 1987, 711.
758. See Mudryk, B.; Makosza, M. Tetrahedron 1988, 44, 209.
759. Makosza, M.; Owczarczyk, Z. J. Org. Chem. 1989, 54, 5094.
760. Ek, F.; Axelsson, O.; Wistrand, L.-G.; Frejd, T. J. Org. Chem. 2002, 67, 6376.
761. See Vorbrüggen, H. Adv. Heterocycl. Chem. 1990, 49, 117; McGill, C.K.; Rappa, A. Adv. Heterocycl. Chem. 1988, 44, 1; Pozharskii, A.F.; Simonov, A.M.; Doron'kin, V.N. Russ. Chem. Rev. 1978, 47, 1042.
762. Wozniak, M.; Baránski, A.; Nowak, K.; van der Plas, H.C. J. Org. Chem. 1987, 52, 5643.
763. Ban, Y.; Wakamatsu, T. Chem. Ind. (London) 1964, 710.
764. See Levitt, L.S.; Levitt, B.W. Chem. Ind. (London) 1975, 520.
765. Katritzky, A.R.; Laurenzo, K.S. J. Org. Chem. 1986, 51, 5039; 1988, 53, 3978.
766. Bakke, J.M.; Svensen, H.; Trevisan, R. J. Chem. Soc. Perkin Trans. 1 2001, 376.
767. Kauffmann, T.; Hansen, J.; Kosel, C.; Schoeneck, W. Liebigs Ann. Chem. 1962, 656, 103.
768. Suzuki, H.; Iwaya, M.; Mori, T. Tetrahedron Lett. 1997, 38, 5647.
769. See Wulfman, D.S. in Patai, S. The Chemistry of Diazonium and Diazo Groups, pt. 1; Wiley, NY, 1978, pp. 286–297.
770. Szele, I.; Zollinger, H. Helv. Chim. Acta 1978, 61, 1721.
771. See Pérez, P. J. Org. Chem. 2003, 68, 5886.
772. See in Patai, S. The Chemistry of Diazonium and Diazo Groups, Wiley, NY, 1978, the articles by Hegarty, A.F. pt. 2, pp. 511–591, and Schank, K. pt. 2, pp. 645–657; Godovikova, T.I.; Rakitin, O.A.; Khmel'nitskii, L.I. Russ. Chem. Rev. 1983, 52, 440; Challis, B.C.; Butler, A.R. in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, pp. 305–320. See Butler, A.R. Chem. Rev. 1975, 75, 241.
773. See Sorriso, S. in Patai, S. The Chemistry of Diazonium and Diazo Groups, pt. 1, Wiley, NY, 1978, pp. 95–105.
774. Rømming, C. Acta Chem. Scand. 1963, 17, 1444; Sorriso, S. in Patai, S. The Chemistry of Diazonium and Diazo Groups, pt. 1, Wiley, NY, 1978, p. 98; Ball, R.G.; Elofson, R.M. Can. J. Chem. 1985, 63, 332.
775. See Wulfman, D.S. in Patai, S. The Chemistry of Diazonium and Diazo Groups, pt. 1, Wiley, NY, 1978, pp. 247–339.
776. Korzeniowski, S.H.; Leopold, A.; Beadle, J.R.; Ahern, M.F.; Sheppard, W.A.; Khanna, R.K.; Gokel, G.W. J. Org. Chem. 1981, 46, 2153, and references cited therein; Bartsch, R.A. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt.1, Wiley, NY, 1983, pp. 889–915.
777. Kornblum, N.; Iffland, D.C. J. Am. Chem. Soc. 1949, 71, 2137.
778. See Regitz, M.; Maas, G. Diazo Compounds, Academic Press, NY, 1986. For reviews, see, in Patai, S. The Chemistry of Diazonium and Diazo Groups, pt. 1, Wiley, NY, 1978, the articles by Regitz, M. pt. 2, pp. 659–708, 751–820, and Wulfman, D.S.; Linstrumelle, G.; Cooper, C.F. pt. 2, pp. 821–976.
779. Takamura, N.; Mizoguchi, T.; Koga, K.; Yamada, S. Tetrahedron 1975, 31, 227.
780. Butler, R.N. in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, p. 305.
781. Challis, B.C.; Larkworthy, L.F.; Ridd, J.H. J. Chem. Soc. 1962, 5203.
782. Hughes, E.D.; Ingold, C.K.; Ridd, J.H. J. Chem. Soc. 1958, 58, 65, 77, 88; Hughes, E.D.; Ridd, J.H. J. Chem. Soc. 1958, 70, 82.
783. See Williams, D.L.H. Nitrosation, Cambridge University Press, Cambridge, 1988, pp. 95–109; Ridd, J.H. Q. Rev. Chem. Soc. 1961, 15, 418, p. 422.
784. Williams, D.L.H. Nitrosation, Cambridge University Press, Cambridge, 1988, pp. 84–93.
785. Zhang, Z.; Zhang, Q.; Zhang, S.; Liu, X.; Zhao, G. Synth. Commun. 2001, 31, 329.
786. Zhang, O.Z.; Zhang, S.; Zhang, J. Synth. Commun. 2001, 31, 1243.
787. Horning, D.E.; Ross, D.A.; Muchowski, J.M. Can. J. Chem. 1973, 51, 2347.
788. Cohen, T.; Dietz, Jr., A.G.; Miser, J.R. J. Org. Chem. 1977, 42, 2053.
789. Dreher, E.; Niederer, P.; Rieker, A.; Schwarz, W.; Zollinger, H. Helv. Chim. Acta 1981, 64, 488.
790. Yoneda, N.; Fukuhara, T.; Mizokami, T.; Suzuki, A. Chem. Lett. 1991, 459.
791. Abeywickrema, A.N.; Beckwith, A.L.J. J. Am. Chem. Soc. 1986, 108, 8227, and references cited therein.
792. See Price, C.C.; Tsunawaki, S. J. Org. Chem. 1963, 28, 1867.
793. Allaire, F.S.; Lyga, J.W. Synth. Commun. 2001, 31, 1857.
794. Arnould, J.C.; Didelot, M.; Cadilhac, C.; Pasquet, M.J. Tetrahedron Lett. 1996, 37, 4523.
795. See Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. Synthesis 2007, 81; Filimonov, V.D.; Semenischeva, N.I.; Krasnokutskaya, E.A.; Tretyakov, A.N.; Hwang, H.Y.; Chi, K.W. Synthesis 2008, 185.
796. Tamura, M.; Shibakami, M.; Sekiya, A. Eur. J. Org. Chem. 1998, 725.
797. Wu, Z.; Moore, J.S. Tetrahedron Lett. 1994, 35, 5539.
798. Carey, J.G.; Millar, I.T. Chem. Ind. (London) 1960, 97.
799. Packer, J.E.; Taylor, R.E.R. Aust. J. Chem. 1985, 38, 991; Abeywickrema, A.N.; Beckwith, A.L.J. J. Org. Chem. 1987, 52, 2568.
800. Thompson, A.L.S.; Kabalka, G.W.; Akula, M.R.; Huffman, J.W. Synthesis 2005, 547.
801. Furuya, T.; Kaiser, H.M.; Ritter, T. Angew. Chem. Int. Ed. 2008, 47, 5993.
802. See Suschitzky, H. Adv. Fluorine Chem. 1965, 4, 1.
803. Doyle, M.P.; Bryker, W.J. J. Org. Chem. 1979, 44, 1572.
804. Sellers, C.; Suschitzky, H. J. Chem. Soc. C 1968, 2317.
805. Rosenfeld, M.N.; Widdowson, D.A. J. Chem. Soc. Chem. Commun. 1979, 914. For another alternative procedure, see Yoneda, N.; Fukuhara, T.; Kikuchi, T.; Suzuki, A. Synth. Commun. 1989, 19, 865.
806. See also, Swain, C.G.; Sheats, J.E.; Harbison, K.G. J. Am. Chem. Soc. 1975, 97, 783, 796; Becker, H.G.O.; Israel, G. J. Prakt. Chem. 1979, 321, 579.
807. Makarova, L.G.; Matveeva, M.K. Bull. Acad. Sci. USSR Div. Chem. Sci. 1958, 548; Makarova, L.G.; Matveeva, M.K.; Gribchenko, E.A. Bull. Acad. Sci. USSR Div. Chem. Sci. 1958, 1399.
808. Swain, C.G.; Rogers, R.J. J. Am. Chem. Soc. 1975, 97, 799.
809. See Boyer, J.H. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, pt. 1, Wiley, NY, 1969, pp. 278–283.
810. Ayyangar, N.R.; Naik, S.N.; Srinivasan, K.V. Tetrahedron Lett. 1989, 30, 7253.
811. Kikukawa, K.; Kono, K.; Wada, F.; Matsuda, T. J. Org. Chem. 1983, 48, 1333.
812. Ek, F.; Wistrand, L.-G.; Frejd, T. J. Org. Chem. 2003, 68, 1911.
813. Sengupta, S.; Bhattacharya, S. J. Chem. Soc. Perkin Trans. 1 1993, 1943.
814. Darses, S.; Genêt, J.-P.; Brayer, J.-L.; Demoute, J.-P. Tetrahedron Lett. 1997, 38, 4393.
815. Darses, S.; Jeffery, T.; Genêt, J.-P.; Brayer, J.-L.; Demoute, J.-P. Tetrahedron Lett. 1996, 37, 3857.
816. Darses, S.; Michaud, G.; Genêt, J.-P. Eur. J. Org. Chem. 1999, 1875.
817. Willis, D.M.; Strongin, R.M. Tetahedron Lett. 2000, 41, 6271.
818. See Ganushchak, N.I.; Obushak, N.D.; Luka, G.Ya. J. Org. Chem. USSR 1981, 17, 765.
819. Dombrovskii, A.V. Russ. Chem. Rev., 1984, 53, 943; Rondestvedt, Jr., C.S. Org. React., 1976, 24, 225.
820. Doyle, M.P.; Siegfried, B.; Elliott, R.C.; Dellaria, Jr., J.F. J. Org. Chem. 1977, 42, 2431.
821. Dickerman, S.C.; Vermont, G.B. J. Am. Chem. Soc. 1962, 84, 4150; Morrison, R.T.; Cazes, J.; Samkoff, N.; Howe, C.A. J. Am. Chem. Soc. 1962, 84, 4152.
822. See Bolton, R.; Williams, G.H. Chem. Soc. Rev., 1986, 15, 261; Hey, D.H. Adv. Free-Radical Chem. 1966, 2, 47. Also see Vernin, G.; Dou, H.J.; Metzger, J. Bull. Soc. Chim. Fr. 1972, 1173.
823. Beadle, J.R.; Korzeniowski, S.H.; Rosenberg, D.E.; Garcia-Slanga, B.J.; Gokel, G.W. J. Org. Chem. 1984, 49, 1594.
824. Kamigata, N.; Kurihara, T.; Minato, H.; Kobayashi, M. Bull. Chem. Soc. Jpn. 1971, 44, 3152.
825. For a review, see Abramovitch, R.A. Adv. Free-Radical Chem. 1966, 2, 87.
826. Elofson, R.M.; Gadallah, F.F. J. Org. Chem. 1971, 36, 1769.
827. Chauncy, B.; Gellert, E. Aust. J. Chem. 1969, 22, 993. See also, Duclos, Jr., R.I.; Tung, J.S.; Rappoport, H. J. Org. Chem. 1984, 49, 5243.
828. Robinson, M.K.; Kochurina, V.S.; Hanna, Jr., J.M. Tetrahedron Lett. 2007, 48, 7687.
829. See Butler, R.N.; O'Shea, P.D.; Shelly, D.P. J. Chem. Soc. Perkin Trans. 1, 1987, 1039.
830. Fillipi, G.; Vernin, G.; Dou, H.J.; Metzger, J.; Perkins, M.J. Bull. Soc. Chim. Fr. 1974, 1075.
831. Eliel, E.L.; Saha, J.G.; Meyerson, S. J. Org. Chem. 1965, 30, 2451.
832. For an alternative method to generate aryl cations, see Milanesi, S.; Fagnoni, M.; Albini, A. J. Org. Chem. 2005, 70, 603.
833. Cadogan, J.I.G.; Murray, C.D.; Sharp, J.T. J. Chem. Soc. Perkin Trans. 2, 1976, 583, and references cited therein.
834. Gragerov, I.P.; Levit, A.F. J. Org. Chem. USSR 1968, 4, 7.
835. See Gadallah, F.F.; Cantu, A.A.; Elofson, R.M. J. Org. Chem. 1973, 38, 2386.
836. See Burri, P.; Zollinger, H. Helv. Chim. Acta 1973, 56, 2204; Eustathopoulos, H.; Rinaudo, J.; Bonnier, J.M. Bull. Soc. Chim. Fr. 1974, 2911; Zollinger, H. Acc. Chem. Res. 1973, 6, 335, p. 338.
837. See Cohen, T.; Lewarchik, R.J.; Tarino, J.Z. J. Am. Chem. Soc. 1974, 96, 7753.
838. Palani, N.; Jayaprakash, K.; Hoz, S. J. Org. Chem. 2003, 68, 4388.
839. See Knudsen, R.D.; Snyder, H.R. J. Org. Chem. 1974, 39, 3343.
840. Suzuki, H.; Yazawa, N.; Yoshida, Y.; Furusawa, O.; Kimura, O. Bull. Chem. Soc. Jpn. 1990, 63, 2010; Effenberger, F.; Streicher, W. Chem. Ber. 1991, 124, 157.
841. Liu, J.-T.; Jang, Y.-J.; Shih, Y.-K.; Hu, S.-R.; Chu, C.-M.; Yao, C.-F. J. Org. Chem. 2001, 66, 6021.
842. For a review, see Shine, H.J. Aromatic Rearrangements, Elsevier, NY, 1967, pp. 326–335.
843. Bunnett, J.F.; Rauhut, M.M. J. Org. Chem. 1956, 21, 934, 944.
844. Rosenblum, M. J. Am. Chem. Soc. 1960, 82, 3796.
845. Ibne-Rasa, K.M.; Koubek, E. J. Org. Chem. 1963, 28, 3240.
846. Samuel, D. J. Chem. Soc. 1960, 1318. For other evidence, see Cullen, E.; L'Ecuyer, P. Can. J. Chem. 1961, 39, 144, 155, 382; Ullman, E.F.; Bartkus, E.A. Chem. Ind. (London) 1962, 93.
847. See Pine, S.H. Org. React., 1970, 18, 403; Lepley, A.R.; Giumanini, A.G. Mech. Mol. Migr. 1971, 3, 297; Wittig, G. Bull. Soc. Chim. Fr. 1971, 1921; Stevens, T.S.; Watts, W.E. Selected Molecular Rearrangements, Van Nostrand–Reinhold, Princeton, 1973, pp. 81–88; Shine, H.J. Aromatic Rearrangements, Elsevier, NY, 1967, pp. 316–326. Also see, Klunder, J.M. J. Heterocyclic Chem. 1995, 32, 1687.
848. Beard, W.Q.; Hauser, C.R. J. Org. Chem. 1960, 25, 334.
849. Jones, G.C.; Beard, W.Q.; Hauser, C.R. J. Org. Chem. 1963, 28, 199.
850. See, however, Nakano, M.; Sato, Y. J. Org. Chem. 1987, 52, 1844; Shirai, N.; Sato, Y. J. Org. Chem. 1988, 53, 194.
851. Wittig, G.; Streib, H. Liebigs Ann. Chem. 1953, 584, 1.
852. See Puterbaugh, W.H.; Hauser, C.R. J. Am. Chem. Soc. 1964, 86, 1105; Pine, S.H.; Sanchez, B.L. Tetrahedron Lett. 1969, 1319; Shirai, N.; Watanabe, Y.; Sato, Y. J. Org. Chem. 1990, 55, 2767.
853. Kantor, S.W.; Hauser, C.R. J. Am. Chem. Soc. 1951, 73, 4122.
854. Pine, S.H. Tetrahedron Lett. 1967, 3393; Pine, S.H. Org. React. 1970, 18, 403, p. 418.
855. Bumgardner, C.L. J. Am. Chem. Soc. 1963, 85, 73.
856. See Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, pp. 118–124.
857. See Shine, H.J. Aromatic Rearrangements, Elsevier, NY, 1967, pp. 182–190.
858. Also see Kohnstam, G.; Petch, W.A.; Williams, D.L.H. J. Chem. Soc. Perkin Trans. 2 1984, 423; Sternson, L.A.; Chandrasakar, R. J. Org. Chem. 1984, 49, 4295, and references cited therein.
859. Fishbein, J.C.; McClelland, R.A. J. Am. Chem. Soc. 1987, 109, 2824.
860. Sundermeier, M.; Zapf, A.; Beller, M. Angew. Chem. Int. Ed. 2003, 42, 1661.
861. Hoshino, Y.; Okuno, M.; Kawamura, E.; Honda, K.; Inoue, S. Chem. Commun 2009, 2281.
862. See Truce, W.E.; Kreider, E.M.; Brand, W.W. Org. React., 1971, 18, 99; Shine, H.J. Aromatic Rearrangements, Elsevier, NY, 1967, pp. 307–316; Stevens, T.S.; Watts, W.E. Selected Molecular Rearrangements, Van Nostrand–Reinhold, Princeton, 1973, pp. 120–126.
863. Grundon, M.F.; Matier, W.L. J. Chem. Soc., B 1966, 266; Schmidt, D.M.; Bonvicino, G.E. J. Org. Chem. 1984, 49, 1664.
864. See Cerfontain, H. Mechanistic Aspects in Aromatic Sulfonation and Desulfonation, Wiley, NY, 1968, pp. 262–274.
865. Truce, W.E.; Robbins, C.R.; Kreider, E.M. J. Am. Chem. Soc. 1966, 88, 4027; Drozd, V.N.; Nikonova, L.A. J. Org. Chem, USSR 1969, 5, 313.
866. Bunnett, J.F.; Okamoto, T. J. Am. Chem. Soc. 1956, 78, 5363.
867. Kent, B.A.; Smiles, S. J. Chem. Soc. 1934, 422.
868. For a stable sulfenic acid, see Nakamura, N. J. Am. Chem. Soc. 1983, 105, 7172.
869. Truce, W.E.; Ray, Jr., W.J.; Norman, O.L.; Eickemeyer, D.B. J. Am. Chem. Soc. 1958, 80, 3625.
870. Erickson, W.R.; McKennon, M.J. Tetrahedron Lett. 2000, 41, 4541.
871. Fukazawa, Y.; Kato, N.; Ito, S. Tetrahedron Lett. 1982, 23, 437.
872. Hirota, T.; Tomita, K.; Sasaki, K.; Okuda, K.; Yoshida, M.; Kashino, S. Heterocycles 2001, 55, 741.
873. Truce, W.E.; Hampton, D.C. J. Org. Chem. 1963, 28, 2276.
874. Mitchell, L.H.; Barvian, N.C. Tetrahedron Lett. 2004, 45, 5669.