March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 15. Addition to Carbon–Carbon Multiple Bonds
There are four fundamental ways in which addition to a double or triple bond can take place. Three of these are two-step processes, with initial attack by a nucleophile, or attack upon an electrophile or a free radical. The second step consists of combination of the resulting intermediate with, respectively, a positive species, a negative species, or a neutral entity. In the fourth type of mechanism, attack at the two carbon atoms of the double or triple bond is simultaneous (concerted). Which of the four mechanisms is operating in any given case is determined by the nature of the substrate, the reagent, and the reaction conditions. Some of the reactions in this chapter can take place by all four mechanistic types.
15.A. Mechanisms
15.A.i. Electrophilic Addition1
In this mechanism, a positive species approaches the double or triple bond and in the first step forms a bond by donation of the π pair of electrons2 to the electrophilic species to form a σ bond:
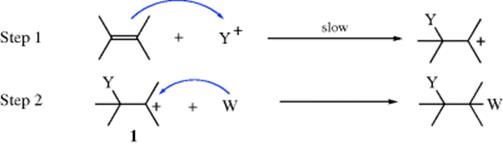
The IUPAC designation for this mechanism is AE + AN (or AH + AN if Y+ = H+). As in electrophilic substitution (Sec. 11.A.i), Y need not actually be a positive ion, but can be the positive end of a dipole or an induced dipole, with the negative part breaking off either during the first step or shortly after. The second step is a combination of 1 with a species carrying an electron pair and often bearing a negative charge. This step is the same as the second step of the SN1 mechanism. Not all electrophilic additions follow the simple mechanism given above. In many bromination reactions it is fairly certain that 1, if formed at all, very rapidly cyclizes to a bromonium ion (2):

This intermediate is similar to those encountered in the neighboring-group mechanism of nucleophilic substitution (see Sec. 10.C). The attack of W: on an intermediate like 2 is an SN2 step. Whether the intermediate is 1 or 2, the mechanism is called AdE2 (electrophilic addition, bimolecular).
The most useful type of information for investigating the mechanism of addition to a double bond, is perhaps, the stereochemistry of the reaction.3 The two carbons of the double bond and the four atoms immediately attached to them are all in a plane (Sec. 1.D); there are thus three possibilities. Both Y and W may enter from the same side of the plane, in which case the addition is stereospecific and syn; they may enter from opposite sides for stereospecific anti addition; or the reaction may be nonstereospecific. In order to determine which of these possibilities is occurring in a given reaction, the following type of experiment is often done: YW is added to the cis and trans isomers of an alkene of the form ABC=CBA. Using the cis-alkene as an example, if the addition is syn, the product will be the erythro dl pair, because each carbon has a 50% chance of being attacked by Y:
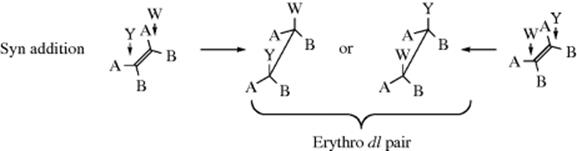
On the other hand, if the addition is anti, the threo dl pair will be formed:
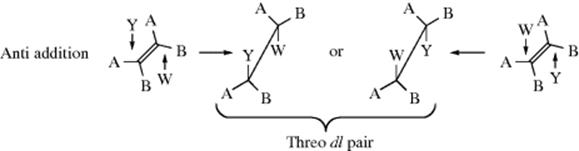
Of course, the trans isomer will give the opposite results: the threo pair if the addition is syn and the erythro pair if it is anti. These diastereomers have different physical properties. In the special case, where Y = W (as in the addition of Br2), the “erythro pair” is a meso compound. In addition to triple-bond compounds of the type AC![]() CA, syn addition results in a cis-alkene and anti addition in a trans-alkene. By the definition given in Section 4.N, addition to triple bonds cannot be stereospecific, although it can be, and often is, stereoselective.
CA, syn addition results in a cis-alkene and anti addition in a trans-alkene. By the definition given in Section 4.N, addition to triple bonds cannot be stereospecific, although it can be, and often is, stereoselective.
It is easily seen that in reactions involving cyclic intermediates like 2, addition must be anti, since the second step is an SN2 step and must occur from the rear. It is not so easy to predict the stereochemistry for reactions involving 1. If 1 has a relatively long life, the addition should be nonstereospecific, since there will be free rotation about the single bond. In addition, the carbocation is planar, so there is no facial bias for nucleophilic attack. On the other hand, if there is some factor that maintains the configuration, the group W may come in from the same or the opposite side, depending on the circumstances. For example, the positive charge might be stabilized by an attraction for Y that does not involve a full bond (see 3). In such a case, Y is said to stabilize the positive center via back-donation.
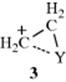
Such stabilization usually leads to anti attack of the second group. This contrasts with a circumstance that would favor syn addition: Formation of an ion pair (4) after the addition of Y4:
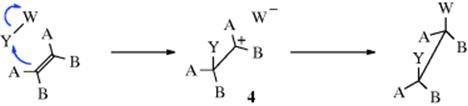
Since W is already on the same side of the plane as Y, collapse of the ion pair leads to syn addition.
Another possibility is that anti addition might, at least in some cases, be caused by the operation of a mechanism in which attack by W and Y are essentially simultaneous, but from opposite sides:
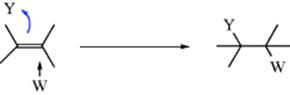
This mechanism, called the AdE3 mechanism (termolecular addition, IUPAC ANAE),5 has the disadvantage that three molecules must come together in the transition state. Note that it is the reverse of the E2 mechanism for elimination, for which the transition state is known to possess this geometry (Sec. 17.A.i).
There is much evidence that when an alkene attacks Br+ (or a carrier of it), bromonium ion 2 is usually the intermediate and the addition is anti, so the reaction is diastereospecific. Such ions have also been isolated in reactions involving addition of a Br+ species to a double bond.6 Treatment of maleic acid with bromine gave the dl pair of 2,3-dibromosuccinic acid, for example, while fumaric acid (the trans isomer) gave the meso compound.7 Many similar experiments have been performed with similar results. Bromination of dicarboxyacetylene gave 70% of the trans isomer.8

There is other evidence for mechanisms involving 2. As seen in Section 10.C, bromonium ions have been isolated in stable solutions in nucleophilic substitution reactions involving bromine as a neighboring group. The following is further evidence. If the two bromines approach the double bond from opposite sides, it is very unlikely that they could come from the same bromine molecule. This means that if the reaction is performed in the presence of nucleophiles, some of these will compete in the second step with the bromide liberated from the bromine. It has been found, indeed, that treatment of ethylene with bromine in the presence of chloride ions gives some 1-chloro-2-bromoethane along with the dibromoethane.9 Similar results are found when the reaction is carried out in the presence of water (Reaction 15-40) or of other nucleophiles.10 Ab initio MO studies show that 2 is more stable than its open isomer 1 (Y = Br).11 There is evidence that formation of 2 is reversible.12
However, a number of examples have been found where addition of bromine is not stereospecifically anti. For example, the addition of Br2 to cis- and trans-1-phenylpropenes in CCl4 was nonstereospecific.13 Furthermore, the stereospecificity of bromine addition to stilbene depends on the dielectric constant of the solvent. In solvents of low dielectric constant, the addition was 90–100% anti, but with an increase in dielectric constant, the reaction became less stereospecific, until at a dielectric constant of ~35, the addition was completely nonstereospecific.14 Likewise in the case of triple bonds, stereoselective anti addition was found in bromination of 3-hexyne, but both cis and trans products were obtained in bromination of phenylacetylene.15 These results indicate that a bromonium ion is not formed where the open cation can be stabilized in other ways (e.g., addition of Br+ to 1-phenylpropene gives the ion PhC+HCHBrCH3, which is a relatively stable benzylic cation). In addition, there is probably a spectrum of mechanisms between complete bromonium ion (2, no rotation) formation and completely open-cation (1, free rotation) formation, with partially bridged bromonium ions (3, restricted rotation) in between.16 Previously seen cases (Sec. 10.C.i, category 4) showed that cations require more stabilization from outside sources as they become intrinsically less stable themselves.17 Further evidence for the open cation mechanism where aryl stabilization is present was reported in an isotope effect study of Br2 addition to ArCH=CHCHAr′ (Ar = p-nitrophenyl, Ar′ = p-tolyl). The ![]() isotope effect for one of the double-bond carbons (the one closer to the NO2 group) was considerably larger than for the other one.18
isotope effect for one of the double-bond carbons (the one closer to the NO2 group) was considerably larger than for the other one.18
When the π bond of an alkene attacks Cl+,19 I+,20 or RS+,21 the result is similar to that when the electrophile is Br+; there is a spectrum of mechanisms between cyclic intermediates and open cations. As might be expected from the discussion in Section 10.C, iodonium ions compete with open carbocations more effectively than bromonium ions, while chloronium ions compete less effectively. There is kinetic and spectral evidence that at least in some cases (e.g., in the addition of Br2 or ICl), the electrophile forms a π complex with the alkene before a covalent bond is formed.22 There is evidence for β-chloro and β-bromocarbenium ions in some reactions.23
When the electrophile is a proton,24 a cyclic intermediate is not possible, and the mechanism is the simple AH + AN process shown before
![]()
This is an A-SE2 mechanism (see Reaction 10-6). There is a great deal of evidence25 for it, including:
1. The reaction is general acid, not specific acid catalyzed, implying rate-determining proton transfer from the acid to the double bond.26
2. The existence of open carbocation intermediates is supported by the contrast in the pattern of alkyl substituent effects27 with that found in brominations, where cyclic intermediates are involved. In the latter case, substitution of alkyl groups on H2C=CH2 causes a cumulative rate acceleration
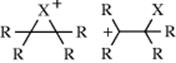
until all four hydrogen atoms have been replaced by alkyl groups, because each group helps to stabilize the positive charge.28 In addition of HX, the effect is not cumulative. Replacement of the two hydrogen atoms on one carbon causes great rate increases (primary → secondary → tertiary carbocation), but additional substitution on the other carbon produces little or no acceleration.29 This is evidence for open cations when a proton is the electrophile.30
3. Open carbocations are prone to rearrange (Chapter 18). Many rearrangements have been found to accompany additions of HX and H2O.31
It may also be recalled that vinylic ethers react with proton donors in a similar manner (see Reaction 10-6).
The stereochemistry of HX addition is varied. Examples are known of predominant syn, anti, and nonstereoselective addition. It was found that treatment of 1,2-dimethylcyclohexene (5) with HBr gave predominant anti addition,32 while addition of water to 5 gave equal amounts of the cis and trans alcohols:33
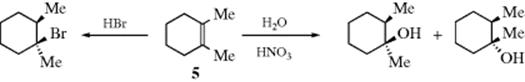
On the other hand, addition of DBr to acenaphthylene and to indene and 1-phenylpropene gave predominant syn addition, as shown:34
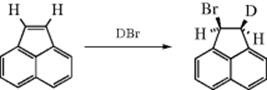
In fact, it has been shown that changing the reaction conditions can control the stereoselectivity of HCl addition. Addition of HCl to 5 in CH2Cl2 at −98 °C gave predominantly syn addition, while in ethyl ether at 0 °C, the addition was mostly anti.35
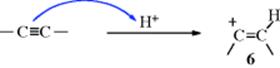
Addition of HX to triple bonds has the same mechanism, although the intermediate in this case is a vinylic cation (6).36
In all these cases (except for the AdE3 mechanism), it was assumed that formation of the intermediate (1, 2, or 3) is the slow step and attack by the nucleophile on the intermediate is rapid. This finding is probably true in most cases. However, some additions have been found in which the second step is rate determining.37
15.A.ii. Nucleophilic Addition38
In the first step of nucleophilic addition, a nucleophile donates a pair of electrons to one carbon atom of the double or triple bond, creating a carbanion, which reacts with a positive species in the second step:
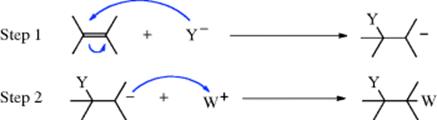
This mechanism is the same as the simple electrophilic one shown in Section 15.A.i except that the charges are reversed (IUPAC AN + AE or AN + AH). When the alkene contains a good leaving group (as defined for nucleophilic substitution), substitution is a side reaction (this is nucleophilic substitution at a vinylic substrate, see Sec. 10.F).
In the special case of addition of HY to a substrate of the form –C=C–Z, where Z = CHO, COR39 (including quinones40), CO2R, CONH2, CN, NO2, SOR, 2R,41 and so on, addition nearly always follows a nucleophilic mechanism,42 with Y− bonding with the carbon away from the Z group, for example,
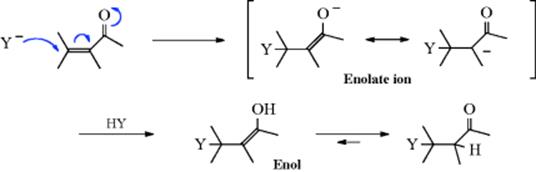
After the reaction with HY, the product is a resonance-stabilized enolate anion. Subsequent protonation of the enolate ion is probably at the oxygen to give an enol, which tautomerizes to the ketone (Sec. 2.N.i). Although the net result of the reaction is addition to a carbon–carbon double bond, the mechanism is 1,4-nucleophilic addition to the C=C–C=O (or similar) system and is thus very similar to the mechanism of addition to carbon–oxygen double and similar bonds (see Chapter 16). This mechanism is essentially that of the Michael reaction discussed in Reaction 15-24, and such reactions are often called Michael reactions or Michael-type reactions. When Z is CN or a C=O group, it is also possible for Y− to attack C of the cyano or carbonyl, which sometimes competes. When it happens, it is called 1,2-addition (see nucleophilic acyl addition in Chapter 16). 1,4-Addition to these substrates is also known as conjugate addition. Attack of the Y− ion at the 3 position is very difficult, since the resulting carbanion would have no resonance stabilization.43 Systems of the type C=C–C=C–Z can give 1,2-, 1,4-, or 1,6-addition.44Michael-type reactions are reversible, and compounds of the type YCH2CH2Z can often be decomposed to YH and CH2=CHZ by heating, either with or without alkali.
If the mechanism for nucleophilic addition is the simple carbanion mechanism outlined above for an alkene, the addition should be nonstereospecific, although it might well be stereoselective (see Sec. 4.N for the distinction). For example, the (E) and (Z) forms of an alkene ABC=CDE would give 7 and 8. If the carbanion has
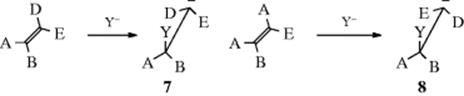
even a short lifetime, 7 and 8 will assume the most favorable conformation before the reaction with W. This is of course the same for both, and reaction with W will give the same product. This will be one of two possible diastereomers, so the reaction will be diastereoselective; but since the cis and trans isomers do not give rise to different isomers, it will not be diastereospecific. Unfortunately, this prediction has not been tested on open-chain alkenes. Except for Michael-type substrates, the stereochemistry of nucleophilic addition to double bonds has been studied only in cyclic systems, where only the cis isomer exists. In these cases, the reaction has been shown to be stereoselective, with syn addition reported in some cases45 and anti addition in others.46 When the reaction is performed on a Michael-type substrate (C=C–Z) the hydrogen does not arrive at the carbon directly, but only through a tautomeric equilibrium. The product naturally assumes the most thermodynamically stable configuration, without relation to the direction of original attack of Y. In one such case (the addition of EtOD and of Me3CSD to trans-MeCH=CHCOOEt), predominant anti addition was found. There is evidence that the stereoselectivity here results from the final protonation of the enolate, and not from the initial attack.47 For obvious reasons, additions to triple bonds cannot be stereospecific. As with electrophilic additions, nucleophilic additions to triple bonds are usually stereoselective and anti,48 although syn addition49 and nonstereoselective addition50 have also been reported.
15.A.iii. Free Radical Addition
The mechanism of free radical addition51 follows the pattern discussed in Section 14.A.i. The method of principal component analysis has been used to analyze polar and enthalpic effects in radical addition reactions.52 A radical is generated by
![]()
or
![]()
For subsequent reaction with an alkene, propagation occurs by
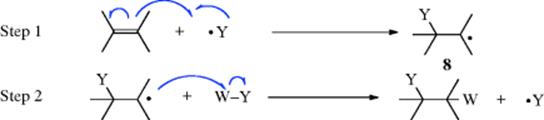
Step 2 is an abstraction (an atom transfer), so W is nearly always univalent, either hydrogen or halogen (Sec. 14.B.i). Termination of the chain can occur in any of the ways discussed in Section 14.A.i. If 9 adds to another alkene molecule, a dimer is formed. This can add to still another alkene unit, and chains, long or short, may be built up. This is the mechanism of free radical polymerization. Short polymeric molecules (called telomers), formed in this manner, are often troublesome side products in free radical addition reactions.
![]()
When free radicals are added to 1,5- or 1,6-dienes, the initially formed radical (10) can add intramolecularly to the other bond, leading to a cyclic product (11).53 When the radical is generated from a precursor that gives vinyl radical 12, however, cyclization leads to 13, which is in equilibrium with cyclopropylcarbinyl radical 14 via a 5-exo-trig reaction.54 A 6-endo-trig reaction (Sec. 6.E) leads to 15, but unless there are perturbing substituent effects, cyclopropanation should be the major process. Radicals of type 10, generated in other ways, also undergo these cyclizations. Both five- and six-membered rings can be formed (Sec. 15.B.i).
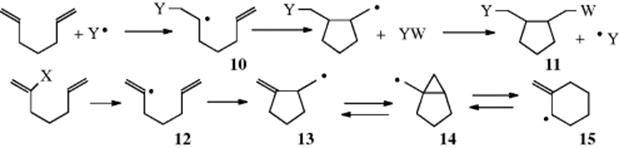
The free radical addition mechanism just outlined predicts that the addition should be nonstereospecific, at least if 9 has any, but an extremely short lifetime. However, the reactions may be stereoselective, for reasons similar to those discussed above for nucleophilic addition to alkenes. A few free radical additions are selective. For example, addition of HBr to 1-bromocyclohexene is regioselective in that it gave only cis-1,2-dibromocyclohexane and none of the trans isomer (anti addition),55 and propyne (at −78 to −60 °C) gave only cis-1-bromopropene (anti addition), making it stereoselective.56 Selectivity was observed in radical cyclization reactions of functionalized alkenes, which proceeded via a trans-ring closure.57 The most important case is probably addition of HBr to 2-bromo-2-butene under free radical conditions at −80 °C. Under these conditions, the cis isomer gave 92% of the meso product, while the trans isomer gave mostly the dl pair.58 This stereospecificity disappeared at room temperature, where both alkenes gave the same mixture of products (~78% of the dl pair and 22% of the meso compound), so the addition was still stereoselective, but no longer stereospecific. The stereospecificity at low temperatures is probably caused by a stabilization of the intermediate radical through the formation of a bridged bromine radical, of the type mentioned in Section 14.A.iv:
![]()
This species is similar to the bromonium ion (2) that is responsible for stereospecific anti addition in the electrophilic mechanism. Further evidence for the existence of such bridged radicals was obtained by addition of Br• to alkenes at 77 K. The ESR spectra of the resulting species were consistent with bridged structures.59
For many radicals, step 1 (C=C + Y• → •C–C–Y) is reversible. In such cases free radicals can cause cis → trans isomerization of a double bond by the pathway60
![]()
15.A.iv. Cyclic Mechanisms
There are some addition reactions where the initial reaction is not at one carbon of the double bond, but both carbons react simultaneously. Some of these are four-center mechanisms, which follow the following pattern:
![]()
In others, there is a five- or a six-membered transition state. In these cases the addition to the double or triple bond must be syn. The most important reaction of this type is the Diels–Alder reaction (15-60).
15.A.v. Addition to Conjugated Systems
When electrophilic addition is carried out on a compound with two double bonds in conjugation, a 1,2-addition product (16) is often obtained, but in most cases there is also a 1,4-addition product (17), often in larger yield:61
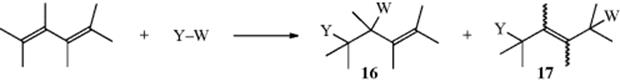
If the diene is unsymmetrical, there may be two 1,2-addition products. The competition between two types of addition product comes about because the carbocation resulting from attack on Y+ is a resonance hybrid, with partial positive charges at the 2 and 4 positions:
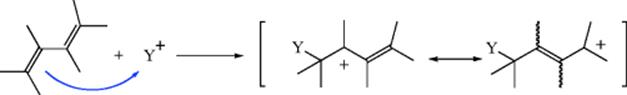
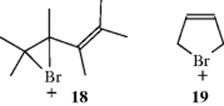
W− may then be attacked by either position. The original reaction with Y+ is always at the end of the conjugated system because reaction at a middle carbon would give a cation unstabilized by resonance. In the case of electrophiles like Br+, which can form cyclic intermediates, both 1,2- and 1,4-addition products can be rationalized as stemming from an intermediate like 18. Direct nucleophilic attack by W− would give the 1,2-product, while the 1,4-product could be formed by attack at the 4 position, by an SN2′-type mechanism (see Sec. 10.E). Intermediates like 19 have been postulated, but ruled out for Br and Cl by the observation that chlorination or bromination of butadiene gives trans 1,4-products.62 If an ion like 19 were the intermediate, the 1,4-products would have to have the cis configuration.
In most cases, more 1,4- than 1,2-addition product is obtained. This may be a consequence of thermodynamic control of products, as against kinetic, and usually temperature dependent. In most cases, under the reaction conditions, 16 is converted to a mixture of 16 and 17 that is richer in 17. That is, either isomer gives the same mixture of both, which contains more 17. It was found that at low temperatures, butadiene and HCl gave only 20–25% 1,4-adduct, while at high temperatures, where attainment of equilibrium is more likely, the mixture contained 75% 1,4-product.63 1,2-Addition predominated over 1,4- in the reaction between DCl and 1,3-pentadiene, where the intermediate was symmetrical (except for the D label).64 Ion pairs were invoked to explain this result, since Cl− would attack a free ion equally well at both positions, except for the very small isotope effect.
![]()
Addition to conjugated systems can also be accomplished by any of the other three mechanisms. In each case, there is competition between 1,2- and 1,4-addition. In the case of nucleophilic or free radical attack,65 the intermediates are resonance hybrids and behave like the intermediate from electrophilic attack. Dienes can similarly give 1,4-addition by a cyclic mechanism:

Other conjugated systems, including trienes, enynes, diynes, and so on, have been studied much less but behave similarly. 1,4-Addition to enynes is an important way of making allenes:
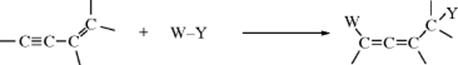
Radical addition to conjugated systems is an important part of chain-propagation reactions. The rate constants for addition of cyclohexyl radical to conjugated amides have been measured, and shown to be faster than addition to styrene.66 In additions to RCH=C(CN)2 systems, where the R group has a stereogenic center, the Felkin–Anh model (Sec. 4.H, category 1) applies and the reaction proceeds with high selectivity.67 Addition of some radicals [e.g., (Me3Si)3Si•], is reversible and this can lead to poor selectivity or isomerization.68