March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 16. Addition to Carbon–Hetero Multiple Bonds
16.A. Mechanism and Reactivity
The reactions considered in this chapter involve addition to the carbon–oxygen, carbon–nitrogen, and carbon–sulfur double bonds, as well as the carbon–nitrogen triple bond. The mechanistic study of these reactions is much simpler than that of the additions to carbon–carbon multiple bonds considered in Chapter 15.1 Since C=O, C=N, and C![]() N bonds are strongly polar, with the carbon always the positive end (except for isocyanides, see Sec. 16.B.iv), there is never any doubt about the orientation of unsymmetrical addition to these bonds (the regiochemical preference). Nucleophilic attacking species always go to the carbon and electrophilic species to the oxygen or nitrogen. Additions to C=S bonds are much less common,2 but in these cases the addition is sometimes in the other direction (reaction at sulfur is called thiophilic addition and addition to the carbon is called carbophilic addition).3 For example, the reaction of phenyllithium with thiobenzophenone (Ph2C=S) gives, after hydrolysis, benzhydryl phenyl sulfide (Ph2CHSPh).4
N bonds are strongly polar, with the carbon always the positive end (except for isocyanides, see Sec. 16.B.iv), there is never any doubt about the orientation of unsymmetrical addition to these bonds (the regiochemical preference). Nucleophilic attacking species always go to the carbon and electrophilic species to the oxygen or nitrogen. Additions to C=S bonds are much less common,2 but in these cases the addition is sometimes in the other direction (reaction at sulfur is called thiophilic addition and addition to the carbon is called carbophilic addition).3 For example, the reaction of phenyllithium with thiobenzophenone (Ph2C=S) gives, after hydrolysis, benzhydryl phenyl sulfide (Ph2CHSPh).4
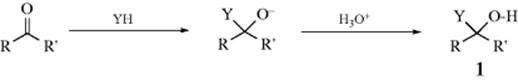
The normal acyl addition of YH to a ketone gives an alkoxide, and hydrolysis gives 1. Note that the product has a stereogenic carbon, but unless there is chirality in R or R′, or YH is optically active, the product must be a racemic mixture because there is no facial bias for addition to the carbonyl. The same holds true for C=N and C=S bonds. The stereochemistry of addition of a single YH to the carbon–nitrogen triple bond could be investigated, since the product can exist in (E) and (Z) forms (Sec. 4.K.i), but these reactions generally give imine products that undergo further reaction. Of course, if R or R′ is chiral, a mixture of diastereomers will result, and the stereochemistry of addition can be studied in such cases. Cram's rule or the Felkin–Anh model (Sec. 4.H) allows the direction of attack of Y to be predicted in many cases.5 However, the relative directions of attack of Y and H are not determined, but only the direction of attack of Y with respect to the rest of the substrate molecule.
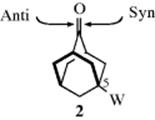
In Section 15.B.iii, electronic effects were shown to play a part in determining which face of a carbon–carbon double bond is attacked. The same applies to additions to carbonyl groups. For example, in 5-substituted adamantanones (2) electron-withdrawing (−I) groups W cause the attack to come from the syn face, while electron-donating groups cause it to come from the anti face.6 In 5,6-disubstituted norborn-2-en-7-one systems, the carbonyl appears to tilt away from the π bond, with reduction occurring from the more hindered face.7 An ab initio study of nucleophilic addition to 4-tert-butylcyclohexanones attempted to predict π-facial selectivity.8
The mechanistic picture is further simplified by the fact that free radical additions to carbon–heteroatom double bonds are not as prevalent (but see Reaction 16-31).9 In most cases, the nucleophile forms the first new bond to carbon, and these reactions are regarded as nucleophilic additions, which can be represented as:
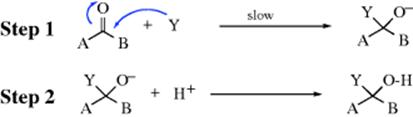
The electrophile shown in step 2 is the proton. In almost all the reactions considered in this chapter the electrophilic atom is either hydrogen or carbon. Note that step 1 is exactly the same as step 1 of the tetrahedral mechanism of nucleophilic substitution at a carbonyl carbon (Sec. 16.A.i), but carbon groups (A, B = H, alkyl aryl, etc.) are poor leaving groups so that substitution does not compete with addition. For carboxylic acid derivatives, there are leaving groups (B = Cl, OR, NH2, etc.) and acyl substitution predominates (Sec. 16.A.i). The nature of A and B determines whether a nucleophilic attack at a carbon–heteroatom multiple bond will lead to substitution or addition.
Both acids and bases can catalyze many of these reactions.10 Bases catalyze the reaction by converting a reagent YH to the more powerful nucleophile Y− (see Sec. 10.G.ii). Acids catalyze it by converting the substrate to an heteroatom-stabilized cation (oxocarbenium ion 3 from C=O) in step 1, thus making it more attractive to nucleophilic attack and making the reverse reaction somewhat less favorable. Similar catalysis can also be achieved with metallic ions (e.g., Ag+), which act as Lewis acids.11
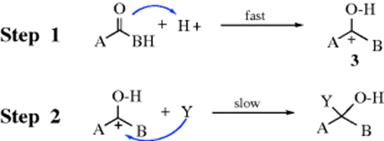
In step 1 of the acid-catalyzed mechanism, the carbonyl reacts as a base with the proton to give 3, which is known as an oxocarbenium ion. In Section 5.A.ii, it was pointed out that oxocarbenium ions are comparatively stable carbocations because the positive charge is delocalized on the oxygen by resonance.12 Intermediate 3 then reacts with the nucleophile in step 2 to give the acyl addition product. The rate-determining step is usually the one involving nucleophilic attack. If one heteroatom (e.g., oxygen) stabilizes an adjacent carbocation, as in 3, a second heteroatom (X) will stabilize an oxocarbenium ion (–X–C+–X–) to a greater extent.13
Reactivity factors for carbon–heteroatom multiple bonds are similar to those for the tetrahedral mechanism of nucleophilic substitution.14 If A and/or B are electron-donating groups (e.g., alkyl groups), rates are decreased. Electron-attracting substituents increase rates. This means that aldehydes are more reactive than ketones. Aryl groups are somewhat deactivating compared to alkyl, because of resonance that stabilizes the substrate molecule, but is lost on going to the intermediate. Double bonds in conjugation with the carbon–heteroatom multiple bond also lower addition rates, for similar reasons but, more important, may provide competition from 1,4-addition (Sec. 15.A.ii). Steric factors are also quite important and contribute to the decreased reactivity of ketones compared with aldehydes. Highly hindered ketones like hexamethylacetone and dineopentyl ketone either do not undergo many of these reactions or require extreme conditions.
16.A.i. Nucleophilic Substitution at an Aliphatic Trigonal Carbon: The Tetrahedral Mechanism
All the mechanisms discussed in previous chapters for substitution take place at a saturated carbon atom. Nucleophilic substitution is also important at trigonal carbons, especially when the carbon is double bonded to an oxygen, a sulfur, or a nitrogen. Substitution at a carbonyl group (or the corresponding nitrogen and sulfur analogues) most often proceeds by a second-order mechanism, which in this book is called the tetrahedral15mechanism.16 The IUPAC designation is AN + DN. The SN1 mechanisms, involving carbocations, are sometimes found with these substrates, especially with essentially ionic substrates (e.g., RCO+ BF4−; there is evidence that in certain cases simple SN2 mechanisms can take place, especially with a very good leaving group (e.g., Cl−);17 and an SET mechanism has also been reported.18 However, the tetrahedral mechanism is by far the most prevalent. Although this mechanism displays second-order kinetics, it is not the same as the SN2 mechanism discussed in Section 10.A.i. In the tetrahedral mechanism, first Y attacks to give an intermediate containing both X and Y (4), and then X leaves. This sequence, impossible at a saturated carbon, is possible at an unsaturated one because the central carbon can release a pair of electrons to the oxygen and so preserve its octet:
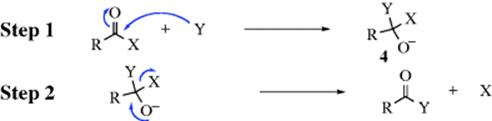
When reactions are carried out in acid solution, there may also be a preliminary and a final step:
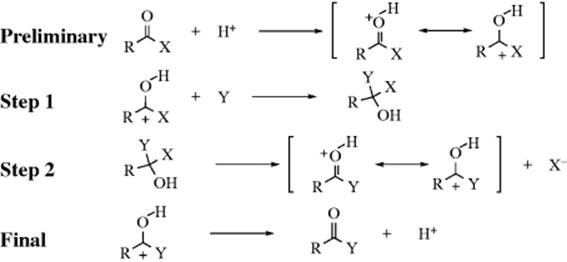
The hydrogen ion is a catalyst. The reaction rate is increased because it is easier for the nucleophile to attack the carbon when the electron density of the latter has been decreased.19
Evidence for the existence of the tetrahedral mechanism is as follows:20
1. The kinetics are first order each in the substrate and in the nucleophile, as predicted by the mechanism.
2. There is other kinetic evidence in accord with a tetrahedral intermediate. For example, the rate “constant” for the reaction between acetamide and hydroxylamine is not constant, but decreases with increasing hydroxylamine concentration.21 This is not a smooth decrease; there is a break in the curve. A straight line is followed at low hydroxylamine concentration and another straight line at high concentration. This means that the identity of the rate-determining step is changing. Obviously, this cannot happen if there is only one step: there must be two steps, and hence an intermediate. Similar kinetic behavior has been found in other cases as well;22 in particular, plots of rate against pH are often bell-shaped.
3. Basic hydrolysis has been carried out on carboxylic esters labeled with 18O in the carbonyl group.23 If this reaction proceeded by the normal SN2 mechanism, all the 18O would remain in the carbonyl group, even if, in an equilibrium process, some of the carboxylic acid formed went back to the starting material:
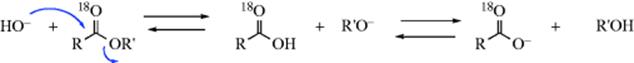
On the other hand, if the tetrahedral mechanism operates
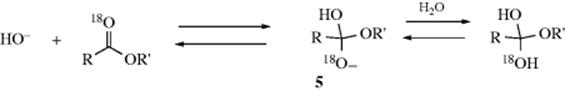
then the intermediate 5 reacts with an acid (e.g., water) and is converted to the conjugate acid, symmetrical intermediate 6. In this intermediate, the OH groups are equivalent, and (except for the small 18O/16O isotope effect) either one can lose a proton with equal facility:
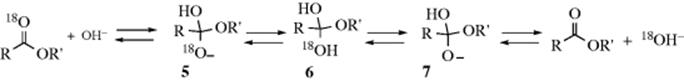
The intermediates 5 and 7 can now lose OR′ to give the carboxylic acid (not shown in the equations given), or they can lose OH to regenerate the carboxylic ester. If 5 reverts back to an ester, the ester will still be labeled, but if 7 reverts to an ester, the 18O will be lost. A test of the two possible mechanisms is to stop the reaction before completion and to analyze the recovered ester for 18O. Experiments by Bender24 found that in alkaline hydrolysis of methyl, ethyl, and isopropyl benzoates, the esters had lost 18O. A similar experiment carried out for acid-catalyzed hydrolysis of ethyl benzoate showed that here too the ester lost 18O. However, alkaline hydrolysis of substituted benzyl benzoates showed no18O loss.24 This result does not necessarily mean that no tetrahedral intermediate is involved in this case. If 5 and 7 do not revert to an ester, but go entirely to an acid, no 18O loss will be found even with a tetrahedral intermediate. In the case of benzyl benzoates, this may very well be happening, because formation of the acid relieves steric strain. Another possibility is that 5 loses OR′ before it can become protonated to 6.25 Even the experiments that do show 18O loss do not prove the existence of the tetrahedral intermediate, since it is possible that 18O is lost by some independent process not leading to ester hydrolysis. To deal with this possibility, Bender and Heck26 measured the rate of 18O loss in the hydrolysis of ethyl trifluorothioloacetate–18O:
![]()
This reaction had previously been shown27 to involve an intermediate by the kinetic methods mentioned above. Bender and Heck26 showed that the rate of 18O loss and the value of the partitioning ratio k2/k3 as determined by the oxygen-exchange technique were exactly in accord with these values as previously determined by kinetic methods. Thus the original 18O exchange measurements showed that there is a tetrahedral species present, but not necessarily on the reaction path, while the kinetic experiments showed that there is some intermediate present, but not necessarily tetrahedral. Bender and Heck's26 results demonstrate that there is a tetrahedral intermediate and that it lies on the reaction pathway.
4. In some cases, tetrahedral intermediates have been isolated28 or detected spectrally.29
Several studies have been made of the directionality of approach by the nucleophile.30 Menger30 has proposed for reactions in general, and specifically for those that proceed by the tetrahedral mechanism, that there is no single definable preferred transition state, but rather a “cone” of trajectories. All approaches within this cone lead to reaction at comparable rates; it is only when the approach comes outside of the cone that the rate falls.
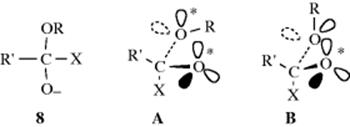
Directionality has also been studied for the second step. Once the tetrahedral intermediate (4) is formed, it loses Y (giving the product) or X (reverting to the starting compound). Deslongchamps has proposed that one of the factors affecting this choice is the conformation of the intermediate; more specifically, the positions of the lone pairs. In this view, a leaving group X or Y can depart only if the other two atoms on the carbon both have an orbital antiperiplanar to the C–X or C–Y bond. For example, consider an intermediate 8 formed by attack of −OR on a substrate R′COX. Cleavage of the C–X bond with loss of X can take place from conformation A, because the two lone-pair orbitals marked ∗ are antiperiplanar to the C–X bond, but not from B because only the O− has such an orbital. If the intermediate is in conformation B, the OR may leave (if X has a lone-pair orbital in the proper position) rather than X. This factor is called stereoelectronic control.31 Free rotation in acyclic intermediates leads to many conformations, but some are preferred, and cleavage reactions may take place faster than rotation, so stereoelectronic control can be a factor in some situations. Much evidence has been presented for this concept.32 More generally, the term stereoelectronic effects refers to any case in which orbital position requirements affect the course of a reaction. The backside attack in the SN2 mechanism is an example of a stereoelectronic effect.
Some nucleophilic substitutions at a carbonyl carbon are catalyzed by nucleophiles.33 There occur, in effect, two tetrahedral mechanisms:
![]()
(For an example, see Reaction 16-58). When this happens internally, it is a neighboring-group mechanism at a carbonyl carbon.34 For example, the hydrolysis of phthalamic acid (9) takes place as follows:
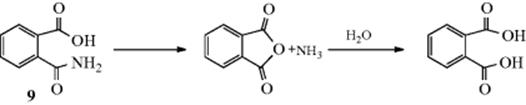
Evidence comes from comparative rate studies.35 Thus 9 was hydrolyzed ~ 105 times faster than benzamide (PhCONH2) at about the same concentration of hydrogen ions. That this enhancement of rate was not caused by the resonance or field effects of COOH (an electron-withdrawing group) was shown by the fact both o-nitrobenzamide and terephthalamic acid (the para isomer of 9) were hydrolyzed more slowly than benzamide. Other examples of neighboring-group participation at a carbonyl carbon have been reported.36 It is likely that nucleophilic catalysis is involved in enzyme catalysis of ester hydrolysis.
The attack of a nucleophile on a carbonyl group can result in substitution or addition, depending on the substituents, but the first step of each mechanism is the same. The main factor that determines the product is the identity of the group X in RCOX. When X is alkyl or hydrogen, addition usually takes place. When X is halogen, OCOR, NH2, and so on, the usual reaction is substitution. When X = OH, protonation to generate OH2+ is usually required before the group can be lost.
In both the SN1 and SN2 mechanisms, the leaving group departs during the rate-determining step and so directly affects the rate. In the tetrahedral mechanism at a carbonyl carbon, the bond between the substrate and leaving group is still intact during the slow step. Nevertheless, the nature of the leaving group still affects the reactivity in two ways: (1) By altering the electron density at the carbonyl carbon, the rate of the reaction is affected. The greater the electron-withdrawing character of X, the greater the partial positive charge on C and the more rapid the attack by a nucleophile. (2) The nature of the leaving group affects the position of equilibrium. In the intermediate 4, there is competition between X and Y as to which group leaves. If X is a poorer leaving group than Y, then Y will preferentially leave and 4 will revert to the starting compounds. Thus there is a partitioning factor between 4 going on to product (loss of X) or back to starting compound (loss of Y). The sum of these two factors causes the sequence of reactivity to be RCOCl > RCOOCOR′ > RCOOAr > RCOOR′ > RCONH2 > RCONR′2 > RCOO−.37 Note that this order is approximately the order of decreasing stability of the leaving-group anion. If the leaving group is bulky, it may exert a steric effect and retard the rate for this reason.
For a list of some of the more important reactions that operate by the tetrahedral mechanism, see Table 16.1, which shows the main reactions that proceed by the tetrahedral mechanism.
Table 16.1 The More Important Synthetic Reactions That Take Place by the Tetrahedral Mechanism.a
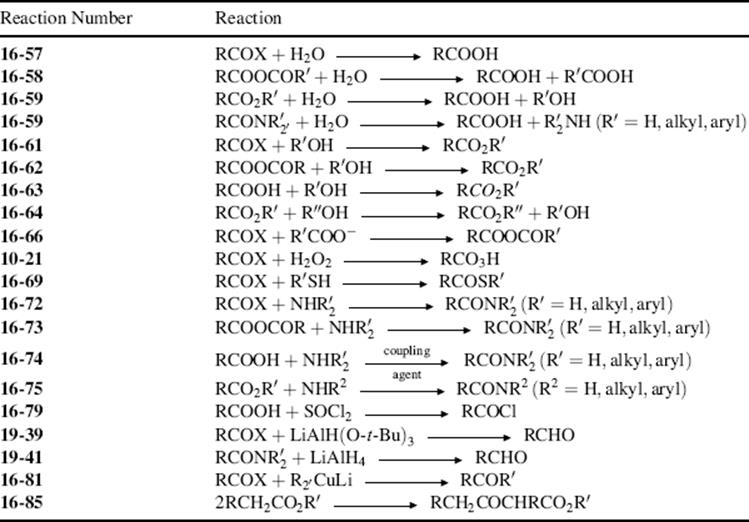
a Catalysts are not shown.