March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 18. Rearrangements
18.F. Reactions
The reactions in this chapter are classified into three main groups and 1,2-shifts are considered first. Within this group, reactions are classified according to (1) the identity of the substrate atoms A and B and (2) the nature of the migrating group W. The cyclic rearrangements are in the second group. The third group consists of rearrangements that cannot be fitted into either of the first two categories.
Reactions in which the migration terminus is on an aromatic ring have been treated under aromatic substitution. These are Reactions 11-27–11-32, 11-36, 13-30-13-32, and, partially, 11-33, 11-38, and 11-39. Double-bond shifts have also been treated in other chapters, although they may be considered rearrangements (Sec. 8.A and Reactions 12-4, and 12-2). Other reactions that may be regarded as rearrangements are the Pummerer (19-83) and Willgerodt(19-84) reactions.
18.F.i. 1,2-Rearrangements
A. Carbon-to-Carbon Migrations of R, H, and Ar
18-1 Wagner–Meerwein and Related Reactions
1/Hydro,1/hydroxy-(2/→1/alkyl)- migro- elimination, and so on
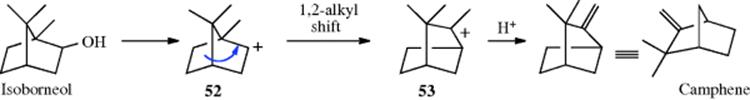
Wagner-Meerwein rearrangements were first discovered in reactions of bicyclic terpenes, and most of the early development of this reaction was with these compounds.96 An example is the conversion of isoborneol to camphene. It fundamentally involves a 1,2-alkyl shift of an intermediate carbocation, (e.g., 52 → 53). When alcohols are treated with acids, simple substitution (e.g., Reaction 10-48) or elimination (Reaction 17-1) usually accounts for most or all of the products. But in many cases, especially where two or three alkyl or aryl groups are on the β carbon, some or all of the product is rearranged. These rearrangements have been called Wagner–Meerwein rearrangements, although this term is nowadays reserved for relatively specific transformations (e.g., isoborneol to camphene and related reactions). As pointed out previously, the carbocation that is a direct product of the rearrangement must stabilize itself, and most often it does this by the loss of a hydrogen β to it, so the rearrangement product is usually an alkene.97 If there is a choice of protons, Zaitsev's rule (Sec. 17.A.i, category 3) governs the direction, as expected. Sometimes a different positive group is lost instead of a proton. Less often, the new carbocation reacts with a nucleophile instead of losing a proton. The nucleophile may be the water that is the original leaving group, in which case the product is a rearranged alcohol; or it may be some other species present (solvent, added nucleophile, etc.).
Rearrangement is usually predominant in neopentyl and neophyl types of substrates, and with these types normal nucleophilic substitution is difficult (normal elimination is of course impossible). Under SN2 conditions, substitution is extremely slow98; and under SN1 conditions, carbocations are formed that rapidly rearrange. However, free radical substitution, unaccompanied by rearrangement, can be carried out on neopentyl systems, although, as seen previously (Sec. 18.C), neophyl systems undergo rearrangement as well as substitution.
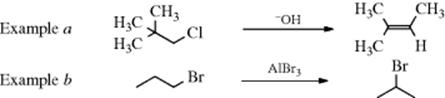
Examples of carbocation rearrangements are found in simpler systems (e.g., neopentyl chloride, example a) and even 1-bromopropane (example b). These examples illustrate the following points:
1. Hydride ion can migrate. In example b, it was hydride that shifted, not bromine:
![]()
2. The leaving group does not have to be H2O, but can be any departing species whose loss creates a carbocation, including N2 from aliphatic diazonium ions99 (see the section on leaving groups in nucleophilic substitution in Sec. 10.A.ii, category 1). Rearrangement may follow when the carbocation is created by addition of a proton or other positive species to a double bond.
3. Example b illustrates that the last step can be substitution instead of elimination.
4. Example a illustrates that the new double bond is formed in accord with Zaitsev's rule.
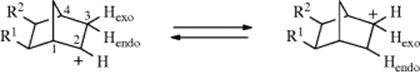
2-Norbornyl cations (see 52), besides displaying the 1,2-shifts of a CH2 group previously illustrated for the isoborneol → camphene conversion, are also prone to rapid hydride shifts from the 3 to the 2 position (known as 3,2-shifts). These 3,2-shifts usually take place from the exo side100; that is, the 3-exo hydrogen migrates to the 2-exo position.101 This stereoselectivity is analogous to the behavior previously seen for norbornyl systems, namely, that nucleophiles attack norbornyl cations from the exo side (Sec. 10.C.i, category 4) and that addition to norbornenes is also usually from the exo direction (Sec. 15.B.iii).
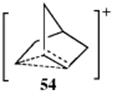
For rearrangements of alkyl carbocations, the direction of rearrangement is usually toward the most stable carbocation (or radical), which is tertiary > secondary > primary, but rearrangements in the other direction have also been found,102 and the product is sometimes a mixture corresponding to an equilibrium mixture of the possible carbocations. The Wagner–Meerwein rearrangement has been observed for a secondary to a secondary carbocation rearrangement, leading to some controversy. Winstein103 described norbornyl cations in terms of the resonance structures represented by the nonclassical ion 54.104 This view was questioned, primarily by Brown,105 who suggested that the facile rearrangements could be explained by a series of fast 1,3-Wagner–Meerwein shifts.106 There is considerable evidence, however, that the norbornyl cation rearranges with σ participation,107 and there is strong NMr evidence for the nonclassical ion in superacids at low temperatures.108
As alluded to above, the term “Wagner–Meerwein rearrangement” is not precise. Some use it to refer to all the rearrangements in this section and in Reaction 18-2. Others use it only when an alcohol is converted to a rearranged alkene. Many use the term only for rearrangements that involve a nonclassical carbocation intermediate. Terpene chemists call the migration of a methyl group the Nametkin rearrangement. The term retropinacol rearrangement is often applied to some or all of these. Fortunately, this disparity in nomenclature does not seem to cause much confusion. Catalytic asymmetric Wagner–Meerwein shifts have been observed.109 An asymmetric, Pd catalyzed Wagner–Meerwein shift has been reported with allenic alcohols.110
Several of these rearrangements sometimes occur in one molecule, either simultaneously or in rapid succession. A spectacular example is found in the triterpene series. Friedelin is a triterpenoid ketone found in cork. Reduction gives 3β-friedelanol (55). When this compound is treated with acid, 13(18)-oleanene (56) is formed.111 In this case, seven 1,2-shifts take place. Loss of H2O from position 3 leaves a positive charge, and
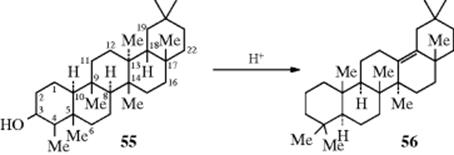
the following shifts occur: hydride from 4 to 3; methyl from 5 to 4; hydride from 10 to 5; methyl from 9 to 10; hydride from 8 to 9; methyl from 14 to 8; and methyl from 13 to 14. This leaves a positive charge at position 13, which is stabilized by loss of the proton at the 18 position to give 56. All these shifts are stereospecific, the group always migrating on the side of the ring system on which it is located; that is, a group above the “plane” of the ring system (indicated by a solid line in 55) moves above the plane, and a group below the plane (dashed line) moves below it. It is probable that the seven shifts are not all concerted, although some of them may be, for intermediate products can be isolated.112 As an illustration of point 2 (see above), it may be mentioned that friedelene, derived from dehydration of 55, also gives 56 on treatment with acid.113
Some alkanes undergo Wagner–Meerwein rearrangements if treated with Lewis acids and a small amount of initiator. An interesting application of this reaction is the conversion of tricyclic molecules to adamantane and its derivatives.114 It has been found that all tricyclic alkanes containing 10 carbons are converted to adamantane by treatment with a Lewis acid, (e.g., AlCl3). If the substrate contains > 10 carbons, alkyl-substituted adamantanes are produced. The IUPAC name for these reactions is Schleyer adamantization. Two examples are the AlCl3-mediated reactions of 57 and 58.
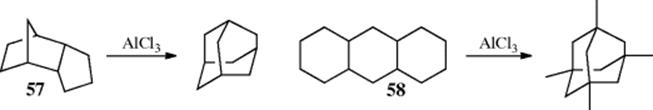
If 14 or more carbons are present, the product may be diamantane or a substituted diamantane.115 These reactions are successful because of the high thermodynamic stability of adamantane, diamantane, and similar diamond-like molecules. The most stable of a set of CnHm isomers (called the stabilomer) will be the end product if the reaction reaches equilibrium.116 Best yields are obtained by the use of “sludge” catalysts117 (i.e., a mixture of AlX3 and tert-butyl bromide or sec-butyl bromide).118 Though it is certain that these adamantane-forming reactions take place by nucleophilic 1,2 shifts, the exact pathways are not easy to unravel because of their complexity.119 Treatment of adamantane-2-14C with AlCl3 results in total carbon scrambling on a statistical basis.120
As already indicated, the mechanism of the Wagner–Meerwein rearrangement is usually nucleophilic. Free radical rearrangements are also known (see Section 18.A), though virtually only with aryl migration. However, carbanion mechanisms (electrophilic) have also been found.94 Thus Ph3CCH2Cl treated with sodium gave Ph2CHCH2Ph along with unrearranged products.121 This is called the Grovenstein-Zimmerman rearrangement. The intermediate is Ph3CCH2−, and the phenyl moves without its electron pair. Only aryl and vinylic,122 and not alkyl, groups migrate by the electrophilic mechanism (see the introductory section preceding Sec. 18.A) and transition states or intermediates analogous to 41 and 42 are likely.123
OS V, 16, 194; VI, 378, 845.
18-2 The Pinacol Rearrangement
1/ O-Hydro,3/hydroxy-(2/→3/alkyl)- migro-elimination
When 1.2-diols (vic-diols; glycols) are treated with acids,124 they rearrange to give aldehydes or ketones, although elimination without rearrangement can also be accomplished. This reaction is called the pinacol rearrangement; the reaction gets its name from a prototype compound pinacol (Me2COHCOHMe2), which is rearranged to pinacolone (Me3CCOCH3).125 In this type of reaction, reduction can compete with rearrangement.126 The reaction has been accomplished many times, with alkyl, aryl, hydrogen, and even ethoxycarbonyl (CO2Et)127 as migrating groups. In most cases, each carbon has at least one alkyl or aryl group, and the reaction is most often carried out with tri- and tetrasubstituted glycols. As mentioned earlier, glycols in which the four R groups are not identical can give rise to more than one product, depending on which group migrates (see Sec. 18.A.iii for a discussion of migratory aptitudes). A noncatalytic reaction is possible in supercritical water.128
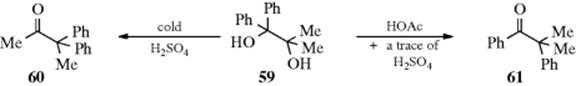
Stereodifferentiation is possible in this reaction.129 When TMSOTf was used to initiate the reaction, it was shown to be highly regioselective.130 Mixtures are often produced, and which group preferentially migrates may depend on the reaction conditions as well as on the nature of the substrate. Thus the action of cold, concentrated sulfuric acid on 59 produces mainly the ketone 60 (methyl migration), while treatment of 59 with acetic acid containing a trace of sulfuric acid gives mostly 61 (phenyl migration).131 If at least one R is hydrogen, aldehydes can be produced as well as ketones. Generally, aldehyde formation is favored by the use of mild conditions (lower temperatures, weaker acids), because under more drastic conditions the aldehydes may be converted to ketones (Reaction 18-4). The reaction has been carried out in the solid state, by treating solid substrates with HCl gas or with a solid organic acid.132
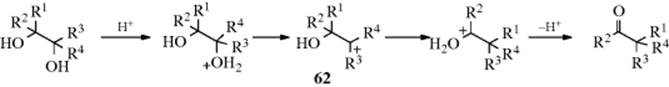
The mechanism involves a simple 1,2-shift. The ion 62 (where all four R groups are Me) has been trapped by the addition of tetrahydrothiophene.133 A migration takes place from the tertiary position because carbocations stabilized by an oxygen atom are even more stable than tertiary alkyl cations (Sec. 5.A.ii). In addition, the new carbocation can immediately stabilize itself by losing a proton.
It is obvious that other compounds in which a positive charge can be placed on a carbon α to one bearing an OH group can also give this rearrangement. This is true for β-amino alcohols, which rearrange on treatment with nitrous acid (this is called the semipinacol rearrangement), for iodohydrins, for which the reagent is mercuric oxide or silver nitrate, for β-hydroxyalkyl selenides [R1R2C(OH)C(SeR5)R3R4],134 and for allylic alcohols,135 which can rearrange on treatment with a strong acid that protonates the double bond. A related rearrangement is the Et2Zn mediated rearrangement of bromohydrins to give ketones.136
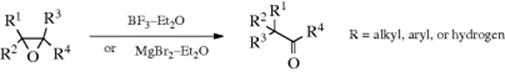
A similar rearrangement is given by epoxides, when treated with acidic reagents (e.g., BF3–etherate or MgBr2–etherate), 5M LiClO4 in ether,137 InCl3,138 Bi(OTf)3,139 or sometimes by heat alone.140 Epoxides are converted to aldehydes or ketones on treatment with certain metallic catalysts141 including treatment with iron complexes,142 IrCl3,143 or with BiOClO4.144 Base-induced rearrangement is also known, but the products are usually different.145
The Meinwald rearrangement converts epoxides to carbonyl compounds.146 Several reagents mediate this transformation, including Cu compounds.147 A closely related reaction of vinyl epoxides gives alkenyl ketones upon treatment with Ga compounds.148 It has been shown that epoxides are intermediates in the pinacol rearrangements of certain glycols.149 Among the evidence for the mechanism given is that Me2COHCOHMe2, Me2COHC(NH2)Me2, and Me2COHCClMe2 gave the reaction at different rates (as expected), but yielded the same mixture of two products, pinacol and pinacolone, indicating a common intermediate.150
A good way to prepare β-diketones consists of heating α,β-epoxy ketones at 80–140 °C in toluene with small amounts of (Ph3P)4Pd and dppe.151 Epoxides are converted to 1,2-diketones with Bi, DMSO, O2 and a catalytic amounts of Cu(OTf)2 at 100 °C.152 α,β-Epoxy ketones are also converted to 1,2-diketones with a Ru catalyst153 or an Fe catalyst.154 Epoxides with an α-hydroxyalkyl substituent give a pinacol rearrangement product in the presence of a ZnBr2155 or Tb(OTf)3156 catalyst to give a γ-hydroxy ketone.
Oxaziridines are converted to ring-expanded lactams under photochemical conditions.157N-Tosyl aziridines with an α-hydroxyalkyl substituent give a pinacol rearrangement product in the presence of Lewis acids (e.g., SmI2), in this case a keto-N-tosyl amide.158
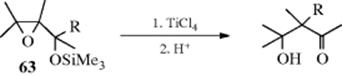
β-Hydroxy ketones can be prepared by treating the silyl ethers (63) of α,β-epoxy alcohols with TiCl4.159
OS I, 462; II, 73, 408; III, 312; IV, 375, 957; V, 326, 647; VI, 39, 320; VII, 129. See also, OS VII, 456.
18-3 Expansion and Contraction of Rings
Demyanov ring contraction; Demyanov ring expansion
![]()
When a positive charge is formed on an alicyclic carbon, migration of an alkyl group can take place to give ring contraction, producing a ring that is one carbon smaller than the original, as in the interconversion of the cyclobutyl cation and the cyclopropylcarbinyl cation (64). Note that this change involves conversion of a
![]()
secondary to a primary carbocation. In a similar manner, when a positive charge is placed on a carbon α to an alicyclic ring, ring expansion can take place.160 The new carbocation, and the old one, can then give products by combination with a nucleophile (e.g., the alcohols shown above), or by elimination, so that this reaction is a special case of 18-1. Often, both rearranged and unrearranged products are formed, so that, for example, cyclobutylamine and cyclopropylmethylamine give similar mixtures of the two alcohols shown above on treatment with nitrous acid (a small amount of 3-buten-1-ol is also produced). When the carbocation is formed by diazotization of an amine, the reaction is called the Demyanov rearrangement,161 but of course similar products are formed when the carbocation is generated in other ways. The expansion reaction has been performed on rings of C3–C8,162 but yields are best with the smaller rings, where relief of small-angle strain provides a driving force for the reaction. Strain is apparently much less of a factor in the cyclobutyl–cyclopropylmethyl interconversion (for a discussion of this interconversion, see Sec. 10.C.i). The influence of substituents on this rearrangement has been examined.163 Note that a hybrid of a [1,2]-sigmatropic hydrogen shift (also See 18-29) and a two-electron-electrocyclic ring opening has been discovered for cyclopropylcarbinyl cations that was labeled as a “hiscotropic” rearrangement.”164 The contraction reaction has been applied to four-membered rings and to rings of C6–C8, but contraction of a cyclopentyl cation to a cyclobutylmethyl system is generally not feasible because of the additional strain involved.
A related rearrangement involves cyclopropyl propargylic alcohols, which gives an alkylidene cyclobutanone in the presence of Ag and Au catalysts,165 or Ru and In catalysts.166 Cyclopropylcarbinyl rearrangements are catalyzed by ionic liquids under solvent-free conditions.167 Methylenecyclopropanes rearrange to cyclobutenes in the presence of 1 atm of CO and Pt catalyst168 or a Pd catalyst, mediated by a Cu catalyst.169 Arylvinylidenecyclopropanes rearrange to bicyclic systems in the presence of a Lewis acid.170

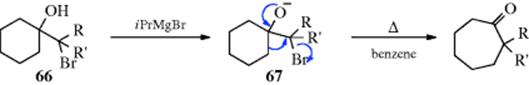
Ring expansions of certain hydroxyamines (e.g., 65) are analogous to the semipinacol rearrangement (Reaction 18-2). This reaction is called the Tiffeneau–Demyanov ring expansion. These have been performed on rings of C4–C8and the yields are better than for the simple Demyanov ring expansion. A similar reaction has been used to expand rings of from five to eight members.171 In this case, a cyclic bromohydrin of the form 66 is treated with a Grignard reagent, which, acting as a base, removes the OH proton to give the alkoxide 67. When 67 is heated to reflux, ring enlargement occurs. The reaction has been done with 66 in which at least one R group is phenyl or methyl,172 but fails when both R groups are hydrogen.173
A positive charge generated on a three-membered ring gives “contraction” to an allylic cation, as shown.174
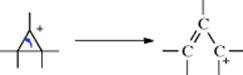
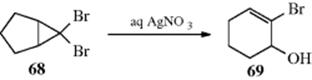
As seen in Section 10.G.i, category 7, this is the reason nucleophilic substitutions are not feasible at a cyclopropyl substrate. The reaction is often used to convert cyclopropyl halides and tosylates to allylic products, especially for the purpose of ring expansion, an example being the conversion of 68 to 69.175 The stereochemistry of these cyclopropyl cleavages is governed by the principle of orbital symmetry conservation (for a discussion, see Reaction 18-27, the Möbius–Hückel method).
Three-membered rings can also be cleaved to unsaturated products in at least two other ways. (1) Upon pyrolysis, cyclopropanes can undergo “contraction” to propenes.176 In the simplest case, cyclopropane gives propene when heated to 400–500 °C. The mechanism is generally regarded177 as involving a diradical
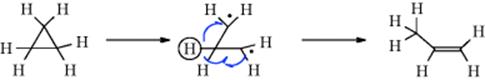
intermediate178 (recall that free radical 1,2-migration is possible for diradicals, Sec. 18.C). (2) The generation of a carbene or carbenoid carbon in a three-membered ring can lead to allenes, and allenes are often prepared in
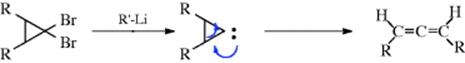
this way.179 Flash vacuum pyrolysis of 1-chlorocyclopropene thermally rearranges to chloroallene.180 One way to generate such a species is treatment of a 1,1-dihalocyclopropane with an alkyllithium compound (Reaction 12-39).181In contrast, the generation of a carbene or carbenoid at a cyclopropylmethyl carbon gives ring expansion.182
![]()
Some free radical ring enlargements are also known, an example being183:
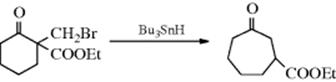
This reaction has been used to make rings of 6, 7, 8, and 13 members. A possible mechanism is
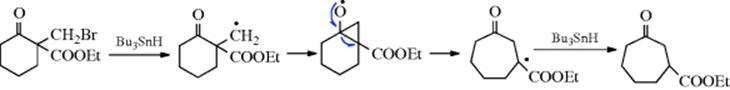
This reaction has been extended to the expansion of rings by three or four carbons, by the use of a substrate containing (CH2)nX (n = 3 or 4) instead of CH2Br.184 By this means, 5-, 6-, and 7-membered rings were enlarged to 18–11-membered rings. A β-keto ester (e.g., 2-carboxyethyl cyclohexanone) is converted to 3-carboxyethyl cylcoheptanone when treated with CF3CO2ZnCH2I.185
OS III, 276; IV, 221, 957; V, 306, 320; VI, 142, 187; VII, 12, 114, 117, 129, 135; VIII, 179, 467, 556, 578.
18-4 Acid-Catalyzed Rearrangements of Aldehydes and Ketones
1/Alkyl,2/alkyl-interchange, and so on
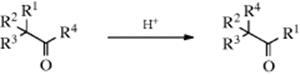
Rearrangements of this type, where a group α to a carbonyl “changes places” with a group attached to the carbonyl carbon, occur when migratory aptitudes are favorable.186 The R2, R3, and R4 groups may be alkyl or hydrogen. Certain aldehydes have been converted to ketones, and ketones to other ketones (though more drastic conditions are required for the latter), but no rearrangement of a ketone to an aldehyde (R1 = H) has so far been reported. There are two mechanisms,187 each beginning with protonation of the oxygen and each involving two migrations. In one pathway, the migrations are in opposite directions188:
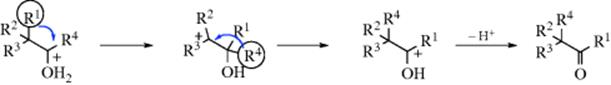
In the other pathway, the migrations are in the same direction. The actual mechanism of this pathway is not certain, but an epoxide (protonated) intermediate189 is one possibility190:
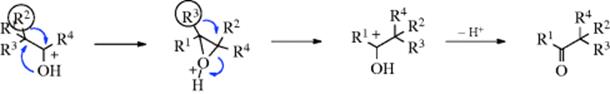
If the reaction is carried out with ketone labeled in the C=O group with ![]() , the first pathway predicts that the product will contain all the
, the first pathway predicts that the product will contain all the ![]() in the C=O carbon, while in the second pathway the label will be in the α carbon (demonstrating migration of oxygen). The results of such experiments191 have shown that in some cases only the C=O carbon was labeled, in other cases only the α carbon, while in still others both carbons bore the label, indicating that in these cases both pathways were in operation. With α-hydroxy aldehydes and ketones, the process may stop after only one migration (this is called the α-ketol rearrangement).
in the C=O carbon, while in the second pathway the label will be in the α carbon (demonstrating migration of oxygen). The results of such experiments191 have shown that in some cases only the C=O carbon was labeled, in other cases only the α carbon, while in still others both carbons bore the label, indicating that in these cases both pathways were in operation. With α-hydroxy aldehydes and ketones, the process may stop after only one migration (this is called the α-ketol rearrangement).
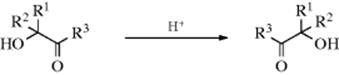
The α-ketol rearrangement can also be brought about by base catalysis, but only if the alcohol is tertiary, since if R1 or R2 = hydrogen, enolization of the substrate is more favored than rearrangement.
18-5 The Dienone–Phenol Rearrangement
2/ C→5/ O-Hydro,1/ C→2/ C-alkyl-bis -migration
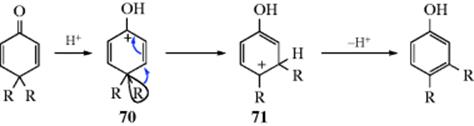
Cyclohexadienone derivatives that have two alkyl groups in the 4 position undergo, on acid treatment,192 1,2-migration of one of these groups from 70 to give the phenol. Note that a photochemical version of this reaction has been observed.193 The driving force in the overall reaction (the dienone–phenol rearrangement) is of course creation of an aromatic system.194 Note that 70 and 71 are arenium ions (Sec. 5.A.ii), the same as those generated by attack of a phenol on an electrophile.195 Sometimes, in the reaction of a phenol with an electrophile, a kind of reverse rearrangement (called the phenol–dienone rearrangement) takes place, though without an actual migration.196 An example is
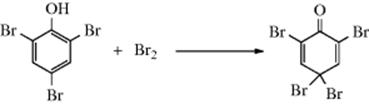
18-6 The Benzil–Benzilic Acid Rearrangement
1/ O-Hydro,3/oxido-(1/→2/aryl)- migro-addition
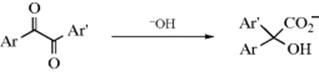
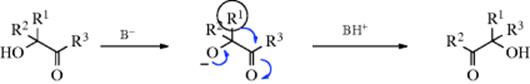
When treated with base, α-diketones rearrange to give the salts of α-hydroxy acids, a reaction known as the benzil–benzilic acid rearrangement (benzil is PhCOCOPh; benzilic acid is Ph2COHCO2H).197 A Rh catalyzed version of this reaction has also been reported.198 Though the reaction is usually illustrated with aryl groups, it can also be applied to aliphatic diketones199 and to α-keto aldehydes. The use of an alkoxide instead of hydroxide gives the corresponding ester directly,200 although alkoxide ions that are readily oxidized (e.g., OEt− or OCHMe2−) are not useful here, since they reduce the benzil to a benzoin. The mechanism is similar to the rearrangements in Reaction 18-1–18-4, but there is a difference: The migrating group does not move to a carbocation. The first step is attack of the base at the carbonyl group, the same as the first step of the tetrahedral mechanism of nucleophilic substitution (Sec. 16.A.i) and of many additions to the C=O bond (Chapter 16):
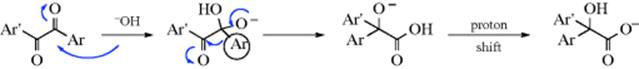
The mechanism has been intensely studied,188 and there is much evidence for it.201 The reaction is irreversible.
OS I, 89.
18-7 The Favorskii Rearrangement
2/Alkoxy-de-chloro(2/→1/alkyl)- migro-substitution
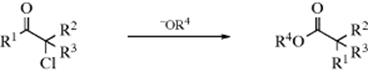
The reaction of α-halo ketones (chloro, bromo, or iodo) with alkoxide ions202 to give rearranged esters is called the Favorskii rearrangement.203 The use of hydroxide ions or amines as bases leads to the free carboxylic acid (salt) or amide, respectively, instead of the ester. Cyclic α-halo ketones give ring contraction, as in the conversion of 72 to 73.
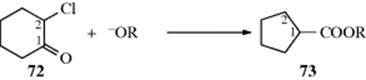
The reaction has also been carried out on α-hydroxy ketones204 and on α,β-epoxy ketones, which give β-hydroxy acids.205 The fact that an epoxide gives a reaction analogous to a halide indicates that the oxygen and halogen are leaving groups in a nucleophilic substitution step.
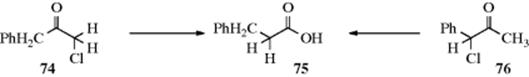
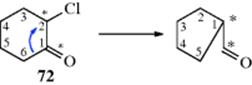
Investigation of the mechanism206 of the Favorskii rearrangement has led to proposals for at least five different mechanisms. However, the finding207 that 74 and 75 both give 76 (this behavior is typical) shows that any mechanism where the halogen leaves and R1 takes its place is invalid, since in such a case 74 would be expected to give 76 (with PhCH2 migrating), but 75 should give PhCHMeCOOH (with CH3 migrating). That is, in the case of 75, it was PhCH that migrated and not methyl. Another important result was determined by radioactive labeling. Chloroketone (72), in which C-1 and C-2 were equally labeled with ![]() , was converted to 73. The product was found to contain 50% of the label on the carbonyl carbon, 25% on C-1, and 25% on C-2.208 Now the carbonyl carbon, which originally carried half of the radioactivity, still had this much, so the rearrangement did not directly affect it. However, if the C-6 carbon had migrated to C-2, the other half of the radioactivity would be only on C-1 of the product:
, was converted to 73. The product was found to contain 50% of the label on the carbonyl carbon, 25% on C-1, and 25% on C-2.208 Now the carbonyl carbon, which originally carried half of the radioactivity, still had this much, so the rearrangement did not directly affect it. However, if the C-6 carbon had migrated to C-2, the other half of the radioactivity would be only on C-1 of the product:
On the other hand, if the migration had gone the other way (if the C-2 carbon had migrated to C-6), then this half of the radioactivity would be found solely on C-2 of the product. The fact that C-1 and C-2 were equally labeled showed that both migrations occurred, with equal probability. Since C-2 and C-6 of 72 are not equivalent, this means that there must be a symmetrical intermediate.209 The type of intermediate that best fits the circumstances is a cyclopropanone,210 and the mechanism (for the general case) is formulated (replacing R1 of our former symbolism with CHR5R6, since it is obvious that for this mechanism an α hydrogen is required on the non-halogenated side of the carbonyl):
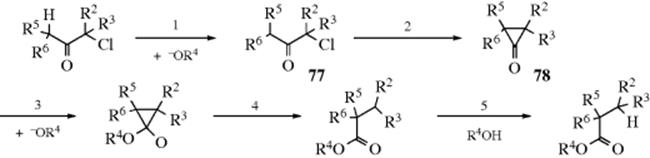
The intermediate corresponding to 78, in the case of 72, is a symmetrical compound, and the three-membered ring can be opened with equal probability on either side of the carbonyl, accounting for the results with 14C. In the general case, 78 is not symmetrical and should open on the side that gives the more stable carbanion.211 This accounts for the fact that 74 and 75 give the same product. The intermediate in both cases is 77, which always opens to give the carbanion stabilized by resonance. The cyclopropanone intermediate (78) has been isolated in the case where R2 = R5 = t-Bu and R3 = R6 = H,212 and it has also been trapped.213 Also, cyclopropanones synthesized by other methods have been shown to give Favorskii products on treatment with NaOMe or other bases.214
The mechanism discussed is in accord with all the facts when the halo ketone contains an α hydrogen on the other side of the carbonyl group. However, ketones that do not have an α hydrogen also rearrange to give the same type of product in what is usually called the quasi-Favorskii rearrangement. The quasi-Favorskii rearrangement cannot take place by the cyclopropanone mechanism. The mechanism that is generally accepted (called the semibenzilic mechanism215) is a base-catalyzed pinacol rearrangement-type mechanism similar to that of 18-6. This mechanism requires inversion at the migration terminus and this has been found.216 It has been shown that even where there is an appropriately situated α hydrogen, the semibenzilic mechanism may still operate.217
An interesting analogue of the Favorskii rearrangement treats a ketone, (e.g., 4-tert-butylcyclohexanone), without an α-halogen with Tl(NO3)3 to give 3-tert-butylcyclopentane-1-carboxylic acid.218
OS IV, 594; VI, 368, 711.
18-8 The Arndt–Eistert Synthesis
![]()
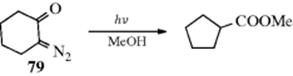
In the Arndt–Eistert synthesis, an acyl halide is converted to a carboxylic acid with one additional carbon.219 The first step of this process is Reaction 16-89. The actual rearrangement occurs in the second step after treatment of the diazo ketone with water and silver oxide or with silver benzoate and triethylamine. This rearrangement is called the Wolff rearrangement.220 It is the best method of increasing a carbon chain by one from a carboxylic acid (see Reaction 10-75 and 16-30). If an alcohol (R′OH) is used instead of water, the ester (RCH2CO2R′) is isolated.221 Similarly, treatment with ammonia gives the amide. Other catalysts are sometimes used (colloidal Pt, Cu, etc.), but occasionally the diazo ketone is simply heated or photolyzed in the presence of water, an alcohol, or ammonia, with no catalyst at all using ultrasound.222 The photolysis method223 often gives better results than the Ag catalysis method. Of course, diazo ketones prepared in any other way also give the rearrangement.224 The reaction is of wide scope. The R group may be alkyl or aryl and may contain many functional groups including unsaturation, but not including groups acidic enough to react with CH2N2 or diazo ketones (e.g., Reaction 10-5 and 10-19). Sometimes the reaction is performed with other diazoalkanes (i.e., R′CHN2) to give RCHR′COOH. The reaction has been used for ring contraction of cyclic diazo ketones225 (e.g., 79).226
An asymmetric variation converted ketones to esters using an azaferrocene catalyst.227
The mechanism is generally regarded as involving formation of a carbene.228 It is the divalent carbon that has the open sextet and to which the migrating group brings its electron pair:
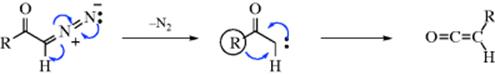
The actual product of the reaction is thus the ketene, which then reacts with water (15-3), an alcohol (15-5), or ammonia or an amine (15-8). Particularly stable ketenes229 (e.g., Ph2C=C=O) have been isolated and others have been trapped in other ways (e.g., as β-lactams,230 reaction 16-96). The purpose of the catalyst is not well understood, though many suggestions have been made. This mechanism is strictly analogous to that of the Curtius rearrangement(Reaction 18-14). Although the mechanism as shown above involves a free carbene and there is much evidence to support this,231 it is also possible that at least in some cases the two steps are concerted and a free carbene is absent.
When the Wolff rearrangement is carried out photochemically, the mechanism is basically the same,223 but another pathway can intervene. Some of the ketocarbene originally formed can undergo a carbene–carbene rearrangement, through an oxirene intermediate.232 This was shown by ![]() labeling experiments, where
labeling experiments, where
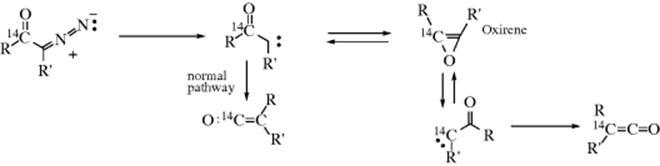
diazoketones labeled in the carbonyl group gave rise to ketenes that bore the label at both C=C carbons.233 In general, the smallest degree of scrambling (and thus of the oxirene pathway) was found when R′ = H. An intermediate believed to be an oxirene has been detected by laser spectroscopy.234 The oxirene pathway is not found in the thermal Wolff rearrangement. It is likely that an excited singlet state of the carbene is necessary for the oxirene pathway to intervene.235 In the photochemical process, ketocarbene intermediates, in the triplet state, have been isolated in an Ar matrix at 10–15 K, where they have been identified by UV–visible, IR, and ESR spectra.236 These intermediates went on to give the rearrangement via the normal pathway, with no evidence for oxirene intermediates.
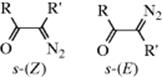
The diazo ketone can exist in two conformations, called s-(E) and s-(Z). Studies have shown that Wolff rearrangement takes place preferentially from the s-(Z) conformation.237
OS III, 356; VI, 613, 840.
18-9 Homologation of Aldehydes and Ketones
Methylene-insertion
![]()
Aldehydes and ketones238 can be converted to their homologues with diazomethane.239 Several other reagents240 are also effective, including Me3SiI, and then silica gel.241 With the diazomethane reaction, formation of an epoxide (16-46) is a side reaction. Superficially, this reaction appears to be similar to the insertion of carbenes into C–H bonds, (Reaction 12-21, and IUPAC names it as an insertion), but the mechanism is quite different. However, it is a true rearrangement and no free carbene is involved. The first step is an addition to the C=O bond:
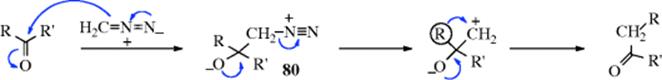
The betaine (80) can sometimes be isolated. As shown in Reaction 16-46, intermediate 80 can also go to the epoxide. The evidence for this mechanism has been summarized in the review by Gutsche.239 Note that this mechanism is essentially the same as in the apparent “insertions” of oxygen (Reaction 18-19) and nitrogen (Reaction 18-16) into ketones.
1,3-Diketones are converted to 1,4-diketones upon treatment with CF3CO2ZnCH2I.242 In a related reaction, alkenes insert into aldehydes in the presence of a Rh catalyst to give the corresponding ketone.243
Aldehydes give fairly good yields of methyl ketones; that is, hydrogen migrates in preference to alkyl. The most abundant side product is not the homologous aldehyde, but the epoxide. However, the yield of aldehyde at the expense of methyl ketone can be increased by the addition of methanol. If the aldehyde contains electron-withdrawing groups, the yield of epoxides is increased and the ketone is formed in smaller amounts, if at all. Ketones give poorer yields of homologous ketones. Epoxides are usually the predominant product here, especially when one or both R groups contain an electron-withdrawing group. The yield of ketones also decreases with increasing length of the chain. The use of a Lewis acid increases the yield of ketone.244 Cyclic ketones,245 three membered246 and larger, behave particularly well and give good yields of ketones with the ring expanded by one.247 Aliphatic diazo compounds (RCHN2 and R2CN2) are sometimes used instead of diazomethane, with the expected results.248 Ethyl diazoacetate can be used analogously, in the presence of a Lewis acid or of triethyloxonium fluoroborate,249 to give a β-keto ester, (e.g., 81).
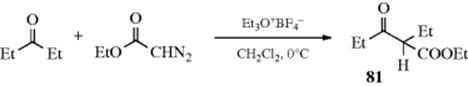
When unsymmetrical ketones were used in this reaction (with BF3 as catalyst), the less highly substituted carbon preferentially migrated.250 The reaction can be made regioselective by applying this method to the α-halo ketone, in which case only the other carbon migrates.251 The ethyl diazoacetate procedure has also been applied to the acetals or ketals of α,β-unsaturated aldehydes and ketones.252
Bicyclic ketones can be expanded to form monocyclic ketones in the presence of certain reagents. Treatment of a bicyclo[4.1.0]hexan-4-one derivative with SmI2 led to a cyclohexanone.253 The SmI2 also converts α-halomethyl cyclic ketones to the next larger ring ketone254 and cyclic ketones to the next larger ring ketone in the presence of CH2I2.255
Another homologation reaction converts an aldehyde to its tosyl hydrazone, and subsequent reaction with an aldehyde and NaOEt/EtOH give a ketone.256 The reaction of an aldehyde with vinyl acetate and Ba(OH)2 gives the homologated conjugated aldehyde.257
OS IV, 225, 780. For homologation of carboxyl acid derivatives, see OS IX, 426
B. Carbon-to-Carbon Migrations of Other Groups
18-10 Migrations of Halogen, Hydroxyl, Amino, and so on
Hydroxy-de-bromo- cine-substitution, and so on
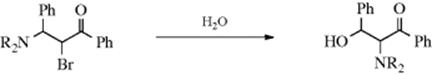
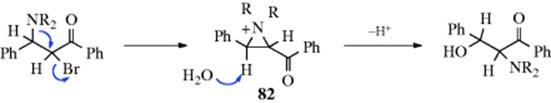
When a nucleophilic substitution is carried out on a substrate that has a neighboring group (Sec. 10.C) on the adjacent carbon, a cyclic intermediate can be generated that is opened on the opposite side, resulting in migration of the neighboring group. In the example shown above (NR2 = morpholino),258 the reaction took place via an aziridinium salt (82) to give an α-amino-β-hydroxy ketone. Sulfonate esters and halides can also migrate in this reaction.259
α-Halo and α-acyloxy epoxides undergo ready rearrangement to α-halo and α-acyloxy ketones, respectively.260 These substrates are prone to rearrange, and often do so upon standing without a catalyst, although an acid catalyst is necessary in some cases. The reaction is essentially the same as the rearrangement of epoxides shown in 18-2, except that halogen or acyloxy is the migrating group (as shown above; however, it is also possible for one of the R groups (alkyl, aryl, or hydrogen) to migrate instead, and mixtures are sometimes obtained). In a related reaction, α-bromoaziridines undergo rearrangement to the isomerized α-bromoaziridine in the presence of MgBr2.261
In the presence of a Cu catalyst, alkenyl epoxides (vinyl oxiranes) rearrange to a 2,5-dihydrofuran.262 Alkenyl thiiranes are similarly converted to 2,5-dihydrothiophenes with a Cu catalyst.263
Allylic alcohols migrate to give a new allylic alcohol in the presence of a Re catalyst. An example is the conversion of 83 to 84.264 Variations using Rh265 or Ir266 catalysts are known, and methanesulfonic acid catalyzes the isomerization.267 In the presence of a Ru catalyst, an allylic alcohol was isomerized to an aliphatic ketone.268 There is a similar Au catalyzed isomerization of allylic acetates.269

The Meyer–Schuster Rearrangement is an acid-catalyzed rearrangement of a propargyl alcohol to a conjugated carbonyl compound.270 Rearrangement was also catalyzed by a cationic Rh–bisphosphane complex.271 An Au catalyzed rearrangement of ethoxyalkynyl carbinols gave α,β-unsaturated esters.272 The base-induced isomerization of a propargylic alcohol gave a conjugated ketone,273 and a combination of Mo–Au led to rapid 1,3-rearrangement of propargyl alcohols.274 An Au–Ag catalyzed reaction with propargyl esters gave a 2-O-pivaloyl conjugated aldehyde.275 A similar reaction with a Pt catalyst converted a 1-ethoxy propargylic ester to a 2-carboethoxy conjugated ketone.276 An Au catalyzed isomerization of allenyl carbinol esters is also known.277
18-11 Migration of Boron
Hydro,dialkylboro-interchange, and so on
![]()
Boranes are prepared by the reaction of BH3 (B2H6) or an alkylborane with an alkene (15-16). When a nonterminal borane is heated at temperatures ranging from 100 to 200 °C, the boron moves toward the end of the chain.278The reaction is catalyzed by small amounts of borane or other species containing B–H bonds. The boron can move past a branch, for example,
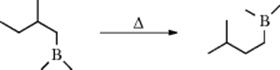
but not past a double branch, for example,
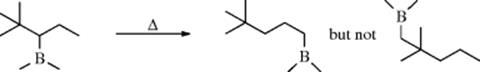
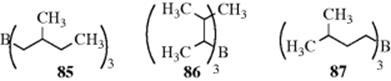
The reaction is an equilibrium: 85, 86, and 87 each gave a mixture containing ~40% 85, 1% 86, and 59% 87. The migration can go quite a long distance, including a migration of 11 positions.279 If the boron is on a cycloalkyl ring, it can move around the ring; if any alkyl chain is also on the ring, the boron may move from the ring to the chain, ending up at the end of the chain.280 The reaction is useful for the migration of double bonds in a controlled way (see 12-2). The mechanism may involve a π complex, at least partially.281
18-12 The Neber Rearrangement
Neber oxime tosylate-amino ketone rearrangement
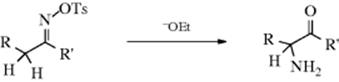
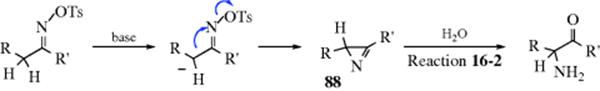
α-Amino ketones can be prepared by treatment of ketoxime tosylates with a base (e.g., ethoxide or pyridine).282 This reaction is called the Neber rearrangement. The R group is usually aryl, although the reaction has been carried out with R = alkyl or hydrogen. The R′ group may be alkyl or aryl, but not hydrogen. The Beckmann rearrangement (Reaction 18-17) and the abnormal Beckmann reaction (elimination to the nitrile, Reaction 17-30) may be side reactions, although these generally occur in acid media. A similar rearrangement is given by N,N-dichloroamines of the type RCH2CH(NCl2)R′, where the product is also RCH(NH2)COR′.283 The mechanism of the Neber rearrangement involves an azirine intermediate (88).284 The best evidence for this mechanism is that the azirine intermediate has been isolated.285, In contrast to the Beckmann rearrangement, this one is sterically indiscriminate:286Both a syn and an anti ketoxime give the same product. The mechanism, as shown above, consists of three steps. However, it is possible that the first two steps are concerted, and it is also possible that what is shown as the second step is actually two steps: loss of OTs to give a nitrene, and formation of the azirine. In the case of the dichloroamines, HCl is first lost to give RCH2C(=NCl)R′, which then behaves analogously.287N-Chloroimines prepared in other ways also give the reaction.288 Indoles have been prepared via a Neber rearrangement.289
OS V, 909; VII, 149.
C. Carbon-to-Nitrogen Migrations of R and Ar
The reactions in this group are nucleophilic migrations from a carbon to a nitrogen atom. In each case, the nitrogen atom either has six electrons in its outer shell (and thus invites the migration of a group carrying an electron pair) or else loses a nucleofuge concurrently with the migration (Sec. 18.A.i). Reactions 18-13–18-16 are used to prepare amines from acid derivatives. Reactions 18-16 and 18-17 are used to prepare amines from ketones. The mechanisms of Reaction 18-13–18-16 (with carboxylic acids) are very similar and follow one of two patterns:
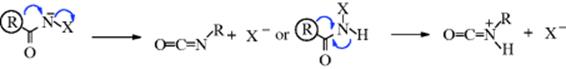
Some of the evidence290 is (1) configuration is retained in R (Sec. 18.A.ii); (2) the kinetics are first order; (3) intramolecular rearrangement is shown by labeling; and (4) no rearrangement occurs within the migrating group, for example, a neopentyl group on the carbon of the starting material is still a neopentyl group on the nitrogen of the product.
In many cases, it is not certain whether the nucleofuge X is lost first, creating an intermediate nitrene291 or nitrenium ion, or whether migration and loss of the nucleofuge are simultaneous, as shown above.292 It is likely that both possibilities can exist, depending on the substrate and reaction conditions.
18-13 The Hofmann Rearrangement
Bishydrogen-(2/→1/ N-alkyl)- migro-detachment (formation of isocyanate)
![]()
In the Hofmann rearrangement, an unsubstituted amide is treated with sodium hypobromite (or sodium hydroxide and bromine, which is essentially the same thing) to give an isocyanate, but this compound is seldom isolated293since it is usually hydrolyzed under the reaction conditions. The final isolated product is a primary amine that has one carbon fewer than the starting amide.294 The R group may be alkyl or aryl, but if it is an alkyl group of more than about six or seven carbons, low yields are obtained unless Br2 and NaOMe are used instead of Br2 and NaOH.295 Another modification uses NBS/NaOMe.296 Under these conditions, the product of addition to the isocyanate is the carbamate (RNHCOOMe, Reaction 16-8), which is easily isolated or can be hydrolyzed to the amine.297 A mixture of NBS and DBU (see Reaction 17-13) in methanol gives the carbamate,298 as does electrolysis in methanol.299
Side reactions when NaOH is the base are formation of ureas (RNHCONHR) and acylureas (RCONHCONHR) by addition, respectively, of RNH2 and RCONH2 to RNCO (16-20). If acylureas are desired, they can be made the main products by using only one-half of the usual quantities of Br2 and NaOH. Another side product, but only from primary R, is the nitrile derived from oxidation of RNH2 (Reaction 19-5).
Imides react to give amino acids, (e.g., phthalimide gives o-aminobenzoic acid). α-Hydroxy and α-halo amides give aldehydes and ketones by way of the unstable α-hydroxy- or α-haloamines. However, a side product with an α-halo amide is a gem-dihalide. Ureas analogously give hydrazines.
The mechanism follows the pattern outlined in the discussion preceding Reaction 18-13.
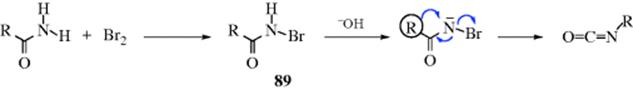
The first step is an example of Reaction 12-52 and intermediate N-halo amides (89) have been isolated. Compound 89 is acidic because of the presence of two electron-withdrawing groups (acyl and halo) on the nitrogen, and in the second step, 89 loses a proton to the base. It is possible that the third step is actually two steps: loss of bromide to form a nitrene, followed by the actual migration, but most of the available evidence favors the concerted reaction.300A similar reaction can be effected by the treatment of amides with lead tetraacetate.301 Among other reagents that convert RCONH2 to RNH2 (R = alkyl, but not aryl) are phenyliodosyl bis(trifluoroacetate) [PhI(OCOCF3)2]302 and hydroxy(tosyloxy)iodobenzene [PhI(OH)OTs].303
A variation of the Hofmann rearrangement treated a β-hydroxy primary amide with PhI(O2CCF3)3 in aq acetonitrile, giving an isocyanate via –CON-I, which reacts with the hydroxyl group intramolecularly to give a cyclic carbamate.304 Note that carbamates are converted to isocyanates by heating with Montmorillonite K-10.305
OS II, 19, 44, 462; IV, 45; VIII, 26, 132.
18-14 The Curtius Rearrangement
Dinitrogen-(2/→1/ N-alkyl)- migro-detachment
![]()
The Curtius rearrangement involves heating acyl azides to yield isocyanates.306 The reaction gives good yields of isocyanates, since no water is present to hydrolyze them to the amine. Of course, they can be subsequently hydrolyzed, and indeed the reaction can be carried out in water or alcohol, in which case the products are amines, carbamates, or acylureas, as in 18-13.307 This is a very general reaction and can be applied to almost any carboxylic acid: aliphatic, aromatic, alicyclic, heterocyclic, unsaturated, and containing many functional groups. Acyl azides can be prepared as in Reaction 10-43 or by treatment of acylhydrazines (hydrazides) with nitrous acid (analogous to Reaction 12-49). The Curtius rearrangement is catalyzed by Lewis acids or protic acids, but these are usually not necessary for good results.
The mechanism is similar to that in Reaction 18-13 to give an isocyanate. Also note the exact analogy between this Reaction and 18-8. However, in this case, there is no evidence for a free nitrene and it is probable that the conversion is concerted.308
![]()
Alkyl azides can be similarly pyrolyzed to give imines, in an analogous reaction309:
![]()
The R groups may be alkyl, aryl, or hydrogen, although if hydrogen migrates, the product is the unstable R2C=NH. The mechanism is essentially the same as that of the Curtius rearrangement. However, in pyrolysis of tertiary alkyl azides, there is evidence that free alkyl nitrenes are intermediates.310 The reaction can also be carried out with acid catalysis, in which case lower temperatures can be used, although the acid may hydrolyze the imine (16-2). Cycloalkyl azides give ring expansion as shown.311
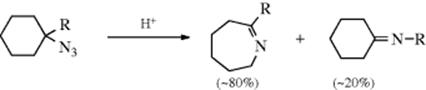
Aryl azides also give ring expansion on heating, for example,312
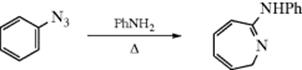
OS III, 846; IV, 819; V, 273; VI, 95, 910. Also see, OS VI, 210.
18-15 The Lossen Rearrangement
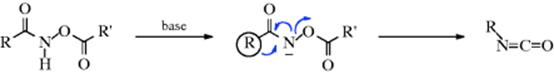
Hydro,acetoxy-(2/→1 N-alkyl)- migro-detachment
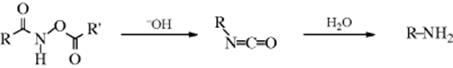
The O-acyl derivatives of hydroxamic acids313 give isocyanates when treated with bases or sometimes even just on heating, in a reaction known as the Lossen rearrangement.314 The mechanism is similar to that of Reaction 18-13and 18-14:
In a similar reaction, aromatic acyl halides are converted to amines in one laboratory step by treatment with hydroxylamine-O-sulfonic acid.315
A chiral Lossen rearrangement is known.316
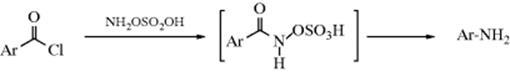
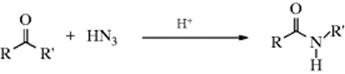
18-16 The Schmidt Reaction
![]()
There are actually three reactions called by the name Schmidt reaction, involving the addition of hydrazoic acid to carboxylic acids, aldehydes and ketones, and alcohols and alkenes.317 The most common is the reaction with carboxylic acids, illustrated above.318 Sulfuric acid is a common catalyst, but Lewis acids have also been used. Good results are obtained for aliphatic R, especially for long chains. When R is aryl, the yields are variable, being best for sterically hindered compounds like mesitoic acid. This method has the advantage over Reaction 18-13 and 18-14 in that there is just one laboratory step from the acid to the amine, but conditions are more drastic.319 Under the acid conditions employed, the isocyanate is virtually never isolated.
The reaction between a ketone and hydrazoic acid is a method for “insertion” of NH between the carbonyl group and one R group, converting a ketone into an amide.320
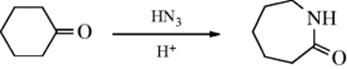
Either or both of the R groups may be aryl. In general, dialkyl ketones and cyclic ketones react more rapidly than alkyl aryl ketones, and these more rapidly than diaryl ketones. The latter require sulfuric acid and do not react in concentrated HCl, which is strong enough for dialkyl ketones. Dialkyl and cyclic ketones react sufficiently faster than diaryl or aryl alkyl ketones or carboxylic acids or alcohols so that these functions may be present in the same molecule without interference. Cyclic ketones give lactams321: With alkyl aryl ketones, it is the aryl group that generally migrates to the nitrogen, except when the alkyl group is bulky.322
The reaction has been applied to a few aldehydes, but rarely. With aldehydes the product is usually the nitrile (Reaction 16-16). Even with ketones, conversion to the nitrile is often a side reaction, especially with the type of ketone that gives 17-30. A useful variation of the Schmidt reaction treats a cyclic ketone with an alkyl azide (RN3)323 in the presence of TiCl4, generating a lactam.324 An intramolecular Schmidt reaction gives bicyclic amines.325 Another variation treats a silyl enol ether of a cyclic ketone with TMSN3 and photolyzes the product with UV light to give a lactam.326 An α–azido cyclic ketone rearrangement to lactams under radical conditions (Bu3SnH/AIBN).327
Alcohols and alkenes react with HN3 to give alkyl azides,328 which in the course of reaction rearrange in the same way as discussed in Reaction 18-14.309 The Mitsunobu Reaction (10-17) can be used to convert alcohols to alkyl azides, and an alternative reagent for azides [(PhO)2PON3], for use in the Mitsunobu is now available.329 In the presence of an Au catalyst, an acetylenic azide was converted to a pyrrole derivative.330 An intramolecular Schmidt reaction gives bicyclic lactams in the presence of MeAlCl2.331
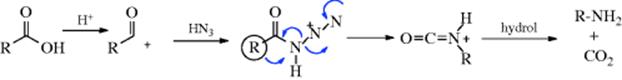
There is evidence that the mechanism with carboxylic acids320 is similar to that of Reaction 18-14, except that it is the protonated azide that undergoes the rearrangement332:
The first step is the same as that of the AAC1 mechanism (Reaction 16-59, which explains why good results are obtained with hindered substrates. The mechanism with ketones involves formation of a nitrilium ion (82), which reacts with water.
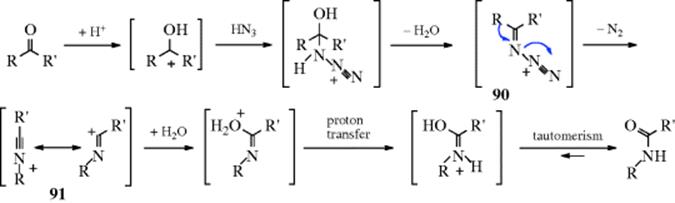
Intermediates (e.g., 90) have been independently generated in aqueous solution.333 Note the similarity of this mechanism to those of “insertion” of CH2 (Reaction 18-9) and of O (Reaction 18-19). The three reactions are essentially analogous, both in products and in mechanism.334,320 Also note the similarity of the latter part of this mechanism to that of the Beckmann rearrangement (Reaction 18-17).
OS V, 408; VI, 368; VII, 254; X, 207. See also, OS V, 623.
18-17 The Beckmann Rearrangement
Beckmann oxime–amide rearrangement
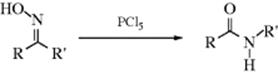
When oximes are treated with PCl5 or a number of other reagents, they rearrange to substituted amides in a reaction called the Beckmann rearrangement.335 Reagents used include concentrated H2SO4 acid, formic acid, liquid SO2, silica gel,336 RuCl3,337 Y(OTf)3,338 I2,339 HgCl2,340 triphosphazene,341 bromodimethylsulfonium bromide–ZnCl2,342 neat with FeCl3,343 cyanuric acid,344 and polyphosphoric acid.345 Simply heating the oxime of benzophenone neat leads to N-phenyl benzamide.346 The reaction has been done in supercritical water347 and in ionic liquids.348 A polymer-bound Beckman rearrangement has been reported.349 Microwave assisted Beckmann rearrangements are known.350 Note that the reaction of an imine with BF3·OEt2 and m-chloroperoxybenzoic acid leads to a formamide.351
The oximes of cyclic ketones give ring enlargement and form the lactam,352 as in the formation of caprolactam (see Reaction 18-16) from the oxime of cyclohexanone. Solvent-free reactions are known.353 Cyclic ketones can be converted directly to lactams in one laboratory step by treatment with NH2OSO2OH and formic acid (Reaction 16-14 takes place first, then the Beckmann rearrangement).354
Of the groups attached to the carbon of the C=N unit, the one that migrates in the Beckman rearrangement is generally the one anti to the hydroxyl, and this is often used as a method of determining the configuration of the oxime. However, it is not unequivocal. It is known that with some oximes the syn group migrates and that with others, especially where R and R′ are both alkyl, mixtures of the two possible amides are obtained. However, this behavior does not necessarily mean that the syn group actually undergoes migration. In most cases, the oxime undergoes isomerization under the reaction conditions before migration takes place.355 The scope of the reaction is quite broad and R and R′ may be alkyl, aryl, or hydrogen. However, hydrogen very seldom migrates, so the reaction is not generally a means of converting aldoximes to unsubstituted amides (RCONH2). This latter conversion can be accomplished, however, by treatment of the aldoxime with nickel acetate under neutral conditions356 or by heating the aldoxime for 60 h at 100 °C after it has been adsorbed onto silica gel.357 As in the case of the Schmidt rearrangement (Reaction 18-16), when the oxime is derived from an alkyl aryl ketone, it is generally the aryl group that preferentially migrates.358
Not only do oximes undergo the Beckmann rearrangement, but so also do esters of oximes with many acids, organic and inorganic. A side reaction with many substrates is the formation of nitriles (the “abnormal” Beckmann rearrangement, Reaction 17-30). The O-carbonates of imines (e.g., Ph2C=N–OCO2Et) react with BF3·OEt2 to give the corresponding amide in this case N-phenyl benzamide.359
In the first step of the mechanism, the OH group is converted by the reagent to a better leaving group, for example, proton acids convert it to OH2+. After that, the mechanism360 follows a course analogous to that for the Schmidt reaction of ketones (18-16) from the formation of nitrilium ion (92) on361: Alternative modes of reaction are possible. For example, when PCl5 is used to induce the reaction, a N-O-PCl4 species is formed, which generates 92. Intermediates of the form 92 have been detected by NMR and UV spectroscopy.362 The rearrangement has also been found to take place by a different mechanism, involving formation of a nitrile by fragmentation, and then addition by a Ritter Reaction (16-91).363 Beckmann rearrangements have also been carried out photochemically.364 A computational study compared converted versus stepwise mechanisms for the Beckmann rearrangement, and found that proton relay between the substrate and the solvent molecules controls the reaction, and migration and N–O bond scission occur simultaneously.365
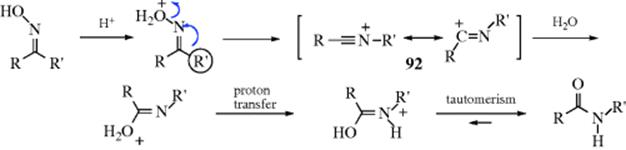
If the rearrangement of oxime sulfonates is induced by organoaluminum reagents,366 the nitrilium ion intermediate (92) is captured by the nucleophile originally attached to the Al. By this means an oxime can be converted to an imine, an imino thioether (R–N=C–SR), or an imino nitrile (R–N=C–CN).367 In the last case, the nucleophile comes from added trimethylsilyl cyanide. In the presence of LiI, 2-benzyloxypyridine is converted to N-benzyl-2-pyridone.368
In a related reaction, treatment of spirocyclic oxaziridines with MnCl(tpp) (tpp = triphenylphosphine, ligand)369 or photolysis370 leads to a lactam.
OS II, 76, 371; VIII, 568.
18-18 Stieglitz and Related Rearrangements
Methoxy-de- N-chloro-(2/→1/ N-alkyl)- migro-substitution, and so on
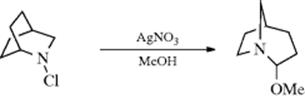
In addition to the reactions discussed at 18-13–18-17, other rearrangements are known in which an alkyl group migrates from C to N. Certain bicyclic N-haloamines (e.g., N-chloro-2-azabicyclo[2.2.2]octane, above), undergo rearrangement when solvolyzed in the presence of silver nitrate.371 This reaction is similar to the Wagner–Meerwein rearrangement (18-1) and is initiated by the silver-catalyzed departure of the chloride ion.372 Similar reactions have been used for ring expansions and contractions, analogous to those discussed for Reaction 18-3.373 An example is the conversion of 1-(N-chloroamino)cyclopropanols to β-lactams.374 Methyl prolinate was converted to the 2-piperidone upon treatment with SmI2 and pivalic acid–THF.375
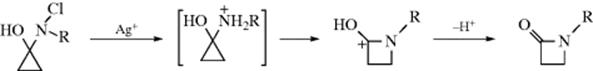
The name Stieglitz rearrangement is generally applied to the rearrangements of trityl N-haloamines and
![]()
hydroxylamines. These reactions are similar to the rearrangements of alkyl azides (18-14), and the name Stieglitz rearrangement is also given to the rearrangement of trityl azides. Another similar reaction is the rearrangement undergone by tritylamines when treated with lead tetraacetate (Ar3CNH2 → Ar2C=NAr.376
D. Carbon-to-Oxygen Migrations of R and Ar
18-19 The Baeyer–Villiger Rearrangement377
Oxy-insertion
![]()
The treatment of ketones with peroxyacids (e.g., peroxybenzoic or peroxyacetic acid or with other peroxy compounds in the presence of acid catalysts, gives carboxylic esters by migration of an alkyl group oxygen378 and the carboxylic acid parent of the peroxyacid as a byproduct. In other words, there is a C → O rearrangement, and the reaction is called the Baeyer–Villiger rearrangement.379 A particularly good reagent is peroxytrifluoroacetic acid. Reactions with this reagent are rapid and clean, giving high yields of product, although it is often necessary to add a buffer (e.g., Na2HPO4), to prevent transesterification of the product with trifluoroacetic acid that is also formed during the reaction. The reaction is often applied to cyclic ketones to give lactones.380 Hydrogen peroxide has been used to convert cyclic ketones to lactones using a catalytic amount of MeReO3381 or a diselenide catalyst.382Heterogeneous catalysts are used for the Baeyer–Villiger reaction.383 Transition metal catalysts have been used with peroxyacids to facilitate the oxidation.384 Polymer-supported peroxy acids have been used,385 and solvent-free Bayer–Villiger reactions are known.386 Potassium peroxomonosulfate supported on acidic silica gel has been used.387
Enantioselective synthesis388 of chiral lactones from achiral ketones has been achieved by the use of enzymes389 and other asymmetric reactions are known.390 Chiral Pd complexes give chiral lactones from cyclic ketones with high enantioselectively.391 Other chiral catalysts include those based on Al.392 Baeyer–Villiger oxidation of chiral substrates with m-chloroperoxybenzoic acid (mcpba) also leads to chiral lactones.393
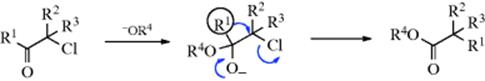
For acyclic compounds, R′ must usually be secondary, tertiary, or vinylic, although primary R′ has been rearranged with peroxytrifluoroacetic acid,394 with I2–H2O2,395 BF3–H2O2,396 and with K2S2O8–H2SO4.397 For unsymmetrical ketones, the approximate order of migration is tertiary alkyl > secondary alkyl, aryl > primary alkyl > methyl. Since the methyl group has a low migrating ability, the reaction provides a means of cleaving a methyl ketone (R′COMe) to produce an alcohol or phenol (R′OH, by hydrolysis of the ester R′OCOMe). The migrating ability of aryl groups is increased by electron-donating and decreased by electron-withdrawing substituents.398 There is a preference of anti- over gauche migration.399
Enolizable β-diketones do not react. α-Diketones can be converted to anhydrides.400 With aldehydes, migration of hydrogen gives the carboxylic acid, and this is a way of accomplishing Reaction 19-23. Migration of the other group would give formates, but this seldom happens, though aryl aldehydes have been converted to formates with H2O2 and a selenium compound401 (see also, the Dakin Reaction in 19-11).
The mechanism402 is similar to those of the analogous reactions with hydrazoic acid (18-16 with ketones) and diazomethane (18-8):
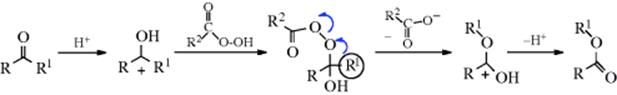
One important piece of evidence for this mechanism was that benzophenone–![]() gave ester entirely labeled in the carbonyl oxygen, with none in the alkoxyl oxygen.403 Carbon-14 isotope-effect studies on acetophenones have shown that migration of aryl groups takes place in the rate-determining step,404 demonstrating that migration of Ar is concerted with departure of OCOR2.405 It is hardly likely that migration would be the slow step if the leaving group departed first to give an ion with a positive charge on an oxygen atom, which would be a highly unstable species.
gave ester entirely labeled in the carbonyl oxygen, with none in the alkoxyl oxygen.403 Carbon-14 isotope-effect studies on acetophenones have shown that migration of aryl groups takes place in the rate-determining step,404 demonstrating that migration of Ar is concerted with departure of OCOR2.405 It is hardly likely that migration would be the slow step if the leaving group departed first to give an ion with a positive charge on an oxygen atom, which would be a highly unstable species.
18-20 Rearrangement of Hydroperoxides
C-Alkyl- O-hydroxy-elimination
![]()
Hydroperoxides (R = alkyl, aryl, or hydrogen) can be cleaved by proton or Lewis acids in a reaction whose principal step is a rearrangement.406 The reaction has also been applied to peroxy esters (R3COOCOR′), but less often. When aryl and alkyl groups are both present, migration of aryl dominates. It is not necessary actually to prepare and isolate hydroperoxides. The reaction takes place when the alcohols are treated with H2O2 and acids. Migration of an alkyl group of a primary hydroperoxide provides a means for converting an alcohol to its next lower homolo (RCH2OOH → CH2=O + ROH).
The mechanism is as follows407:
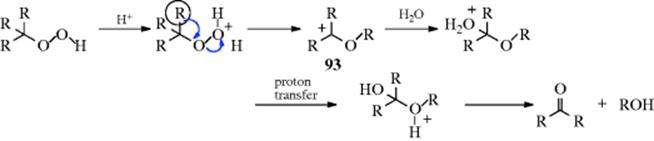
The last step is hydrolysis of the unstable hemiacetal. Alkoxycarbocation intermediates (93, R = alkyl) have been isolated in superacid solution408 at low temperatures, and their structures proved by NMr.409 The protonated hydroperoxides could not be observed in these solutions, evidently reacting immediately on formation.
OS V, 818.
E. Nitrogen-to-Carbon, Oxygen-to-Carbon, and Sulfur-to-Carbon Migration
18-21 The Stevens Rearrangement
Hydron-(2/ N→1/alkyl)- migro-detachment
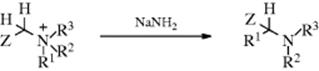
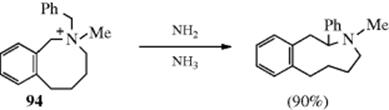
In the Stevens rearrangement,410 a quaternary ammonium salt containing an electron-withdrawing group Z on one of the carbons attached to the nitrogen is treated with a strong base (e.g., NaOR or NaNH2) to give a rearranged tertiary amine. The Z group may be RCO, ROOC, or phenyl.411 The most common migrating groups are allylic, benzylic, benzhydryl, 3-phenylpropargyl, and phenacyl, though even methyl migrates to a sufficiently negative center. Migration of aryl is rare, but has been reported.412 When an allylic group migrates, it may or may not involve an allylic rearrangement within the migrating group (see Reaction 18-35), depending on the substrate and reaction conditions. The reaction has been used for ring enlargement,413 illustrated by the rearrangement of 94.
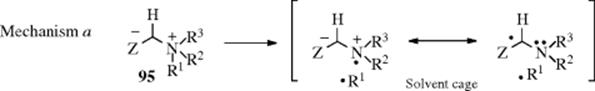
The mechanism has been the subject of much study.414 The rearrangement is intramolecular, as shown by cross-over experiments, by ![]() labeling,415 and by the fact that retention of configuration is found at R1.416 The first step is loss of the acidic proton to give the ylid (95), which has been isolated.417 The finding418 that CIDNP is observed419 in many instances shows that in these cases the product is formed directly from a free-radical precursor. The following radical pair mechanism was proposed420:
labeling,415 and by the fact that retention of configuration is found at R1.416 The first step is loss of the acidic proton to give the ylid (95), which has been isolated.417 The finding418 that CIDNP is observed419 in many instances shows that in these cases the product is formed directly from a free-radical precursor. The following radical pair mechanism was proposed420:
The radicals do not drift apart because the solvent cage holds them together. According to this mechanism, the radicals must recombine rapidly in order to account for the fact that R1 does not racemize. Other evidence in favor of mechanism a is that in some cases small amounts of coupling products (R1–R1) have been isolated,421 which would be expected if some √R1 leaked from the solvent cage. However, not all the evidence is easily compatible with mechanism a.422 It is possible that another mechanism (b), similar to mechanism a, but
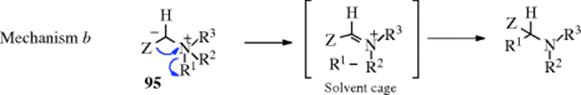
involving ion pairs in a solvent cage instead of radical pairs, operates in some cases. A third possible mechanism would be a concerted 1,2-shift,423 but the orbital symmetry principle requires that this take place with inversion at R1.424 (see Reaction 18-30 and [1,5]-migration). Since the actual migration takes place with retention, it cannot, according to this argument, proceed by a concerted mechanism. However, in the case where the migrating group is allylic, a concerted mechanism can also operate (Reaction 18-35). An interesting finding compatible with all three mechanisms is that optically active allylbenzylmethylphenylammonium iodide (asymmetric nitrogen, see Sec. 4.C, category 3) gave an optically active product425:
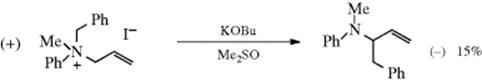
The Sommelet–Hauser rearrangement competes when Z is an aryl group (see Reaction 13-31). Hofmann elimination competes when one of the R groups contains a β hydrogen atom (Reaction 17-7 and 17-8).

Sulfur ylids containing a Z group give an analogous rearrangement (see the reaction), sometimes referred to as a Stevens rearrangement.426 In this case too, there is much evidence (including CIDNP) that a radical-pair cage mechanism is operating,427 except that when the migrating group is allylic, the mechanism may be different (see Reaction 18-35).
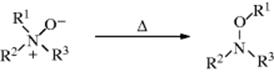
Another reaction with a similar mechanism428 is the Meisenheimer rearrangement,429 in which certain tertiary amine oxides rearrange on heating to give substituted hydroxylamines (see reaction).430 The migrating group R1 is almost always allylic or benzilic.431 Both R2 and R3 may be alkyl or aryl, but if one of the R groups contains a β hydrogen, Cope elimination (Reaction 17-9) often competes. In a related reaction, when 2-methylpyridine N-oxides are treated with trifluoroacetic anhydride, the Boekelheide reaction occurs to give 2-hydroxymethylpyridines.432
Certain tertiary benzylic amines, when treated with BuLi, undergo a rearrangement analogous to the Wittig rearrangement (Reaction 18-22, e.g., PhCH2NPh2 → Ph2CHNHPh).433 Only aryl groups migrate in this reaction.
Isocyanides, when heated in the gas phase or in nonpolar solvents, undergo a 1,2-intramolecular rearrangement to nitriles (RNC → RCN).434 In polar solvents, the mechanism is different.435
18-22 The Wittig Rearrangement436
Hydron-(2/ O→1/alkyl)- migro-detachment
![]()
The rearrangement of ethers upon treatment with alkyllithium reagents is called the Wittig rearrangement (not to be confused with the Wittig Reaction, 16-44) and is similar to 18-21.411 However, a stronger base is required (e.g., phenyllithium or sodium amide). The R and R′ groups may be alkyl,437 aryl, or vinylic.438 Also, one of the hydrogen atoms may be replaced by an alkyl or aryl group, in which case the product is the salt of a tertiary alcohol. Migratory aptitudes here are allylic, benzylic > ethyl > methyl > phenyl.439 The stereospecificity of the 1,2-Wittig rearrangement has been discussed.440 The following radical-pair mechanism441 (similar to mechanism a of Reaction 18-21) is likely, after removal of the proton by the base. One of the
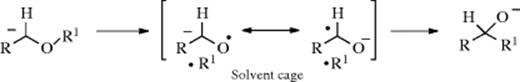
the radical pair is a ketyl. Evidence for this mechanism includes (1) the rearrangement is largely intramolecular; (2) migratory aptitudes are in the order of free-radical stabilities, not of carbanion stabilities442 (which rules out an ion-pair mechanism similar to mechanism b of Reaction 18-21); (3) aldehydes are obtained as side products443; (4) partial racemization of R′ has been observed444 (the remainder of the product retained its configuration); (5) cross-over products have been detected445; and (6) when ketyl radicals and R√ radicals from different precursors were brought together, similar products resulted.446 However, there is evidence that at least in some cases the radical-pair mechanism accounts for only a portion of the product, and some kind of concerted mechanism can also take place.447 Most of the above investigations were carried out with systems where R′ is alkyl, but a radical-pair mechanism has also been suggested for the case where R′ is aryl.448 When R′ is allylic a concerted mechanism can operate (Reaction 18-35).
When R is vinylic it is possible, by using a combination of an alkyllithium and t-BuOK, to get migration to the γ carbon (as well as to the α carbon), producing an enolate that, on hydrolysis, gives an aldehyde449:
![]()
An aza-Wittig rearrangement is also known.450 Other [2,3]-rearrangements are discussed in Reaction 18-35.
There are no OS references, but see OS VIII, 501, for a related reaction.
F. Boron-to-Carbon Migrations451
For another reaction involving boron-to-carbon migration, see 10-73.
18-23 Conversion of Boranes to Alcohols
![]()
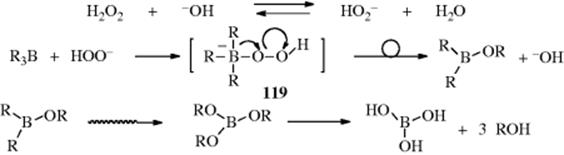
Oxidation of trialkylboranes (see Reaction 15-16) uses NaOH and H2O2, which react to give the hydroperoxide anion, (HOO−). Reaction of the organoborane with basic H2O2 (via HOO−) leads to an ate-complex, and subsequent B → O rearrangement of an alkyl group on boron to a peroxy oxygen, with expulsion of hydroxide, leads to a borate, and then an alcohol after hydrolysis. The proposed mechanism452 is shown in which a trialkylborane is converted to 3 molar equivalents of the alcohol, along with boric acid.
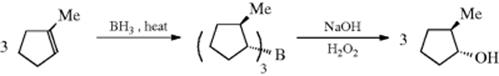
Using the hydroboration reaction in 15-16, this procedure converts alkenes to an anti-Markovnikov borane, and oxidation leads to the anti-Markovnikov alcohol. An example is the conversion of methylcyclopentene to trans-2-methylcyclopentanol.453 Formation of the organoborane proceeds via a cis-addition of B–H, placing the boron trans to the methyl group, and stereoselective oxidation and B → O rearrangement leads to retention of configuration in the alcohol, as shown.
Trialkylboranes can be prepared from alkenes by Reaction 15-16, and they react with carbon monoxide454 at 100–125 °C in the presence of ethylene glycol to give the 2-bora-1,3-dioxolanes (96), which are easily oxidized (Reaction 12-27) to tertiary alcohols.455 The R groups may be primary, secondary, or tertiary, and may be the same or different.456 Yields are high and the reaction is quite useful, especially for the preparation of sterically hindered alcohols (e.g., tricyclohexylcarbinol, 97 and tri-2-norbornylcarbinol, 98), which are difficult to prepare by Reaction 16-24. Heterocycles in which boron is a ring atom react similarly (except that high CO pressures are required), and cyclic alcohols can be obtained from these substrates.457 The preparation of such heterocyclic boranes was discussed at Reaction 15-16. The overall conversion of a diene or triene to a cyclic alcohol has been described by H.C. Brown as “stitching” with boron and “riveting” with carbon.
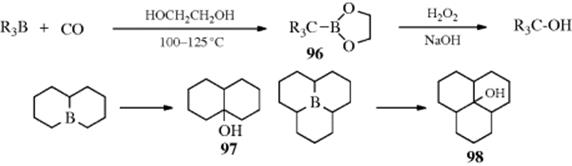
The mechanism has been shown to be intramolecular by the failure to find cross-over products when mixtures of boranes are used.458 The following scheme, involving three boron-to-carbon migrations, to 99 and then to 100 has been suggested.
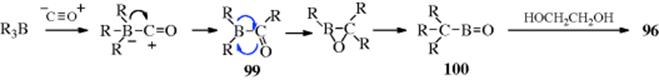
The purpose of the ethylene glycol is to intercept the boronic anhydride (100), which otherwise forms polymers that are difficult to oxidize. As will be seen in Reaction 18-23 and 18-24, it is possible to stop the reaction after only one or two migrations have taken place.
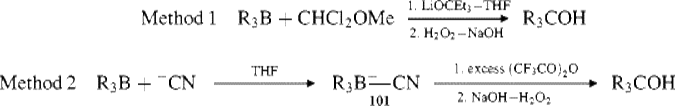
There are two other methods for achieving the conversion R3B → R3COH, which often give better results: (1) treatment with α,α-dichloromethyl methyl ether and the base lithium triethylcarboxide459 (2) treatment with a suspension of sodium cyanide in THF followed by reaction of the resulting trialkylcyanoborate (101) with an excess (>2 equiv) of trifluoroacetic anhydride.460 All the above migrations take place with retention of configuration at the migrating carbon.461
Several other methods for the conversion of boranes to tertiary alcohols are also known.462
If the reaction between trialkylboranes and carbon monoxide (18-23) is carried out in the presence of water followed by addition of NaOH, the product is a secondary alcohol. If H2O2 is added along with the NaOH, the corresponding ketone is obtained instead.463 Various functional groups (e.g., OAc, COOR, CN) may be present in R without being affected,464 although if they are in the α or β position relative to the boron atom, difficulties
![]()
may be encountered. The use of an equimolar amount of trifluoroacetic anhydride leads to the ketone rather than the tertiary alcohol.465 By this procedure thexylboranes (RR′R2B, where R2 = thexyl) can be converted to unsymmetrical ketones (RCOR′).466 Variations of this methodology have been used to prepare optically active alcohols.467 For another conversion of trialkylboranes to ketones see Reaction 18-26.468
OS VII, 427. Also see, OS VI, 137.
18-24 Conversion of Boranes to Primary Alcohols, Aldehydes, or Carboxylic Acids
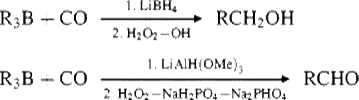
When the reaction between a trialkylborane and carbon monoxide (18-23) is carried out in the presence of a reducing agent (e.g., lithium borohydride or potassium triisopropoxyborohydride), the reduction agent intercepts the intermediate (99), so that only one boron→carbon migration takes place, and the product is hydrolyzed to a primary alcohol or oxidized to an aldehyde.469 This procedure wastes two of the three R groups, but this problem can be avoided by the use of B-alkyl-9-BBN derivatives (see Reaction 15-16). Since only the 9-alkyl group migrates, this method permits the conversion in high yield of an alkene to a primary alcohol or aldehyde containing one more carbon.470 When B-alkyl-9-BBN derivatives are treated with CO and lithium tri-tert-butoxyaluminum hydride,471 other functional groups (e.g., CN and ester) can be present in the alkyl group without being reduced.472 Boranes can be directly converted to carboxylic acids by reaction with the dianion of phenoxyacetic acid.473
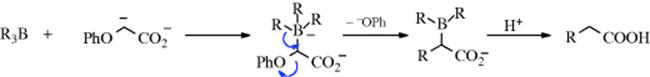
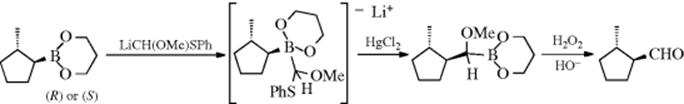
Boronic esters [RB(OR′)2] react with methoxy(phenylthio)methyllithium [LiCH(OMe)SPh] to give salts, which, after treatment with HgCl2, and then H2O2, yield aldehydes.474 This synthesis has been made enantioselective, with high ee values (>99%), by the use of an optically pure boronic ester,475 for example:
18-25 Conversion of Vinylic Boranes to Alkenes
![]()
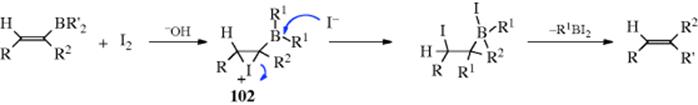
The reaction between trialkylboranes and iodine to give alkyl iodides was mentioned at 12-31. When the substrate contains a vinylic group, the reaction takes a different course,476 with one of the R′ groups migrating to the carbon, to give alkenes.477 The reaction is stereospecific in two senses: (1) if the groups R and R″ are cis in the starting compound, they will be trans in the product; (2) there is retention of configuration within the migrating group R′.478Since vinylic boranes can be prepared from alkynes (Reaction 15-16), this is a method for the addition of R′ and H to a triple bond. If R2 = H, the product is a (Z)-alkene. The mechanism is believed to involve an iodonium intermediate, (e.g., 102) and attack by iodide on boron. When R′ is vinylic, the product is a conjugated diene.479
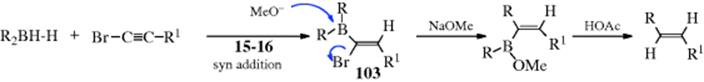
In another procedure, the addition of a dialkylborane to a 1-haloalkyne produces an α-halo vinylic borane (103).480 Treatment of 103 with NaOMe gives the rearrangement shown, and protonolysis of the product produces the (E)-alkene.478 If R is a vinylic group, the product is a 1,3-diene.481 If one of the groups is thexyl, the other migrates.482 A combination of both of the procedures described above results in the preparation of trisubstituted alkenes.483
18-26 Formation of Alkynes, Alkenes, and Ketones from Boranes and Acetylides
![]()
A hydrogen directly attached to a triple-bond carbon can be replaced in high yield by an alkyl or an aryl group, by treatment of the lithium acetylide with a trialkyl- or triarylborane, followed by reaction of the lithium alkynyltrialkylborate (104) with iodine.484 The R′ group may be primary or secondary alkyl as well as aryl, so the reaction has a broader scope than the older Reaction 10-74.485 The R group may be alkyl, aryl, or hydrogen, although in the last-mentioned case satisfactory yields are obtained only if lithium acetylide-ethylenediamine is used as the starting compound.486 Optically active alkynes can be prepared by using optically active thexylborinates (RR2BOR′, R2 = thexyl, where R is chiral) and LiC![]() CSiMe3.487 The reaction can be adapted to the preparation of alkenes488 by treatment of 104 with an electrophile (e.g., propanoic acid489 or tributyltin chloride).490 The reaction with Bu3SnCl produces the (Z)-alkene stereoselectively.
CSiMe3.487 The reaction can be adapted to the preparation of alkenes488 by treatment of 104 with an electrophile (e.g., propanoic acid489 or tributyltin chloride).490 The reaction with Bu3SnCl produces the (Z)-alkene stereoselectively.
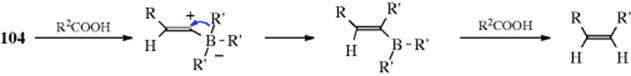
Treatment of 104 with electrophiles (e.g., methyl sulfate, allyl bromide, or triethyloxonium borofluoride), followed by oxidation of the resulting vinylic borane, gives a ketone (illustrated for methyl sulfate)491:
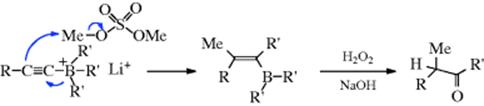
Note that there are reactions that involve N → O rearrangements, including those mediated by silicon.492
18.F.ii. Non-1,2 Rearrangements
A. Electrocyclic Rearrangements
18-27 Electrocyclic Rearrangements of Cyclobutenes and 1,3-Cyclohexadienes
(4) seco-1/4/Detachment; (4) cyclo-1/4/Attachment
(6) seco-1,6/Detachment; (6) cyclo-1/6/Attachment
Cyclobutenes and 1,3-dienes can be interconverted by treatment with UV light or with heat.493 These are 4π-electrocyclizations. The thermal reaction is generally not reversible (although exceptions494 are known), and many cyclobutenes have been converted to 1,3-dienes by heating at temperatures between 100 and 200 °C.495 Benzocyclobutenes also undergo electrocyclic ring opening,496 as do benzocyclobutanones.497 The photochemical conversion can in principle be carried out in either direction, but most often 1,3-dienes are converted to cyclobutenes rather than the reverse, because the dienes are stronger absorbers of light at the wavelengths used.498 In a similar reaction, 1,3-cyclohexadienes interconvert with 1,3,5-trienes, but in this case the ring-closing process is generally favored thermally and the ring-opening process photochemically, but exceptions are known in both directions.499 Substituent effects can lead to acceleration of the electrocyclization process.500 Torquoselectivity in cyclobutene ring-opening reaction has been examined.501
Examples of these types of reactions include:
Ref. 502
An interesting example of 1,3-cyclohexadiene→1,3,5-triene interconversion is the reaction of norcaradienes to give cycloheptatrienes.503 This is a 6π-electrocyclization, and it has been catalyzed by Lewis acids.504 Norcaradienes give this reaction so readily (because they are cis-1,2-divinylcyclopropanes, see Reaction 18-32) that they cannot generally be isolated, though some exceptions are known505 (see also, 15-64).
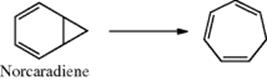
These reactions, called electrocyclic rearrangements,506 take place by pericyclic mechanisms. The evidence comes from stereochemical studies, which show a remarkable stereospecificity whose direction depends on whether the reaction is induced by heat or light. For example, it was found for the thermal reaction that cis-3,4-dimethylcyclobutene gave only cis,trans-2,4-hexadiene, while the trans isomer gave only the trans–trans-diene507:
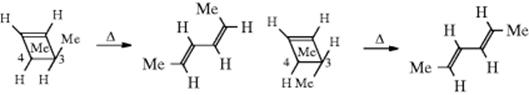
This is evidence for a four-membered cyclic transition state and arises from conrotatory motion about the C-3–C-4 bond.508 It is called conrotatory because both movements are clockwise (or both counterclockwise). Because both rotate in the same direction, the cis isomer gives the cis–trans-diene.509 The other possibility (disrotatory motion) would have one moving clockwise while the other moves counterclockwise; the cis isomer would have given the cis–cis-diene (shown) or the trans–trans-diene. If the motion had been disrotatory, this would still have been evidence for a cyclic mechanism. If the mechanism were a diradical or some other kind of noncyclic process, it is likely that no stereospecificity of either kind would have been observed. The reverse
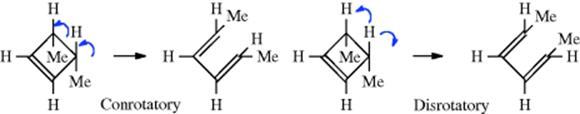
reaction is also conrotatory. In contrast, the photochemical cyclobutene: 1,3-diene interconversion is disrotatory in either direction.510 On the other hand, the cyclohexadiene: 1,3,5-triene interconversion shows precisely the opposite behavior. The thermal process is disrotatory, while the photochemical process is conrotatory (in either direction). These startling results are a consequence of the symmetry rules mentioned in Section 15-60, the FOM.511 As in the case of cycloaddition reactions, we will use the FOM and Möbius–Hückel approaches.512
The Frontier Orbital Method (FOM)513
As applied to these reactions, the FOM may be expressed: A σ bond will open in such a way that the resulting p orbitals will have the symmetry of the highest occupied π orbital of the product. In the case of cyclobutenes, the HOMO of the product in the thermal reaction is the χ2 orbital (Fig. 18.1). Therefore, in a thermal process, the cyclobutene must open so that on one side the positive lobe lies above the plane, and on the other side below it. Thus the substituents are forced into conrotatory motion (Fig. 18.2). On the other hand, in the photochemical process, the HOMO of the product is now the χ3 orbital (Fig. 18.1), and in order for the p orbitals to achieve this symmetry (the two plus lobes on the same side of the plane), the substituents are forced into disrotatory motion.
Fig. 18.1 Symmetries of the X2 and X3∗ orbitals of a conjugated diene
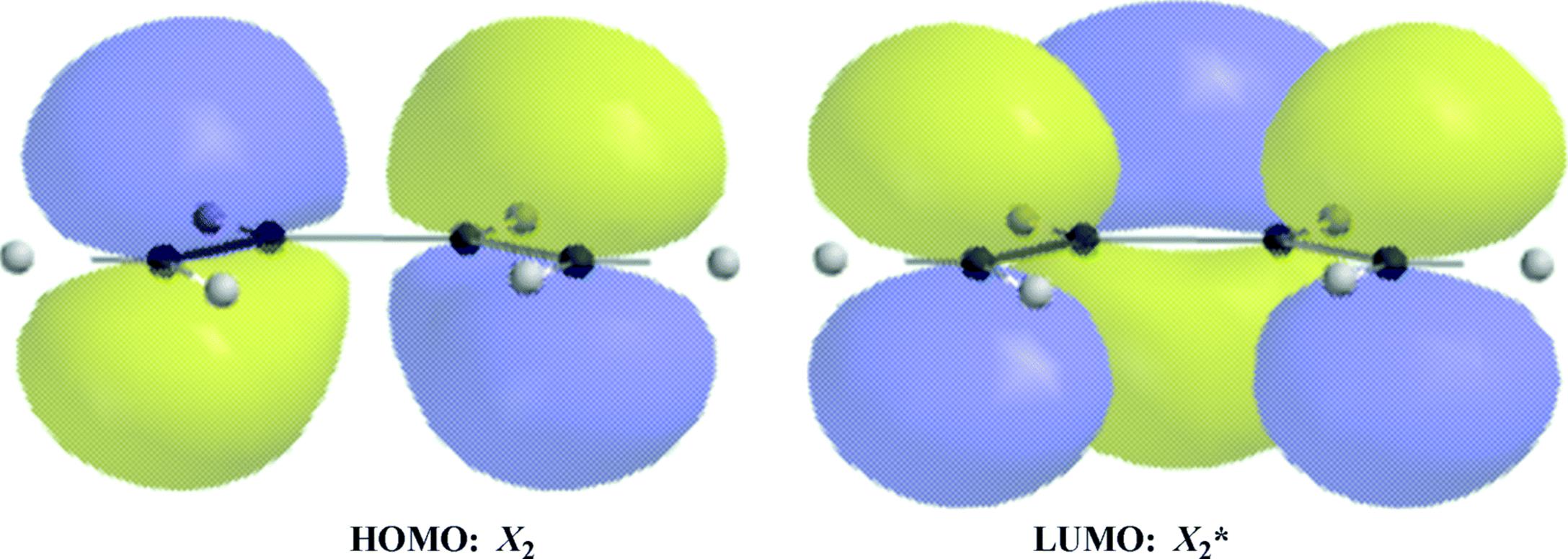
Fig. 18.2 Thermal opening of 1,2-diethylcyclobutene. The two hydrogens and two methyls are forced into conrotatory motion so that the resulting p-orbitals have the symmetry of the HOMO of the diene.
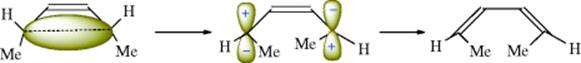
This reaction may be considered from the opposite direction (ring closing). For this direction, the rule is that those lobes of orbitals that overlap (in the HOMO) must be of the same sign. For thermal cyclization of butadienes, this requires conrotatory motion (Fig. 18.3). In the photochemical process, the HOMO is the χ3 orbital, so that disrotatory motion is required for lobes of the same sign to overlap.
Fig. 18.3 Thermal ring closing of a 1,3-diene. Conrotatory motion is required for two + lobes to overlap.
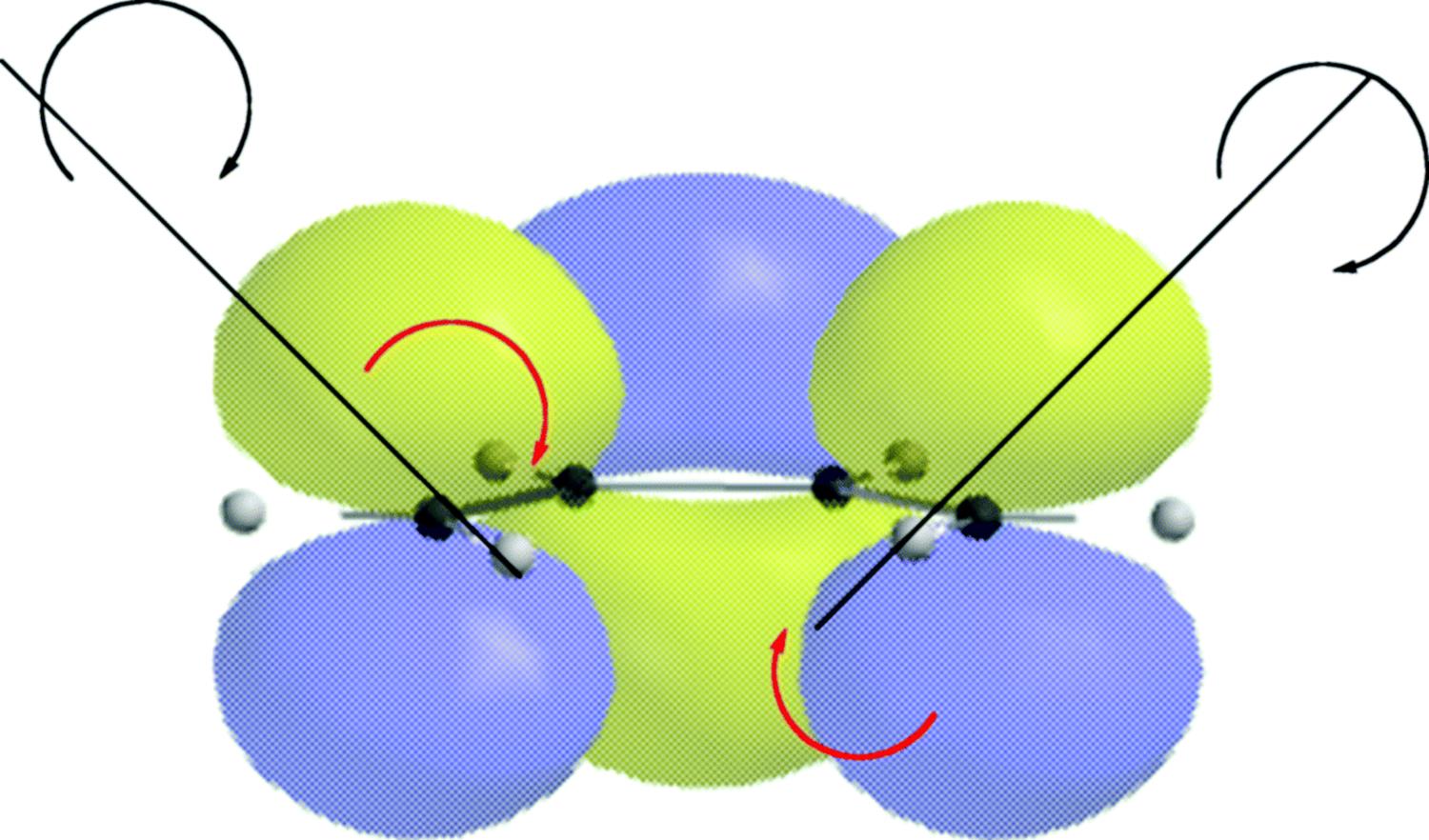
The Möbius–Hückel Method
As seen in Reaction 15-60, the Möbius–Hückel Method, a basis set of p orbitals is chosen and inspected for sign inversions in the transition state. Figure 18.4 shows a basis set for a 1,3-diene. It is seen that disrotatory ring closing (Fig. 18.4a) results in overlap of plus lobes only, while in conrotatory closing (Fig. 18.4b) there is one overlap of a plus with a minus lobe. In the first case, there are zero sign inversions, while in the second there is one sign inversion. With zero (or an even number of) sign inversions, the disrotatory transition state is a Hückel system, and so is allowed thermally only if the total number of electrons is 4n + 2 (Sec. 15-60, the Möbius–Hückel Method). Since the total here is 4, the disrotatory process is not allowed. On the other hand, the conrotatory process, with one sign inversion, is a Möbius system, which is thermally allowed if the total number is 4n. The conrotatory process is therefore allowed thermally. For the photochemical reactions, the rules are reversed: A reaction with 4n electrons requires a Hückel system, so only the disrotatory process is allowed.
Fig. 18.4 The 1,3-diene–cyclobutene interconversion. The orbitals shown are not molecular orbitals, but a basis set of p atomic orbitals. (a) Disrotatory ring closure gives zero sign inversion. (b) Conrotatory ring closure gives one sign inversion. We could have chosen to show any other basis set (another basis set would have two plus lobes above the plane and two below, etc.). This would change the number of sign inversion, but the disrotatory mode would still have an even number of sign inversions, and the conrotatory mode an odd number, whichever basis set was chosen.
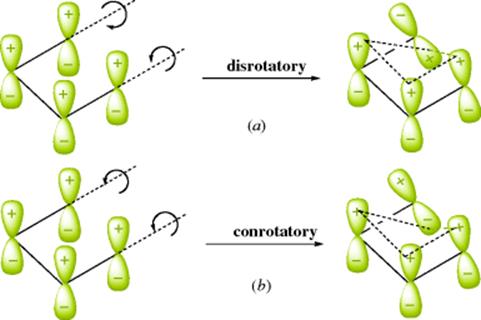
Both the FOM and the Möbius–Hückel methods can also be applied to the cyclohexadiene: 1,3,5-triene reaction514; in either case the predicted result is that for the thermal process, only the disrotatory pathway is allowed, and for the photochemical process, only the conrotatory. For example, for 1,3,5-hexatriene, the symmetry of the HOMO is
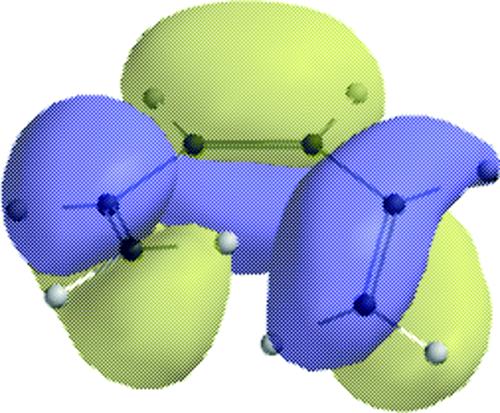
In the thermal cleavage of cyclohexadienes, then, the positive lobes must lie on the same side of the plane, requiring disrotatory motion:
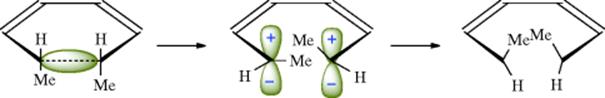
Disrotatory motion is also necessary for the reverse reaction, in order that the orbitals that overlap may be of the same sign:
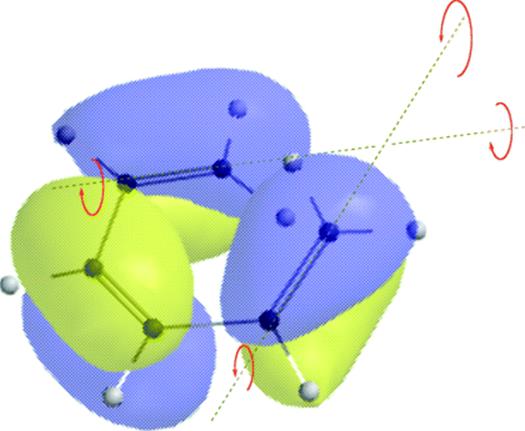
All these directions are reversed for photochemical processes, because in each case a higher orbital, with inverted symmetry, is occupied.
In the Möbius–Hückel approach, diagrams similar to Fig. 18.4 can be drawn for this case. Here too, the disrotatory pathway is a Hückel system and the conrotatory pathway a Möbius system, but since six electrons are now involved, the thermal reaction follows the Hückel pathway and the photochemical reaction the Möbius pathway.
In the most general case, four possible products can arise from a given cyclobutene or cyclohexadiene: two from the conrotatory and two from the disrotatory pathway. For example, conrotatory ring opening of 105 gives either 106 or 107, while disrotatory opening gives either 108 or 109. The orbital-symmetry rules indicate when a given reaction will operate by the conrotatory and when by the disrotatory mode, but not which of the two possible conrotatory or disrotatory pathways will be followed. Often, however, it is possible to make such
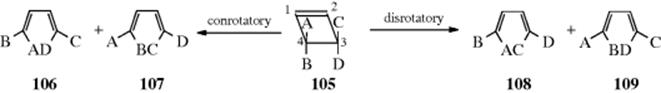
predictions on steric grounds. For example, in the opening of 105 by the disrotatory pathway, 108 arises when groups A and C swing in toward each other (clockwise motion around C-4, counterclockwise around C-3), while 109 is formed when groups B and D swing in and A and C swing out (clockwise motion around C-3, counterclockwise around C-4). This observation leads to a prediction that when A and C are larger than B and D, the predominant or exclusive product will be 109 rather than 108. Predictions of this kind have largely been borne out.515 There is evidence, however, that steric effects516 are not the only factor, and that electronic effects also play a role, and their role may be even greater.517 An electron-donating group stabilizes the transition state when it rotates outward, because it mixes with the LUMO; if it rotates inward, it mixes with the HOMO, destabilizing the transition state.518 The compound 3-formylcyclobutene provided a test. Steric factors would cause the CHO (an electron-withdrawing group) to rotate outward; electronic effects would cause it to rotate inward. The experiment showed inward rotation.519
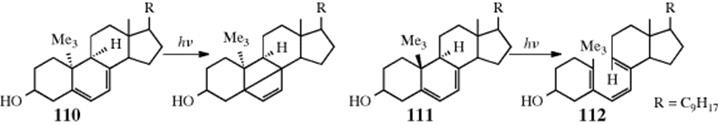
Cyclohexadienes are of course 1,3-dienes, and in certain cases it is possible to convert them to cyclobutenes instead of to 1,3,5-trienes.520 An interesting example is found in the pyrocalciferols. Photolysis of the syn isomer (110) (or of the other syn isomer, not shown) leads to the corresponding cyclobutene,521 while photolysis of the anti isomers (one of them is 111) gives the ring-opened 1,3,5-triene (112). This difference in behavior is at first sight remarkable, but is easily explained by the orbital-symmetry rules. Photochemical ring opening to a 1,3,5-triene must be conrotatory. If 110 were to react by this pathway, the product would be the triene 112, but this compound would have to contain a trans-cyclohexene ring (either the methyl group or the hydrogen would have to be directed inside the ring). On the other hand, photochemical conversion to a cyclobutene must be disrotatory, but if 111 were to give this reaction, the product would have to have a trans-fused ring junction. Compounds with such ring junctions are known (Sec. 4.K.iii), but are very strained. Stable trans-cyclohexenes are unknown (Sec. 4.Q.iii). Thus, 110 and 111give the products they do owing to a combination of orbital-symmetry rules and steric influences.
A related process is the Bergmann cyclization,522 where an ene–diyne cyclizes to a biradical (103) and then aromatizes as shown. Simply heating the en–diyne will usually lead to aromatization via this pathway.523 Quinones can be formed via Bergman cyclization524 and there are other synthetic applications.525 The role of vinyl substitution has been examined.526 An aza-Bergman cyclization is known.527
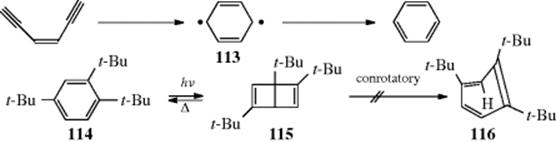
The 1,3-diene-cyclobutene interconversion can even be applied to benzene rings. For example,528 photolysis of 1,2,4-tri-tert-butylbenzene (114) gives 1,2,5-tri-tert-butyl[2.2.0]hexadiene (115, a Dewar benzene).529 The reaction owes its success to the fact that once 115 is formed, it cannot, under the conditions used, revert to 114 by either a thermal or a photochemical route. The orbital-symmetry rules prohibit thermal conversion of 115 to 114 by a pericyclic mechanism, because thermal conversion of a cyclobutene to a 1,3-diene must be conrotatory, and conrotatory reaction of 115 would result in a 1,3,5-cyclohexatriene containing one trans double bond (116), which is of course too strained to exist. Compound 115 cannot revert to 114 by a photochemical pathway either, because light of the frequency used to excite 114 would not be absorbed by 115. This is another example of a molecule that owes its stability to the orbital-symmetry rules (see Reaction 15-63). Pyrolysis of 115 does give 114, probably by a diradical mechanism.530 In the case of 117 and 118, the Dewar benzene is actually more stable than the benzene. Compound 117 rearranges to 118 in 90% yield at 120 °C.531 In this case, thermolysis of the benzene gives the Dewar benzene (rather than the reverse), because of the strain of four adjacent tert-butyl groups on the ring.
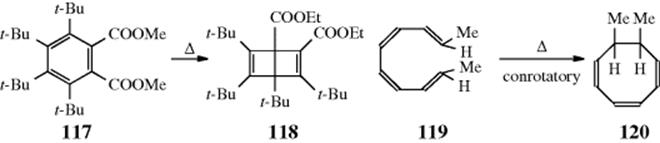
A number of electrocyclic reactions have been carried out with systems of other sizes, [(e.g., conversion of the 1,3,5,7-octatetraene (119) to the cyclooctatriene (120)].532 The stereochemistry of these reactions can be predicted in a similar manner. The results of such predictions can be summarized according to whether the number of electrons involved in the cyclic process is of the form 4n or 4n + 2 (where n is any integer including zero).
Thermal Reaction
Photochemical Reaction
4n
Conrotatory
Disrotatory
4n + 2
Disrotatory
Conrotatory
Although the orbital-symmetry rules predict the stereochemical results in almost all cases, it is necessary to recall (Reaction 15-60, the Möbius–Hückel Method) that they only say what is allowed and what is forbidden, but the fact that a reaction is allowed does not necessarily mean that the reaction takes place, and if an allowed reaction does take place, it does not necessarily follow that a concerted pathway is involved, since other pathways of lower energy may be available.533 Furthermore, a “forbidden” reaction might still be made to go, if a method of achieving its high activation energy can be found. This was, in fact, done for the cyclobutene–butadiene interconversion (cis-3,4-dichlorocyclobutene gave the forbidden cis,cis- and trans,trans-1,4-dichloro-1,3-butadienes, as well as the allowed cis, trans isomer) by the use of IR laser light.534 This is a thermal reaction. The laser light excites the molecule to a higher vibrational level (Sec. 7.A.i), but not to a higher electronic state.
As is the case for [2 + 2]-cycloaddition reactions (15-63), certain forbidden electrocyclic reactions can be made to take place by the use of metallic catalysts.535 An example is the silver ion-catalyzed conversion of tricyclo[4.2.0.02.5]octa-3,7-diene to cyclooctatetraene536:
This conversion is very slow thermally (i.e., without the catalyst) because the reaction must take place by a disrotatory pathway, which is disallowed thermally.537 In another example, the major thermal product from the barrelene anion is a rearranged allyl anion that is formed by disrotatory cleavage of the cyclopropyl ring, a formally Woodward–Hoffmann-forbidden process.538
The ring opening of cyclopropyl cations (Sec. 10.G.i, category 7 and Reaction 18-3) is an electrocyclic reaction and is governed by the orbital symmetry rules.539 For this case, the rule is invoked that the σ bond opens in such a way that the resulting p orbitals have the symmetry of the highest occupied orbital of the product, in this case,
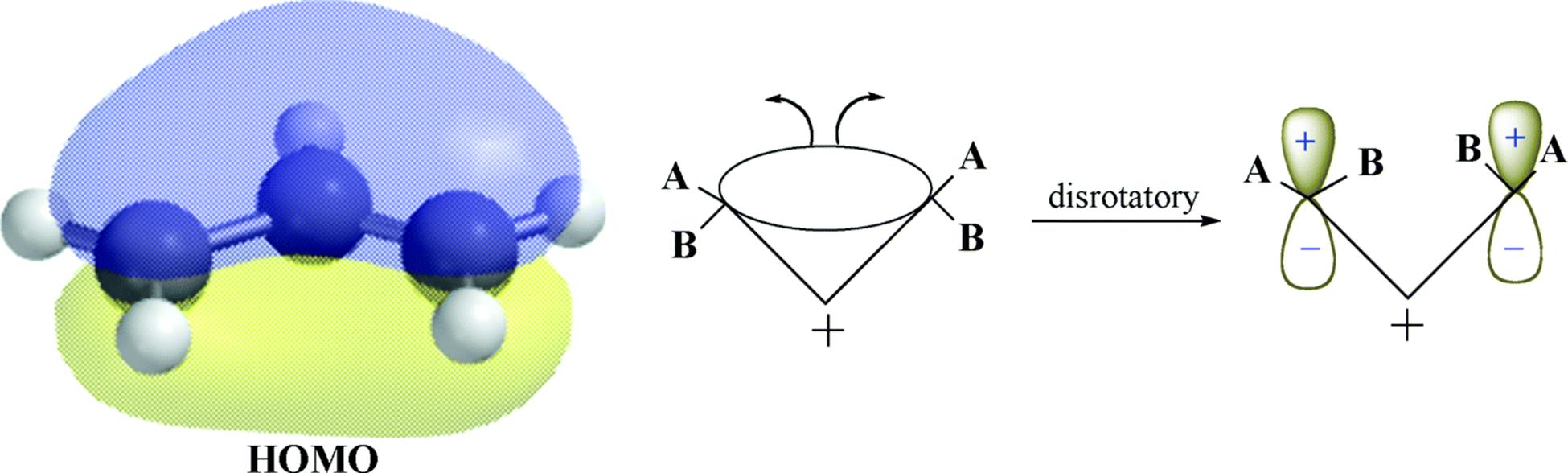
an allylic cation. Recall that an allylic system has three molecular orbitals (Sec. 2.C, category 3). For the cation, with only two electrons, the highest occupied orbital is the one of lowest energy (HOMO). Thus, the cyclopropyl cation must undergo a disrotatory ring opening in order to maintain the symmetry. Note that, in contrast, ring opening of the cyclopropyl anion must be conrotatory,540 since in this case it is the next orbital of the allylic system that is the highest occupied, and this has the opposite symmetry.541 However, it is difficult to generate a free cyclopropyl cation (Sec. 10.G.i, category 7), and it is likely that in most cases, cleavage of the σ bond is concerted with departure of the leaving group in the original cyclopropyl substrate. This, of course, means that the σ bond provides anchimeric assistance to the removal of the leaving group (an SN2 type process), and we would expect that such assistance should come from the back side. This has an important effect on the direction of ring opening. The orbital-symmetry rules require that the ring opening is disrotatory, but as seen above, there are two disrotatory pathways and the rules do not indicate which is preferred. But the fact that the σ orbital provides assistance from the backside means that the two substituents that are trans to the leaving group must move outward, not inward.542 Thus, the disrotatory pathway that is followed is the one shown in B, not the one shown in B′, because the former puts the electrons of the σ bond on the side opposite
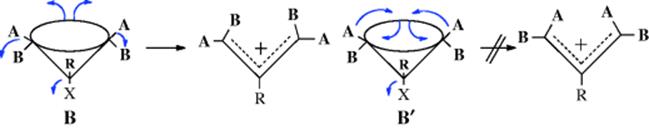
that of the leaving group.543 Strong confirmation of this picture544 comes from acetolysis of endo- (121) and exo-bicyclo[3,1,0]hexyl-6-tosylate (112). The groups trans to the tosylate must move outward. For 121, this means that the two hydrogen atoms can go outside the framework of the six-membered ring, but for 122 they
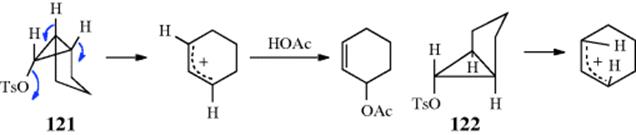
are forced to go inside. Consequently, it is not surprising that the rate ratio for solvolysis of 121/122 was found to be >2.5 × 106 and that at 150 °C, 122 did not solvolyze at all.545 This evidence is kinetic. Unlike the cases of the cyclobutene (1,3-diene and cyclohexadiene) 1,3,5-triene interconversions, the direct product here is a cation, which is not stable but reacts with a nucleophile and loses some of its steric integrity in the process, so that much of the evidence has been of the kinetic type rather than from studies of product stereochemistry. However, it has been shown by investigations in superacids (Sec. 5.A.ii), where it is possible to keep the cations intact and to study their structures by NMR, that in all cases studied the cation that is predicted by these rules is in fact formed.546
OS V, 235, 277, 467; VI, 39, 145, 196, 422, 427, 862; IX, 180.
18-28 Conversion of One Aromatic Compound to Another
(6) cyclo-de-hydrogen-coupling (Overall transformation)
Stilbenes can be converted to phenanthrenes by irradiation with UV light547 in the presence of an oxidizing agent, (e.g., dissolved molecular oxygen, FeCl3, or iodine).548 The reaction is a photochemically allowed conrotatory549conversion of a 1,3,5-hexatriene to a cyclohexadiene, followed by removal of two hydrogen atoms by the oxidizing agent. The intermediate dihydrophenanthrene has been isolated.550 The actual reacting species must be the cis-stilbene, but trans-stilbenes can often be used, because they are isomerized to the cis isomers under the reaction conditions. The reaction can be extended to the preparation of many fused aromatic systems, for example551:
though not all such systems give a reaction.552 The use of substrates containing heteroatoms (e.g., PhN=NPh) allows the formation of heterocyclic ring systems.
Isomerization of biphenylene to benzo[a]pentalene553 is a well-known benzene ring contraction rearrangement,554 driven by relief of strain in the four-membered ring. Related to this process is the flash vacuum pyrolysis (FVP) of the alternant polycyclic aromatic hydrocarbon benzo[b]biphenylene at 1100 °C, which gives fluoranthene, a nonalternant polycyclic aromatic hydrocarbon, as the major product at 1100 °C in the gas phase.555 The mechanism used explain that this isomerization involves equilibrating diradicals of 2-phenylnaphthalene, which rearrange by the net migration of a phenyl group to give equilibrating diradicals of 1-phenylnaphthalene, one isomer of which then cyclizes to fluoranthene.
Another transformation of one aromatic compound to another is the Stone–Wales rearrangement of pyracyclene (123),556 which is a bond-switching reaction. The rearrangement of bifluorenylidene (124) to dibenzo[g,p]chrysene (125) occurs at temperatures as low as 400 °C and is accelerated in the presence of decomposing iodomethane, a convenient source of methyl radicals.557 This result suggested a radical rearrangement. This rearrangement is believed to occur by a radical-promoted mechanism consisting of a sequence of homoallyl-cyclopropylcarbinyl rearrangement steps.558
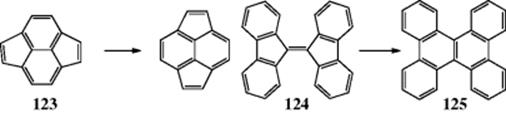
B. Sigmatropic Rearrangements
A sigmatropic rearrangement is defined559 as migration, in an uncatalyzed intramolecular process, of a σ bond, adjacent to one or more π systems, to a new position in a molecule, with the π systems becoming reorganized in the process. Examples are
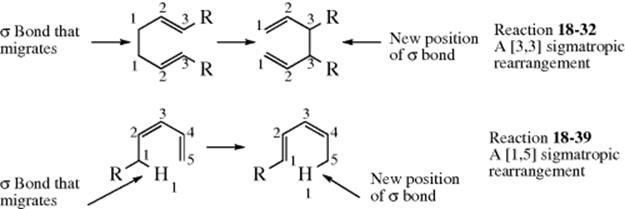
The order of a sigmatropic rearrangement is expressed by two numbers set in brackets: [i,j]. These numbers can be determined by counting the atoms over which each end of the σ bond has moved. Each of the original termini is given the number 1. Thus in the first example above, each terminus of the σ bond has migrated from C-1 to C-3, so the order is [3,3]. In the second example, the carbon terminus has moved from C-1 to C-5, but the hydrogen terminus has not moved at all, so the order is [1,5].
18-29 [1,j]-Sigmatropic Migrations of Hydrogen
1/→3/Hydrogen-migration; 1/→5/Hydrogen-migration
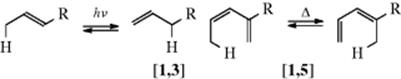
Many examples of thermal or photochemical rearrangements in which a hydrogen atom migrates from one end of a system of π bonds to the other have been reported,560 although the reaction is subject to geometrical constraints. Isotope effects play a role in sigmatropic rearrangements, and there is evidence for a kinetic silicon isotope effect.561 Pericyclic mechanisms are involved,562 and the hydrogen must, in the transition state, be in contact with both ends of the chain at the same time. This means that for [1,5] and longer rearrangements, the molecule must be able to adopt the cisoid conformation. Furthermore, there are two geometrical pathways by which any sigmatropic rearrangement can take place, illustrated for the case of a [1,5]-sigmatropic rearrangement,563 starting with a substrate of the form 126, where the migration origin is an asymmetric carbon atom and U ≠ V. In one of the two pathways, the hydrogen moves along the top or bottom face of the π system. This is called suprafacial migration. In the other pathway, the hydrogen moves across the π system, from top to bottom, or vice versa. This is antarafacialmigration. Altogether, a single isomer like 126 (different rotamers) can give four products. In a suprafacial migration, H can move across the top of the π system (as drawn above) to give the (R,Z)-isomer, or it can rotate 180 ° and move across the bottom of the π system to give the (S,E)- isomer.564 The antarafacial migration can similarly lead to two diastereomers, in this case the (S,Z)- and (R,E)-isomers.
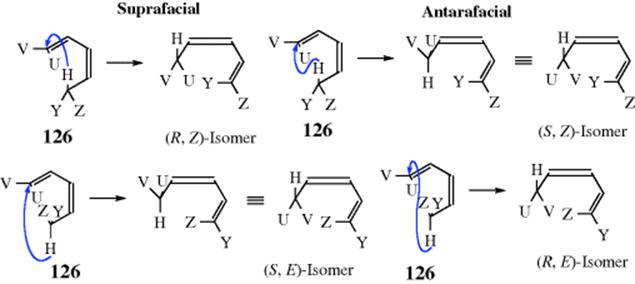
In any given sigmatropic rearrangement, only one of the two pathways is allowed by the orbital-symmetry rules; the other is forbidden. To analyze this situation, first use a modified frontier orbital approach.565 Imagine that in the transition state C, the migrating H atom breaks away from the rest of the system, which is treated as if it were a free radical.
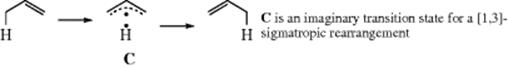
Note that this is not what actually takes place; it is imagined in order to analyze the process. In a [1,3]-sigmatropic rearrangement, the imaginary transition state consists of a hydrogen atom and an allyl radical. The latter species (Sec. 2.C, category 3) has three π orbitals, but the only one that is of concern, the HOMO, which, in a thermal rearrangement is D. The electron of the hydrogen atom is of course in a 1s orbital, which has only one lobe. The rule governing sigmatropic migration of hydrogen is the H must move from a plus to a plus or from a minus to a minus lobe, of the HOMO; it cannot move to a lobe of opposite sign.566 The only way this can happen in a thermal [1,3]-sigmatropic rearrangement is by an antarafacial migration. Consequently, the rule predicts that antarafacial thermal [1,3]-sigmatropic rearrangements are allowed, but the suprafacial pathway is forbidden. However, in a photochemical reaction, promotion of an electron means that E is now the HOMO; the suprafacial pathway is now allowed and the antarafacial pathway is forbidden.
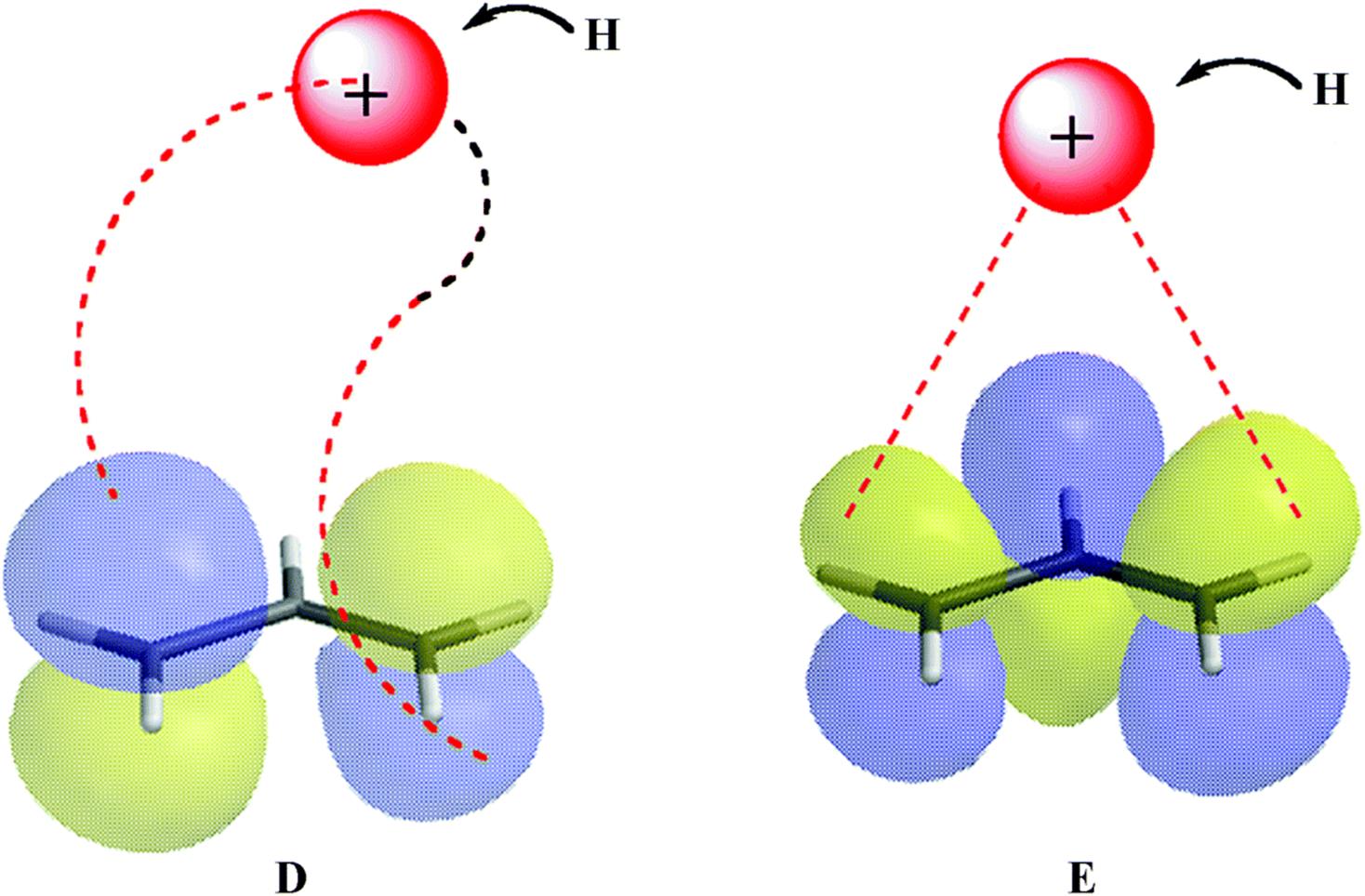
A similar analysis of [1,5]-sigmatropic rearrangements shows that in this case the thermal reaction must be suprafacial and the photochemical process antarafacial. For the general case, with odd-numbered j, [1,j]-suprafacial migrations are allowed thermally when j is of the form 4n + 1, and photochemically when j has the form 4n − 1; the opposite is true for antarafacial migrations.
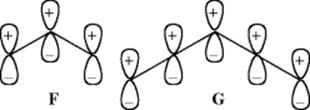
As expected, the Möbius–Hückel method leads to the same predictions. Here, examine the basis set of orbitals shown in F and G for [1,3]- and [1,5]-rearrangements, respectively. A [1,3]-shift involves four electrons, so an allowed thermal pericyclic reaction must be a Möbius system (15-60, the Möbius–Hückel Method) with one or an odd number of sign inversions. As can be seen in F, only an antarafacial migration can achieve this. A [1,5]-shift, with six electrons, is allowed thermally only when it is a Hückel system with zero or an even number of sign inversions; hence it requires a suprafacial migration.567
The actual reported results bear out this analysis. Thus a thermal [1,3]-migration is allowed to take place only antarafacially, but such a transition state would be extremely strained, and thermal [1,3]-sigmatropic migrations of hydrogen are unknown.568 On the other hand, the photochemical pathway allows suprafacial [1,3]-shifts, and a few such reactions are known, an example being the photochemical rearrangement of 127 to 128.569 Substituents influence the efficacy of the [1,3]-hydrogen shift.570
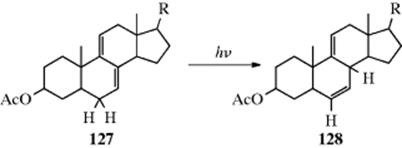
The situation is reversed for [1,5]-hydrogen shifts. In this case, the thermal rearrangements, being suprafacial, are quite common, while photochemical rearrangements are rare.571 Two examples of the thermal reaction are
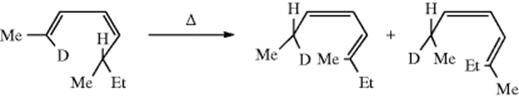
Ref. 572
![]()
Ref. 573
Note that the first example bears out the stereochemical prediction made earlier. Only the two isomers shown were formed. In the second example, migration can continue around the ring. Migrations of this kind are called circumambulatory rearrangements,574 and such migrations are known for cyclopentadiene,575 pyrrole, and phosphole derivatives.576 Geminal bond participation has been observed in pentadienes,577 the effects of phenyl substituents have been studied,578 and the kinetics and activation parameters of [1,5]-hydrogen shifts have been examined.579 The [1,5]-hydrogen shifts are also known with vinyl aziridines.580 A Ru catalyzed cycloisomerization of en-1-ynes leads to cyclic dienes.581
The rare [1,4]-hydrogen transfer has been observed in radical cyclizations.582 With respect to [1,7]-hydrogen shifts, the rules predict the thermal reaction to be antarafacial.583 Unlike the case of [1,3]-shifts, the transition state is not too greatly strained, and an example of such rearrangements is the formation of 129 and 130.584 Photochemical [1,7]-shifts are suprafacial and, not surprisingly, many of these have been observed.585
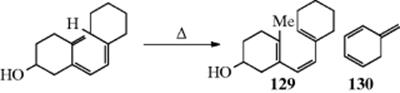
The orbital symmetry rules also help to explain the unexpected stability of certain compounds (see Reaction 15-63, preceding Reaction 15-64 and 18-27, the Möbius–Hückel Method). Thus, 130 could, by a thermal [1,3]-sigmatropic rearrangement, easily convert to toluene, which of course is far more stable because it has an aromatic sextet. Yet 130 has been prepared and is stable at dry ice temperature and in dilute solutions.586
Analogues of sigmatropic rearrangements in which a cyclopropane ring replaces one of the double bonds are also known, for example,587
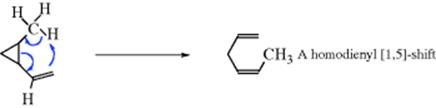
The reverse reaction has also been reported.588 2-Vinylcycloalkanols589 undergo an analogous reaction, as do cyclopropyl ketones (see 18-33, preceding 18-34 for this reaction).
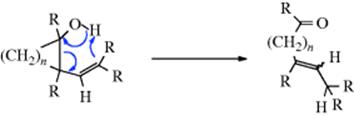
18-30 [1,j]-Sigmatropic Migrations of Carbon
[1,3] migration of alkyl
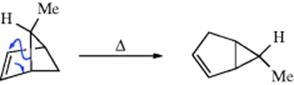
Ref. 590
[1,5] migration of phenyl 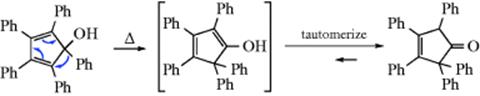
Ref. 591
Sigmatropic migrations of alkyl or aryl groups592 are less common than the corresponding hydrogen migrations.593 When they do take place, there is an important difference. Unlike a hydrogen atom, whose electron is in a 1sorbital with only one lobe, a carbon free radical has its odd electron in a p orbital that has two lobes of opposite sign. Therefore, if the imaginary transition states for this case are drawn (see above), a thermal suprafacial [1,5]-process (Fig. 18.5) is observed, and symmetry can be conserved only if the migrating carbon moves in such a way that the lobe that was originally attached to the π system remains attached to the π system.
Fig. 18.5 Hypothetical orbital movement for a thermal [1,5]-sigmatropic migration of carbon. To move from one negative lobe, the migrating carbon uses only its own negative lobe, retaining its configuration.
This can happen only if configuration is retained within the migrating group. On the other hand, thermal suprafacial [1,3]-migration (Fig. 18.6) can take place if the migrating carbon switches lobes. If the migrating carbon was originally bonded by its minus lobe, it must now use its plus lobe to form the new C–C bond. Thus, configuration in the migrating group will be inverted. From these considerations, suprafacial [1,j]-sigmatropic rearrangements in which carbon is the migrating group should always be allowed, both thermally and photochemically, but thermal [1,3]-migrations594 will proceed with inversion and thermal [1,5]-migrations with retention of configuration within the migrating group. More generally, suprafacial [l,j]-migrations of carbon in systems where j = 4n − 1 proceed with inversion thermally and retention photochemically, while systems where j = 4n + 1 show the opposite behavior. Where antarafacial migrations take place, all these predictions are of course reversed.
Fig. 18.6 Hypothetical orbital movement for a thermal [1,3]-sigmatropic migration of carbon. The migrating carbon moves a negative to a positive lobe, requiring it to switch its own bonding lobe from negative to positive, inverting its configuration.
The first laboratory test of these predictions was the pyrolysis of deuterated endo-bicyclo[3.2.0]hept-2-en-6-yl acetate (131), which gave the exo-deuterio-exo-norbornyl acetate (132).595 Thus, as predicted by the orbital symmetry rules, this thermal suprafacial [1,3]-sigmatropic reaction took place with complete inversion at C-7. Similar results have been obtained in a number of other cases.596 However, similar studies of the pyrolysis of the parent hydrocarbon of 131, labeled with D at C-6 and C-7, showed that while most of the product was formed with inversion at C-7, a significant fraction (11–29%) was formed with retention.597 Other cases of lack of complete inversion are also known.598 A diradical mechanism has been invoked to explain such cases.599 There is strong evidence for a radical mechanism for some [1,3]-sigmatropic rearrangements.600 Photochemical suprafacial [1,3]-migrations of carbon have been shown to proceed with retention, as predicted.601
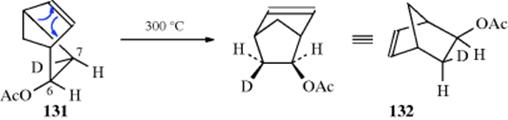
Although allylic vinylic ethers generally undergo [3,3]-sigmatropic rearrangements (Reaction 18-33), they can be made to give the [1,3] kind, to give aldehydes, for example,
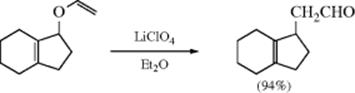
by treatment with LiClO4 in diethyl ether.602 In this case, the C–O bond undergoes a 1,3-migration from the O to the end vinylic carbon. When the vinylic ether is of the type ROCR′=CH2, ketones (RCH2COR′) are formed. There is evidence that this [1,3]-sigmatropic rearrangement is not concerted, but involves dissociation of the substrate into ions.602
Thermal suprafacial [1,5]-migrations of carbon have been found to take place with retention,603 but also with inversion.604 A diradical mechanism has been suggested for the latter case.604
Simple nucleophilic, electrophilic, and free radical 1,2-shifts can also be regarded as sigmatropic rearrangements (in this case, [1,2]-rearrangements). As previously discussed (see discussion in introductory section preceding 18.A) similar principles applied to such rearrangements show that nucleophilic 1,2-shifts are allowed, but the other two types are forbidden unless the migrating group has some means of delocalizing the extra electron or electron pair. The mechanism of the forbidden [3s,5s]-sigmatropic shift has been examined.605
18-31 Conversion of Vinylcyclopropanes to Cyclopentenes
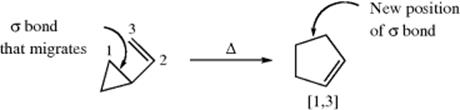
The thermal expansion of a vinylcyclopropane to a cyclopentene ring606 is a special case of a [1,3]-sigmatropic migration of carbon, although it can also be considered an internal [π2 + σ2]-cycloaddition reaction (see 15-63). It is known as a vinylcyclopropane rearrangement.607 The reaction has been carried out on many vinylcyclopropanes bearing various substituents in the ring608 or on the vinyl group and has been extended to 1,1-dicyclopropylethene609and (both thermally610 and photochemically611) to vinylcyclopropenes. This
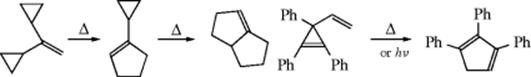
rearrangement can be catalyzed by Rh and Ag compounds, and has been used to form rings.612 Two competing reactions are the homodienyl [1,5]-shift (if a suitable H is available, see 18-29), and simple cleavage of the cyclopropane ring, leading in this case to a diene (see 18-3).
Flash vacuum pyrolysis of the trimethylsilyl ether of cyclopropylcarbinyl alcohols gives ring expanded ketones.613 Various heterocyclic analogues614 are also known, as in the rearrangement of aziridinyl amides (133).615Cyclopropyl ketones can be treated with tosylamine and a Zr catalyst, which converts the imine formed in situ to a pyrroline.616 N-Cyclopropylimines undergo rearrangement to cyclic imines (pyrrolines) under photochemical conditions.617P-Vinyl phosphiranes (the P analogue of cyclopropanes with P in the ring) under a similar rearrangement, and the mechanism has been studied.618
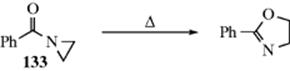
Vinylcyclobutanes can be converted to cyclohexenes,619 but larger ring compounds do not generally give the reaction.620 Tricyclo[4.1.0.02.5]heptanes rearrange to give nonconjugated cycloheptadienes.621 The bicyclo[2.1.0]pentane derivatives undergo this reaction.
The reaction rate has also been greatly increased by the addition of a one-electron oxidant tris-(4-bromophenyl)aminium hexafluoroantimonate (Ar3N√+ SbF6−, Ar = p-bromophenyl).622 This reagent converts the substrate to a cation radical, which undergoes ring expansion much faster.623
The mechanisms of these ring expansions are not certain. Both concerted624 and diradical625 pathways have been proposed,626 and it is possible that both pathways operate, in different systems.
For the conversion of a vinylcyclopropane to a cyclopentene in a different way, see OS 68, 220.
18-32 The Cope Rearrangement
(3/4/)→(1/6/)- sigma-Migration
![]()
When 1,5-dienes are heated, a [3,3]-sigmatropic rearrangement known as the Cope rearrangement (not to be confused with the Cope elimination Reaction, 17-9) occurs to generate an isomeric 1,5-diene.627 When the diene is symmetrical about the 3,4-bond, the reaction gives a product identical with the starting material628:
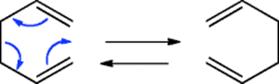
Therefore, a Cope rearrangement can be detected only when the diene is not symmetrical about this bond. Any 1,5-diene gives the rearrangement; for example, 3-methyl-1,5-hexadiene heated to 300 °C gives 1,5-heptadiene.629However, the reaction takes place more easily (lower temperature required) when there is a group on the C-3 or C-4 with leads to the new double bond being substituted. The reaction is reversible630 and produces an equilibrium mixture of the two 1,5-dienes, which is richer in the thermodynamically more stable isomer. However, the equilibrium can be shifted to the right for 3-hydroxy-1,5-dienes,631 because the product tautomerizes to the ketone or aldehyde:
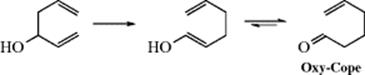
This reaction of 3-hydroxy-1,5-dienes is called the oxy-Cope rearrangement,632 and has proved highly useful in synthesis.633 The oxy-Cope rearrangement is greatly accelerated (by factors of 1010–1017) if the alkoxide is used rather than the alcohol (the anionic oxy-Cope rearrangement),634 where the direct product is the enolate ion, which is hydrolyzed to the ketone. A metal-free reaction using a phosphazene base has been reported.635 The silyloxy-Cope rearrangement has proven to be quite useful.636 An antibody-catalyzed oxy-Cope reaction is known,637 and the mechanism and origins of catalysis for this reaction have been studied.638 Sulfur substituents also lead to rate enhancement of the oxy-Cope rearrangement.639 Note that 2-oxonia Cope rearrangements have been implicated in Prins cyclization reactions (16-54).640 A highly diastereoselective oxonia-Cope rearrangement proceeded using a chiral aldehyde with a chiral conjugated ester.641
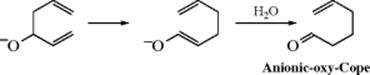
aza-Cope rearrangements are also known.642 There is an enantioselective aza-Cope rearrangement.643 There is also a 1,2-oxaza-Cope rearrangement. Involving esters and alkyl nitrites.644 In amino-Cope rearrangements, the solvent plays a role in the regioselectivity of the reaction.645 It has been suggested that this latter reaction does not proceed solely by a concerted [3.3]-sigmatropic rearrangement.646
The 1,5-diene system may be inside a ring or part of an allenic system647 (the example shown illustrates both of these situations).648 However, the reaction does not take place when one of the double bonds is part of

an aromatic system (e.g., 4-phenyl-1-butene).649 When the two double bonds are in vinylic groups attached to adjacent ring positions, the product is a ring four carbons larger. This has been applied to divinylcyclopropanes and divinylcyclobutanes, as shown.650 Indeed, cis-1,2-divinylcyclopropanes give this rearrangement so rapidly
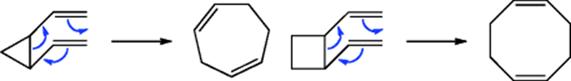
that they generally cannot be isolated at room temperature,651 but exceptions are known.652 Note that divinyloxiranes, divinylphosphiranes, and divinylthiiranes undergo similar rearrangements.653 When heated, 1,5-diynes are converted to 3,4-dimethylenecyclobutenes (134).654 A rate-determining Cope rearrangement is followed by a very rapid electrocyclic (18-27) reaction.
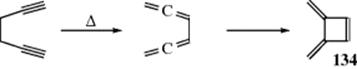
The interconversion of 1,3,5-trienes and cyclohexadienes (in Reaction 18-27) is very similar to the Cope rearrangement, but in 18-27, the 3,4-bond goes from a double to a single bond rather than from a single bond to no bond. Like [2 + 2]-cycloadditions (Reaction 15-63), Cope rearrangements of simple 1,5-dienes can be catalyzed by certain transition metal compounds. For example, the addition of a Pd catalyst causes the reaction to take place at room temperature.655
As indicated with the arrows, the mechanism of the uncatalyzed Cope rearrangement is a simple six-centered pericyclic process.656 Since the mechanism is so simple, it has been possible to study some rather subtle points, among them the question of whether the six-membered transition state is in the boat or the chair form.657 For the case of 3,4-dimethyl-1,5-hexadiene, it was demonstrated conclusively that the transition state is in the chair form. This was shown by the stereospecific nature of the reaction: The meso isomer gave the cis–trans product, while the (±) diastereomer gave the trans–trans-diene.658 If the transition state is in the chair form (taking the meso isomer, e.g.), one methyl must be “axial” and the other “equatorial” and the product must be the
cis–trans-alkene. There are two possible boat forms for the transition state of the meso isomer. One leads to a trans–trans product; the other to a cis–cis-alkene. For the (±) pair, the predictions are just the opposite: There is just one boat form, and it leads to the cis–trans-alkene, while one chair form (“diaxial” methyls) leads to the cis–cis product and the other (“diequatorial” methyls) predicts the trans–trans product. Thus the nature of the products obtained demonstrates that the transition state is a chair and not a boat.659 While 3,4-dimethyl-1,5-hexadiene is free to assume either the chair or boat (it prefers the chair), other compounds are not so free. Thus 1,2-divinylcyclopropane (see above) can react only in the boat form, demonstrating that such reactions are possible.660
Because of the nature of the transition state661 in the pericyclic mechanism, optically active substrates with a stereogenic carbon at C-3 or C-4 transfer the chirality to the product (see Reaction 18-33 for an example in the mechanistically similar Claisen rearrangement).662 There are many examples of asymmetric [3,3]-sigmatropic rearrangements.663
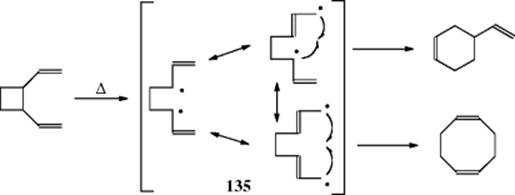
Not all Cope rearrangements proceed by the cyclic six-centered mechanism.664 Thus cis-1,2-divinylcyclobutane (see above) rearranges smoothly to 1,5-cyclooctadiene, since the geometry is favorable. The trans isomer also gives this product, but the main product is 4-vinylcyclohexene (resulting from Reaction 18-31). This reaction can be rationalized as proceeding by a diradical mechanism (see 135),665 although it is possible that at least part of the cyclooctadiene produced comes from a prior epimerization of the trans- to the cis-divinylcyclobutane followed by Cope rearrangement of the latter.666
It has been suggested that another type of diradical two-step mechanism may be preferred by some substrates.667 Indeed, a nonconcerted Cope rearrangement has been reported.668 In this pathway,669 the 1,6-bond is formed before the 3,4-bond breaks:
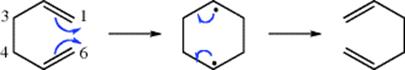
This is related to the Bergman cyclization that was introduced in Reaction 18-27.
It was pointed out earlier that a Cope rearrangement of the symmetrical 1,5-hexadiene gives 1,5-hexadiene. This is a degenerate Cope rearrangement (Sec. 18.A.ii). Bicyclo[5.1.0]octadiene (136) undergoes a similar rearrangement.670 At room temperature, the NMR spectrum of this compound is in accord with the structure shown on the left. At 180 °C, it is converted by a Cope reaction to a compound equivalent to itself. The interesting thing is that at 180 °C the NMR spectrum shows that what exists is an equilibrium mixture of the two
structures. That is, at this temperature the molecule rapidly (faster than 103 times per second) changes back and forth between the two structures. This is called valence tautomerism and is quite distinct from resonance, even though only electrons shift671 (see Sec. 2.N for other types of tautomerism). The positions of the nuclei are not the same in the two structures. Molecules like 136 that exhibit valence tautomerism (in this case, at 180 °C) are said to have fluxional structures. It may be recalled that cis-1,2-divinylcyclopropane does not exist at room temperature because it rapidly rearranges to 1,4-cycloheptadiene (see above), but in 136 the cis-divinylcyclopropane structure is frozen into the molecule in both structures. Several other compounds with this structural feature are also known. Of these, bullvalene (137) is especially interesting. The Cope rearrangement shown for 137 changes the position of the cyclopropane ring from 4,5,10 to 1,7,8. But the molecule could also have undergone rearrangements to put this ring at 1,2,8 or 1,2,7. Any of these could then undergo several Cope
rearrangements. In all, there are ![]() or >1.2 million tautomeric forms, and the cyclopropane ring can be at any three carbons that are adjacent. Since each of these tautomers is equivalent to all the others, this has been called an infinitely degenerate Cope rearrangement. Bullvalene has been synthesized and its
or >1.2 million tautomeric forms, and the cyclopropane ring can be at any three carbons that are adjacent. Since each of these tautomers is equivalent to all the others, this has been called an infinitely degenerate Cope rearrangement. Bullvalene has been synthesized and its ![]() NMR spectrum was determined.672 At −25 °C, there are two peaks with an area ratio of 6 : 4. This is in accord with a single nontautomeric structure. The six protons are the vinylic protons and the four protons are the allylic ones. But at 100 °C, the compound shows only one NMR peak, indicating that the compound rapidly interchanges its structure among 1.2 million equivalent forms.673 The
NMR spectrum was determined.672 At −25 °C, there are two peaks with an area ratio of 6 : 4. This is in accord with a single nontautomeric structure. The six protons are the vinylic protons and the four protons are the allylic ones. But at 100 °C, the compound shows only one NMR peak, indicating that the compound rapidly interchanges its structure among 1.2 million equivalent forms.673 The ![]() NMR spectrum of bullvalene also shows only one peak at 100 °C.674
NMR spectrum of bullvalene also shows only one peak at 100 °C.674
Another compound for which degenerate Cope rearrangements result in equivalence for all the carbons is hypostrophene (138).675 In the case of the compound barbaralane (139)676 (bullvalene in which one CH=CH has been replaced by a CH2), there are only 2 equiv tautomers.677 However, NMR spectra indicate that even at room temperature a rapid interchange of both tautomers is present, although by about −100 °C this has slowed to the point where the spectrum is in accord with a single structure. In the case of semibullvalene (140) (barbaralane in which the CH2 has been removed), not only is there a rapid interchange at room temperature, but even at −110 °C.678 Compound 140 has the lowest energy barrier of any known compound capable of undergoing the Cope rearrangement.679
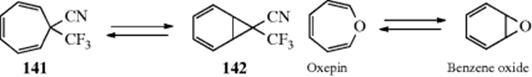
The molecules taking part in a valence tautomerization need not be equivalent. Thus, NMR spectra indicate that a true valence tautomerization exists at room temperature between the cycloheptatriene (141) and the norcaradiene (142).680 In this case, one isomer (142) has the cis-1,2-divinylcyclopropane structure, while the other does not. In an analogous interconversion, benzene oxide681 and oxepin exist in a tautomeric equilibrium at room temperature.682
Bullvalene and hypostrophene are members of a group of compounds all of whose formulas can be expressed by the symbol (CH)10.683 Many other members of this group are known. Similar groups of (CH)n compounds exist for other even-numbered values of “n”.685 For example, there are 20 possible (CH)8684 compounds,685 and five possible (CH)6 compounds,686 all of which are known: benzene, prismane (Sec. 4.Q.i), Dewar benzene (see Reaction 18-27, the Möbius–Hückel Method), bicyclopropenyl,687 and benzvalene.688
An interesting example of valence tautomerism is the case of 1,2,3-tri-tert-butylcyclobutadiene (Sec. 2.K.ii). There are two isomers, both rectangular, and ![]() NMR spectra show that they exist in a dynamic equilibrium, even at −185 °C.689
NMR spectra show that they exist in a dynamic equilibrium, even at −185 °C.689
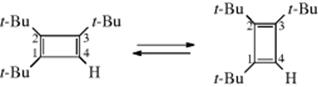
18-33 The Claisen Rearrangement690
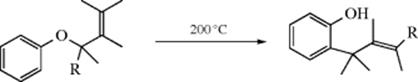
Allylic aryl ethers, when heated, rearrange to o-allylphenols in a reaction called the Claisen rearrangement.691 If both ortho positions are filled, the allylic group migrates to the para position (this is often called the para-Claisen rearrangement).692 There is no reaction when the para and both ortho positions are filled. Migration to the meta position has not been observed. In the ortho migration, the allylic group always undergoes an allylic shift. That is, as shown above, a substituent α to the oxygen is now γ to the ring (and vice versa). On the other hand, in the para migration there is never an allylic shift: The allylic group is found exactly as it was in the original ether. Compounds with propargylic groups (i.e., groups with a triple bond in the appropriate position) do not generally give the corresponding products.
The mechanism is a concerted pericyclic [3,3]-sigmatropic rearrangement693 and accounts for all these facts. For the ortho rearrangement,
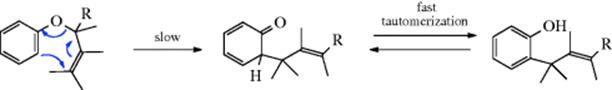
Evidence is the lack of a catalyst, the fact that the reaction is first order in the ether, the absence of cross-over products when mixtures are heated, and the presence of the allylic shift, which is required by this mechanism. The allylic shift for the ortho rearrangement (and the absence of one for the para) has been demonstrated by 14C labeling, even when no substituents are present. Studies of the transition state geometry have shown that, like the Cope rearrangement, the Claisen rearrangement usually prefers a chair-like transition state.694 A retro-Claisen rearrangement is known and its mechanism has been examined.695
When the ortho positions have no hydrogen, a second [3,3]-sigmatropic migration (a Cope reaction) follows:
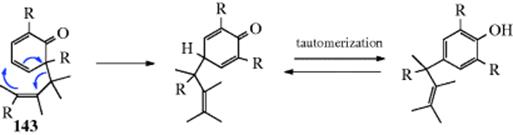
and the migrating group is restored to its original structure. Intermediates of structure 143 have been trapped by means of a Diels–Alder reaction (15-60).696 The rearrangement of aryl allyl ethers is facilitated by Ag–KI in hot acetic acid,697 and by AlMe3 in water.698 A solid-phase reaction of polymer-bound substrate undergoes the Claisen rearrangement with microwave irradiation.699
Allylic ethers of enols (allylic vinylic ethers, 144) also undergo the Claisen rearrangement700; in fact, it was discovered with these compounds first.701 In these cases of course, the final tautomerization does not take place even when R′ = H, since there is no aromaticity to restore, and ketones are more stable than enols.702 Catalytic Claisen rearrangements of allyl vinyl ethers are well known.703 The use of water as solvent accelerates the reaction.704Microwave induced reactions on silica gel705 and in ionic liquids706 are known The mechanism is similar to that with allylic aryl ethers.707 In the presence of a chiral Cu complex, Claisen rearrangements proceed with good enantioselectivity.708 N-Heterocyclic carbenes catalyzed an enantioselective Claisen rearrangement.709 A chiral hydrogen-bond donor has been used for enantioselective Claisen rearrangements.710 A chiral allylic ether gave an enantioselective Claisen rearrangement with an Ir catalyst.711
Since the Claisen rearrangement mechanism does not involve ions, it should not be greatly dependent on the presence or absence of substituent groups on the ring.712 This is the case. Electron-donating groups increase the rate and electron-withdrawing groups decrease it, but the effect is small, with the p-amino compound reacting only ~10–20 times faster than the p-nitro compound.713 However, solvent effects714 are greater: Rates varied over a 300-fold range when the reaction was run in 17 different solvents.715 An especially good solvent is trifluoroacetic acid, in which the reaction can be carried out at room temperature.716 Most Claisen rearrangements are performed without a catalyst, but AlCl3 or BF3 are sometimes used.717 In this case, it may become a Friedel–Crafts reaction, with the mechanism no longer cyclic,718 and ortho, meta, and para products may be obtained.
Allyl allene ethers undergo a Claisen rearrangement when heated in DMF to give the expected diene with a conjugated aldehyde unit.719 Butenolides with a β-allylic ether unit undergo Claisen rearrangement–Conia reaction720cascade to give an oxaspiroheptane with β-keto lactone comprising the five-membered ring.721 Allylic esters of β-keto acids undergo a Claisen rearrangement in what is known as the Carroll rearrangement722 (also called the Kimel–Cope rearrangement723), and the reaction can be catalyzed by a Ru complex.724 An asymmetric Carroll rearrangement was catalyzed by a chiral Pd complex.725
Heating an allylic alcohol with N,N-dimethylacetamide dimethyl acetal yields a transient intermediate, and subsequent Claisen rearrangement gives an amide in a sequence that is known as the Eschenmoser variant or the Eschenmoser–Claisen rearrangement.726 This reaction has also been called the Meerwein–Eschenmoser Claisen rearrangement.727 An enantioselective version has been reported using a chiral Pd complex.728
The enolate anions (145) of allylic esters (formed by treatment of the esters with LICA) rearrange to γ,δ-unsaturated acids.729 Alternatively, the silylketene acetal [R3R2C=C(OSiR3)OCH2CH=CHR1] may be used instead of 145.730This rearrangement also proceeds at room temperature. By either procedure, the reaction is called the Ireland–Claisen rearrangement.731 Note the presence of the negative charge in 145. As with the oxy-Cope rearrangement (in Reaction 18-34), negative charges generally accelerate the Claisen reaction,732 although the extent of the acceleration can depend on the identity of the positive counterion.733 The reaction proceeds with good syn selectivity in many cases.734 The Ireland–Claisen rearrangement has been made enantioselective by converting 145 to an enol borinate in which the boron is attached to a chiral group.735 The Ireland–Claisen rearrangement can be done with amide derivatives also.736
As just mentioned, asymmetric Claisen rearrangement reactions are well known.737 Chiral Lewis acids have been designed for this purpose.738 In general, asymmetric [3,3]-sigmatropic rearrangements are well known.739
A number of analogues of the Claisen rearrangement are known, for example, rearrangement of ArNHCH2CH=CH2,740 of N-allylic enamines (R2C=CRNRCR2CR=CR2)741 of allylic imino esters [RC(OCH2CH=CH2)=NR]742(these have often been rearranged with transition metal catalysts743), and of RCH=NRCHRCH2CH=CH2. These rearrangements of nitrogen-containing compounds can be called aza-Claisen rearrangements,744 but are sometimes called aza-Cope rearrangements,745 as described in Reaction 18-32. A Pd catalyzed aza-Claisen has been reported.746 An important contribution to this variation is the rearrangement of trichloroacetimidate derivatives of prochiral (Z)-2-alken-1-ols, usually with a Pd catalyst, to give chiral allylic esters.747 There is a catalytic enantioselective aza-Cope rearrangement.748 A so-called amine-Claisen rearrangement was reported for N-allyl indoles, when heated in the presence of BF3√OEt2.749 An azo-Cope rearrangement, CH2=CHCR2′)CR2′N=NAr → R2′C=CHCH2NArN=CR22, has been reported.750 In a related reaction, allylic phorphorimidates undergo [3,3]-sigmatropic rearrangement.751
The conversion of allylic aryl thioethers, (ArSCH2CH=CH2) to o-allylic thiophenols is not feasible, because the latter are not stable,752 but react to give bicyclic compounds.753 However, many allylic vinylic sulfides do give the rearrangement (the thio-Claisen rearrangement).754 Allylic vinylic sulfones (e.g., H2C=CRCH2–SO2–CH=CH2) rearrange, when heated in the presence of ethanol and pyridine, to unsaturated sulfonate salts (CH2=CRCH2CH2CH2SO3−), produced by reaction of the reagents with the unstable sulfene intermediates CH2=CRCH2CH2CH=SO2.755 Allylic vinylic sulfoxides rapidly rearrange at room temperature or below.756 Chiral vinyl sulfoxides undergo Claisen rearrangement with good enantioselectivity.757
Ethers with an alkyl group in the γ position (ArO–C–C=C–R systems) sometimes give abnormal products, with the β carbon becoming attached to the ring758:
It has been established that these abnormal products do not arise directly from the starting ether, but are formed by a further rearrangement of the normal product759:
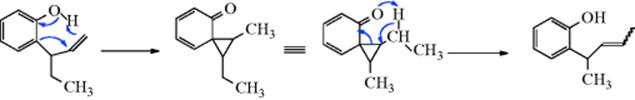
This rearrangement, which has been called an enolene rearrangement, a homodienyl [1,5]-sigmatropic hydrogen shift (see Reaction 18-29), and a [1,5]-homosigmatropic rearrangement, involves a shift of three electron pairs over seven atoms. It has been found that this “abnormal” Claisen rearrangement is general and can interconvert the enol forms of systems of the types 146 and 147 through the cyclopropane intermediate (148).760
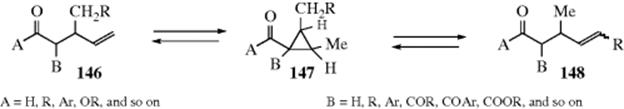
OS III, 418; V, 25; VI, 298, 491, 507, 584, 606; VII, 177; VIII, 251, 536.
18-34 The Fischer Indole Synthesis
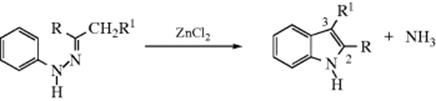
When arylhydrazones of aldehydes or ketones are treated with a catalyst, elimination of ammonia takes place and an indole is formed, in the Fischer indole synthesis.761 Zinc chloride is a commonly used catalyst, but dozens of others, including other metal halides, proton and Lewis acids, and certain transition metals have also been used. Microwave irradiation has been used to facilitate this reaction.762 The reaction has been done using an AlCl3 complex as an ionic liquid,763 and solid-phase Fischer indole syntheses are known.764 Aniline derivatives react with α-diazoketones, in the presence of a Rh catalyst, to give indoles as well.765 Arylhydrazones are easily prepared by the treatment of aldehydes or ketones with phenylhydrazine (Reaction 16-2) or by aliphatic diazonium coupling (Reaction 12-7). However, it is not necessary to isolate the arylhydrazone. The aldehyde or ketone can be treated with a mixture of phenylhydrazine and the catalyst; this is now common practice. In order to obtain an indole, the aldehyde or ketone must be of the form RCOCH2R′ (R = alkyl, aryl, or hydrogen). Vinyl ethers (e.g., dihydrofuran) serve as an aldehyde surrogate when treated with phenylhydrazine and a catalytic amount of aq H2SO4 to give an 3-substituted indole.766
At first glance, the reaction does not seem to be a rearrangement. However, the key step of the mechanism767 is a [3,3]-sigmatropic rearrangement768:
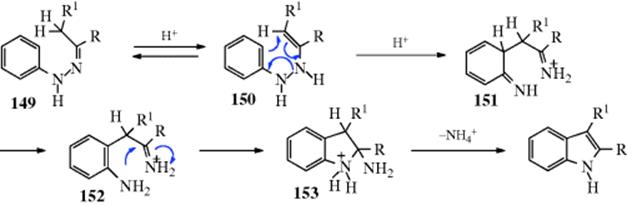
There is much evidence for this mechanism, for example, (1) the isolation of 153,769 (2) the detection of 152 by ![]() and
and ![]() NMR,770 (3) the isolation of side products that could only have come from 151,771 and (4)
NMR,770 (3) the isolation of side products that could only have come from 151,771 and (4) ![]() -labeling experiments showing that it was the nitrogen farther from the ring that is eliminated as ammonia.772 The main function of the catalyst seems to be to speed the conversion of 149 to 150. The reaction can be performed without a catalyst.
-labeling experiments showing that it was the nitrogen farther from the ring that is eliminated as ammonia.772 The main function of the catalyst seems to be to speed the conversion of 149 to 150. The reaction can be performed without a catalyst.
OS III, 725; IV, 884. Also see, OS IV, 657.
18-35 [2,3]-Sigmatropic Rearrangements
(2/ S-3/)→(1/5/)- sigma-Migration

Sulfur ylids bearing an allylic group are converted to unsaturated sulfides on heating.773 This is a concerted [2,3]-sigmatropic rearrangement774 and has also been demonstrated for the analogous cases of nitrogen ylids775 and the conjugate bases of allylic ethers (in the last case it is called a [2,3]-Wittig rearrangement).776 It has been argued that the [2,3]-Wittig rearrangement demands severe deformation of the molecule in order to proceed.777 It has been shown that SmI2 induces a [2,3]-Wittig rearrangement.778 The reaction has been extended to certain other systems,779 even to an all-carbon system.780
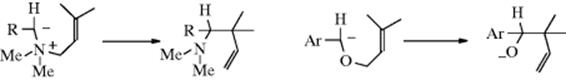
Treatment of an α-(N-allylic amino) ketone with NaH led to 2-allylic α-amino ketones via a [2,3]-rearrangement.781 In the presence of a chiral ligand on nitrogen,781 or with a chiral additive,782 good asymmetric induction is possible. Vinylaziridines undergo [2.3]-sigmatropic rearrangement.783
Since the reactions involve migration of an allylic group from a sulfur, nitrogen, or oxygen atom to an adjacent negatively charged carbon atom, they are special cases of the Stevens or Wittig rearrangements (18-21 and 18-22). However, in this case the migrating group must be allylic (in 18-21 and 18-22 other groups can also migrate). Thus, when the migrating group is allylic, there are two possible pathways: (1) the radical-ion or ion-pair mechanisms (18-21 and 18-22) and (2) the concerted pericyclic [2,3]-sigmatropic rearrangement. These are easily distinguished since the latter always involves an allylic shift (as in the Claisen rearrangement), while the former pathway does not.

Of these reactions, the [2,3]-Wittig rearrangement in particular has often been used as a means of transferring chirality. The product of this reaction has potential stereogenic centers at C-3 and C-4 (if R5 ≠ R6), and if the starting ether is optically active because of a stereogenic center at C-1, the product may be optically active as well. Many examples are known in which optically active ethers were converted to a product that was optically active because of chirality at C-3, C-4, or both.784 If a suitable stereogenic center is present in R1 (or if a functional group in R1 can be so converted), then stereocontrol over three contiguous stereogenic centers can be achieved. Stereocontrol of the new double bond (E or Z) has also been accomplished.
If an OR or SR group is attached to the negative carbon, the reaction becomes a method for the preparation of β,γ-unsaturated aldehydes, because the product is easily hydrolyzed.785
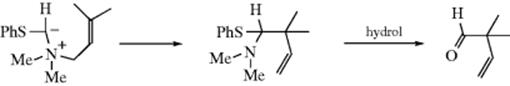
Another [2,3]-sigmatropic rearrangement converts allylic sulfoxides to rearranged allylic alcohols by treatment with a thiophilic reagent (e.g., trimethyl phosphite).786 This is the Mislow–Evans rearrangement. In this case, the migration is from sulfur to oxygen. [2,3]-Oxygen-to-sulfur migrations are also known.787 The Sommelet–Hauser rearrangement discussed in Reaction 13-31 is also a [2,3]-sigmatropic rearrangement.
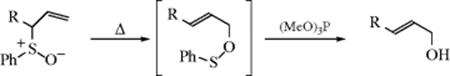
OS VIII, 427.
18-36 The Benzidine Rearrangement
When hydrazobenzene is treated with acids, it rearranges to give ~70% 4,4′-diaminobiphenyl (154, benzidine) and ~30% 2,4′-diaminobiphenyl. This reaction is called the benzidine rearrangement and is general for N,N′-diarylhydrazines.788 Usually, the major product is the 4,4′-diaminobiaryl, but four other products may also be produced: the 2,4′-diaminobiaryl, already referred to, the 2,2′-diaminobiaryl, and the o- and p-arylaminoanilines (called semidines). The 2,2′- and p-arylaminoaniline compounds are formed less often and in smaller amounts than the other two side products. Usually, the 4,4′-diaminobiaryl predominates, except when one or both para positions of the diarylhydrazine are occupied. However, the 4,4′-diamine may still be produced even if the para positions are occupied. If SO3H, CO2H, or Cl (but not R, Ar, or NR2) is present in the para position, it may be ejected. With dinaphthylhydrazines, the major products are not the 4,4′-diaminobinaphthyls, but the 2,2′ isomers. Another side reaction is disproportionation to ArNH2 and ArN=NAr. For example, p,p′-PhC6H4NHNHC6H4Ph gives 88% disproportionation products at 25 °C.789
The mechanism has been exhaustively studied and several mechanisms have been proposed.790 At one time, it was believed that NHAr broke away from ArNHNHAr and became attached to the para position to give the semidine, which then went on to product. The fact that semidines could be isolated lent this argument support, as did the fact that this would be analogous to the rearrangements considered in Chapter 11 (Reaction 11-28–11-32). However, this theory was proved incorrect when it was discovered that semidines could not be converted to benzidines under the reaction conditions. Cleavage into two independent pieces (either ions or radicals) has been ruled out by many types of cross-over experiments, which always showed that the two rings of the starting material are in the product; that is, ArNHNHAr′ gives no molecules (of any of the five products) containing two Ar groups or two Ar′ groups, and mixtures of ArNHNHAr and Ar′NHNHAr′ give no molecules containing both Ar and Ar′. An important discovery was the fact that, although the reaction is always first order in substrate, it can be either first791 or second792 order in [H+]. With some substrates the reaction is entirely first order in [H+], while with others it is entirely second order in [H+], regardless of the acidity. With still other substrates, the reaction is first order in [H+] at low acidities and second order at higher acidities. With the latter substrates fractional orders can often be observed,793 because at intermediate acidities, both processes take place simultaneously. These kinetic results seem to indicate that the actual reacting species can be either the monoprotonated substrate ArNHNH2Ar or the diprotonated ArNH2NH2Ar.
Most of the proposed mechanisms794 attempted to show how all five products could be produced by variations of a single process. An important breakthrough was the discovery that the two main products are formed in entirely different ways, as shown by isotope-effect studies.795 When the reaction was run with hydrazobenzene labeled with ![]() at both nitrogen atoms, the isotope effect was 1.022 for formation of 154, but 1.063 for formation of 2,4′-diaminobiphenyl. This showed that the N–N bond is broken in the rate-determining step in both cases, but the steps themselves are obviously different. When the reaction was run with hydrazobenzene labeled with
at both nitrogen atoms, the isotope effect was 1.022 for formation of 154, but 1.063 for formation of 2,4′-diaminobiphenyl. This showed that the N–N bond is broken in the rate-determining step in both cases, but the steps themselves are obviously different. When the reaction was run with hydrazobenzene labeled with ![]() at a para position, there was an isotope effect of 1.028 for formation of 154, but essentially no isotope effect (1.001) for formation of 2,4′-diaminobiphenyl. This can only mean that for 154 formation of the new C–C bond and breaking of the N–N bond both take place in the rate-determining step; in other words, the mechanism is concerted. The following [5.5]-sigmatropic rearrangement accounts for this796:
at a para position, there was an isotope effect of 1.028 for formation of 154, but essentially no isotope effect (1.001) for formation of 2,4′-diaminobiphenyl. This can only mean that for 154 formation of the new C–C bond and breaking of the N–N bond both take place in the rate-determining step; in other words, the mechanism is concerted. The following [5.5]-sigmatropic rearrangement accounts for this796:
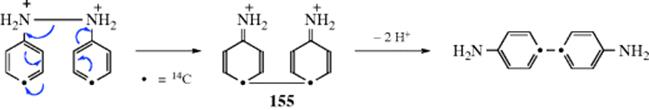
The diion 155 was obtained as a stable species in superacid solution at −78 °C by treatment of hydrazobenzene with FSO3H–SO2 (SO2ClF).797 Although the results just given were obtained with hydrazobenzene, which reacts by the diprotonated pathway, monoprotonated substrates have been found to react by the same [5,5]-sigmatropic mechanism.798 Some of the other rearrangements in this section are also sigmatropic. Thus, formation of the p-semidine takes place by a [1,5]-sigmatropic rearrangement,799 and the conversion of 2,2′-hydrazonaphthalene to 2,2′-diamino-1,1′-binaphthyl by a [3,3] sigmatropic rearrangement.800
2,4′-Diaminobiphenyl is formed by a completely different mechanism, though the details are not known. There is rate-determining breaking of the N–N bond, but the C–C bond is not formed during this step.801 The formation of the o-semidine also takes place by a nonconcerted pathway.802 Under certain conditions, benzidine rearrangements have been found to go through radical cations.803
C. Other Cyclic Rearrangements
18-37 Metathesis of Alkenes (Alkene or Olefin Metathesis)804
Alkene metathesis
![]()
When alkenes are treated with certain catalysts they are converted to other alkenes in a reaction in which one set of alkylidene groups (R1R2C=) have become interchanged with other alkylidene groups (R3R4C=) by a process schematically illustrated by the equilibrium reaction shown below. In an early example shown above, 2-pentene (either cis, trans, or a cis–trans mixture) is converted to a mixture of ~50% 2-pentene, 25% 2-butene, and 25% 3-hexene. Nowadays, superior catalysts and experimental procedures have made this reaction synthetically useful (see below). The reaction is reversible805 and the alkene starting material and products exist in equilibrium, so the same mixture can be obtained by starting with equimolar quantities of 2-butene and 3-hexene.806 The reaction is called metathesis of alkenes or alkene metathesis (olefin metathesis).807 In general, the reaction can be applied to a single unsymmetrical alkene, giving a mixture of itself and two other alkenes, or to a mixture of two alkenes, in which case the number of different molecules in the product depends on the symmetry of the reactants. In the example, a mixture of R1R2C=CR1R2 and R3R4C=CR3R4 gives rise to only one new alkene (R1R2C=CR3R4), while in the most general case, the reaction of two alkenes (R1R2C=CR3R4 and R5R6C=CR7R8) can give a mixture of 10 alkenes: the original 2 + 8 new ones. In early work, W, Mo,808 or Re complexes were used, and with simple alkenes the proportions of products are generally statistical,809 which limited the synthetic utility of the reaction since the yield of any one product is low. In some cases, one alkene may be more or less thermodynamically stable than the rest, so that the proportions are not statistical in all cases.
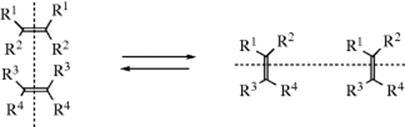
It is possible to shift the equilibrium to favor certain products. For example, 2-methyl-1-butene gives rise to ethylene and 3,4-dimethyl-3-hexene. By allowing the gaseous ethylene to escape, the yield of 3,4-dimethyl-3-hexene can be raised to 95%.810 This example shows that it is possible to tailor the substrate to include two terminal alkenes that lead to ethylene as a product, whose escape from the reaction drives the equilibrium to product.
The development of better catalysts has revolutionized this reaction,811 making it one of the most important methods available for modern synthesis. Both homogeneous812 and heterogeneous813 catalysts have been used for this reaction. Of the many homogeneous catalysts, Ru complexes are the most important,814 and important heterogeneous catalysts include oxides of Mo, W, and Re deposited on alumina or silica gel.815 The major breakthrough in these catalysts was the development of catalysts that are relatively air stable. Three important catalysts are metal carbene complexes 156816 and 157817 (Grubbs catalyst I and II; Mes = mesityl, respectively), and 158 (the Shrock catalyst).818 Catalyst 157 can be generated in situ from air stable precursors.819
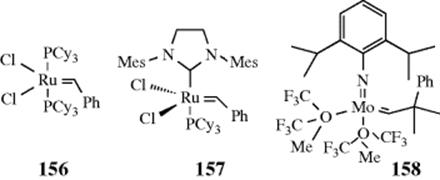
The synthetic importance820 of ring-closing and ring-opening metathesis reactions has, in part, led to the ongoing development of new catalysts.821 Catalysts have been developed that are compatible with both water and methanol.822 The reaction is compatible with the presence of other functional groups823 (e.g., other alkene units,824 carbonyl units,825 the alkene unit of conjugated esters,826 butenolides827 and other lactones,828 amines,829 amides,830sulfones,831 phosphine oxides,832 sulfonate esters,833 and sulfonamides,834 see 149).835 Ether groups,836 including vinyl ethers,837 vinyl halides,838 vinyl silanes,839 vinyl sulfones,840 allylic ethers,841 and thioethers842 are also compatible.
Asymmetric ring-closing metathesis reactions have been reported,843 and chiral metathesis catalysts are continually being developed.844 The enantioselective synthesis of bicyclic lactams from dienyl lactams used a chiral Mo catalyst.845 Asymmetric ring-opening metathesis has also been reported.846
Recyclable catalysts have been developed,847 and the reaction has been done in ionic liquids,848 supercritical CO2849 (Sec. 9.D.ii), and in aqueous media.850 Microwave-induced ring-closing metathesis851 and also cross-metathesis reactions852 are known. Polymer-bound Ru853 and Mo catalysts854 have been used, and catalyst 157 has been immobilized on PEG.855 Efficient methods have been developed for the removal of Ru byproducts from metathesis reactions that include the use of a scavenger resin,856 and removal by aqueous extraction.857
By choice of the proper catalyst, the reaction has been applied to terminal and internal alkenes, straight chain or branched. The effect of substitution on the ease of reaction is CH2= > RCH2CH= > R2CHCH= > R2C=.858 Note that isomerization of the C=C unit can occur after metathesis,859 but methods have been developed to prevent this, including addition of 2,6-dichlorobenzoquinone.860 Cross-metathesis861 (or symmetrical homo-metathesis862) of alkenes to give new alkenes can be accomplished. Monosubstituted alkenes react faster than disubstituted alkenes.863 A double metathesis reaction of a diene (also called domino864 or tandem metathesis865) with conjugated aldehydes has been reported,866 and a triple metathesis was reported to for a dihydropyran with two dihydropyran substituents.867 Cross-metathesis of vinylcyclopropanes leads to an alkene with two cyclopropyl substituents.868 Vinylcyclopropane-alkyne metathesis reactions have been reported.869 Cyclic alkenes can be opened, usually with polymerization using metathesis catalysts. Ring-opening metathesis generates dienes from cyclic alkenes.870 Allenes undergo a metathesis reaction to give symmetrical allenes.871 An interesting variation reacts an α,ω-diene with a cyclic alkene. The combination of ring-opening metathesis and ring-closing cross-metathesis leads to ring expansion to give a macrocyclic nonconjugated diene.872 Note that an alkane metathesis reaction is known.873
Dienes can react intermolecularly or intramolecularly.874 Intramolecular reactions generate rings, including small rings,875 usually alkenes or dienes. Alkene metathesis can be used to form very large rings, including 21-membered lactone rings.876 Diynes undergo both cross-metathesis and ring-closing metathesis.877 Diynes can also react intramolecularly to give large-ring alkynes.878 Metathesis with vinyl-cyclopropyl-alkynes is also known, producing a ring-expanded product (see 159).879 Vinyl halides undergo metathesis reactions.880
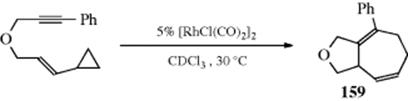
Two cyclic alkenes react to give dimeric dienes,881 for example,
![]()
With many catalysts, the products can then react with additional monomers and with each other, so that polymers are produced, and the cyclic dienes are obtained only in low yield. The reaction between a cyclic and a linear alkene can give a ring-opened diene882:

The reaction has also been applied to internal alkynes883:
![]()
and some success has been reported for terminal triple bonds.884 As noted above, molecules with a terminal alkene and a terminal alkyne react quite well (ene-yne metathesis).885 Intramolecular reactions of a double bond with a triple bond are known886 and a tetracyclic tetraene has been prepared from a poly-yne-diene.887 Cross-metathesis of terminal alkynes and terminal alkenes (en-ynes)888 to give a diene has also been reported.889 Enyne metathesis generates 1,3-dienes.890
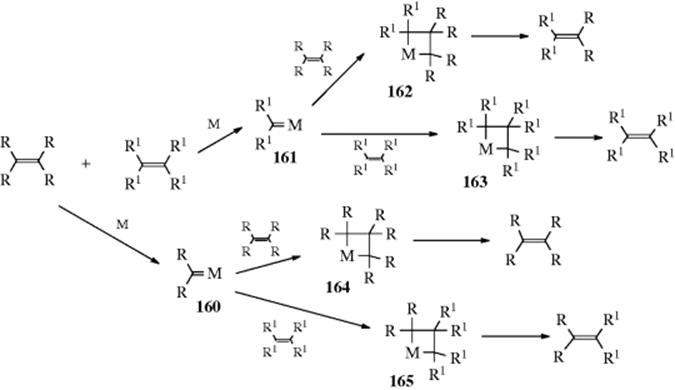
The generally accepted mechanism is a chain mechanism,891 involving the intervention of a metal–carbene complex (160 and 161)892 and a four-membered ring containing a metal893 (162–165).894 In the cross-metathesis reaction shown as an example, R2C=CR2 reacts with R12C=CR12 in the presence of a metal catalyst (M). Initial reaction with the catalyst leads to the two expected metal carbenes, (160 and 161). Metal carbene (161) can react with both alkenes to form metallocyclobutanes 162 and 163. Each of these intermediates loses the metal to form the alkenes, the product of metathesis (R2C=CR12) and one of the original alkenes. In a likewise manner, 160 reacts with each alkene to form metallocyclobutanes 164 and 165, which decomposes to R2C=CR2 and the metathesis product. It has been shown that the phosphine-containing methylidene complexes decompose to methylphosphonium salts,895
OS 80, 85; 81, 1.
18-38 Metal-Ion-Catalyzed σ-Bond Rearrangements
Many highly strained cage molecules undergo rearrangement when treated with metallic ions [e.g., Ag+, Rh(I), or Pd(II)].896 The bond rearrangements observed can be formally classified into two main types: (Type 1)
[2 + 2]-ring openings of cyclobutanes and (Type 2) conversion of a bicyclo[2.2.0] system to a bicyclopropyl system. The molecule cubane supplies an example of each type (see above). Treatment with Rh(I) complexes converts cubane to tricyclo[4.2.0.02.5]octa-3,7-diene (166),897 an example of type 1, while Ag+ or Pd(II) causes the second type of reaction, producing cuneane.898 Other examples are the conversion of 167 to 168, and formation of 159, the 9,10-dicarbomethyoxy derivative of snoutane (pentacyclo[3.3.2.02,4.03,7.06,8]decane).
Type 1

Ref. 899
Type 2900
Ref. 901
The mechanisms of these reactions are not completely understood, although relief of strain undoubtedly supplies the driving force. The reactions are thermally forbidden by the orbital-symmetry rules, and the role of the catalyst is to provide low-energy pathways so that the reactions can take place. Type 1 reactions are the reverse of the catalyzed [2 + 2] ring closures discussed at Reaction 15-63. The following mechanism, in which Ag+ attacks one of the edge bonds, has been suggested for the conversion of 167 to 168.902
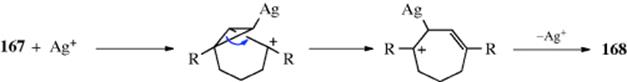
Simpler bicyclobutanes can also be converted to dienes, but in this case the products usually result from cleavage of the central bond and one of the edge bonds.903 For example, treatment of 170 with AgBF4,904
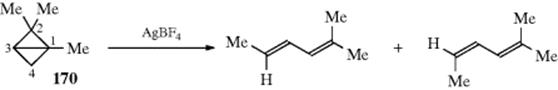
or [(π-allyl)PdCl]2905 gives a mixture of the two dienes shown, resulting from a formal cleavage of the C-1–C-3 and C-1–C-2 bonds (note that a hydride shift has taken place). Dienes can also be converted to bicyclobutanes under photochemical conditions.906
18-39 The Di-π-methane and Related Rearrangements
Di-π-methane rearrangement
1,4-Dienes carrying alkyl or aryl substituents on C-3907 can be photochemically rearranged to vinylcyclopropanes in a reaction called the di-π-methane rearrangement.908 An example is conversion of 171 to 172.909 For most 1,4-dienes, it is only the singlet excited state that gives the reaction; triplet states generally take other pathways.910 For unsymmetrical dienes, the reaction is regioselective. For example, 173 gave 174, not 175:911
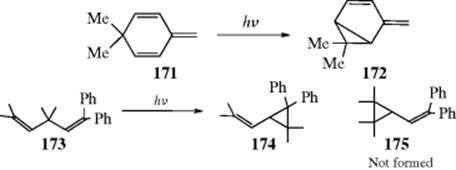
The mechanism can be described by the diradical pathway given912 (the C-3 substituents act to stabilize the radical), although the species shown are not necessarily intermediates, but may represent transition states. It has been shown, for the case of certain substituted substrates, that configuration is retained at C-1 and C-5 and inverted at C-3.913
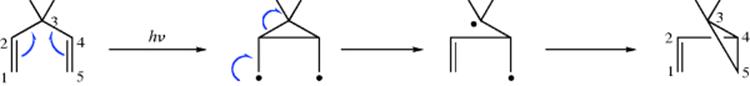
The reaction has been extended to allylic benzenes914 (in this case C-3 substituents are not required), to β,γ-unsaturated ketones915 (the latter reaction, which is called the oxa-di-π-methane rearrangement,916 generally occurs only from the triplet state), to β,γ-unsaturated imines,917 and to triple-bond systems.918
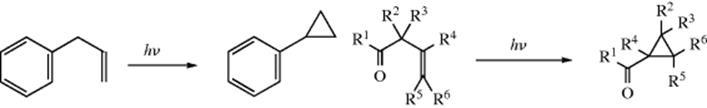
When photolyzed, 2,5-cyclohexadienones can undergo a number of different reactions, one of which is formally the same as the di-π-methane rearrangement.919 In this reaction, photolysis of the substrate 176 gives the bicyclo[3.1.0]hexenone (181). Although the reaction is formally the same (note the conversion of 171 to 172),
the mechanism is different from that of the di-π-methane rearrangement, because irradiation of a ketone can cause an n → π∗ transition, which is of course not possible for a diene lacking a carbonyl group. The mechanism920 in this case has been formulated as proceeding through the excited triplet states 178 and 179. In step 1, the molecule undergoes an n → π∗ excitation to the singlet species 177, which cross to the triplet 178. Step 3 is a rearrangement from one excited state to another. Step 4 is a π∗→ n electron demotion (an intersystem crossing from T1 → S0, see Sec. 7.A.vi, category 4). The conversion of 180 to 181 consists of two 1,2-alkyl migrations (a one-step process would be a 1,3-migration of alkyl to a carbocation center): The old C-6–C-5 bond becomes the new C-6–C-4 bond and the old C-6–C-1 bond becomes the new C-6–C-5 bond.921
2,4-Cyclohexadienones also undergo photochemical rearrangements, but the products are different, generally involving ring opening.922
18-40 The Hofmann–Löffler and Related Reactions
A common feature of the reactions in this section923 is that they serve to introduce functionality at a position remote from functional groups already present. As such, they have proved very useful in synthesizing many compounds, especially in the steroid field (see also, Reactions 19-2 and 19-17). When N-haloamines in which one alkyl group has a hydrogen in the 4 or 5 position are heated with sulfuric acid, pyrrolidines, or piperidines are formed, in a reaction known as the Hofmann–Löffler reaction (also called the Hofmann–Löffler–Freytag reaction).924 The R′ group is normally alkyl, but the reaction has been extended to R′ = H by the use of concentrated sulfuric
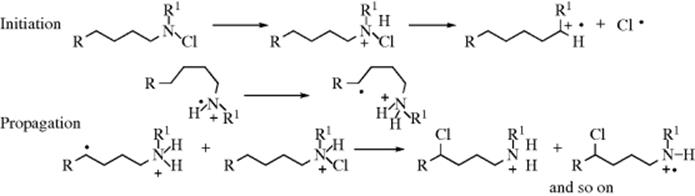
acid solution and ferrous salts.925 The first step of the reaction is a rearrangement, with the halogen migrating from the nitrogen to the 4 or 5 position of the alkyl group. It is possible to isolate the resulting haloamine salt, but usually this is not done, and the second step, the ring closure (Reaction 10-31), takes place. The reaction is most often induced by heat, but this is not necessary, and irradiation and chemical initiators (e.g., peroxides) have been used instead. The mechanism is of a free radical type, with the main step involving an internal hydrogen abstraction.926
A similar reaction has been carried out on N-halo amides, which give γ-lactones927:
Another related reaction is the Barton reaction,928 by which a methyl group in a unique position relative to an OH group can be oxidized to a CHO group. The alcohol is first converted to the nitrite ester. Photolysis of the nitrite results in conversion of the nitrite group to the OH group and nitrosation of the methyl group. Formation of a radical and with the methyl group in the appropriate position, hydrogen-atom transfer via a six-center transition state leads to a nitrite. Hydrolysis of the oxime tautomer gives the aldehyde, for example929
This reaction takes place only when the methyl group is in a favorable steric position.930 The mechanism is similar to that of the Hofmann–Löffler reaction.931
This is one of the few known methods for effecting substitution at an angular methyl group. Not only CH3 groups but also alkyl groups of the form RCH2 and R2CH can give the Barton reaction if the geometry of the system is favorable. An RCH2 group is converted to the oxime R(C=NOH), which is hydrolyzable to a ketone, or to a nitroso dimer, while an R2CH group gives a nitroso compound [R2C(NO)]. With very few exceptions, the only carbons that become nitrosated are those in the position δ to the original OH group, indicating that a six-membered transition state is necessary for the hydrogen abstraction.932
OS III, 159.
D. Noncyclic Rearrangements
18-41 Hydride Shifts
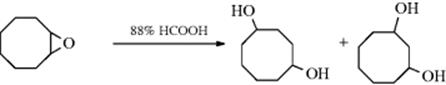
The example shown is typical of a transannular hydride shift. The 1,2-diol is formed by a normal epoxide hydrolysis reaction (10-7).933 For a discussion of 1,3 and longer hydride shifts, see Sec. 18.B.
18-42 The Chapman Rearrangement
1/ O→3/ N-Aryl-migration
In the Chapman rearrangement, N,N-diaryl amides are formed when aryl imino esters are heated.934 Best yields are obtained in refluxing tetraethylene glycol dimethyl ether (tetraglyme),935 although the reaction can also be carried out without any solvent at all. Many groups may be present in the rings (e.g., alkyl, halo, OR, CN, and CO2R). Aryl migrates best when it contains electron-withdrawing groups. On the other hand, electron-withdrawing groups in Ar2 or Ar3 decrease the reactivity. The products can be hydrolyzed to
diarylamines, and this is a method for preparing these compounds. The mechanism probably involves an intramolecular936 aromatic nucleophilic substitution, resulting in a 1,3 oxygen-to-nitrogen shift via a species (e.g., 182). Aryl imino esters can be prepared from N-aryl amides by reaction with PCl5, followed by treatment
of the resulting imino chloride (183) with an aroxide ion.937 Imino esters with any or all of the three groups being alkyl also rearrange, but they require catalysis by H2SO4 or a trace of methyl iodide or methyl sulfate.938 The mechanism is different, involving an intermolecular process.939 This is also true for derivatives for formamide (Ar2 = H).
18-43 The Wallach Rearrangement
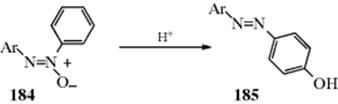
The conversion of azoxy compounds (e.g., 184), upon acid treatment, to p-hydroxy azo compounds (e.g., 185, or sometimes the o-hydroxy isomers940) is called the Wallach rearrangement.941 When both para positions are occupied, the o-hydroxy product may be obtained, but ipso substitution at one of the para positions is also possible.942 The following facts are known for the proposed mechanism943: (1) The para rearrangement is intermolecular.944(2) When the reaction was carried out with an azoxy compound in which the N–O nitrogen was labeled with ![]() , both nitrogens of the product carried the label equally,945 demonstrating that the oxygen did not have a preference for migration to either the near or the far ring. This shows that there is a symmetrical intermediate. (3) Kinetic studies show that two protons are normally required for the reaction.946 The following mechanism,947 involving the symmetrical intermediate (187), has been proposed to explain the facts.948 It has proved possible to obtain 186 and 187 as stable species in superacid solutions.797 Another mechanism, involving an intermediate with only one positive charge, has been proposed for certain substrates at low acidities.949
, both nitrogens of the product carried the label equally,945 demonstrating that the oxygen did not have a preference for migration to either the near or the far ring. This shows that there is a symmetrical intermediate. (3) Kinetic studies show that two protons are normally required for the reaction.946 The following mechanism,947 involving the symmetrical intermediate (187), has been proposed to explain the facts.948 It has proved possible to obtain 186 and 187 as stable species in superacid solutions.797 Another mechanism, involving an intermediate with only one positive charge, has been proposed for certain substrates at low acidities.949
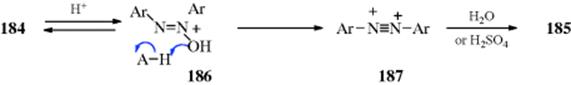
A photochemical Wallach rearrangement950 is also known: The product is the o-hydroxy azo compound, the OH group is found in the farther ring, and the rearrangement is intramolecular.951
18-44 Dyotropic Rearrangements
1/ C-Trialkylsilyl,2/ O-trialkylsilyl-interchange
A dyotropic rearrangement952 is an uncatalyzed process in which two σ bonds simultaneously migrate intramolecularly.953 There are two types. The above is an example of type 1, which consists of reactions in which the two σ bonds interchange positions. In type 2, the two σ bonds do not interchange positions. An example is
Some other examples are
Type 1
![]()
Ref. 954
Type 1
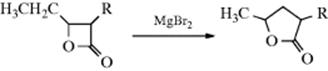
Ref. 955
Type 2
Ref. 956
A useful type 1 example is the Brook rearrangement,957 a stereospecific intramolecular migration of silicon from carbon to oxygen that occurs for (α-hydroxybenzyl)trialkylsilanes (188) in the presence of a catalytic amount of base.958 Formation of a Si–O bond rather than the Si–C bond drives the rearrangement, which is believed to proceed via formation of 189, and does proceed with inversion of configuration at carbon and retention of configuration at silicon.959 A reverse Brook rearrangement is also known.960 The reaction has been extended to other systems. A homo-Brook rearrangement has also been reported.961 Another variation is the aza-Brook rearrangement of α-(silylallyl)amines.962 The Brook rearrangement has been used in synthesis involving silyl dithianes.963 A Brook rearrangement mediated [6 + 2]-annulation has been used for the construction of eight-membered carbocycles.964
The Brook rearrangement has been used in two important synthetic applications, a multicomponent coupling protocol initiated by a Brook rearrangement involving silyl dithianes as mentioned, and anion relay chemistry (ARC) involving a Brook rearrangement. An example of the former is the conversion of the 2-silyl dithiane (190) to the anion with tert-butyllithium followed by ring opening of an epoxide to give 191.965 Treatment with HMPA triggers a solvent-controlled Brook rearrangement that gives a new dithiane anion (192), which then reacts with a different epoxide to give the final product 193. An example of the anion relay chemistry treats dithiane (194) with n-butyllithium, and then 195 to give 196.966 Subsequent treatment with a variety of electrophiles (e.g., allyl bromide, in HMPA), leads to 197 via a Brook rearrangement, and then alkylation of the resultant dithiane anion. This reaction can be initiated by nucleophiles other than dithiane anion.
Notes
1. de Mayo, P. Rearrangements in Ground and Excited States, 3 Vols., Academic Press, NY, 1980; Stevens, T.S.; Watts, W.E. Selected Molecular Rearrangements, Van Nostrand-Reinhold, Princeton, 1973; Collins, C.J.; Eastham, J.F. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 761–821. See also, the series Mechanisms of Molecular Migrations.
2. Vogel, P. Carbocation Chemistry; Elsevier, NY, 1985, pp. 323–372; Shubin, V.G. Top. Curr. Chem. 1984, 116/117, 267; Saunders, M.; Chandrasekhar, J.; Schleyer, P.v.R. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 1, Academic Press, NY, 1980, pp. 1–53; Kirmse, W. Top. Curr. Chem. 1979, 80, 89. For reviews of rearrangements in vinylic cations, see Shchegolev, A.A.; Kanishchev, M.I. Russ. Chem. Rev. 1981, 50, 553; Lee, C.C. Isot. Org. Chem. 1980, 5, 1.
3. It was first postulated by Whitmore, F.C. J. Am. Chem. Soc. 1932, 54, 3274.
4. The IUPAC designations depend on the nature of the steps. For the rules, see Guthrie, R.D. Pure Appl. Chem. 1989, 61, 23, pp. 44–45.
5. Dostrovsky, I.; Hughes, E.D. J. Chem. Soc. 1946, 166.
6. Borodkin, G.I.; Shakirov, M.M.; Shubin, V.G.; Koptyug, V.A. J. Org. Chem. USSR 1978, 14, 290, 924.
7. Brouwer, D.M.; Hogeveen, H. Prog. Phys. Org. Chem. 1972, 9, 179, see pp. 203–237; Olah, G.A.; Olah, J.A. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, pp. 751–760, 766–778. For a discussion of the rates of these reactions, see Sorensen, T.S. Acc. Chem. Res. 1976, 9, 257.
8. Brouwer, D.M. Recl. Trav. Chim. Pays-Bas 1968, 87, 210; Saunders, M.; Hagen, E.L. J. Am. Chem. Soc. 1968, 90, 2436.
9. Ahlberg, P.; Jonsäll, G.; Engdahl, C. Adv. Phys. Org. Chem. 1983, 19, 223; Leone, R.E.; Barborak, J.C.; Schleyer, P.v.R. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 4, Wiley, NY, 1970, pp. 1837–1939; Leone, R.E.; Schleyer, P.v.R. Angew. Chem. Int. Ed. 1970, 9, 860.
10. Campbell, A.; Kenyon, J. J. Chem. Soc. 1946, 25, and references cited therein.
11. See Kirmse, W.; Gruber, W.; Knist, J. Chem. Ber. 1973, 106, 1376; Borodkin, G.I.; Panova, Y.B.; Shakirov, M.M.; Shubin, V.G. J. Org. Chem. USSR 1983, 19, 103.
12. See Cram, D.J. in Newman, M.S. Steric Effects in Organic Chemistry, Wiley, NY, 1956; pp. 251–254; Wheland, G.W. Advanced Organic Chemistry, 3rd ed., Wiley, NY, 1960, pp. 597–604.
13. Bernstein, H.I.; Whitmore, F.C. J. Am. Chem. Soc. 1939, 61, 1324. For other examples, see Tsuchihashi, G.; Tomooka, K.; Suzuki, K. Tetrahedron Lett. 1984, 25, 4253.
14. See Meerwein, H.; van Emster, K. Ber. 1920, 53, 1815; 1922, 55, 2500; Meerwein, H.; Gérard, L. Liebigs Ann. Chem. 1923, 435, 174.
15. See Winstein, S.; Morse, B.K. J. Am. Chem. Soc. 1952, 74, 1133.
16. Collins, C.J.; Benjamin, B.M. J. Org. Chem. 1972, 37, 4358, and references cited therein.
17. Mosher, H.S. Tetrahedron 1974, 30, 1733. See also, Guthrie, R.D. J. Am. Chem. Soc. 1967, 89, 6718.
18. Shiner, Jr., V.J.; Imhoff, M.A. J. Am. Chem. Soc. 1985, 107, 2121.
19. Racho![]() , J.; Goedken, V.; Walborsky, H.M. J. Org. Chem. 1989, 54, 1006. An opposing view: Kirmse, W.; Feyen, P. Chem. Ber. 1975, 108, 71; Kirmse, W.; Plath, P.; Schaffrodt, H. Chem. Ber. 1975, 108, 79.
, J.; Goedken, V.; Walborsky, H.M. J. Org. Chem. 1989, 54, 1006. An opposing view: Kirmse, W.; Feyen, P. Chem. Ber. 1975, 108, 71; Kirmse, W.; Plath, P.; Schaffrodt, H. Chem. Ber. 1975, 108, 79.
20. Skell, P.S.; Starer, I.; Krapcho, A.P. J. Am. Chem. Soc. 1960, 82, 5257.
21. Karabatsos, G.J.; Orzech, Jr., C.E.; Meyerson, S. J. Am. Chem. Soc. 1964, 86, 1994.
22. Saunders, M.; Vogel, P.; Hagen, E.L.; Rosenfeld, J. Acc. Chem. Res. 1973, 6, 53; Lee, C.C. Prog. Phys. Org. Chem. 1970, 7, 129; Collins, C.J. Chem. Rev. 1969, 69, 543. See also, Cooper, C.N.; Jenner, P.J.; Perry, N.B.; Russell-King, J.; Storesund, H.J.; Whiting, M.C. J. Chem. Soc. Perkin Trans. 2 1982, 605.
23. Karabatsos, G.J.; Orzech, Jr., C.E.; Fry, J.L.; Meyerson, S. J. Am. Chem. Soc. 1970, 92, 606.
24. Lee, C.C.; Cessna, A.J.; Ko, E.C.F.; Vassie, S. J. Am. Chem. Soc. 1973, 95, 5688. See also, Lee, C.C.; Reichle, R. J. Org. Chem. 1977, 42, 2058 and references cited therein.
25. Karabatsos, G.J.; Hsi, N.; Meyerson, S. J. Am. Chem. Soc. 1970, 92, 621. See also, Karabatsos, G.J.; Anand, M.; Rickter, D.O.; Meyerson, S. J. Am. Chem. Soc. 1970, 92, 1254.
26. Karabatsos, G.J.; Fry, J.L.; Meyerson, S. J. Am. Chem. Soc. 1970, 92, 614. See also, Lee, C.C.; Zohdi, H.F. Can. J. Chem. 1983, 61, 2092.
27. Saunders, M.; Jaffe, M.H.; Vogel, P. J. Am. Chem. Soc. 1971, 93, 2558; Saunders, M.; Vogel, P. J. Am. Chem. Soc. 1971, 93, 2559, 2561; Kirmse, W.; Loosen, K.; Prolingheuer, E. Chem. Ber. 1980, 113, 129.
28. Kirmse, W.; Ratajczak, H.; Rauleder, G. Chem. Ber. 1977, 110, 2290.
29. Brouwer, D.M.; Hogeveen, H. Recl. Trav. Chim. Pays-Bas 1970, 89, 211; Majerski, Z.; Schleyer, P.v.R.; Wolf, A.P. J. Am. Chem. Soc. 1970, 92, 5731.
30. See Koptyug, V.A.; Shubin, V.G. J. Org. Chem. USSR 1980, 16, 1685; Wheland, G.W. Advanced Organic Chemistry, 3rd ed., Wiley, NY, 1960, pp. 573–597.
31. See Cram, D.J. in Newman, M.S. Steric Effects in Organic Chemistry, Wiley, NY, 1956, pp. 270–276. For an interesting example, see Nickon, A.; Weglein, R.C. J. Am. Chem. Soc. 1975, 97, 1271.
32. See McCall, M.J.; Townsend, J.M.; Bonner, W.A. J. Am. Chem. Soc. 1975, 97, 2743; Brownbridge, P.; Hodgson, P.K.G.; Shepherd, R.; Warren, S. J. Chem. Soc. Perkin Trans. 1 1976, 2024.
33. Grimaud, J.; Laurent, A. Bull. Soc. Chim. Fr. 1967, 3599.
34. See Fischer, A.; Henderson, G.N. J. Chem. Soc., Chem. Commun. 1979, 279, and references cited therein. See also, Marx, J.N.; Hahn, Y.P. J. Org. Chem. 1988, 53, 2866.
35. See Pilkington, J.W.; Waring, A.J. J. Chem. Soc. Perkin Trans. 2 1976, 1349; Korchagina, D.V.; Derendyaev, B.G.; Shubin, V.G.; Koptyug, V.A. J. Org. Chem. USSR 1976, 12, 378; Wistuba, E.; Rüchardt, C. Tetrahedron Lett.1981, 22, 4069; Jost, R.; Laali, K.; Sommer, J. Nouv. J. Chim. 1983, 7, 79.
36. Bachmann, W.E.; Ferguson, J.W. J. Am. Chem. Soc. 1934, 56, 2081.
37. Le Drian, C.; Vogel, P. Helv. Chim. Acta 1987, 70, 1703; Tetrahedron Lett. 1987, 28, 1523.
38. For a review, see Berson, J.A. Angew. Chem. Int. Ed. 1968, 7, 779.
39. Berson, J.A.; Poonian, M.S.; Libbey, W.J. J. Am. Chem. Soc. 1969, 91, 5567; Berson, J.A.; Donald, D.S.; Libbey, W.J. J. Am. Chem. Soc. 1969, 91, 5580; Berson, J.A.; Wege, D.; Clarke, G.M.; Bergman, R.G. J. Am. Chem. Soc. 1969, 91, 5594, 5601.
40. See Collins, C.J. Acc. Chem. Res. 1971, 4, 315; Collins, J.A.; Glover, I.T.; Eckart, M.D.; Raaen, V.F.; Benjamin, B.M.; Benjaminov, B.S. J. Am. Chem. Soc. 1972, 94, 899; Svensson, T. Chem. Scr. 1974, 6, 22.
41. See Collins, C.J. Chem. Soc. Rev. 1975, 4, 251.
42. See Kirmse, W.; Günther, B. J. Am. Chem. Soc. 1978, 100, 3619.
43. Skell, P.S.; Reichenbacher, P.H. J. Am. Chem. Soc. 1968, 90, 2309.
44. Reineke, C.E.; McCarthy, Jr., J.R. J. Am. Chem. Soc. 1970, 92, 6376; Smolina, T.A.; Gopius, E.D.; Gruzdneva, V.N.; Reutov, O.A. Doklad. Chem. 1973, 209, 280.
45. See Fry, J.L.; Karabatsos, G.J. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, p. 527.
46. See Kirmse, W.; Knist, J.; Ratajczak, H. Chem. Ber. 1976, 109, 2296.
47. Skell, P.S.; Maxwell, R.J. J. Am. Chem. Soc. 1962, 84, 3963. See also, Skell, P.S.; Starer, I. J. Am. Chem. Soc. 1962, 84, 3962.
48. Hudson, H.R.; Koplick, A.J.; Poulton, D.J. Tetrahedron Lett. 1975, 1449; Fry, J.L.; Karabatsos, G.J. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, p. 527.
49. Saunders, M.; Stofko, Jr., J.J. J. Am. Chem. Soc. 1973, 95, 252.
50. See Cope, A.C.; Martin, M.M.; McKervey, M.A. Q. Rev. Chem. Soc. 1966, 20, 119.
51. Prelog, V.; Küng, W. Helv. Chim. Acta 1956, 39, 1394.
52. For an apparent exception, see Farcasiu, D.; Seppo, E.; Kizirian, M.; Ledlie, D.B.; Sevin, A. J. Am. Chem. Soc. 1989, 111, 8466.
53. Cope, A.C.; Burton, P.E.; Caspar, M.L. J. Am. Chem. Soc. 1962, 84, 4855.
54. Beckwith, A.L.J.; Ingold, K.U. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 1, Academic Press, NY, 1980, pp. 161–310; Wilt, J.W. in Kochi, J.K. Free Radicals, Vol. 1, Wiley, NY, 1973, pp. 333–501; Stepukhovich, A.D.; Babayan, V.I. Russ. Chem. Rev. 1972, 41, 750; Nonhebel, D.C.; Walton, J.C. Free-Radical Chemistry, Cambridge University Press, London, 1974, pp. 498–552; Huyser, E.S. Free-Radical Chain Reactions, Wiley, NY, 1970, pp. 235–255; Freidlina, R.Kh. Adv. Free-Radical Chem. 1965, 1, 211-278; Pryor, W.A. Free Radicals, McGraw-Hill, NY, 1966, pp. 266–284.
55. Antunes, C.S.A.; Bietti, M.; Ercolani, G.; Lanzalunga, O.; Salamone, M. J. Org. Chem. 2005, 70, 3884.
56. Seubold Jr., F.H. J. Am. Chem. Soc. 1953, 75, 2532. For the observation of this rearrangement by ESR, see Hamilton, Jr., E.J.; Fischer, H. Helv. Chim. Acta 1973, 56, 795.
57. See Walter, D.W.; McBride, J.M. J. Am. Chem. Soc. 1981, 103, 7069, 7074. For a review, see Studer, A.; Bossart, M. Tetrahedron 2001, 57, 9649.
58. Prévost, N.; Shipman, M. Org. Lett. 2001, 3, 2383.
59. He, X.; Ortiz de Montellano, P.R. J. Org. Chem. 2004, 69, 5684.
60. Crich, D.; Filzen, G.F. J. Org. Chem. 1995, 60, 4834.
61. Beckwith, A.L.J.; Duggan, P.J. J. Chem. Soc. Perkin Trans. 2 1992, 1777; 1993, 1673.
62. Crich, D.; Yao, Q. Tetrahedron Lett. 1993, 34, 5677. See Ganapathy, S.; Cambron R.T.; Dockery, K.P.; Wu, Y.-W.; Harris, J.M.; Bentrude, W.G. Tetrahedron Lett. 1993, 34, 5987.
63. Brooks, M.A.; Scott, L.T. J. Am. Chem. Soc. 1999, 121,5444.
64. Several unsuccessful attempts: Slaugh, L.H.; Magoon, E.F.; Guinn, V.P. J. Org. Chem. 1963, 28, 2643.
65. Seubold Jr., F.H. J. Am. Chem. Soc. 1954, 76, 3732.
66. Cristol, S.J.; Brindell, G.D. J. Am. Chem. Soc. 1954, 76, 5699.
67. Eaton, P.E.; Yip, Y. J. Am. Chem. Soc. 1991, 113, 7692.
68. Brown, H.C.; Russel, G.A. J. Am. Chem. Soc. 1952, 74, 3995. See also, Desai, V.R.; Nechvatal, A.; Tedder, J.M. J. Chem. Soc. B 1970, 386.
69. See Freidlina, R.Kh.; Terent'ev, A.B. Russ. Chem. Rev. 1974, 43, 129.
70. McKnight, C.; Rowland, F.S. J. Am. Chem. Soc. 1966, 88, 3179. See Gajewski, J.J.; Burka, L.T. J. Am. Chem. Soc. 1972, 94, 8857, 8860, 8865; Adam, W.; Aponte, G.S. J. Am. Chem. Soc. 1971, 93, 4300.
71. For MO calcualtions indicating that 45 is an intermediate, see Yamabe, S. Chem. Lett. 1989, 1523.
72. Edge, D.J.; Kochi, J.K. J. Am. Chem. Soc. 1972, 94, 7695.
73. Olah, G.A.; Krishnamurthy, V.V.; Singh, B.P.; Iyer, P.S. J. Org. Chem. 1983, 48, 955. 45 has been detected as an intemediate in a different reaction: Effio, A.; Griller, D.; Ingold, K.U.; Scaiano, J.C.; Sheng, S.J. J. Am. Chem. Soc. 1980, 102, 6063; Leardini, R.; Nanni, D.; Pedulli, G.F.; Tundo, A.; Zanardi, G.; Foresti, E.; Palmieri, P. J. Am. Chem. Soc. 1989, 111, 7723.
74. See Martin, M.M. J. Am. Chem. Soc. 1962, 84, 1986; Rüchardt, C.; Hecht, R. Chem. Ber. 1965, 98, 2460, 2471; Rüchardt, C.; Trautwein, H. Chem. Ber. 1965, 98, 2478.
75. See Newcomb, M.; Glenn, A.G.; Williams, W.G. J. Org. Chem. 1989, 54, 2675.
76. See Lewis, S.N.; Miller, J.J.; Winstein, S. J. Org. Chem. 1972, 37, 1478.
77. See Montgomery, L.K.; Matt, J.W. J. Am. Chem. Soc. 1967, 89, 934, 6556; Giese, B.; Heinrich, N.; Horler, H.; Koch, W.; Schwarz, H. Chem. Ber. 1986, 119, 3528.
78. Barclay, L.R.C.; Lusztyk, J.; Ingold, K.U. J. Am. Chem. Soc. 1984, 106, 1793.
79. Baldwin, J.E.; Burrell, R.C.; Shukla, R. Org. Lett. 2002, 4, 3305.
80. See Freidlina, R.Kh.; Terent'ev, A.B. Russ. Chem. Rev. 1979, 48, 828; Freidlina, R.Kh. Adv. Free-Radical Chem. 1965, 1, 211, pp. 231–249.
81. See Chen, K.S.; Tang, D.Y.H.; Montgomery, L.K.; Kochi, J.K. J. Am. Chem. Soc. 1974, 96, 2201.
82. Lindsay, D.A.; Lusztyk, J.L.; Ingold, K.U. J. Am. Chem. Soc. 1984, 106, 7087.
83. See Dannenberg, J.J.; Dill, K. Tetrahedron Lett. 1972, 1571.
84. See Freidlina, R.Kh.; Terent'ev, A.B. Acc. Chem. Res. 1977, 10, 9.
85. Traynham, J.G.; Couvillon, T.M. J. Am. Chem. Soc. 1967, 89, 3205.
86. See Baird, M.S. Chem. Rev. 2003, 103, 1271.
87. de Meijere, A.; Kozhushkov, S.I.; Faber, D.; Bagutskii, V.; Boese, R.; Haumann, T.; Walsh, R. Eur. J. Org. Chem. 2001, 3607.
88. Nickon, A.; Stern, A.G.; Ilao, M.C. Tetrahedron Lett. 1993, 34, 1391.
89. Merrer, D.C.; Moss, R.A.; Liu, M.T.H.; Banks, J.-T.; Ingold, K.U. J. Org. Chem. 1998, 63, 3010.
90. Moss, R.A.; Ho, C.-J.; Liu, W.; Sierakowski, C. Tetrahedron Lett. 1992, 33, 4287.
91. Hayes, R.L.; Fattal, E.; Govind, N.; Carter, E.A. J. Am. Chem. Soc. 2001, 123, 641.
92. Gilbert, J.C.; Kirschner, S. Tetrahedron Lett. 1993, 34, 599, 603.
93. Moss, R.A.; Zheng, F.; Krough-Jespersen, K. Org. Lett. 2001, 3, 1439.
94. See Hunter, D.H.; Stothers, J.B.; Warnhoff, E.W. in de Mayo, P. Rearrangments in Ground and Excited States, Vol. 1, Academic Press, NY, 1980, pp. 391–470; Grovenstein Jr., E. Angew. Chem. Int. Ed. 1978, 17, 313; Jensen, F.R.; Rickborn, B. Electrophilic Substitution of Organomercurials, McGraw-Hill, NY, 1968, pp. 21–30; Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965, pp. 223–243.
95. Borosky, G.L. J. Org. Chem. 1998, 63, 3337.
96. See Hogeveen, H.; van Kruchten, E.M.G.A. Top. Curr. Chem. 1979, 80, 89; Arbuzov, B.A.; Isaeva, Z.G. Russ. Chem. Rev. 1976, 45, 673; Banthorpe, D.V.; Whittaker, D. Q. Rev. Chem. Soc. 1966, 20, 373.
97. See Kaupp, G. Top. Curr. Chem. 1988, 146, 57.
98. See, however, Lewis, R.G.; Gustafson, D.H.; Erman, W.F. Tetrahedron Lett. 1967, 401; Paquette, L.A.; Philips, J.C. Tetrahedron Lett. 1967, 4645; Anderson, P.H.; Stephenson, B.; Mosher, H.S. J. Am. Chem. Soc. 1974, 96, 3171.
99. See, in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, the articles by White, E.H.; Woodcock, D.J. pp. 407–497 (473–483) and by Banthorpe, D.V. pp. 585–667 (pp. 586–612).
100. See Berson, J.A.; Hammons, J.H.; McRowe, A.W.; Bergman, R.G.; Remanick, A.; Houston, D. J. Am. Chem. Soc. 1967, 89, 2590.
101. For an example of a 3,2-endo shift, see Wilder, Jr., P.; Hsieh, W. J. Org. Chem. 1971, 36, 2552.
102. See Cooper, C.N.; Jenner, P.J.; Perry, N.B.; Russell-King, J.; Storesund, H.J.; Whiting, M.C. J. Chem. Soc. Perkin Trans. 2 1982, 605.
103. See Winstein, S. Quart. Rev. Chem. Soc. 1969, 23, 141.
104. Berson, J.A. in de Mayo, P. Molecular Rearrangements, Vol. 1, Academic Press, NY, 1980, p. 111; Sargent, G.D. Quart. Rev. Chem. Soc. 1966, 20, 301; Olah, G.A. Acc. Chem. Res. 1976, 9, 41; Scheppele, S.E. Chem. Rev.1972, 72, 511.
105. Brown, H.C. The Non–Classical Ion Problem, Plenum, New York, 1977.; Brown, H.C. Tetrahedron 1976, 32, 179; Brown, H.C.; Kawakami, J.H. J. Am. Chem. Soc. 1970, 92, 1990. See also, Story, R.R.; Clark, B.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, New York, 1972, p. 1007.
106. Brown, H.C.; Ravindranathan, M. J. Am. Chem. Soc. 1978, 100, 1865.
107. Coates, R.M.; Fretz, E.R. J. Am. Chem. Soc. 1977, 99, 297; Brown, H.C.; Ravindranathan, M. J. Am. Chem. Soc. 1977, 99, 299.
108. Olah, G.A. Carbocations and Electrophilic Reactions, Verlag Chemie/Wiley, New York, 1974, pp. 80–89; Olah, G.A.; White, A.M.; DeMember, J.R.; Commeyras, A.; Lui, C.Y. J. Am. Chem. Soc. 1970, 92, 4627.
109. Trost, B.M.; Yasukata, T. J. Am. Chem. Soc. 2001, 123, 7162.
110. Trost, B.M.; Xie, J. J. Am. Chem. Soc. 2006, 128, 6044.
111. Corey, E.J.; Ursprung, J.J. J. Am. Chem. Soc. 1956, 78, 5041.
112. See Whitlock Jr., H.W.; Olson, A.H. J. Am. Chem. Soc. 1970, 92, 5383.
113. Dutler, H.; Jeger, O.; Ruzicka, L. Helv. Chim. Acta 1955, 38, 1268; Brownlie, G.; Spring, F.S.; Stevenson, R.; Strachan, W.S. J. Chem. Soc. 1956, 2419; Coates, R.M. Tetrahedron Lett. 1967, 4143.
114. See McKervey, M.A.; Rooney, J.J. in Olah, G.A. Cage Hydrocarbons, Wiley, NY, 1990, pp. 39–64; McKervey, M.A. Tetrahedron 1980, 36, 971; Chem. Soc. Rev. 1974, 3, 479; Greenberg, A.; Liebman, J.F. Strained Organic Molecules, Academic Press, NY, 1978, pp. 178–202; Bingham, R.C.; Schleyer, P.v.R. Fortschr. Chem. Forsch. 1971, 18, 1, pp. 3–23.
115. See Gund, T.M.; Osawa, E.; Williams, Jr., V.Z.; Schleyer, P.v.R. J. Org. Chem. 1974, 39, 2979.
116. See Godleski, S.A.; Schleyer, P.v.R.; Osawa, E.; Wipke, W.T. Prog. Phys. Org. Chem. 1981, 13, 63.
117. Schneider, A.; Warren, R.W.; Janoski, E.J. J. Org. Chem. 1966, 31, 1617; Williams, Jr., V.Z.; Schleyer, P.v.R.; Gleicher, G.J.; Rodewald, L.B. J. Am. Chem. Soc. 1966, 88, 3862; Robinson, M.J.T.; Tarratt, H.J.F. Tetrahedron Lett. 1968, 5.
118. See Olah, G.A.; Wu, A.; Farooq, O.; Prakash, G.K.S. J. Org. Chem. 1989, 54, 1450.
119. See Klester, A.M.; Ganter, C. Helv. Chim. Acta 1985, 68, 734.
120. Majerski, Z.; Liggero, S.H.; Schleyer, P.v.R.; Wolf, A.P. Chem. Commun. 1970, 1596.
121. Grovenstein, Jr., E.; Williams Jr., L.P. J. Am. Chem. Soc. 1961, 83, 412; Zimmerman, H.E.; Zweig, A. J. Am. Chem. Soc. 1961, 83, 1196. See also, Grovenstein, Jr., E.; Cheng, Y. J. Am. Chem. Soc. 1972, 94, 4971.
122. See Grovenstein, Jr., E.; Black, K.W.; Goel, S.C.; Hughes, R.L.; Northrop, J.H.; Streeter, D.L.; VanDerveer, D. J. Org. Chem. 1989, 54, 1671, and references cited therein.
123. Bertrand, J.A.; Grovenstein, Jr., E.; Lu, P.; VanDerveer, D. J. Am. Chem. Soc. 1976, 98, 7835.
124. See Lopez, L.; Mele, G.; Mazzeo, C. J. Chem. Soc. Perkin Trans. 1 1994, 779; de Sanabia, J.A.; Carrión, A.E. Tetrahedron Lett. 1993, 34, 7837; Harada, T.; Mukaiyama, T. Chem. Lett. 1992, 81.
125. Bartók, M.; Molnár, A. in Patai, S. The Chemistry of Functional Groups, Supplement E, Wiley, NY, 1980, pp. 722–732; Collins, C.J.; Eastham, J.F. in Patai, S. The Chemistry of the Carbonyl Group,Vol. 1, Wiley, NY, 1966, pp. 762–771.
126. Grant, A.A.; Allukian, M.; Fry, A.J. Tetrahedron Lett. 2002, 43, 4391.
127. Kagan, J.; Agdeppa Jr., D.A.; Mayers, D.A.; Singh, S.P.; Walters, M.J.; Wintermute, R.D. J. Org. Chem. 1976, 41, 2355. Also see Berner, D.; Cox, D.P.; Dahn, H. J. Am. Chem. Soc. 1982, 104, 2631.
128. Ikushima, Y.; Hatakeda, K.; Sato, O.; Yokoyama, T.; Arai, M. J. Am. Chem. Soc. 2000, 122, 1908.
129. Paquette, L.A.; Lanter, J.C.; Johnston, J.N. J. Org. Chem. 1997, 62, 1702.
130. Kudo, K.; Saigo, K.; Hashimoto, Y.; Saito, K.; Hasegawa, M. Chem. Lett. 1992, 1449.
131. Ramart-Lucas, P.; Salmon-Legagneur, F. C. R. Acad. Sci. 1928, 188, 1301.
132. Toda, F.; Shigemasa, T. J. Chem. Soc. Perkin Trans. 1 1989, 209.
133. Bosshard, H.; Baumann, M.E.; Schetty, G. Helv. Chim. Acta 1970, 53, 1271.
134. For a review, see Krief, A.; Laboureur, J.L.; Dumont, W.; Labar, D. Bull. Soc. Chim. Fr. 1990, 681.
135. See Wang, B.M.; Song, Z.L.; Fan, C.A.; Tu, Y.Q.; Chen, W.M. Synlett 2003, 1497; Hurley, P.B.; Dake, G.R. Synlett 2003, 2131.
136. Li, L.; Cai, P.; Guo, Q.; Xue, S. J. Org. Chem. 2008, 73, 3516.
137. Sankararaman, S.; Nesakumar, J.E. J. Chem. Soc., Perkin Trans. 1 1999, 3173.
138. Ranu, B.C.; Jana, U. J. Org. Chem. 1998, 63, 8212.
139. Bhatia, K.A.; Eash, K.J.; Leonard, N.M.; Oswald, M.C.; Mohan, R.S. Tetrahedron Lett. 2001, 42, 8129.
140. For a list of reagents with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1277–1280.
141. See Prandi, J.; Namy, J.L.; Menoret, G.; Kagan, H.B. J. Organomet. Chem. 1985, 285, 449; Miyashita, A.; Shimada, T.; Sugawara, A.; Nohira, H. Chem. Lett. 1986, 1323; Maruoka, K.; Nagahara, S.; Ooi, T.; Yamamoto, H. Tetrahedron Lett. 1989, 30, 5607.
142. Suda, K.; Baba, K.; Nakajima, S.-I.; Takanami, T. Tetrahedron Lett. 1999, 40, 7243.
143. Karamé, I.; Tommasino, M.L.; LeMaire, M. Tetrahedron Lett. 2003, 44, 7687.
144. Anderson, A.M.; Blazek, J.M.; Garg, P.; Payne, B.J.; Mohan, R.S. Tetrahedron Lett. 2000, 41, 1527.
145. See Yandovskii, V.N.; Ershov, B.A. Russ. Chem. Rev. 1972, 41, 403, 410. Also see Hodgson, D.M.; Robinson, L.A.; Jones, M.L. Tetrahedron Lett. 1999, 40, 8637.
146. See Meinwald, J.; Labana, S. S.; Chadha, M. S. J. Am. Chem. Soc. 1963, 85, 582.
147. Robinson, M.W.C.; Pillinger, K.S.; Graham, A.E. Tetrahedron Lett. 2006, 47, 5919; Robinson, M.W.C.; Pillinger, K.S.; Mabbett, I.; Timms, D.A.; Graham, A.E. Tetrahedron 2010, 66, 8377.
148. Deng, X.-M.; Sun, X.-L.; Tang, Y. J. Org. Chem. 2005, 70, 6537.
149. See Pocker, Y.; Ronald, B.P. J. Am. Chem. Soc. 1970, 92, 3385; J. Org. Chem. 1970, 35, 3362; Tamura, K.; Moriyoshi, T. Bull. Chem. Soc. Jpn. 1974, 47, 2942.
150. Pocker, Y. Chem. Ind. (London), 1959, 332. See also, Herlihy, K.P. Aust. J. Chem. 1981, 34, 107.
151. Suzuki, M.; Watanabe, A.; Noyori, R. J. Am. Chem. Soc. 1980, 102, 2095.
152. Antoniotti, S.; Duñach, E. Chem. Commun. 2001, 2566.
153. Chang, C.-L.; Kumar, M.P.; Liu, R.-S. J. Org. Chem. 2004, 69, 2793.
154. Suda, K.; Baba, K.; Nakajima, S.; Takanami, T. Chem. Commun. 2002, 2570.
155. Tu, Y.Q.; Fan, C.A.; Ren, S.K.; Chan, A.S.C. J. Chem. Soc., Perkin Trans. 1 2000, 3791.
156. Bickley, J.F.; Hauer, B.; Pena, P.C.A.; Roberts, S.M.; Skidmore, J. J. Chem. Soc., Perkin Trans. 1 2001, 1253.
157. Bourguet, E.; Baneres, J.-L.; Girard, J.-P.; Parello, J.; Vidal, J.-P.; Lusinchi, X.; Declerzq, J.-P. Org. Lett. 2001, 3, 3067.
158. Wang, B.M.; Song, Z.L.; Fan, C.A.; Tu, Y.Q.; Shi, Y. Org. Lett. 2002, 4, 363.
159. Maruoka, K.; Hasegawa, M.; Yamamoto, H.; Suzuki, K.; Shimazaki, M.; Tsuchihashi, G. J. Am. Chem. Soc. 1986, 108, 3827.
160. See Hesse, M. Ring Enlargement in Organic Chemistry, VCH, NY, 1991; Gutsche, C.D.; Redmore, D. Carbocyclic Ring Expansion Reactions, Academic Press, NY, 1968; Baldwin, J.E.; Adlington, R.M.; Robertson, J. Tetrahedron 1989, 45, 909; Salaün, J. in Rappoport, Z. The Chemistry of the Cyclopropyl Group, pt. 2, Wiley, NY, 1987, pp. 809–878; Conia, J.M.; Robson, M.J. Angew. Chem. Int. Ed. 1975, 14, 473. For a list of ring expansions and contractions, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1283–1302.
161. See Smith, P.A.S.; Baer, D.R. Org. React. 1960, 11, 157. See also, Chow, L.; McClure, M.; White, J. Org. Biomol. Chem. 2004, 2, 648.
162. See Wong, H.N.C.; Hon, M.; Tse, C.; Yip, Y.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165, see pp. 182–186; Breslow, R. in Mayo, P. Molecular Rearrangements, Vol. 1, Wiley, NY, 1963, pp. 233–294.
163. Wiberg, K.B.; Shobe, D.; Nelson, G.C. J. Am. Chem. Soc. 1993, 115, 10645.
164. Nouri, D.H.; Tantillo, D.J. J. Org. Chem. 2006, 71, 3686.
165. Markham, J.P.; Staben, S.T.; Toste, F.D. J. Am. Chem. Soc. 2005, 127, 9708.
166. Trost, B.M.; Xie, J.; Maulide, N. J. Am. Chem. Soc. 2008, 130, 17258.
167. Ranu, B.C.; Banerjee, S.; Das, A. Tetrahedron Lett. 2006, 47, 881.
168. Fürstner, A.; Aïssa, C. J. Am. Chem. Soc. 2006, 128, 6306.
169. Shi, M.; Liu, L.-P.; Tang, J. J. Am. Chem. Soc. 2006, 128, 7430.
170. Xu, G.-C.; Liu, L.-P.; Lu, J.-M.; Shi, M. J. Am. Chem. Soc. 2005, 127, 14552.
171. Sisti, A.J. J. Org. Chem. 1968, 33, 453. See also, Sisti, A.J.; Vitale, A.C. J. Org. Chem. 1972, 37, 4090.
172. Sisti, A.J.; Rusch, G.M. J. Org. Chem. 1974, 39, 1182.
173. Sisti, A.J. J. Org. Chem. 1968, 33, 3953.
174. Marvell, E.N. Thermal Electrocylic Reactions, Academic Press, NY, 1980, pp. 23–53; Sorensen, T.S.; Rauk, A. in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions, Vol. 2, Academic Press, NY, 1977, pp. 1–78.
175. Skell, P.S.; Sandler, S.R. J. Am. Chem. Soc. 1958, 80, 2024.
176. See Berson, J.A. in de Mayo, P. Rearrangaements in Ground and Excited States, Vol. 1, Academic Press, NY, 1980, pp. 324–352; Ann. Rev. Phys. Chem. 1977, 28, 111; Bergman, R.G. in Kochi, J.K. Free Radicals, Vol. 1, Wiley, NY, 1973, pp. 191–237; Frey, H.M. Adv. Phys. Org. Chem. 1966, 4, 147, see pp. 148–170. Also see Baldwin, J.E.; Day, L.S.; Singer, S.R. J. Am. Chem. Soc. 2005, 127, 9370.
177. See Baldwin, J.E.; Grayston, M.W. J. Am. Chem. Soc. 1974, 96, 1629, 1630.
178. See Bergman, R.G.; Carter, W.L. J. Am. Chem. Soc. 1969, 91, 7411.
179. See Schuster, H.F.; Coppola, G.M. Allenes in Organic Synthesis, Wiley, NY, 1984, pp. 20–23; Kirmse, W. Carbene Chemistry, 2nd ed., Academic Press, NY, 1971, pp. 462–467.
180. Billups, W.E.; Bachman, R.E. Tetrahedron Lett. 1992, 33, 1825.
181. See Baird, M.S.; Baxter, A.G.W. J. Chem. Soc. Perkin Trans. 1 1979, 2317, and references cited therein.
182. See Gutsche, C.D.; Redmore, D. Carbocyclic Ring Expansion Reactions, Academic Press, NY, 1968, pp. 111–117.
183. Dowd, P.; Choi, S. Tetrahedron 1991, 47, 4847. For a related ring expansion, see Baldwin, J.E.; Adlington, R.M.; Robertson, J. J. Chem. Soc., Chem. Commun. 1988, 1404.
184. Dowd, P.; Choi, S. J. Am. Chem. Soc. 1987, 109, 6548; Tetrahedron Lett. 1991, 32, 565.
185. Xue, S.; Liu, Y.-K.; Li, L.-Z.; Guo, Q.-X. J. Org. Chem. 2005, 70, 8245.
186. See Fry, A. Mech. Mol. Migr. 1971, 4, 113; Collins, C.J.; Eastham, J.F. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 771–790.
187. Favorskii, A.; Chilingaren, A. C. R. Acad. Sci. 1926, 182, 221.
188. Collins, C.J.; Bowman, N.S. J. Am. Chem. Soc. 1959, 81, 3614.
189. Zook, H.D.; Smith, W.E.; Greene, J.L. J. Am. Chem. Soc. 1957, 79, 4436.
190. Some such pathway is necessary to account for the migration of oxygen that is found. It may involve a protonated epoxide, a 1,2-diol, or simply a 1,2-shift of an OH group.
191. See Fry, A.; Oka, M. J. Am. Chem. Soc. 1979, 101, 6353.
192. See Chalais, S.; Laszlo, P.; Mathy, A. Tetrahedron Lett. 1986, 27, 2627.
193. Guo, Z.; Schultz, A.G. Org. Lett. 2001, 3, 1177.
194. Perkins, M.J.; Ward, P. Mech. Mol. Migr. 1971, 4, 55, pp. 90–103; Miller, B. Mech. Mol. Migr. 1968, 1, 247; Shine, H.J. Aromatic Rearrangements; Elsevier, NY, 1967, pp. 55–68; Waring, A.J. Adv. Alicyclic Chem. 1966, 1,129, pp. 207–223. See Miller, B. Acc. Chem. Res. 1975, 8, 245.
195. See Vitullo, V.P.; Grossman, N. J. Am. Chem. Soc. 1972, 94, 3844; Planas, A.; Tomás, J.; Bonet, J. Tetrahedron Lett. 1987, 28, 471.
196. See Ershov, V.V.; Volod'kin, A.A.; Bogdanov, G.N. Russ. Chem. Rev. 1963, 32, 75.
197. See Selman, S.; Eastham, J.F. Q. Rev. Chem. Soc. 1960, 14, 221.
198. Shimizu, I.; Tekawa, M.; Maruyama, Y.; Yamamoto, A. Chem. Lett. 1992, 1365.
199. For an example, see Schaltegger, A.; Bigler, P. Helv. Chim. Acta 1986, 69, 1666.
200. Doering, W. von E.; Urban, R.S. J. Am. Chem. Soc. 1956, 78, 5938.
201. However, see Screttas, C.G.; Micha-Screttas, M.; Cazianis, C.T. Tetrahedron Lett. 1983, 24, 3287.
202. See Giordano, C.; Castaldi, G.; Casagrande, F.; Abis, L. Tetrahedron Lett. 1982, 23, 1385.
203. Boyer, L.E.; Brazzillo, J.; Forman, M.A.; Zanoni, B. J. Org. Chem. 1996, 61, 7611; Hunter, D.H.; Stothers, J.B.; Warnhoff, E.W. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 1, Academic Press, NY, 1980, pp. 437–461; Rappe, C. in Patai, S. The Chemistry of the Carbon–Halogen Bond, pt. 2, Wiley, NY, 1973, pp. 1084–1101; Redmore, D.; Gutsche, C.D. Carbocylcic Ring Expansion Reactions, Academic Press, NY, 1968, pp. 46–69; Satoh, T.; Motohashi, S.; Kimura, S.; Tokutake, N.; Yamakawa, K. Tetrahedron Lett. 1993, 34, 4823.
204. Craig, J.C.; Dinner, A.; Mulligan, P.J. J. Org. Chem. 1972, 37, 3539.
205. See Mouk, R.W.; Patel, K.M.; Reusch, W. Tetrahedron 1975, 31, 13.
206. See Baretta, A.; Waegell, B. React. Intermed. (Plenum) 1982, 2, 527. For a theoretical study, see Hamblin, G.D.; Jimenez, R.P.; Sorensen, T.S. J. Org. Chem. 2007, 72, 8033.
207. Bordwell, F.G.; Scamehorn, R.G.; Springer, W.R. J. Am. Chem. Soc. 1969, 91, 2087.
208. Loftfield, R.B. J. Am. Chem. Soc. 1951, 73, 4707.
209. A preliminary migration of the chlorine from C-2 to C-6 was ruled out by the fact that recovered 72 had the same isotopic distribution as the starting 72.
210. See Wasserman, H.H.; Clark, G.M.; Turley, P.C. Top. Curr. Chem. 1974, 47, 73; Turro, N.J. Acc. Chem. Res. 1969, 2, 25.
211. See Rappe, C.; Knutsson, L.; Turro, N.J.; Gagosian, R.B. J. Am. Chem. Soc. 1970, 92, 2032.
212. Pazos, J.F.; Pacifici, J.G.; Pierson, G.O.; Sclove, D.B.; Greene, F.D. J. Org. Chem. 1974, 39, 1990.
213. See Baldwin, J.E.; Cardellina, J.H.I. Chem. Commun. 1968, 558.
214. Wharton, P.S.; Fritzberg, A.R. J. Org. Chem. 1972, 37, 1899.
215. Tchoubar, B.; Sackur, O. C. R. Acad. Sci. 1939, 208, 1020.
216. Baudry, D.; Bégué, J.; Charpentier-Morize, M. Bull. Soc. Chim. Fr. 1971, 1416.
217. See Salaun, J.R.; Garnier, B.; Conia, J.M. Tetrahedron 1973, 29, 2895.
218. Ferraz, H.M.; Silva, Jr., J.F. Tetrahedron Lett. 1997, 38, 1899.
219. Meier, H.; Zeller, K. Angew. Chem. Int. Ed. 1975, 14, 32; Kirmse, W. Carbene Chemistry, 2nd ed., Academic Press, NY, 1971, pp. 475–493; Whittaker, D. in Patai, S. The Chemistry of Diazonium and Diazo Compounds, pt. 2, Wiley, NY, 1978, pp. 593–644.
220. Kirmse, W. Eur. J. Org. Chem. 2002, 2193. See Sudrik, S.G.; Sharma, J.; Chavan, V.B.; Chaki, N.K.; Sonawane, H.R.; Vijayamohanan, K.P. Org. Lett. 2006, 8, 1089.
221. Winum, J.-Y.; Kamal, M.; Leydet, A.; Roque, J.-P.; Montero, J.-L. Tetrahedron Lett. 1996, 37, 1781.
222. For a list of methods, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1850–1851.
223. See Regitz, M.; Maas, G. Diazo Compounds, Academic Press, NY, 1986, pp. 185–195; Ando, W. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966,78, pp. 458–475.
224. See Aoyama, T.; Shioiri, T. Tetrahedron Lett. 1980, 21, 4461.
225. Redmore, D.; Gutsche, C.D. Carbocyclic Ring Expansion Reactions, Academic Press, NY, 1968, pp. 125–136.
226. Korobitsyna, I.K.; Rodina, L.L.; Sushko, T.P. J. Org. Chem. USSR 1968, 4, 165; Jones, Jr., M.; Ando, W. J. Am. Chem. Soc. 1968, 90, 2200. See Lee, Y.R.; Suk, J.Y.; Kim, B.S. Tetrahedron Lett. 1999, 40, 8219.
227. Wiskur, S.L.; Fu, G.C. J. Am. Chem. Soc. 2005, 127, 6176.
228. See Scott, A.P.; Platz, M.S.; Radom, L. J. Am. Chem. Soc. 2001, 123, 6069.
229. See Farlow, R.A.; Thamatloor, D.A.; Sunoj, R.B.; Hadad, C.M. J. Org. Chem. 2002, 67, 3257.
230. Kirmse, W.; Horner, L. Chem. Ber. 1956, 89, 2759. Also see, Horner, L.; Spietschka, E. Chem. Ber. 1956, 89, 2765.
231. See Kirmse, W. Carbene Chemistry, 2nd ed., Academic Press, NY, 1971, pp. 476–480. See also, Torres, M.; Ribo, J.; Clement, A.; Strausz, O.P. Can. J. Chem. 1983, 61, 996; Tomoika, H.; Hayashi, N.; Asano, T.; Izawa, Y. Bull. Chem. Soc. Jpn. 1983, 56, 758.
232. See Lewars, Y. Chem. Rev. 1983, 83, 519.
233. Fenwick, J.; Frater, G.; Ogi, K.; Strausz, O.P. J. Am. Chem. Soc. 1973, 95, 124; Zeller, K. Chem. Ber. 1978, 112, 678. See also, Majerski, Z.; Redvanly, C.S. J. Chem. Soc., Chem. Commun. 1972, 694.
234. Tanigaki, K.; Ebbesen, T.W. J. Am. Chem. Soc. 1987, 109, 5883. See also, Bachmann, C.; N'Guessan, T.Y.; DebÛ, F.; Monnier, M.; Pourcin, J.; Aycard, J.; Bodot, H. J. Am. Chem. Soc. 1990, 112, 7488.
235. Csizmadia, I.G.; Gunning, H.E.; Gosavi, R.K.; Strausz, O.P. J. Am. Chem. Soc. 1973, 95, 133.
236. McMahon, R.J.; Chapman, O.L.; Hayes, R.A.; Hess, T.C.; Krimmer, H. J. Am. Chem. Soc. 1985, 107, 7597.
237. Tomioka, H.; Okuno, H.; Izawa, Y. J. Org. Chem. 1980, 45, 5278.
238. See Yamamoto, M.; Nakazawa, M.; Kishikawa, K.; Kohmoto, S. Chem. Commun. 1996, 2353.
239. See Gutsche, C.D. Org. React. 1954, 8, 364.
240. See Aoyama, T.; Shioiri, T. Synthesis 1988, 228.
241. Lemini, C.; Ordoñez, M.; Pérez–Flores, J.; Cruz-Almanza, R. Synth. Commun. 1995, 25, 2695.
242. Xue, S.; Li, L.-Z.; Liu, Y.-K.; Guo, Q.-X. J. Org. Chem. 2006, 71, 215.
243. Aïssa, C.; Fürstner, A. J. Am. Chem. Soc. 2007, 129, 14836.
244. See Müller, E.; Kessler, H.; Zeeh, B. Fortschr. Chem. Forsch. 1966, 7, 128, see pp. 137–150.
245. See Krief, A.; Laboureur, J.L. Tetrahedron Lett. 1987, 28, 1545; Krief, A.; Laboureur, J.L.; Dumont, W. Tetrahedron Lett. 1987, 28, 1549; Abraham, W.D.; Bhupathy, M.; Cohen, T. Tetrahedron Lett. 1987, 28, 2203; Trost, B.M.; Mikhail, G.K. J. Am. Chem. Soc. 1987, 109, 4124.
246. See Turro, N.J.; Gagosian, R.B. J. Am. Chem. Soc. 1970, 92, 2036.
247. See Gutsche, C.D.; Redmore, D. Carbocyclic Ring Expansion Reactions, Academic Press, NY, 1968, pp. 81–98. For a review pertaining to bridged bicyclic ketones, see Krow, G.R. Tetrahedron 1987, 43, 3.
248. See Loeschorn, C.A.; Nakajima, M.; Anselme, J. Bull. Soc. Chim. Belg. 1981, 90, 985.
249. Mock, W.L.; Hartman, M.E. J. Org. Chem. 1977, 42, 459, 466; Baldwin, S.W.; Landmesser, N.G. Synth. Commun. 1978, 8, 413.
250. Liu, H.J.; Majumdar, S.P. Synth. Commun. 1975, 5, 125.
251. Dave, V.; Warnhoff, E.W. J. Org. Chem. 1983, 48, 2590.
252. Doyle, M.P.; Trudell, M.L.; Terpstra, J.W. J. Org. Chem. 1983, 48, 5146.
253. Lee, P.H.; Lee, J. Tetrahedron Lett. 1998, 39, 7889.
254. Hasegawa, E.; Kitazume, T.; Suzuki, K.; Tosaka, E. Tetrahedron Lett. 1998, 39, 4059.
255. Fukuzawa, S.; Tsuchimoto, T. Tetrahedron Lett. 1995, 36, 5937.
256. Angle, S.R.; Neitzel, M.L. J. Org. Chem. 2000, 65, 6458.
257. Mahata, P.K.; Barun, O.; Ila, H.; Junjappa, H. Synlett 2000, 1345.
258. Southwick, P.L.; Walsh, W.L. J. Am. Chem. Soc. 1955, 77, 405. See also, Kiss, L.; Mangelinckx, S.; Fülöp, F.; De Kimpe, N. Org. Lett. 2007, 9, 4399.
259. See Peterson, P.E. Acc. Chem. Res. 1971, 4, 407. See also, Brusova, G.P.; Gopius, E.D.; Smolina, T.A.; Reutov, O.A. Doklad. Chem. 1980, 253, 334; Kobrina, L.S.; Kovtonyuk, V.N. Russ. Chem. Rev. 1988, 57, 62; Warren, S. Acc. Chem. Res. 1978, 11, 403. See also, Aggarwal, V.K.; Warren, S. J. Chem. Soc. Perkin Trans. 1 1987, 2579.
260. For a review, see McDonald, R.N. Mech. Mol. Migr. 1971, 3, 67.
261. Karikomi, M.; Takayama, T.; Haga, K.; Hiratani, K. Tetrahedron Lett. 2005, 46, 6541.
262. Batory, L.A.; McInnis, C.E.; Njardarson, J.T. J. Am. Chem. Soc. 2006, 128, 16054.
263. Rogers, E.; Araki, H.; Batory, L.A.; McInnis, C.E.; Njardarson, J.T. J. Am. Chem. Soc. 2007, 129, 2768.
264. Morrill, C.; Grubbs, R.H. J. Am. Chem. Soc. 2005, 127, 2842; Morrill, C.; Beutner, G.L.; Grubbs, R.H. J. Org. Chem. 2006, 71, 7813.
265. Boeda, F.; Mosset, P.; Crévisy, C. Tetrahedron Lett. 2006, 47, 5021.
266. Mantilli, L.; Gérard, D.; Torche, S.; Besnard, C.; Mazet, C. Pure Appl. Chem. 2010, 82, 1461.
267. Leleti, R.R.; Hu, B.; Prashad, M.; Repi![]() , O. Tetrahedron Lett. 2007, 48, 8505.
, O. Tetrahedron Lett. 2007, 48, 8505.
268. Ito, M.; Kitahara, S.; Ikariya, T. J. Am. Chem. Soc. 2005, 127, 6172.
269. Marion, N.; Gealageas, R.; Nolan, S.P. Org. Lett. 2007, 9, 2653.
270. Meyer, K.H.; Schuster, K. Ber. 1922, 55, 819; Swaminathan, S.; Narayan, K.V. Chem. Rev. 1971, 71, 429.
271. Tanaka, K.; Shoji, T.; Hirano, M. Eur. J. Org. Chem. 2007, 2687.
272. Lopez, S.S.; Engel, D.A.; Dudley, G.B. Synlett 2007, 949.
273. Sonye, J.P.; Koide, K. J. Org. Chem. 2007, 72, 1846.
274. Egi, M.; Yamaguchi, Y.; Fujiwara, N.; Akai, S. Org. Lett. 2008, 10, 1867.
275. Witham, C.A.; Mauleón, P.; Shapiro, N.D.; Sherry, B.D.; Toste, F.D. J. Am. Chem. Soc. 2007, 129, 5838.
276. Barluenga, J.; Riesgo, L.; Vicente, R.; López, L.A.; Tomás, M. J. Am. Chem. Soc. 2007, 129, 7772.
277. Buzas, A.K.; Istrate, F.M.; Gagosz, F. Org. Lett. 2007, 9, 985.
278. Brown, H.C. Hydroboration, W. A. Benjamin, NY, 1962, pp. 136–149, Brown, H.C.; Zweifel, G. J. Am. Chem. Soc. 1966, 88, 1433. See also, Brown, H.C.; Racherla, U.S. J. Organomet. Chem. 1982, 241, C37.
279. Logan, T.J. J. Org. Chem. 1961, 26, 3657.
280. Brown, H.C.; Zweifel, G. J. Am. Chem. Soc. 1967, 89, 561.
281. See Wood, S.E.; Rickborn, B. J. Org. Chem. 1983, 48, 555; Field, L.D.; Gallagher, S.P. Tetrahedron Lett. 1985, 26, 6125.
282. For a review, see Conley, R.T.; Ghosh, S. Mech. Mol. Migr. 1971, 4, 197, pp. 289–304.
283. Baumgarten, H.E.; Petersen, H.E. J. Am. Chem. Soc. 1960, 82, 459, and references cited therein.
284. Cram, D.J.; Hatch, M.J. J. Am. Chem. Soc. 1953, 75, 33; Hatch, M.J.; Cram, D.J. J. Am. Chem. Soc. 1953, 75, 38.
285. Neber, P.W.; Burgard, A. Liebigs Ann. Chem. 1932, 493, 281; Parcell, R.F. Chem. Ind. (London) 1963, 1396. Also see Ref. 284.
286. House, H.O.; Berkowitz, W.F. J. Org. Chem. 1963, 28, 2271.
287. See Nakai, M.; Furukawa, N.; Oae, S. Bull. Chem. Soc. Jpn. 1969, 42, 2917.
288. Baumgarten, H.E.; Petersen, J.M.; Wolf, D.C. J. Org. Chem. 1963, 28, 2369.
289. Taber, D.F.; Tian, W. J. Am. Chem. Soc. 2006, 128, 1058.
290. Smith, P.A.S. in de Mayo, P. Molecular Rearrangements, Vol. 1, Wiley, NY, 1963, Vol. 1, pp. 258–550.
291. See Boyer, J.H. Mech. Mol. Migr. 1969, 2, 267.
292. The question is discussed by Lwowski, W. in Lwowski, W. Nitrenes, Wiley, NY, 1970, pp. 217–221.
293. See Sy, A.O.; Raksis, J.W. Tetrahedron Lett. 1980, 21, 2223.
294. See Wallis, E.S.; Lane, J.F. Org. React. 1946, 3, 267.
295. See Radlick, P.; Brown, L.R. Synthesis 1974, 290.
296. Huang, X.; Keillor, J.W. Tetrahedron Lett. 1997, 38, 313.
297. See Gogoi, P.; Konwar, D. Tetrahedron Lett. 2007, 48, 531.
298. Huang, X.; Seid, M.; Keillor, J.W. J. Org. Chem. 1997, 62, 7495.
299. Matsumura, Y.; Maki, T.; Satoh, Y. Tetrahedron Lett. 1997, 38, 8879.
300. Imamoto, T.; Tsuno, Y.; Yukawa, Y. Bull. Chem. Soc. Jpn. 1971, 44, 1632, 1639, 1644; Imamoto, T.; Kim, S.; Tsuno, Y.; Yukawa, Y. Bull. Chem. Soc. Jpn. 1971, 44, 2776.
301. Baumgarten, H.E.; Smith, H.L.; Staklis, A. J. Org. Chem. 1975, 40, 3554.
302. Loudon, G.M.; Radhakrishna, A.S.; Almond, M.R.; Blodgett, J.K.; Boutin, R.H. J. Org. Chem. 1984, 49, 4272; Boutin, R.H.; Loudon, G.M. J. Org. Chem. 1984, 49, 4277; Pavlides, V.H.; Chan, E.D.; Pennington, L.; McParland, M.; Whitehead, M.; Coutts, I.G.C. Synth. Commun. 1988, 18, 1615.
303. Vasudevan, A.; Koser, G.F. J. Org. Chem. 1988, 53, 5158.
304. Yu, C.; Jiang, Y.; Liu, B.; Hu, L. Tetrahedron Lett. 2001, 42, 1449.
305. Uriz, P.; Serra, M.; Salagre, P.; Castillon, S.; Claver, C.; Fernandez, E. Tetrahedron Lett. 2002, 43, 1673.
306. See Banthorpe, D.V. in Patai, S. The Chemistry of the Azido Group, Wiley, NY, 1971, pp. 397–405.
307. See Pfister, J.R.; Wyman, W.E. Synthesis 1983, 38. See also, Lebel, H.; Leogane, O. Org. Lett. 2005, 7, 4107. See also, Ma, B.; Lee, W.-C. Tetrahedron Lett. 2010, 51, 385.
308. See, Smalley, R.K.; Bingham, T.E. J. Chem. Soc. C 1969, 2481.
309. See Scriven, E.F.V. Azides and Nitrenes, Academic Press, NY, 1984; Stevens, T.S.; Watts, W.E. Selected Molecular Rearrnagements, Van Nostrand-Reinhold, Princeton, 1973, pp. 45–52; in Lwowski, W. Nitrenes, Wiley, NY, 1970, the chapters by Lewis, F.D.; Saunders Jr., W.H. pp. 47–97, pp. 47–78 and by Smith, P.A.S. pp. 99–162.
310. Montgomery, F.C.; Saunders, Jr., W.H. J. Org. Chem. 1976, 41, 2368.
311. Smith, P.A.S.; Lakritz, J. cited in Smith, P.A.S. in de Mayo, P. Molecular Rearrangments, Vol. 1, Wiley, NY, 1963, p. 474.
312. Huisgen, R.; Vossius, D.; Appl, M. Chem. Ber. 1958, 91, 1,12.
313. See Bauer, L.; Exner, O. Angew. Chem. Int. Ed. 1974, 13, 376.
314. See Salomon, C.J.; Breuer, E. J. Org. Chem, 1997, 62, 3858.
315. Wallace, R.G.; Barker, J.M.; Wood, M.L. Synthesis 1990, 1143.
316. Chandrasekhar, S.; Sridhar, M. Tetrahedron Asymmetry 2000, 11, 3467.
317. See Banthorpe, D.V. in Patai, S. The Chemistry of the Azido Group, Wiley, NY, 1971, pp. 405–434.
318. See Koldobskii, G.I.; Ostrovskii, V.A.; Gidaspov, B.V. Russ. Chem. Rev. 1978, 47, 1084.
319. See Smith, P.A.S. Org. React. 1946, 3, 337, pp. 363–366.
320. See Koldobskii, G.I.; Tereschenko, G.F.; Gerasimova, E.S.; Bagal, L.I. Russ. Chem. Rev. 1971, 40, 835; Beckwith, A.L.J. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 137–145.
321. See Krow, G.R. Tetrahedron 1981, 37, 1283.
322. Exceptions to this statement are found in Bhalerao, U.T.; Thyagarajan, G. Can. J. Chem. 1968, 46, 3367; Tomita, M.; Minami, S.; Uyeo, S. J. Chem. Soc. C 1969, 183.
323. See Furness, K.; Aubé, J. Org. Lett. 1999, 1, 495.
324. Sahasrabudhe, K.; Gracias, V.; Furness, K.; Smith, B.T.; Katz, C.E.; Reddy, D.S.; Aubé, J. J. Am. Chem. Soc. 2003, 125, 7914. See Mossman, C.J.; Aubé, J. Tetrahedron, 1996, 52, 3403.
325. Pearson, W.H.; Hutta, D.A.; Fang, W.-k. J. Org. Chem. 2000, 65, 8326. See also, Wrobleski, A.; Aubé, J. J. Org. Chem. 2001, 66, 886.
326. Evans, P.A.; Modi, D.P. J. Org. Chem. 1995, 60, 6662.
327. Benati, L.; Nanni, D.; Sangiorgi, C.; Spagnolo, P. J. Org. Chem. 1999, 64, 7836.
328. See Kumar, H.M.S.; Reddy, B.V.S.; Anjaneyulu, S.; Yadav, J.S. Tetrahedron Lett. 1998, 39, 7385. Also see, Saito, A.; Saito, K.; Tanaka, A.; Oritani, T. Tetrahedron Lett. 1997, 38, 3955.
329. Thompson, A.S.; Humphrey, G.R.; DeMarco, A.M.; Mathre, D.J.; Grabowski, E.J.J. J. Org. Chem. 1993, 58, 5886.
330. Gorin, D.J.; Davis, N.R.; Toste, F.D. J. Am. Chem. Soc. 2005, 127, 11260.
331. Yao, L.; Aubé, J. J. Am. Chem. Soc. 2007, 129, 2766.
332. This mechanism has been controversial, see Vogler, E.A.; Hayes, J.M. J. Org. Chem. 1979, 44, 3682.
333. Amyes, T.L.; Richard, J.P. J. Am. Chem. Soc. 1991, 113, 1867.
334. See Ostrovskii, V.A.; Koshtaleva, T.M.; Shirokova, N.P.; Koldobskii, G.I.; Gidaspov, B.V. J. Org. Chem. USSR 1974, 10, 2365 and references cited therein.
335. Gawley, R.E. Org. React. 1988, 35, 1; McCarty, C.G. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 408–439. Also see, Nguyen, M.T.; Raspoet, G.; Vanquickenborne, L.G. J. Am. Chem. Soc. 1997, 119, 2552.
336. Costa, A.; Mestres, R.; Riego, J.M. Synth. Commun. 1982, 12, 1003.
337. De, S.K. Synth. Commun. 2004, 34, 3431.
338. De, S.K. Org. Prep. Proceed. Int. 2004, 36, 383.
339. Ganguly, N.C.; Mondal, P. Synthesis 2010, 3705.
340. Ramalingan, C.; Park, Y.-T. J. Org. Chem. 2007, 72, 4536.
341. Hashimoto, M.; Obora, Y.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2008, 73, 2894.
342. Yadav, L.D.S.; Patel, R.; Srivastava, V.P. Synthesis 2010, 1771. See also Yadav, L.D.S.; Garima, Srivastava, V.P. Tetrahedron Lett. 2010, 51, 739.
343. Khodaei, M.M.; Meybodi, F.A.; Rezai, N.; Salehi, P. Synth. Commun. 2001, 31, 2047.
344. Furuya, Y.; Ishihara, K.; Yamamoto, H. J. Am. Chem. Soc. 2005, 127, 11240. In ionic liquids, see Betti, C.; Landini, D.; Maia, A.; Pasi, M. Synlett 2008, 908.
345. See Beckwith, A.L.J. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 131–137.
346. Chandrasekhar, S.; Gopalaiah, K. Tetrahedron Lett. 2001, 42, 8123.
347. Boero, M.; Ikeshoji, T.; Liew, C.C.; Terakura, K.; Parrinello, M. J. Am. Chem. Soc. 2004, 126, 6280.
348. Peng, J.; Deng, Y. Tetrahedron Lett. 2001, 42, 403; Ren, R.X.; Zueva, L.D.; Ou, X. Tetrahedron Lett. 2001, 42, 8441.
349. His, S.; Meyer, C.; Cossy, J.; Emeric, G.; Greiner, A. Tetrahedron Lett. 2003, 44, 8581.
350. Thakur, A.J.; Boruah, A.; Prajapati, D.; Sandhu, J.S. Synth. Commun. 2000, 30, 2105; On silica with microwave irradiation, see Loupy, A.; Régnier, S. Tetrahedron Lett. 1999, 40, 6221.
351. An, G.-i.; Kim, M.; Kim. J.Y.; Rhee, H. Tetrahedron Lett. 2003, 44, 2183.
352. Vinnik, M.I.; Zarakhani, N.G. Russ. Chem. Rev. 1967, 36, 51; Krow, G.R. Tetrahedron 1981, 37, 1283.
353. Sharghi, H.; Hosseini, M. Synthesis 2002, 1057; Eshghi, H.; Gordi, Z. Synth. Commun. 2003, 33, 2971; Moghaddam, F.M.; Rad, A.A.R.; Zali-Boinee, H. Synth. Commun. 2004, 34, 2071.
354. Olah, G.A.; Fung, A.P. Synthesis 1979, 537. See also, Novoselov, E.F.; Isaev, S.D.; Yurchenko, A.G.; Vodichka, L.; Trshiska, Ya. J. Org. Chem. USSR 1981, 17, 2284.
355. See Lansbury, P.T.; Mancuso, N.R. Tetrahedron Lett. 1965, 2445.
356. Field, L.; Hughmark, P.B.; Shumaker, S.H.; Marshall, W.S. J. Am. Chem. Soc. 1961, 83, 1983. See also, Leusink, A.J.; Meerbeek, T.G.; Noltes, J.G. Recl. Trav. Chim. Pays-Bas 1976, 95, 123; 1977, 96, 142.
357. Chattopadhyaya, J.B.; Rama Rao, A.V. Tetrahedron 1974, 30, 2899.
358. See Arisawa, M.; Yamaguchi, M. Org. Lett. 2001, 3, 311.
359. Anilkumar, R.; Chandrasekhar, S. Tetrahedron Lett. 2000, 41, 5427.
360. See Nguyen, M.T.; Vanquickenborne, L.G. J. Chem. Soc. Perkin Trans. 2 1993, 1969.
361. Donaruma, L.G.; Heldt, W.Z. Org. React. 1960, 11, 1, pp. 5–14; Smith, P.A.S. in de Mayo, P. Molecular Rearrangments, Vol. 1, Wiley, NY, 1963, 483–507, pp. 488–493.
362. Gregory, B.J.; Moodie, R.B.; Schofield, K. J. Chem. Soc. B 1970, 338.
363. Palmere, R.M.; Conley, R.T.; Rabinowitz, J.L. J. Org. Chem. 1972, 37, 4095.
364. See, Suginome, H.; Yagihashi, F. J. Chem. Soc. Perkin Trans. 1 1977, 2488.
365. Yamabe, S.; Tsuchida, N.; Yamazaki, S. J. Org. Chem. 2005, 70, 10638.
366. For a review, see Maruoka, K.; Yamamoto, H. Angew. Chem. Int. Ed. 1985, 24, 668.
367. Maruoka, K.; Miyazaki, T.; Ando, M.; Matsumura, Y.; Sakane, S.; Hattori, K.; Yamamoto, H. J. Am. Chem. Soc. 1983, 105, 2831; Maruoka, K.; Nakai, S.; Yamamoto, H. Org. Synth. 66, 185.
368. Lanni, E.L.; Bosscher, M.A.; Ooms, B.; Shandro, C.A.; Ellsworth, B.A.; Anderson, C.E. J. Org. Chem. 2008, 73, 6425.
369. Suda, K.; Sashima, M.; Izutsu, M.; Hino, F. J. Chem. Soc., Chem. Commun. 1994, 949.
370. Post, A.J.; Nwaukwa, S.; Morrison, H. J. Am. Chem. Soc. 1994, 116, 6439.
371. Gassman, P.G.; Fox, B.L. J. Am. Chem. Soc. 1967, 89, 338. See also, Davies, J.W.; Malpass, J.R.; Walker, M.P. J. Chem. Soc., Chem. Commun. 1985, 686; Hoffman, R.V.; Kumar, A.; Buntain, G.A. J. Am. Chem. Soc. 1985, 107, 4731.
372. See Kovacic, P.; Lowery, M.K.; Roskos, P.D. Tetrahedron 1970, 26, 529.
373. Hoffman, R.V.; Buntain, G.A. J. Org. Chem. 1988, 53, 3316.
374. Wasserman, H.H.; Glazer, E.A.; Hearn, M.J. Tetrahedron Lett. 1973, 4855.
375. Honda, T.; Ishikawa, F. Chem. Commun. 1999, 1065.
376. Sisti, A.J.; Milstein, S.R. J. Org. Chem. 1974, 39, 3932.
377. For a review, see Renz, M.; Meunier, B. Eur. J. Org. Chem. 1999, 737. For a review of green procedures, see ten Brink, G.-J.; Arends, I.W.C.E.; Sheldon, R.A. Chem. Rev. 2004, 104, 4105.
378. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1665–1667.
379. Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, 1990, pp. 186–195; Plesnicar, B. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. C, Academic Press, NY, 1978, pp. 254–267; House, H.O. Modern Synthetic Reactions, 2nd ed.; W.A. Benjamin, NY, 1972, pp. 321–329; Lewis, S.N. in Augustine, R.L. Oxidation, Vol. 1, Marcel Dekker, NY, 1969, pp. 237–244. Also see, Carlqvist, P.; Eklund, R.; Brinck, T. J. Org. Chem. 2001, 66, 1193.
380. See Krow, G.R. Tetrahedron 1981, 37, 2697.
381. Phillips, A.M.F.; Romão, C. Eur. J. Org. Chem. 1999, 1767. In an ionic liquid, see Bernini, R.; Coratti, A.; Fabrizi, G.; Goggiamani, A. Tetrahedron Lett. 2003, 44, 8991.
382. ten Brink, G.-J.; Vis, J.-M.; Arends, I.W.C.E.; Sheldon, R.A. J. Org. Chem. 2001, 66, 2429.
383. For a review, see Jiménez-Sanchidrián, C.; Ruiz, J.R. Tetrahedron 2008, 64, 2011.
384. Alam, M.M.; Varala, R.; Adapa, S.R. Synth. Commun. 2003, 33, 3035.
385. Lambert, A.; Elings, J.A.; Macquarrie, D.J.; Carr, G.; Clark, J.H. Synlett 2000, 1052. See Hagiwara, H.; Nagatomo, H.; Yoshii, F.; Hoshi, T.; Suzuki, T.; Ando, M. J. Chem. Soc., Perkin Trans. 1 2000, 2645.
386. Yakura, T.; Kitano, T.; Ikeda, M.; Uenishi, J. Tetrahedron Lett. 2002, 43, 6925.
387. González-Núñez, M.E.; Mello, R.; Olmos, A.; Asensio, G. J. Org. Chem. 2005, 70, 10879. In suspercritical CO2, see González-Núñez, M.E.; Mello, R.; Olmos, A.; Asensio, G. J. Org. Chem. 2006, 71, 6432.
388. See Bolm, C.; Frison, J.-C.; Zhang, Y.; Wulff, W.D. Synlett 2004, 1619.
389. See Mihovilovic, M.D.; Müller, B.; Kayser, M.M.; Stewart, J.D.; Stanetty, P. Synlett 2002, 703. See Clouthier, C.M.; Kayser, M.M.; Reetz, M.T. J. Org. Chem. 2006, 71, 8431; Pazmiño, D.E.T.; Snajdrova, R.; Baas, B.-J.; Ghobrial, M.; Mihovilovic, M.D.; Fraaije, M.W. Angew. Chem. Int. Ed. 2008, 47, 2275.
390. See Watanabe, A.; Uchida, T.; Ito, K.; Katsuki, T. Tetrahedron Lett. 2002, 43, 4481; Murhashi, S.-I.; Ono, S.; Imada, Y. Angew. Chem. Int. Ed. 2002, 41, 2366.
391. Malkov, A.V.; Friscourt, F.; Bell, M.; Swarbrick, M.E.; Koovský, P. J. Org. Chem. 2008, 73, 3996
392. Frison, J.-C.; Palazzi, C.; Bolm, C. Tetrahedron 2006, 62, 6700.
393. Hunt, KW.; Grieco, P.A. Org. Lett. 2000, 2, 1717.
394. Emmons, W.D.; Lucas, G.B. J. Am. Chem. Soc. 1955, 77, 2287.
395. Gaikwad, D.D.; Dake, S.A.; Kulkarni, R.S.; Jadhav, W.N.; Kakde, S.B.; Pawar, R.P. Synth. Commun. 2007, 37, 4093.
396. McClure, J.D.; Williams, P.H. J. Org. Chem. 1962, 27, 24.
397. Deno, N.C.; Billups, W.E.; Kramer, K.E.; Lastomirsky, R.R. J. Org. Chem. 1970, 35, 3080. See Chrobok, A. Tetrahedron 2010, 66, 6212.
398. See Noyori, R.; Sato, T.; Kobayashi, H. Bull. Chem. Soc. Jpn. 1983, 56, 2661.
399. Snowden, M.; Bermudez, A.; Kelly, D.R.; Radkiewicz-Poutsma, J.L. J. Org. Chem. 2004, 69, 7148.
400. See Cullis, P.M.; Arnold, J.R.P.; Clarke, M.; Howell, R.; DeMira, M.; Naylor, M.; Nicholls, D. J. Chem. Soc., Chem. Commun. 1987, 1088.
401. Syper, L. Synthesis 1989, 167. See also, Godfrey, I.M.; Sargent, M.V.; Elix, J.A. J. Chem. Soc. Perkin Trans. 1 1974, 1353.
402. Proposed by Criegee, R. Liebigs Ann. Chem. 1948, 560, 127.
403. Doering, W. von E.; Dorfman, E. J. Am. Chem. Soc. 1953, 75, 5595. Also see Smith, P.A.S. in de Mayo, P. Molecular Rearrangements Vol. 1, Wiley, NY, 1963, pp. 578–584.
404. Palmer, B.W.; Fry, A. J. Am. Chem. Soc. 1970, 92, 2580. See Mitsuhashi, T.; Miyadera, H.; Simamura, O. Chem. Commun. 1970, 1301; Winnik, M.A.; Stoute, V.; Fitzgerald, P. J. Am. Chem. Soc. 1974, 96, 1977.
405. Also see Ogata, Y.; Sawaki, Y. J. Org. Chem. 1972, 37, 2953.
406. Yablokov, V.A. Russ. Chem. Rev. 1980, 49, 833; Lee, J.B.; Uff, B.C. Q. Rev. Chem. Soc. 1967, 21, 429, 445–449.
407. See Wistuba, E.; Rüchardt, C. Tetrahedron Lett. 1981, 22, 3389.
408. See Olah, G.A.; Parker, D.G.; Yoneda, N. Angew. Chem. Int. Ed. 1978, 17, 909.
409. Sheldon, R.A.; van Doorn, J.A. Tetrahedron Lett. 1973, 1021.
410. For syntheses, see Vanecko, J.A.; Wan, H.; West, F.G. Tetrahedron 2006, 62, 1043.
411. Lepley, A.R.; Giumanini, A.G. Mech. Mol. Migr. 1971, 3, 297; Pine, S.H. Org. React. 1970, 18, 403; Stevens, T.S.; Watts, W.E. Selected Molecular Rearrangements, Van Nostrand-Reinhold, Princeton, NJ, 1973, pp. 81–116; Wilt, J.W. in Kochi, J.K. Free Radicals, Vol. 1, Wiley, NY, 1973, pp. 448–458; Iwai, I. Mech. Mol. Migr. 1969, 2, 73, see pp. 105–113; Stevens, T.S. Prog. Org. Chem. 1968, 7, 48.
412. Heaney, H.; Ward, T.J. Chem. Commun. 1969, 810; Truce, W.E.; Heuring, D.L. Chem. Commun. 1969, 1499.
413. Elmasmodi, A.; Cotelle, P.; Barbry, D.; Hasiak, B.; Couturier, D. Synthesis 1989, 327.
414. See Heard, G.L.; Yates, B.F. Aust. J. Chem. 1994, 47, 1685.
415. Stevens, T.S. J. Chem. Soc. 1930, 2107; Johnstone, R.A.W.; Stevens, T.S. J. Chem. Soc. 1955, 4487.
416. Brewster, J.H.; Kline, M.W. J. Am. Chem. Soc. 1952, 74, 5179; Schöllkopf, U.; Ludwig, U.; Ostermann, G.; Patsch, M. Tetrahedron Lett. 1969, 3415.
417. Jemison, R.W.; Mageswaran, S.; Ollis, W.D.; Potter, S.E.; Pretty, A.J.; Sutherland, I.O.; Thebtaranonth, Y. Chem. Commun. 1970, 1201.
418. See Lepley, A.R.; Becker, R.H.; Giumanini, A.G. J. Org. Chem. 1971, 36, 1222.
419. For a review of the application of CIDNP to rearrangement reactions, see Lepley, A.R. in Lepley, A.R.; Closs, G.L. Chemically Induced Magnetic Polarization, Wiley, NY, 1973, pp. 323–384.
420. Ollis, W.D.; Rey, M.; Sutherland, I.O. J. Chem. Soc. Perkin Trans. 1 1983, 1009, 1049.
421. Hennion, G.F.; Shoemaker, M.J. J. Am. Chem. Soc. 1970, 92, 1769.
422. See, for example, Pine, S.H.; Catto, B.A.; Yamagishi, F.G. J. Org. Chem. 1970, 35, 3663.
423. For evidence against this mechanism, see Jenny, E.F.; Druey, J. Angew. Chem. Int. Ed. 1962, 1, 155.
424. Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry, Academic Press, NY, 1970, p. 131.
425. Hill, R.K.; Chan, T. J. Am. Chem. Soc. 1966, 88, 866.
426. Olsen, R.K.; Currie, Jr., J.O. in Patai, S. The Chemistry of The Thiol Group, pt. 2, Wiley, NY, 1974, pp. 561–566. See Okazaki, Y.; Asai, T.; Ando, F.; Koketsu, J. Chem. Lett. 2006, 35, 98.
427. See Iwamura, H.I.; Iwamura, M.; Nishida, T.; Yoshida, M.; Nakayama, T. Tetrahedron Lett. 1971, 63.
428. See Ostermann, G.; Schöllkopf, U. Liebigs Ann. Chem. 1970, 737, 170; Lorand, J.P.; Grant, R.W.; Samuel, P.A.; O'Connell, E.; Zaro, J. Tetrahedron Lett. 1969, 4087.
429. Johnstone, R.A.W. Mech. Mol. Migr. 1969, 2, 249. See Buston, J.E.H.; Coldham, I.; Mulholland, K.R. J. Chem. Soc., Perkin Trans. 1 1999, 2327.
430. See Buston, J.E.H.; Coldham, I.; Mulholland, K.R. Tetrahedron Asymmetry, 1998, 9, 1995.
431. See Khuthier, A.; Al-Mallah, K.Y.; Hanna, S.Y.; Abdulla, N.I. J. Org. Chem. 1987, 52, 1710, and references cited therein.
432. Fontenas, C.; Bejan, E.; Haddon, H.A.; Balavoine, G.G.A. Synth. Commun. 1995, 25, 629.
433. Eisch, J.J.; Kovacs, C.A.; Chobe, P. J. Org. Chem. 1989, 54, 1275.
434. See Pakusch, J.; Rüchardt, C. Chem. Ber. 1991, 124, 971 and references cited therein.
435. Meier, M.; Rüchardt, C. Chimia 1986, 40, 238.
436. See Hiersemann, M.; Abraham, L.; Pollex, A. Synlett 2003, 1088.
437. See Bailey, W.F.; England, M.D.; Mealy, M.J.; Thongsornkleeb, C.; Teng, L. Org. Lett. 2000, 2, 489.
438. For migration of vinyl, see Rautenstrauch, V.; Büchi, G.; Wüest, H. J. Am. Chem. Soc. 1974, 96, 2576. For rearrangment of an α-trimethylsilyl allyl ether see Maleczka, Jr., R.E.; Geng, F. Org. Lett. 1999, 1, 1115.
439. Wittig, G. Angew. Chem. 1954, 66, 10; Solov'yanov, A.A.; Ahmed, E.A.A.; Beletskaya, I.P.; Reutov, O.A. J. Chem. Soc., Chem. Commun. 1987, 23, 1232.
440. Maleczka Jr., R.E.; Geng, F. J. Am. Chem. Soc. 1998, 120, 8551.
441. See Schöllkopf, U. Angew. Chem. Int. Ed. 1970, 9, 763.
442. See Schäfer, H.; Schöllkopf, U.; Walter, D. Tetrahedron Lett. 1968, 2809.
443. See Cast, J.; Stevens, T.S.; Holmes, J. J. Chem. Soc. 1960, 3521.
444. Hebert, E.; Welvart, Z. J. Chem. Soc., Chem. Commun. 1980, 1035; Nouv. J. Chim. 1981, 5, 327.
445. Lansbury, P.T.; Pattison, V.A. J. Org. Chem. 1962, 27, 1933; J. Am. Chem. Soc. 1962, 84, 4295.
446. Garst, J.F.; Smith, C.D. J. Am. Chem. Soc. 1973, 95, 6870.
447. Garst, J.F.; Smith, C.D. J. Am. Chem. Soc. 1976, 98, 1526. For evidence against this, see Hebert, E.; Welvart, Z.; Ghelfenstein, M.; Szwarc, H. Tetrahedron Lett. 1983, 24, 1381.
448. Eisch, J.J.; Kovacs, C.A.; Rhee, S. J. Organomet. Chem. 1974, 65, 289.
449. Schlosser, M.; Strunk, S. Tetrahedron 1989, 45, 2649.
450. Anderson, J.C.; Siddons, D.C.; Smith, S.C.; Swarbrick, M.E. J. Chem. Soc., Chem. Commun. 1995, 1835; Ahman, J.; Somfai, P. J. Am. Chem. Soc. 1994, 116, 9781.
451. Matteson, D.S. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1984, pp. 307–409, pp. 346–387; Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, pp. 256–301; Negishi, E.; Idacavage, M.J. Org. React. 1985, 33, 1; Suzuki, A. Top. Curr. Chem. 1983, 112, 67; Pelter, A. Chem. Soc. Rev. 1982, 11, 191; Cragg, G.M.L.; Koch, K.R. Chem. Soc. Rev. 1977, 6, 393; Weill-Raynal, J. Synthesis 1976, 633; Cragg, G.M.L. Organoboranes in Organic Synthesis, Marcel Dekker, NY, 1973, pp. 249–300; Paetzold, P.I.; Grundke, H. Synthesis 1973, 635.
452. Brown, H.C. Hydroboration, W.A. Benjamin, New York, 1962. See Kuivila, H.G. J. Am. Chem. Soc. 1954, 76, 870; 1955, 77, 4014; Kuivila, H.G.; Wiles, R.A. J. Am. Chem. Soc. 1955, 77, 4830; Kuivila, H.G.; Armour, A.G. J. Am. Chem. Soc. 1957, 79, 5659; Wechter, W.J. Chem. & Ind. (London) 1959, 294.
453. Zweifel, G.; Brown, H.C. Org. React. 1964, 13, 1.
454. See Negishi, E. Intra-Sci. Chem. Rep. 1973, 7(1), 81; Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithica, NY, 1972, pp. 343–371; Acc. Chem. Res. 1969, 2, 65.
455. See Brown, H.C.; Cole, T.E.; Srebnik, M.; Kim, K. J. Org. Chem. 1986, 51, 4925.
456. Negishi, E.; Brown, H.C. Synthesis 1972, 197.
457. Brown, H.C.; Negishi, E.; Dickason, W.C. J. Org. Chem. 1985, 50, 520, and references cited therein.
458. Brown, H.C.; Rathke, M.W. J. Am. Chem. Soc. 1967, 89, 4528.
459. Brown, H.C.; Carlson, B.A. J. Org. Chem. 1973, 38, 2422; Brown, H.C.; Katz, J.; Carlson, B.A. J. Org. Chem. 1973, 38, 3968.
460. Pelter, A.; Hutchings, M.G.; Smith, K.; Williams, D.J. J. Chem. Soc. Perkin Trans. 1 1975, 145, and references cited therein.
461. See, however, Pelter, A.; Maddocks, P.J.; Smith, K. J. Chem. Soc., Chem. Commun. 1978, 805.
462. See Pelter, A.; Rao, J.M. J. Organomet. Chem. 1985, 285, 65; Junchai, B.; Hongxun, D. J. Chem. Soc., Chem. Commun. 1990, 323.
463. Brown, H.C.; Rathke, M.W. J. Am. Chem. Soc. 1967, 89, 2738.
464. Brown, H.C.; Kabalka, G.W.; Rathke, M.W. J. Am. Chem. Soc. 1967, 89, 4530.
465. Pelter, A.; Smith, K.; Hutchings, M.G.; Rowe, K. J. Chem. Soc. Perkin Trans. 1 1975, 129; See also, Mallison, P.R.; White, D.N.J.; Pelter, A.; Rowe, K.; Smith, K. J. Chem. Res. (S), 1978, 234. See also, Ref. 460.
466. See Brown, H.C.; Bakshi, R.K.; Singaram, B. J. Am. Chem. Soc. 1988, 110, 1529.
467. See Matteson, D.S. Mol. Struct. Energ. 1988, 5, 343; Acc. Chem. Res. 1988, 21, 294; Synthesis 1986, 973, pp. 980–983.
468. See Pelter, A.; Rao, J.M. J. Organomet. Chem. 1985, 285, 65; Brown. H.C.; Bhat, N.G.; Basavaiah, D. Synthesis 1983, 885; Narayana, C.; Periasamy, M. Tetrahedron Lett. 1985, 26, 6361.
469. Brown, H.C.; Hubbard, J.L.; Smith, K. Synthesis 1979, 701, and references cited therein. See Hubbard, J.L.; Smith, K. J. Organomet. Chem. 1984, 276, C41.
470. Brown, H.C.; Knights, E.F.; Coleman, R.A. J. Am. Chem. Soc. 1969, 91, 2144.
471. Brown, H.C.; Coleman, R.A. J. Am. Chem. Soc. 1969, 91, 4606.
472. See Negishi, E.; Yoshida, T.; Silveira, Jr., A.; Chiou, B.L. J. Org. Chem. 1975, 40, 814.
473. Hara, S.; Kishimura, K.; Suzuki, A.; Dhillon, R.S. J. Org. Chem. 1990, 55, 6356. See also, Brown, H.C.; Imai, T. J. Org. Chem. 1984, 49, 892.
474. Brown, H.C.; Imai, T. J. Am. Chem. Soc. 1983, 105, 6285. For a related method that produces primary alcohols, see Brown, H.C.; Imai, T.; Perumal, P.T.; Singaram, B. J. Org. Chem. 1985, 50, 4032.
475. Brown, H.C.; Imai, T.; Desai, M.C.; Singaram, B. J. Am. Chem. Soc. 1985, 107, 4980.
476. Basavaiah, D.; Kulkarni, S.U.; Bhat, N.G.; Vara Prasad, J.V.N. J. Org. Chem. 1988, 53, 239.
477. For a list of methods of preparing alkenes using boron reagents, with references, see Larock, R.C. Comprehensive Organic Transformations; 2nd ed., Wiley–VCH, NY, 1999, pp. 421–427.
478. Zweifel, G.; Fisher, R.P.; Snow, J.T.; Whitney, C.C. J. Am. Chem. Soc. 1971, 93, 6309.
479. Hyuga, S.; Takinami, S.; Hara, S.; Suzuki, A. Tetrahedron Lett. 1986, 27, 977.
480. For improvements in this method, see Brown, H.C.; Basavaiah, D.; Kulkarni, S.U.; Lee, H.D.; Negishi, E.; Katz, J. J. Org. Chem. 1986, 51, 5270.
481. Negishi, E.; Yoshida, T. J. Chem. Soc. Chem.Commun. 1973, 606; See also, Negishi, E.; Yoshida, T.; Abramovitch, A.; Lew, G.; Williams, R.H. Tetrahedron 1991, 47, 343.
482. Corey, E.J.; Ravindranathan, T. J. Am. Chem. Soc. 1972, 94, 4013; Negishi, E.; Katz, J.; Brown, H.C. Synthesis 1972, 555.
483. Zweifel, G.; Fisher, R.P. Synthesis 1972, 557.
484. Suzuki, A.; Miyaura, N.; Abiko, S.; Itoh, M.; Brown, H.C.; Sinclair, J.A.; Midland, M.M. J. Org. Chem. 1986, 51, 4507; Sikorski, J.A.; Bhat, N.G.; Cole, T.E.; Wang, K.K.; Brown, H.C. J. Org. Chem. 1986, 51, 4521. For a review of reactions of organoborates, see Suzuki, A. Acc. Chem. Res. 1982, 15, 178.
485. For a study of the relative migratory aptitudes of R', see Slayden, S.W. J. Org. Chem. 1981, 46, 2311.
486. Midland, M.M.; Sinclair, J.A.; Brown, H.C. J. Org. Chem. 1974, 39, 731.
487. Brown, H.C.; Mahindroo, V.K.; Bhat, N.G.; Singaram, B. J. Org. Chem. 1991, 56, 1500.
488. See Larock, R.C. Comprehensive Organic Transformations; 2nd ed., Wiley–VCH, NY, 1999, pp. 218–222.
489. Pelter, A.; Gould, K.J.; Harrison, C.R. Tetrahedron Lett. 1975, 3327.
490. Wang, K.K.; Chu, K. J. Org. Chem. 1984, 49, 5175.
491. Pelter, A.; Drake, R.A. Tetrahedron Lett. 1988, 29, 4181.
492. Talami, S.; Stirling, C.J.M. Can. J. Chem. 1999, 77, 1105.
493. See Dolbier, Jr., W.R.; Koroniak, H.; Houk, K.N.; Sheu, C. Acc. Chem. Res. 1996, 29, 471; Niwayama, S.; Kallel, E.A.; Spellmeyer, D.C.; Sheu, C.; Houk, K.N. J. Org. Chem. 1996, 61, 2813. The effect of pressure on this reaction has been discussed, see Jenner, G. Tetrahedron 1998, 54, 2771.
494. See Steiner, R.P.; Michl, J. J. Am. Chem. Soc. 1978, 100, 6413 and references cited therein.
495. See Um, J.M.; Xu, H.; Houk, K.N.; Tang, W. J. Am. Chem. Soc. 2009, 131, 6664.
496. Matsuya, Y.; Ohsawa, N.; Nemoto, H. J. Am. Chem. Soc. 2006, 128, 412.
497. Matsuya, Y.; Ohsawa, N.; Nemoto, H. J. Am. Chem. Soc. 2006, 128, 13072.
498. See Dauben, W.G.; Haubrich, J.E. J. Org. Chem. 1988, 53, 600.
499. See Dauben, W.G.; McInnis, E.L.; Michno, D.M. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 3, Academic Press, NY, 1980, pp. 91–129. For an ab initio study see Rodríguez-Otero, J. J. Org. Chem.1999, 64, 6842.
500. Tanaka, K.; Mori, H.; Yamamoto, M.; Katsumura, S. J. Org. Chem. 2001, 66, 3099. See Beaudry, C.M.; Malerich, J.P.; Trauner, D. Chem. Rev. 2005, 105, 4757.
501. Yasui, M.; Naruse, Y.; Inagaki, S. J. Org. Chem. 2004, 69, 7246.
502. Chapman, O.L.; Pasto, D.J.; Borden, G.W.; Griswold, A.A. J. Am. Chem. Soc. 1962, 84, 1220.
503. See Maier, G. Angew. Chem. Int. Ed. 1967, 6, 402; Vogel, E. Pure Appl. Chem. 1969, 20, 237.
504. Bishop, L.M.; Barbarow, J.E.; Bergman, R.G.; Trauner, D. Angew. Chem. Int. Ed. 2008, 47, 8100.
505. See Ciganek, E. J. Am. Chem. Soc. 1967, 89, 1454.; Iyoda, M.; Oda, M. Angew. Chem. Int. Ed. 1987, 26, 559.
506. See Gajewski, J.J. Hydrocarbon Thermal Isomerizations, Academic Press, NY, 1981; Marvell, E.N. Thermal Electrocyclic Reactions, Academic Press, NY, 1980; Laarhoven, W.H. Org. Photochem. 1987, 9, 129; George, M.V.; Mitra, A.; Sukumaran, K.B. Angew. Chem. Int. Ed. 1980, 19, 973; Jutz, J.C. Top. Curr. Chem. 1978, 73, 125; Gilchrist, T.L.; Storr, R.C. Organic Reactions and Orbital Symmetry, Cambridge University Press, Cambridge, 1972, pp. 48–72; Criegee, R. Angew. Chem. Int. Ed. 1968, 7, 559. See Schultz, A.G.; Motyka, L. Org. Photochem. 1983, 6, 1.
507. Winter, R.E.K. Tetrahedron Lett. 1965, 1207; Criegee, R.; Noll, K. Liebigs Ann. Chem. 1959, 627, 1.
508. Baldwin, J.E.; Gallagher, S.S.; Leber, P.A.; Raghavan, A.S.; Shukla, R. J. Org. Chem. 2004, 69, 7212.
509. See Woodward, R.B.; Hoffmann, R. J. Am. Chem. Soc. 1965, 87, 395.
510. See Leigh, W.J.; Zheng, K. J. Am. Chem. Soc. 1991, 113, 4019; Leigh, W.J.; Zheng, K.; Nguyen, N.; Werstiuk, N.H.; Ma, J. J. Am. Chem. Soc. 1991, 113, 4993, and references cited therein.
511. Woodward, R.B.; Hoffmann, R. J. Am. Chem. Soc. 1965, 87, 395. Also see, Longuet-Higgins, H.C.; Abrahamson, E.W. J. Am. Chem. Soc. 1965, 87, 2045; Fukui, K. Tetrahedron Lett. 1965, 2009.
512. See Jones, R.A.Y. Physical and Mechanistic Organic Chemistry, 2nd ed., Cambridge University Press, Cambridge, 1984, pp. 352–359; Yates, K. Hückel Molecular Orbital Theory, Academic Press, NY, 1978, pp. 250–263. Also see, Zimmerman, H.E. in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions, Vol. 2, Academic Press, NY, 1977, pp. 53–107; Acc. Chem. Res. 1971, 4, 272; Dewar, M.J.S. Angew. Chem. Int. Ed. 1971, 10, 761; Jefford, C.W.; Burger, U. Chimia 1971, 25, 297; Herndon, W.C. J. Chem. Educ. 1981, 58, 371.
513. Fukui, K. Fortschr. Acc. Chem. Res. 1971, 4, 57; Houk, K.N. Acc. Chem. Res. 1975, 8, 361. See also, Chu, S. Tetrahedron 1978, 34, 645; Fleming, I. Pericyclic Reactions, Oxford Univ. Press, Oxford, 1999; Fukui, K. Angew. Chem. Int. Ed. 1982, 21, 801; Houk, K.N., in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions, Vol. 2, Academic Press, NY, 1977, pp. 181–271.
514. For a discussion of the transition structures and energy, see Zora, M. J. Org. Chem. 2004, 69, 1940.
515. See Gesche, P.; Klinger, F.; Riesen, A.; Tschamber, T.; Zehnder, M.; Streith, J. Helv. Chim. Acta 1987, 70, 2087.
516. Leigh, W.J.; Postigo, J.A. J. Am. Chem. Soc. 1995, 117, 1688.
517. Dolbier Jr., W.R.; Gray, T.A.; Keaffaber, J.J.; Celewicz, L.; Koroniak, H. J. Am. Chem. Soc. 1990, 112, 363; Hayes, R.; Ingham, S.; Saengchantara, S.T.; Wallace, T.W. Tetrahedron Lett. 1991, 32, 2953.
518. See Kallel, E.A.; Wang, Y.; Spellmeyer, D.C.; Houk, K.N. J. Am. Chem. Soc. 1990, 112, 6759.
519. Piers, E.; Lu, Y.-F. J. Org. Chem. 1989, 54, 2267.
520. See Dauben, W.G.; Kellogg, M.S.; Seeman, J.I.; Vietmeyer, N.D.; Wendschuh, P.H. Pure Appl. Chem. 1973, 33, 197.
521. Dauben, W.G.; Fonken, G.J. J. Am. Chem. Soc. 1959, 81, 4060.
522. Bergman, R.G. Accts. Chem. Res. 1973, 6, 25; Adam, W.; Krebs, O. Chem. Rev. 2003, 103, 4131. See Lewis, K.D.; Matzger, A.J. J. Am. Chem. Soc. 2005, 127, 9968; Zeidan, T.A; Manoharan, M.; Alabugin, I.V. J. Org. Chem.2006, 71, 954; Zeidan, T.A.; Kovalenko, S.V.; Manoharan, M.; Alabugin, I.V. J. Org. Chem 2006, 71, 962.
523. See Tanaka, H.; Yamada, H.; Matsuda, A.; Takahashi, T. Synlett 1997, 381.
524. Jones, G.B.; Warner, P.M. J. Org. Chem. 2001, 66, 8669.
525. Bowles, D.M.; Palmer, G.J.; Landis, C.A.; Scott, J.L.; Anthony, J.E. Tetrahedron 2001, 57, 3753.
526. Jones, G.B.; Warner, P.M. J. Am. Chem. Soc. 2001, 123, 2134.
527. Feng, L.; Kumar, D.; Kerwin, S.M. J. Org. Chem. 2003, 68, 2234.
528. See Ward, H.R.; Wishnok, J.S. J. Am. Chem. Soc. 1968, 90, 1085; Bryce-Smith, D.; Gilbert, A.; Robinson, D.A. Angew. Chem. Int. Ed. 1971, 10, 745. Also see Barlow, M.G.; Haszeldine, R.N.; Hubbard, R. Chem. Commun.1969, 202; Lemal, D.M.; Staros, J.V.; Austel, V. J. Am. Chem. Soc. 1969, 91, 3373.
529. van Tamelen, E.E. Acc. Chem. Res. 1972, 5, 186. See Schäfer, W.; Criegee, R.; Askani, R.; Grüner, H. Angew. Chem. Int. Ed. 1967, 6, 78), Dewar benzenes can be photolyzed further to give prismanes.
530. See Wingert, H.; Irngartinger, H.; Kallfass, D.; Regitz, M. Chem. Ber. 1987, 120, 825.
531. Maier, G.; Schneider, K. Angew. Chem. Int. Ed. 1980, 19, 1022. See also, Wingert, H.; Maas, G.; Regitz, M. Tetrahedron 1986, 42, 5341.
532. Marvell, E.N.; Seubert, J. J. Am. Chem. Soc. 1967, 89, 3377; Huisgen, R.; Dahmen, A.; Huber, H. Tetrahedron Lett. 1969, 1461; Dahmen, A.; Huber, H. Tetrahedron Lett. 1969, 1465.
533. See Baldwin, J.E.; Andrist, A.H.; Pinschmidt Jr., R.K. Acc. Chem. Res. 1972, 5, 402.
534. Mao, C.; Presser, N.; John, L.; Moriarty, R.M.; Gordon, R.J. J. Am. Chem. Soc. 1981, 103, 2105.
535. Pettit, R.; Sugahara, H.; Wristers, J.; Merk, W. Discuss. Faraday Soc. 1969, 47, 71. See also, Labunskaya, V.I.; Shebaldova, A.D.; Khidekel, M.L. Russ. Chem. Rev. 1974, 43, 1; Mango, F.D. Top. Curr. Chem. 1974, 45, 39; Mango, F.D.; Schachtschneider, J.H. J. Am. Chem. Soc. 1971, 93, 1123.
536. Merk, W.; Pettit, R. J. Am. Chem. Soc. 1967, 89, 4788.
537. See Pinhas, A.R.; Carpenter, B.K. J. Chem. Soc., Chem. Commun. 1980, 15.
538. Leivers, M.; Tam, I.; Groves, K.; Leung, D.; Xie, Y.; Breslow, R. Org. Lett. 2003, 5, 3407.
539. See DePuy, C.H. Acc. Chem. Res. 1968, 1, 33; Schöllkopf, U. Angew. Chem. Int. Ed. 1968, 7, 588.
540. See Boche, G. Top. Curr. Chem. 1988, 146, 1.
541. See Coates, R.M.; Last, L.A. J. Am. Chem. Soc. 1983, 105, 7322. For a review of the analogous ring opening of epoxides, see Huisgen, R. Angew. Chem. Int. Ed. 1977, 16, 572.
542. DePuy, C.H.; Schnack, L.G.; Hausser, J.W.; Wiedemann, W. J. Am. Chem. Soc. 1965, 87, 4006.
543. It has been suggested that the pathway shown in C is possible in certain cases: Hausser, J.W.; Grubber, M.J. J. Org. Chem. 1972, 37, 2648; Hausser, J.W.; Uchic, J.T. J. Org. Chem. 1972, 37, 4087.
544. Also see Reese, C.B.; Shaw, A. J. Am. Chem. Soc. 1970, 92, 2566; Dolbier, Jr., W.R.; Phanstiel, O. Tetrahedron Lett. 1988, 29, 53, and references cited therein.
545. Schöllkopf, U.; Fellenberger, K.; Patsch, M.; Schleyer, P.v.R.; Su, T.M.; Van Dine, G.W. Tetrahedron Lett. 1967, 3639.
546. Schleyer, P.v.R.; Su, T.M.; Saunders, M.; Rosenfeld, J.C. J. Am. Chem. Soc. 1969, 91, 5174.
547. Mallory, F.B.; Mallory, C.W. Org. React. 1984, 30, 1; Blackburn, E.V.; Timmons, C.J. Q. Rev. Chem. Soc. 1969, 23, 482. See Laarhoven, W.H. Org. Photochem. 1989, 10, 163.
548. See Liu, L.; Yang, B.; Katz, T.J.; Poindexter, M.K. J. Org. Chem. 1991, 56, 3769.
549. Cuppen, T.J.H.M.; Laarhoven, W.H. J. Am. Chem. Soc. 1972, 94, 5914.
550. Doyle, T.D.; Benson, W.R.; Filipescu, N. J. Am. Chem. Soc. 1976, 98, 3262.
551. Sato, T.; Shimada, S.; Hata, K. Bull. Chem. Soc. Jpn. 1971, 44, 2484.
552. See Laarhoven, W.H. Recl. Trav. Chim. Pays-Bas 1983, 102, 185, pp. 185–204.
553. Wiersum, U.E.; Jenneskens, L.W. Tetrahedron Lett. 1993, 34, 6615; Brown, R.F.C.; Choi, N.; Coulston, K.J.; Eastwood, F.W.; Wiersum, U.E.; Jenneskens, L.W. Tetrahedron Lett. 1994, 35, 4405.
554. See Brown, R F.C.; Eastwood, F.W.; Wong, N.R. Tetrahedron Lett. 1993, 34, 3607.
555. Preda, D.V.; Scott, L.T. Org. Lett. 2000, 2, 1489.
556. Stone, A.J.; Wales, D.J. Chem. Phys. Lett. 1986, 128, 501.
557. Alder, R.W.; Whittaker, G. J. Chem. Soc., Perkin Trans. 2 1975, 712
558. Alder, R. W.; Harvey, J. N. J. Am. Chem. Soc. 2004, 126, 2490.
559. Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry, Academic Press, NY, 1970, p. 114.
560. Gajewski, J.J. Hydrocarbon Thermal Isomerizations, Academic Press, NY, 1981; Mironov, V.A.; Fedorovich, A.D.; Akhrem, A.A. Russ. Chem. Rev. 1981, 50, 666; Spangler, C.W. Chem. Rev. 1976, 76, 187.
561. Lin, Y.-L.; Turos, E. J. Am. Chem. Soc. 1999, 121, 856.
562. Moss, S.; King, B.T.; de Meijere, A.; Kozhushkov, S.I.; Eaton, P.E.; Michl, J. Org. Lett. 2001, 3, 2375.
563. Note that a [1,5]-sigmatropic rearrangement of hydrogen is also an internal ene synthesis (Reaction 15-20).
564. The designations U, V, Y, and Z are arbitrary, so which isomer is(R,Z) and which is (S,E) is arbitrary.
565. See Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry, Academic Press, NY, 1970, pp. 114–140.
566. This follows from the principle that bonds are formed only by overlap of orbitals of the same sign. Since this is a concerted reaction, the hydrogen orbital in the transition state must overlap simultaneously with one lobe from the migration origin and one from the terminus. It is obvious that both of these lobes must have the same sign.
567. See Kless, A.; Nendel, M.; Wilsey, S.; Houk, K.N. J. Am. Chem. Soc. 1999, 121, 4524.
568. See, however, Yeh, M.; Linder, L.; Hoffman, D.K.; Barton, T.J. J. Am. Chem. Soc. 1986, 108, 7849. See also, Pasto, D.J.; Brophy, J.E. J. Org. Chem. 1991, 56, 4554.
569. Dauben, W.G.; Wipke, W.T. Pure Appl. Chem. 1964, 9, 539, p. 546. See Kropp, P.J.; Fravel, Jr., H.G.; Fields, T.R. J. Am. Chem. Soc. 1976, 98, 840.
570. Hudson, C.E.; McAdoo, D.J. J. Org. Chem. 2003, 68, 2735.
571. See Kiefer, E.F.; Tanna, C.H. J. Am. Chem. Soc. 1969, 91, 4478; Dauben, W.G.; Poulter, C.D.; Suter, C. J. Am. Chem. Soc. 1970, 92, 7408.
572. Roth, W.R.; König, J.; Stein, K. Chem. Ber. 1970, 103, 426.
573. See Klärner, F. Top. Stereochem. 1984, 15, 1; Hess, Jr., B.A.; Baldwin, J.E. J. Org. Chem. 2002, 67, 6025.
574. Childs, R.F. Tetrahedron 1982, 38, 567. See also, Minkin, V.I.; Mikhailov, I.E.; Dushenko, G.A.; Yudilevich, J.A.; Minyaev, R.M.; Zschunke, A.; Mügge, K. J. Phys. Org. Chem. 1991, 4, 31. For a study of [1,5]-sigmatropic shiftamers, see Tantillo, D.J.; Hoffmann, R. Acc. Chem. Res. 2006, 39, 477.
575. Shelton, G.R.; Hrovat, D.A.; Borden, W.T. J. Am. Chem. Soc. 2007, 129, 164.
576. Bachrach, S.M. J. Org. Chem. 1993, 58, 5414.
577. Ikeda, H.; Ushioda, N.; Inagaki, S. Chem. Lett. 2001, 166.
578. Hayase, S.; Hrovat, D.A.; Borden, W.T. J. Am. Chem. Soc. 2004, 126, 10028.
579. Baldwin, J.E.; Raghavan, A.S.; Hess, Jr., B.A.; Smentek, L. J. Am. Chem. Soc. 2006, 128, 14854; Baldwin, J.E.; Chapman, B.R. J. Org. Chem. 2005, 70, 377. See also von E. Doering, W.; Keliher, E.J. J. Am. Chem. Soc.2007, 129, 2488; Peles, D.N.; Thoburn, J.D. J. Org. Chem. 2008, 73, 3135.
580. Somfai, P.; Åhman, J. Tetrahedron Lett. 1995, 36, 1953.
581. Datta, S.; Odedra, A.; Liu, R.-S. J. Am. Chem. Soc. 2005, 127, 11606.
582. Journet, M.; Malacria, M. Tetrahedron Lett. 1992, 33, 1893.
583. See Hess Jr., B.A. J. Org. Chem. 2001, 66, 5897.
584. Gurskii, M.E.; Gridnev, I.D.; Il'ichev, Y.V.; Ignatenko, A.V.; Bubnov, Y.N. Angew. Chem. Int. Ed. 1992, 31, 781; Baldwin, J.E.; Reddy, V.P. J. Am. Chem. Soc. 1988, 110, 8223.
585. See ter Borg, A.P.; Kloosterziel, H. Recl. Trav. Chim. Pays-Bas 1969, 88, 266; Tezuka, T.; Kimura, M.; Sato, A.; Mukai, T. Bull. Chem. Soc. Jpn. 1970, 43, 1120.
586. Bailey, W.J.; Baylouny, R.A. J. Org. Chem. 1962, 27, 3476.
587. See Parziale, P.A.; Berson, J.A. J. Am. Chem. Soc. 1990, 112, 1650; Pegg, G.G.; Meehan, G.V. Aust. J. Chem. 1990, 43, 1009, 1071.
588. Roth, W.R.; König, J. Liebigs Ann. Chem. 1965, 688, 28. See, Grimme, W. Chem. Ber. 1965, 98, 756.
589. Arnold, R.T.; Smolinsky, G. J. Am. Chem. Soc. 1960, 82, 4918; Leriverend, P.; Conia, J.M. Tetrahedron Lett. 1969, 2681; Conia, J.M.; Barnier, J.P. Tetrahedron Lett. 1969, 2679.
590. Roth, W.R.; Friedrich, A. Tetrahedron Lett. 1969, 2607.
591. Youssef, A.K.; Ogliaruso, M.A. J. Org. Chem. 1972, 37, 2601.
592. See Mironov, V.A.; Fedorovich, A.D.; Akhrem, A.A. Russ. Chem. Rev. 1981, 50, 666; Spangler, C.W. Chem. Rev. 1976, 76, 187.
593. See Shen, K.; McEwen, W.E.; Wolf, A.P. Tetrahedron Lett. 1969, 827; Miller, L.L.; Greisinger, R.; Boyer, R.F. J. Am. Chem. Soc. 1969, 91, 1578.
594. See Baldwin, J.E.; Leber, P.A. Org. Biomol. Chem. 2008, 6, 35.
595. Berson, J.A. Acc. Chem. Res. 1968, 1, 152.
596. See Berson, J.A. Acc. Chem. Res. 1972, 5, 406; Klärner, F.; Adamsky, F. Angew. Chem. Int. Ed. 1979, 18, 674.
597. Baldwin, J.E.; Belfield, K.D. J. Am. Chem. Soc. 1988, 110, 296; Klärner, F.; Drewes, R.; Hasselmann, D. J. Am. Chem. Soc. 1988, 110, 297.
598. See Pikulin, S.; Berson, J.A. J. Am. Chem. Soc. 1988, 110, 8500.
599. See Pikulin, S.; Berson, J.A. J. Am. Chem. Soc. 1988, 110, 8500. See also, Berson, J.A. Chemtracts: Org. Chem. 1989, 2, 213.
600. See Dolbier, W.B.; Phanstiel IV, O. J. Am. Chem. Soc. 1989, 111, 4907.
601. Cookson, R.C.; Hudec, J.; Sharma, M. Chem. Commun. 1971, 107, 108.
602. Grieco, P.A.; Clark, J.D.; Jagoe, C.T. J. Am. Chem. Soc. 1991, 113, 5488; Palani, N.; Balasubramanian, K.K. Tetrahedron Lett. 1995, 36, 9527.
603. Boersma, M.A.M.; de Haan, J.W.; Kloosterziel, H.; van de Ven, L.J.M. Chem. Commun. 1970, 1168.
604. See Gajewski, J.J.; Gortva, A.M.; Borden, J.E. J. Am. Chem. Soc. 1986, 108, 1083; Baldwin, J.E.; Broline, B.M. J. Am. Chem. Soc. 1982, 104, 2857.
605. Leach, A.G.; Catak, S.; Houk, K.N. Chem. Eur. J. 2002, 8, 1290.
606. See Baldwin, J.E. Chem. Rev. 2003, 103, 1197; Wong, H.N.C.; Hon, M.; Tse, C.; Yip, Y.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165, pp. 169–172; Hudlicky, T.; Kutchan, T.M.; Naqvi, S.M. Org. React. 1985, 33, 247; Hudlicky, T.; Reed, J.W. Angew. Chem. Int. Ed. 2010, 49, 4864. See Tanko, J.M.; Li, X.; Chahma, M.; Jackson, W.F.; Spencer, J.N. J. Am. Chem. Soc. 2007, 129, 4181.
607. See Armesto, D.; Ramos, A.; Mayoral, E.P.; Ortiz, M.J.; Agarrabeitia, A.R. Org. Lett. 2000, 2, 183.
608. For a study of substituent effects, see McGaffin, G.; Grimm, B.; Heinecke, U.; Michaelsen, H.; de Meijere, A.; Walsh, R. Eur. J. Org. Chem. 2001, 3559.
609. Ketley, A.D. Tetrahedron Lett. 1964, 1687; Branton, G.R.; Frey, H.M. J. Chem. Soc. A 1966, 1342.
610. Small, A.; Breslow, R. cited in Breslow, R. in de Mayo, P. Molecular Rearrangments, Vol. 1, Wiley, NY, 1963, p. 236.
611. Zimmerman, H.E.; Kreil, D.J. J. Org. Chem. 1982, 47, 2060.
612. Wender, P.A.; Husfeld, C.O.; Langkopf, E.; Love, J.A. J. Am. Chem. Soc. 1998, 120, 1940.
613. Rüedi, G.; Nagel, M.; Hansen, H.-J. Org. Lett. 2004, 6, 2989.
614. See Boeckman Jr., R.K.; Walters, M.A. Adv. Heterocycl. Nat. Prod. Synth. 1990, 1, 1.
615. Heine, H.W. Mech. Mol. Migr. 1971, 3, 145; Dermer, O.C.; Ham, G.E. Ethylenimine and Other Aziridines, Academic Press, NY, 1969, pp. 282–290. See also, Wong, H.N.C.; Hon, M.; Tse, C.; Yip, Y.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165, pp. 190–192.
616. Shi, M.; Yang, Y.-H.; Xu, B. Synlett 2004, 1622.
617. Campos, P.J.; Soldevilla, A.; Sampedro, D.; Rodrguez, M.A. Org. Lett. 2001, 3, 4087.
618. Mátrai, J.; Dransfeld, A.; Veszprém, T.; Nguyen, M.T. J. Org. Chem. 2001, 66, 5671.
619. See Baldwin, J.E.; Fedé, J.-M. J. Am. Chem. Soc. 2006, 128, 5608; Northrop, B.H.; Houk, K.N. J. Org. Chem. 2006, 71, 3; Leber, P.A.; Baldwin, J.E. Acc. Chem. Res. 2002, 35, 279.
620. See Thies, R.W. J. Am. Chem. Soc. 1972, 94, 7074.
621. Deak, H.L.; Stokes, S.S.; Snapper, M.L. J.. Am. Chem. Soc. 2001, 123, 5152.
622. Dinnocenzo, J.P.; Conlan, D.A. J. Am. Chem. Soc. 1988, 110, 2324.
623. See Bauld, N.L. Tetrahedron 1989, 45, 5307. For a rearrangement of a housane cation radical, see Gerken, J.B.; Wang, S.C.; Preciado, A.B.; Park, Y.S.; Nishiguchi, G.; Tantillo, D.J.; Little, R.D. J. Org. Chem. 2005, 70, 4598.
624. See Gajewski, J.J.; Olson, L.P. J. Am. Chem. Soc. 1991, 113, 7432.
625. See Zimmerman, H.E.; Fleming, S.A. J. Am. Chem. Soc. 1983, 105, 622; Klumpp, G.W.; Schakel, M. Tetrahedron Lett. 1983, 24, 4595; McGaffin, G.; de Meijere, A.; Walsh, R. Chem. Ber. 1991, 124, 939. See Roth, W.R.; Lennartz, H.; Doering, W. von E.; Birladeanu, L.; Guyton, C.A.; Kitagawa, T. J. Am. Chem. Soc. 1990, 112, 1722 and references cited therein.
626. See Gajewski, J.J.; Olson, L.P.; Willcott III, M.R. J. Am. Chem. Soc. 1996, 118, 299. For a discussion of the mechanism of this reaction, see Su, M.-D. Tetrahedron 1995, 51, 5871.
627. Bartlett, P.A. Tetrahedron 1980, 36, 2, pp. 28–39; Rhoads, S.J.; Raulins, N.R. Org. React. 1975, 22, 1; Smith, G.G.; Kelly, F.W. Prog. Phys. Org. Chem. 1971, 8, 75, pp. 153–201; DeWolfe, R.H., in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 455–461.
628. Note that the same holds true for [1,j]-sigmatropic reactions of symmetrical substrates (18-28 and 18-29).
629. Levy, H.; Cope, A.C. J. Am. Chem. Soc. 1944, 66, 1684.
630. See Cooper, N.J.; Knight, D.W. Tetrahedron 2004, 60, 243.
631. See Elmore, S.W.; Paquette, L.A. Tetrahedron Lett. 1991, 32, 319.
632. Paquette, L.A. Angew. Chem. Int. Ed. 1990, 29, 609; Marvell, E.N.; Whalley, W. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 2, Wiley, NY, 1971, pp. 738–743; Warrington, J.M.; Yap, G.P.A.; Barriault, L. Org. Lett.2000, 2, 663; Ovaska, T.V.; Roses, J.B. Org. Lett. 2000, 2, 2361; Jung, M.E.; Nishimura, N.; Novack, A.R. J. Am. Chem. Soc. 2005, 127, 11206.
633. For a list of references, see Larock, R.C. Comprehensive Organic Transformations; 2nd ed., Wiley–VCH, NY, 1999, pp. 1306–1307.
634. See Gajewski, J.J.; Gee, K.R. J. Am. Chem. Soc. 1991, 113, 967. See also, Wender, P.A.; Ternansky, R.J.; Sieburth, S.M. Tetrahedron Lett. 1985, 26, 4319. Also see Schulze, S.M.; Santella, N.; Grabowski, J.J.; Lee, J.K. J. Org. Chem. 2001, 66, 7247.
635. Mamdani, H.T.; Hartley, R.C. Tetrahedron Lett. 2000, 41, 747.
636. For a review, see Schneider, C. Synlett 2001, 1079.
637. Braisted, A.C.; Schultz, P.G. J. Am. Chem. Soc. 1994, 116, 2211.
638. Black, K.A.; Leach, A.G.; Kalani, Y.S.; Houk, K.N. J. Am. Chem. Soc. 2004, 126, 9695.
639. Paquette, L.A.; Reddy, Y.R.; Vayner, G.; Houk, K.N. J. Am. Chem. Soc. 2000, 122, 10788.
640. See Jasti, R.; Anderson, C.D.; Rychnovsky, S.D. J. Am. Chem. Soc. 2005, 127, 9939.
641. Chen, Y.-H.; McDonald, F.E. J. Am. Chem. Soc. 2006, 128, 4568.
642. Beholz, L.G.; Stille, J.R. J. Org. Chem. 1993, 58, 5095; Sprules, T.J.; Galpin, J.D.; Macdonald, D. Tetrahedron Lett. 1993, 34, 247; Yadav, J.S.; Reddy, B.V.S.; Rasheed, M.A.; Kumar, H.M.S. Synlett 2000, 487.
643. Rueping, M.; Antonchick, A.P. Angew. Chem. Int. Ed. 2008, 47, 10090.
644. Zakarian, A.; Lu, C.-D. J. Am. Chem. Soc. 2006, 128, 5356.
645. Dobson, H.K.; LeBlanc, R.; Perrier, H.; Stephenson, C.; Welch, T.R.; Macdonald, D. Tetrahedron Lett. 1999, 40, 3119.
646. Allin, S.M.; Button, M.A.C. Tetrahedron Lett. 1999, 40, 3801.
647. Duncan, J.A.; Azar, J.K.; Beatle, J.C.; Kennedy, S.R.; Wulf, C.M. J. Am. Chem. Soc. 1999, 121, 12029.
648. Harris, Jr., J.F. Tetrahedron Lett. 1965, 1359.
649. See Newcomb, M.; Vieta, R.S. J. Org. Chem. 1980, 45, 4793. Also see Jung, M.E.; Hudspeth, J.P. J. Am. Chem. Soc. 1978, 100, 4309; Yasuda, M.; Harano, K.; Kanematsu, K. J. Org. Chem. 1980, 45, 2368.
650. Wong, H.N.C.; Hon, M.; Tse, C.; Yip, Y.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165, see pp. 172–174; Mil'vitskaya, E.M; Tarakanova, A.V.; Plate, A.F. Russ. Chem. Rev. 1976, 45, 469, see pp. 475–476.
651. See Schneider, M.P.; Rebell, J. J. Chem. Soc., Chem. Commun. 1975, 283.
652. See Schneider, M.P.; Rau, A. J. Am. Chem. Soc. 1979, 101, 4426.
653. Zora, M. J. Org. Chem. 2005, 70, 6018.
654. Viola, A.; Collins, J.J.; Filipp, N. Tetrahedron 1981, 37, 3765; Théron F.; Verny, M.; Vessière, R. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 381–445, pp. 428–430; Huntsman, W.D. Intra-Sci. Chem. Rep., 1972, 6, 151.
655. Siebert, M.R.; Tantillo, D.J. J. Am. Chem. Soc. 2007, 129, 8686. Overman, L.E. Angew. Chem. Int. Ed. 1984, 23, 579; Lutz, R.P. Chem. Rev. 1984, 84, 205. See Overman, L.E.; Renaldo, A.F. J. Am. Chem. Soc. 1990, 112,3945.
656. See Poupko, R.; Zimmermann, H.; Müller, K.; Luz, Z. J. Am. Chem. Soc. 1996, 118, 7995.
657. See Shea, K.J.; Stoddard, G.J.; England, W.P.; Haffner, C.D. J. Am. Chem. Soc, 1992, 114, 2635. See also, Tantillo, D.J.; Hoffmann, R. J. Org. Chem. 2002, 67, 1419.
658. Doering, W. von E.; Roth, W.R. Tetrahedron 1962, 18, 67. See also, Gajewski, J.J.; Benner, C.W.; Hawkins, C.M. J. Org. Chem. 1987, 52, 5198; Paquette, L.A.; DeRussy, D.T.; Cottrell, C.E. J. Am. Chem. Soc. 1988, 110, 890.
659. See Hoffmann, R.; Woodward, R.B. J. Am. Chem. Soc. 1965, 87, 4389; Fukui, K.; Fujimoto, H. Tetrahedron Lett. 1966, 251.
660. See Gajewski, J.J.; Jimenez, J.L. J. Am. Chem. Soc. 1986, 108, 468.
661. See Özkan, I.; Zora, M. J. Org. Chem. 2003, 68, 9635.
662. See Hill, R.K. in Morrison, J.D. Asymmetric Synthesis, Vol. 3, Academic Press, NY, 1984, pp. 503–572, 503–545.
663. For a review, see Nubbemeyer, U. Synthesis 2003, 961.
664. See Navarro-Vázquez, A.; Prall, M.; Schreiner, P.R. Org. Lett. 2004, 6, 2981.
665. Hammond, G.S.; De Boer, C.D. J. Am. Chem. Soc. 1964, 86, 899; Trecker, D.J.; Henry, J.P. J. Am. Chem. Soc. 1964, 86, 902. Also see, Kessler, H.; Ott, W. J. Am. Chem. Soc. 1976, 98, 5014. Also see Berson, J.A. in de Mayo, P. Rearrangements in Ground and Excited States, Academic Press, NY, 1980, pp. 358–372.
666. See Baldwin, J.E.; Gilbert, K.E. J. Am. Chem. Soc. 1976, 98, 8283. For a similar result in the 1,2-divinylcyclopropane series, see Baldwin, J.E.; Ullenius, C. J. Am. Chem. Soc. 1984, 96, 1542.
667. Kaufmann, D.; de Meijere, A. Chem. Ber. 1984, 117, 1128; Dewar, M.J.S.; Jie, C. J. Am. Chem. Soc. 1987, 109, 5893; J. Chem. Soc., Chem. Commun. 1989, 98. For evidence against this view, see Halevi, E.A.; Rom, R. Isr. J. Chem. 1989, 29, 311; Owens, K.A.; Berson, J.A. J. Am. Chem. Soc. 1990, 112, 5973.
668. Roth, W.R.; Gleiter, R.; Paschmann, V.; Hackler, U.E.; Fritzsche, G.; Lange, H. Eur. J. Org. Chem. 1998, 961; Roth, W.R.; Schaffers, T.; Heiber, M. Chem. Ber. 1992, 125, 739.
669. For a report of still another mechanism, see Gompper, R.; Ulrich, W. Angew. Chem. Int. Ed. 1976, 15, 299. See McGuire, M.J.; Piecuch, P. J. Am. Chem. Soc. 2005, 127, 2608.
670. Doering, W. von E.; Roth, W.R. Tetrahedron 1963, 19, 715.
671. See Decock-Le Révérend, B.; Goudmand, P. Bull. Soc. Chim. Fr. 1973, 389; Gajewski, J.J. Mech. Mol. Migr. 1971, 4, 1, see pp. 32–49; Paquette, L.A. Angew. Chem. Int. Ed. 1971, 10, 11; Schröder, G.; Oth, J.F.M.; Merényi, R. Angew. Chem. Int. Ed. 1965, 4, 752.
672. Schröder, G. Chem. Ber. 1964, 97, 3140; Merényi, R.; Oth, J.F.M.; Schröder, G. Chem. Ber. 1964, 97, 3150. For a review of bullvalenes, see Schröder, G.; Oth, J.F.M. Angew. Chem. Int. Ed. 1967, 6, 414.
673. See Paquette, L.A.; Malpass, J.R.; Krow, G.R.; Barton, T.J. J. Am. Chem. Soc. 1969, 91, 5296.
674. Oth, J.F.M.; Müllen, K.; Gilles, J.; Schröder, G. Helv. Chim. Acta 1974, 57, 1415; Nakanishi, H.; Yamamoto, O. Tetrahedron Lett. 1974, 1803; Günther, H.; Ulmen, J. Tetrahedron 1974, 30, 3781. See Luger, P.; Buschmann, J.; McMullan, R.K.; Ruble, J.R.; Matias, P.; Jeffrey, G.A. J. Am. Chem. Soc. 1986, 108, 7825.
675. McKennis, J.S.; Brener, L.; Ward, J.S.; Pettit, R. J. Am. Chem. Soc. 1971, 93, 4957; Paquette, L.A.; Davis, R.F.; James, D.R. Tetrahedron Lett. 1974, 1615.
676. For a study of sigmatropic shiftamers in extended barbaralanes, see Tantillo, D.J.; Hoffmann, R.; Houk, K.N.; Warner, P.M.; Brown, E.C.; Henze, D.K. J. Am. Chem. Soc. 2004, 126, 4256.
677. Biethan, U.; Klusacek, H.; Musso, H. Angew. Chem. Int. Ed. 1967, 6, 176; Doering, W. von E.; Ferrier, B.M.; Fossel, E.T.; Hartenstein, J.H.; Jones, Jr., M.; Klumpp, G.W.; Rubin, R.M.; Saunders, M. Tetrahedron 1967, 23,3943; Henkel, J.G.; Hane, J.T. J. Org. Chem. 1983, 48, 3858.
678. Meinwald, J.; Schmidt, D. J. Am. Chem. Soc. 1969, 91, 5877; Zimmerman, H.E.; Binkley, R.W.; Givens, R.S.; Grunewald, G.L.; Sherwin, M.A. J. Am. Chem. Soc. 1969, 91, 3316. See Seefelder, M.; Heubes, M.; Quast, H.; Edwards, W.D.; Armantrout, J.R.; Williams, R.V.; Cramer, C.J.; Goren, A.C.; Hrovat, D.A.; Borden, W.T. J. Org. Chem. 2005, 70, 3437.
679. Moskau, D.; Aydin, R.; Leber, W.; Günther, H.; Quast, H.; Martin, H.-D.; Hassenrück, K.; Miller, L.S.; Grohmann, K. Chem. Ber. 1989, 122, 925. Are semibullvalenes homoaromatic? See Williams, R.V.; Gadgil, V.R.; Chauhan, K.; Jackman, L.M.; Fernandes, E. J. Org. Chem. 1998, 63, 3302.
680. Ciganek, E. J. Am. Chem. Soc. 1965, 87, 1149. See Neidlein, R.; Radke, C.M. Helv. Chim. Acta 1983, 66, 2626; Takeuchi, K.; Kitagawa, T.; Ueda, A.; Senzaki, Y.; Okamoto, K. Tetrahedron 1985, 41, 5455.
681. See Shirwaiker, G.S.; Bhatt, M.V. Adv. Heterocycl. Chem. 1984, 37, 67.
682. Vogel, E. Pure Appl. Chem. 1969, 20, 237. See also, Boyd, D.R.; Stubbs, M.E. J. Am. Chem. Soc. 1983, 105, 2554.
683. See Balaban, A.T.; Banciu, M. J. Chem. Educ. 1984, 61, 766; Greenberg, A.; Liebman, J.F. Strained Organic Molecules, Academic Press, NY, 1978, pp. 203–215; Scott, L.T.; Jones, Jr., M. Chem. Rev. 1972, 72, 181. See also, Maier, G.; Wiegand, N.H.; Baum, S.; Wüllner, R. Chem. Ber. 1989, 122, 781.
684. See Hassenrück, K.; Martin, H.; Walsh, R. Chem. Rev. 1989, 89, 1125.
685. See Balaban, A.T; Banziu, M. J. Chem. Educ. 1984, 61, 766; Banciu, M.; Popa, C.; Balaban, A.T. Chem. Scr., 1984, 24, 28.
686. Kobayashi, Y.; Kumadaki, I. Top. Curr. Chem. 1984, 123, 103; Bickelhaupt, F.; de Wolf, W.H. Recl. Trav. Chim. Pays-Bas 1988, 107, 459.
687. See Davis, J.H.; Shea, K.J.; Bergman, R.G. J. Am. Chem. Soc. 1977, 99, 1499.
688. See Christl, M. Angew. Chem. Int. Ed. 1981, 20, 529; Burger, U. Chimia, 1979, 147.
689. Maier, G.; Kalinowski, H.; Euler, K. Angew. Chem. Int. Ed. 1982, 21, 693.
690. Castro, A.M.M. Chem. Rev. 2004, 104, 2939; Hiersemann, M.; Nubbemeyer, U The Claisen Rearrangement: Methods and Applications. Wiley–VCH, 2007.
691. Fleming, I. Pericyclic Reactions, Oxford University Press, Oxford, 1999, pp. 71–83; Moody, C.J. Adv. Heterocycl. Chem. 1987, 42, 203; Bartlett, P.A. Tetrahedron 1980, 36, 2, see pp. 28–39; Ziegler, F.E. Acc. Chem. Res.1977, 10, 227; Bennett, G.B. Synthesis 1977, 589; Rhoads, S.J.; Raulins, N.R. Org. React. 1975, 22, 1; Shine, H.J. Aromatic Rearrangements, Elsevier, NY, 1969, pp. 89–120; Smith, G.G.; Kelly, F.W. Prog. Phys. Org. Chem.1971, 8, 75, pp. 153–201; Hansen, H.; Schmid, H. Chimia, 1970, 24, 89; Jefferson, A.; Scheinmann, F. Q. Rev. Chem. Soc. 1968, 22, 391.
692. See Gozzo, F.C.; Fernandes, S.A.; Rodrigues, D.C.; Eberlin, M.N.; Marsaioli, A.J. J. Org. Chem. 2003, 68, 5493.
693. See Kupczyk-Subotkowska, L.; Saunders, Jr., W.H.; Shine, H.J. J. Am. Chem. Soc. 1988, 110, 7153.
694. Copley, S.D.; Knowles, J.R. J. Am. Chem. Soc. 1985, 107, 5306. Also see, Yoo, H.Y.; Houk, K.N. J. Am. Chem. Soc. 1994, 116, 12047.
695. Boeckman, Jr., R.K.; Shair, M.D.; Vargas, J.R.; Stolz, L.A. J. Org. Chem. 1993, 58, 1295.
696. Conroy, H.; Firestone, R.A. J. Am. Chem. Soc. 1956, 78, 2290.
697. Sharghi, H.; Aghapour, G. J. Org. Chem. 2000, 65, 2813.
698. Wipf, P.; Ribe, S. Org. Lett. 2001, 3, 1503.
699. Kumar, H.M.S.; Anjaneyulu, S.; Reddy, B.V.S.; Yadav, J.C. Synlett 2000, 1129.
700. See Ziegler, F.E. Chem. Rev. 1988, 88, 1423.
701. Claisen, L. Ber. 1912, 45, 3157.
702. See Boeckman, Jr., R.K.; Flann, C.J.; Poss, K.M. J. Am. Chem. Soc. 1985, 107, 4359.
703. For reviews, see Hiersemann, M.; Abraham, L. Eur. J. Org. Chem. 2002, 1461; Majumdar, K.C.; Alam, S.; Chattopadhyay, B. Tetrahedron 2008, 64, 597.
704. Grieco, P.A.; Brandes, E.B.; McCann, S.; Clark, J.D. J. Org. Chem. 1989, 54, 5849. See Guest, J.M.; Craw, J.S.; Vincent, M.A.; Hillier, I.H. J. Chem. Soc. Perkin Trans. 2 1997, 71; Sehgal, A.; Shao, L.; Gao, J. J. Am. Chem. Soc. 1995, 117, 11337.
705. Kotha, S.; Mandal, K.; Deb, A.C.; Banerjee, S. Tetrahedron Lett. 2004, 45, 9603.
706. Lin, Y.-L.; Cheng, J.-Y.; Chu, Y.-H. Tetrahedron 2007, 63, 10949.
707. See Dewar, M.J.S.; Jie, C. J. Am. Chem. Soc. 1989, 111, 511.
708. Balta, B.; Öztürk, C.; Aviyente, V.; Vincent, M.A.; Hillier, I.H. J. Org. Chem. 2008, 73, 4800.
709. Kaeobamrung, J.; Mahatthananchai, J.; Zheng, P.; Bode, J.W. J. Am. Chem. Soc. 2010, 132, 8810.
710. Uyeda, C.; Jacobsen, E.N. J. Am. Chem. Soc. 2008, 130, 9228.
711. Nelson, S.G.; Kan Wang, K. J. Am. Chem. Soc. 2006, 128, 4232.
712. There are substituent effects, see Aviyente, V.; Yoo, H.Y.; Houk, K.N. J. Org. Chem. 1997, 62, 6121.
713. White, W.N.; Slater, C.D. J. Org. Chem. 1962, 27, 2908; Zahl, G.; Kosbahn, W.; Kresze, G. Liebigs Ann. Chem. 1975, 1733. See also, Desimoni, G.; Faita, G.; Gamba, A.; Righetti, P.P.; Tacconi, G.; Toma, L. Tetrahedron1990, 46, 2165; Gajewski, J.J.; Gee, K.R.; Jurayj, J. J. Org. Chem. 1990, 55, 1813.
714. See Gajewski, J.J. Accts. Chem. Res. 1997, 30, 219.
715. White, W.N.; Wolfarth, E.F. J. Org. Chem. 1970, 35, 2196. See also, Brandes, E.; Greico, P.A.; Gajewski, J.J. J. Org. Chem. 1989, 54, 515.
716. Svanholm, U.; Parker, V.D. J. Chem. Soc. Perkin Trans. 2 1974, 169.
717. See Lutz, R.P. Chem. Rev. 1984, 84, 205.
718. See Yagodin, V.G.; Bunina-Krivorukova, L.I.; Bal'yan, Kh.V. J. Org. Chem. USSR 1971, 7, 1491.
719. Parsons, P.J.; Thomson, P.; Taylor, A.; Sparks, T. Org. Lett. 2000, 2, 571.
720. For a review of the Conia-ene reaction, see Conia, J.M.; Le Perchec, P. Synthesis 1975, 1.
721. Schobert, R.; Siegfried, S.; Gordon, G.; Nieuwenhuyzen, M.; Allenmark, S. Eur. J. Org. Chem. 2001, 1951.
722. Carroll, M.F. J. Chem. Soc. 1941, 507; Ziegler, F.E. Chem. Rev. 1988, 88, 1423.
723. Kimel, W.; Cope, A.C. J. Am. Chem. Soc. 1943, 65, 1992.
724. Burger, E.C.; Tunge, J.A. Org. Lett. 2004, 6, 2603.
725. Kuwano, R.; Ishida, N.; Murakami, M. Chem. Commun. 2005, 3951.
726. Felix, D.; Gschwend-Steen, K.; Wick, A.E.; Eschenmoser, A. Helv. Chim. Acta 1969, 52, 1030.
727. Gradl, S. N.; Trauner, D. in The Meerwein–Eschenmoser–Claisen Rearrangement. In The Claisen Rearrangement, Hiersemann, M.; Nubbemeyer, U., Eds., Wiley–VCH, Weinheim, 2007, pp 367–396.
728. Linton, E.C.; Kozlowski, M.C. J. Am. Chem. Soc. 2008, 130, 16162.
729. Gajewski, J.J.; Emrani, J. J. Am. Chem. Soc. 1984, 106, 5733; Cameron A.G.; Knight, D.W. J. Chem. Soc. Perkin Trans. 1 1986, 161. See Wilcox, C.S.; Babston, R.E. J. Am. Chem. Soc. 1986, 108, 6636.
730. Ireland, R.E.; Wipf, P.; Armstrong, III, J.D. J. Org. Chem. 1991, 56, 650. See also Ref. 714.
731. See Chai, Y.; Hong, S.-p.; Lindsay, H.A.; McFarland, C.; McIntosh, M.C. Tetrahedron 2002, 58, 2905.
732. See Denmark, S.E.; Harmata, M.A.; White, K.S. J. Am. Chem. Soc. 1989, 111, 8878.
733. Koreeda, M.; Luengo, J.I. J. Am. Chem. Soc. 1985, 107, 5572; Kirchner, J.J.; Pratt, D.V.; Hopkins, P.B. Tetrahedron Lett. 1988, 29, 4229.
734. Mohamed, M.; Brook, M.A. Tetrahedron Lett. 2001, 42, 191. See Khaledy, M.M.; Kalani, M.Y.S.; Khuong, K.S.; Houk, K.N.; Aviyente, V.; Neier, R.; Soldermann, N.; Velker, J. J. Org. Chem. 2003, 68, 572.
735. Corey, E.J.; Lee, D. J. Am. Chem. Soc. 1991, 113, 4026.
736. Tsunoda, T.; Tatsuki, S.; Shiraishi, Y.; Akasaka, M.; Itô, S. Tetrahedron Lett. 1993, 34, 3297; Walters, M.A.; Hoem, A.B.; Arcand, H.R.; Hegeman, A.D.; McDonough, C.S. Tetrahedron Lett. 1993, 34, 1453.
737. See Zumpe, F.L.; Kazmaier, U. Synlett 1998, 434; Ito, H.; Sato, A.; Taguchi, T. Tetrahedron Lett. 1997, 38, 4815; Kazmaier, U.; Krebs, A. Angew. Chem. Int. Ed. 1995, 34, 2012. For asymmetric induction in the thio-Claisen rearrangement, see Reddy, K.V.; Rajappa, S. Tetrahedron Lett. 1992, 33, 7957.
738. Maruoka, K.; Saito, S.; Yamamoto, J. J. Am. Chem. Soc. 1995, 117, 1165. See Hiersemann, M.; Abraham, L. Org. Lett. 2001, 3, 49; Miller, S.P.; Morken, J.P. Org. Lett. 2002, 4, 2743.
739. For a review, see Enders, D.; Knopp, M.; Schiffers, R. Tetrahedron Asymmetry, 1996, 7, 1847.
740. Jolidon, S.; Hansen, H. Helv. Chim. Acta 1977, 60, 978.
741. Anderson, J.C.; Flaherty, A.; Swarbrick, M.E. J. Org. Chem. 2000, 65, 9152. See Wu, P.; Fowler, F.W. J. Org. Chem. 1988, 53, 5998.
742. See Metz, P.; Mues, C. Tetrahedron 1988, 44, 6841. Also see Gradl, S.N.; Kennedy-Smith, J.J.; Kim, J.; Trauner, D. Synlett 2002, 411.
743. See Schenck, T.G.; Bosnich, B. J. Am. Chem. Soc. 1985, 107, 2058, and references cited therein; Anderson, C.E.; Overman, L.E. J. Am. Chem. Soc. 2003, 125, 12412.
744. See Kirsch, S.F.; Overman, L.F.; Watson, M.P. J. Org. Chem. 2004, 69, 8101. For a review, see Majumdar, K.C.; Bhattacharyya, T.; Chattopadhyay, B.; Sinha, B. Synthesis 2009, 2117. See also, Forte, L.; Lafortune, M.C.; Bierzynski, I.R.; Duncan, J.A. J. Am. Chem. Soc. 2010, 132, 2196.
745. Blechert, S. Synthesis 1989, 71; Heimgartner, H.; Hansen, H.; Schmid, H. Adv. Org. Chem. 1979, 9, pt. 2, 655.
746. Uozumi, Y.; Kato, K.; Hayashi, T. Tetrahedron Asymmetry, 1998, 9, 1065. See Gilbert, J.C.; Cousins, K.R. Tetrahedron 1994, 50, 10671.
747. Kirsch, S.F.; Overman, L.E. J. Am. Chem. Soc. 2005, 127, 2866; Watson, M.P.; Overman, L.E.; Bergman, R.G. J. Am. Chem. Soc. 2007, 129, 5031.
748. Rueping, M.; Antonchick, A.P. Angew. Chem. Int. Ed. 2008, 47, 10090.
749. Anderson, W.K.; Lai, G. Synthesis 1995, 1287.
750. Mitsuhashi, T. J. Am. Chem. Soc. 1986, 108, 2400.
751. Chen, B.; Mapp, A.K. J. Am. Chem. Soc. 2005, 127, 6712.
752. They have been trapped: See, for example, Mortensen, J.Z.; Hedegaard, B.; Lawesson, S. Tetrahedron 1971, 27, 3831; Kwart, H.; Schwartz, J.L. J. Org. Chem. 1974, 39, 1575.
753. Kwart, H.; Cohen, M.H. J. Org. Chem. 1967, 32, 3135; Chem. Commun. 1968, 319; Makisumi, Y.; Murabayashi, A. Tetrahedron Lett. 1969, 1971, 2449.
754. For a review, see Majumdar, K.C.; Ghosh, S.; Ghosh, M. Tetrahedron 2003, 59, 7251.
755. King, J.F.; Harding, D.R.K. J. Am. Chem. Soc. 1976, 98, 3312.
756. Block, E.; Ahmad, S. J. Am. Chem. Soc. 1985, 107, 6731.
757. de la Pradilla, R.F.; Montero, C.; Tortosa, M.; Viso, A. Chemistry: Eur. J. 2009, 15, 697.
758. For abnormal Claisen rearrangements, see Hansen, H. Mech. Mol. Migr. 1971, 3, 177; Marvell, E.N.; Whalley, W. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 2, Wiley, NY, 1971, pp. 743–750.
759. Lauer, W.M.; Johnson, T.A. J. Org. Chem. 1963, 28, 2913; Fráter, G.; Schmid, H. Helv. Chim. Acta 1966, 49, 1957; Marvell, E.N.; Schatz, B. Tetrahedron Lett. 1967, 67.
760. Watson, J.M.; Irvine, J.L.; Roberts, R.M. J. Am. Chem. Soc. 1973, 95, 3348.
761. Robinson, B. The Fischer Indole Synthesis, Wiley, NY, 1983; Grandberg, I.I.; Sorokin, V.I. Russ. Chem. Rev. 1974, 43, 115; Shine, H.J. Aromatic Rearrangements, Elsevier, NY, 1969, pp. 190–207; Sundberg, R.J. The Chemistry of Indoles, Academic Press, NY, 1970, pp. 142–163; Robinson, B. Chem. Rev. 1969, 69, 227. For reviews of so-called abnormal Fischer indole syntheses, see Ishii, H. Acc. Chem. Res. 1981, 14, 275; Fusco, R.; Sannicolo, F. Tetrahedron 1980, 36, 161.
762. Lipinska, T.; Guibé-Jampel, E.; Petit, A.; Loupy, A. Synth. Commun. 1999, 29, 1349.
763. Rebeiro, G.LO.; Khadilkar, B.M. Synthesis 2001, 370.
764. Rosenbaum, C.; Katzka, C.; Marzinzik, A.; Waldmann, H. Chem. Commun. 2003, 1822.
765. Moody, C.J.; Swann, E. Synlett 1998, 135.
766. Campos, K.R.; Woo, J.C.S.; Lee, S.; Tillyer, R.D. Org. Lett. 2004, 6, 79.
767. For a mechanistic study, see Hughes, D.L.; Zhao, D. J. Org. Chem. 1993, 58, 228.
768. This mechanism was proposed by Robinson, G.M.; Robinson, R. J. Chem. Soc. 1918, 113, 639.
769. See Forrest, T.P.; Chen, F.M.F. J. Chem. Soc., Chem. Commun. 1972, 1067.
770. Douglas, A.W. J. Am. Chem. Soc. 1978, 100, 6463; 1979, 101, 5676.
771. Bajwa, G.S.; Brown, R.K. Can. J. Chem. 1969, 47, 785; 1970, 48, 2293 and references cited therein.
772. Clausius, K.; Weisser, H.R. Helv. Chim. Acta 1952, 35, 400.
773. See Ma, M.; Peng, L.; Li, C.; Zhang, X.; Wang, J. J. Am. Chem. Soc. 2005, 127, 15106. For a review as applied to ring expansions, see Vedejs, E. Acc. Chem. Res. 1984, 17, 358.
774. See Hoffmann, R.W. Angew. Chem. Int. Ed. 1979, 18, 563.
775. See Mander, L.N.; Turner, J.V. J. Org. Chem. 1973, 38, 2915; Honda, K.; Inoue, S.; Sato, K. J. Am. Chem. Soc. 1990, 112, 1999.
776. Nakai, T.; Mikami, K. Chem. Rev. 1986, 86, 885. See Schöllkopf, U.; Fellenberger, K.; Rizk, M. Liebigs Ann. Chem. 1970, 734, 106; Rautenstrauch, V. Chem. Commun. 1970, 4. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1063–1067.
777. You, Z.; Koreeda, M. Tetrahedron Lett. 1993, 34, 2597.
778. Kunishima, M.; Hioki, K.; Kono, K.; Kato, A.; Tani, S. J. Org. Chem. 1997, 62, 7542. Also see, Hioki, K.; Kono, K.; Tani, S.; Kunishima, M. Tetrahedron Lett. 1998, 39, 5229. For an enantioselective [2,3]-Wittig rearrangement, see Fujimoto, K.; Nakai, T. Tetrahedron Lett. 1994, 35, 5019.
779. See Murata, Y.; Nakai, T. Chem. Lett. 1990, 2069. Also see Reich, H.J. in Liotta, D.C. Organoselenium Chemistry, Wiley, NY, 1987, pp. 365–393; Reich, H.J. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. C, Academic Press, NY, 1978, pp. 102–111.
780. Baldwin, J.E.; Urban, F.J. Chem. Commun. 1970, 165.
781. Workman, J.A.; Garrido, N.P.; Sançon, J.; Roberts, E.; Wessel, H.P.; Sweeney, J.B. J. Am. Chem. Soc. 2005, 127, 1066.
782. Blid, J.; Panknin, O.; Somfai, P. J. Am. Chem. Soc. 2005, 127, 9352.
783. Somfai, P.; Panknin, O. Synlett 2007, 1190.
784. Mikami, K.; Nakai, T. Synthesis 1991, 594; Nakai, T.; Mikami, K. Chem. Rev. 1986, 86, 885, pp. 888–895. See also, Scheuplein, S.W.; Kusche, A.; Brückner, R.; Harms, K. Chem. Ber. 1990, 123, 917; Wu, Y.; Houk, K.N.; Marshall, J.A. J. Org. Chem. 1990, 55, 1421; Marshall, J.A.; Wang, X. J. Org. Chem. 1990, 55, 2995.
785. Huynh, C.; Julia, S.; Lorne, R.; Michelot, D. Bull. Soc. Chim. Fr. 1972, 4057.
786. Evans, D.A.; Andrews, G.C. Acc. Chem. Res. 1974, 7, 147; Hoffmann, R.W. Angew. Chemie. Int. Ed., Engl., 1979, 18, 563; Sato, T.; Otera, J.; Nozaki, H. J. Org. Chem. 1989, 54, 2779.
787. See Tamaru, Y.; Nagao, K.; Bando, T.; Yoshida, Z. J. Org. Chem. 1990, 55, 1823.
788. Patai, S. The Chemistry of the Hydrazo, Azo, and Azoxy Groups, pt. 2, Wiley, NY, 1975, the reviews by Cox, R.A.; Buncel, E. pp. 775–807; Koga, G.; Koga, N.; Anselme, J. pp. 914–921; Williams, D.L.H. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, Vol. 13, 1972, pp. 437–448; Shine, H.J. Mech. Mol. Migr. 1969, 2, 191; Banthorpe, D.V. Top. Carbocyclic Chem. 1969, 1, 1.
789. Shine, H.J.; Stanley, J.P. J. Org. Chem. 1967, 32, 905. For investigations of the mechanism of the disproportionation reactions, see Rhee, E.S.; Shine, H.J. J. Am. Chem. Soc. 1986, 108, 1000; 1987, 109, 5052.
790. For a history, see Shine, H.J. J. Phys. Org. Chem. 1989, 2, 491.
791. Shine, H.J.; Chamness, J.T. J. Org. Chem. 1963, 28, 1232; Banthorpe, D.V.; O'Sullivan, M. J. Chem. Soc. B 1968, 627.
792. Banthorpe, D.V.; Cooper, A.; O'Sullivan, M. J. Chem. Soc. B 1971, 2054.
793. Banthorpe, D.V.; Ingold, C.K.; Roy, J. J. Chem. Soc. B 1968, 64; Banthorpe, D.V.; Ingold, C.K.; O'Sullivan, M. J. Chem. Soc. B 1968, 624.
794. Banthorpe, D.V.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1964, 2864; Dewar, M.J.S. in de Mayo, P. Molecular Rearrangments, Vol. 1, Wiley, NY, 1963, pp. 323–344.
795. Shine, H.J.; Zmuda, H.; Park, K.H.; Kwart, H.; Horgan, A.J.; Brechbiel, M. J. Am. Chem. Soc. 1982, 104, 2501.
796. This step was also part of the “polar-transition-state mechanism”. See also Ref. 793.
797. Olah, G.A.; Dunne, K.; Kelly, D.P.; Mo, Y.K. J. Am. Chem. Soc. 1972, 94, 7438.
798. Shine, H.J.; Park, K.H.; Brownawell, M.L.; San Filippo, Jr., J. J. Am. Chem. Soc. 1984, 106, 7077.
799. See Shine, H.J.; Zmuda, H.; Kwart, H.; Horgan, A.G.; Brechbiel, M. J. Am. Chem. Soc. 1982, 104, 5181.
800. Shine, H.J.; Gruszecka, E.; Subotkowski, W.; Brownawell, M.; San Filippo, Jr., J. J. Am. Chem. Soc. 1985, 107, 3218.
801. See Rhee, E.S.; Shine, H.J. J. Am. Chem. Soc. 1986, 108, 1000; 1987, 109, 5052.
802. Rhee, E.S.; Shine, H.J. J. Org. Chem. 1987, 52, 5633.
803. See Nojima, M.; Ando, T.; Tokura, N. J. Chem. Soc. Perkin Trans. 1 1976, 1504.
804. Grubbs, R.H. Tetrahedron 2004, 60, 7117; Wakamatsu, H.; Blechert, S. Angew. Chem. Int. Ed. 2002, 41, 2403; Schrock, R.R.; Hoveyda, A.H. Angew. Chem. Int. Ed. 2003, 42, 4592. See Astruc, D. New J. Chem. 2005, 29, 42; Chauvin, Y. Angew. Chem. Int. Ed. 2006, 45, 3740; Schrock, R.R. Angew. Chem. Int. Ed. 2006, 45, 3748; Grubbs, R.H. Angew. Chem. Int. Ed. 2006, 45, 3760.
805. Smith, III, A.B.; Adams, C.M.; Kozmin, S.A. J. Am. Chem. Soc. 2001, 123, 990.
806. See Wang, J.; Menapace, H.R. J. Org. Chem. 1968, 33, 3794; Hughes, W.B. J. Am. Chem. Soc. 1970, 92, 532.
807. Dragutn, V.; Balaban, A.T.; Dimonie, M. Olefin Metathesis and Ring-Opening Polymerization of Cyclo-Olefins, Wiley, NY, 1985; Ivin, K.J. Olefin Metathesis, Academic Press, NY, 1983; Feast, W.J.; Gibson, V.C. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 5, Wiley, NY, 1989, pp. 199–228; Schrock, R.R. J. Organomet. Chem. 1986, 300, 249; Grubbs, R.H. in Wilkinson, G Comprehensive Organometallic Chemistry, Vol. 8,Pergamon, Elmsford, NY, 1982, pp. 499–551; Basset, J.M.; Leconte, M. CHEMTECH 1980, 762; Banks, R.L. Fortschr. Chem. Forsch. 1972, 25, 39; Calderon N.; Lawrence, J.P.; Ofstead, E.A. Adv. Organomet. Chem. 1979, 17,449; Grubbs, R.H. Prog. Inorg. Chem. 1978, 24, 1; Calderon N. in Patai, S. The Chemistry of Functional Groups: Supplement A pt. 2, Wiley, NY, 1977, pp. 913–964; Acc. Chem. Res. 1972, 5, 127; Katz, T.J. Adv. Organomet. Chem. 1977, 16, 283; Haines, R.J.; Leigh, G.J. Chem. Soc. Rev. 1975, 4, 155.
808. See Crowe, W.E.; Zhang, Z.J. J. Am. Chem. Soc. 1993, 115, 10998.; Fu, G.C.; Grubbs, R.H. J. Am. Chem. Soc. 1993, 115, 3800.
809. Calderon N.; Ofstead, E.A.; Ward, J.P.; Judy, W.A.; Scott, K.W. J. Am. Chem. Soc. 1968, 90, 4133.
810. Knoche, H. Ger. Pat.(Offen.) 2024835, 1970 [Chem. Abstr., 1971, 74, 44118b]. See also, Ichikawa, K.; Fukuzumi, K. J. Org. Chem. 1976, 41, 2633; Baker, R.; Crimmin, M.J. Tetrahedron Lett. 1977, 441.
811. See Conrad, J.C.; Parnas, H.H.; Snelgrove, J.L.; Fogg, D.E. J. Am. Chem. Soc. 2005, 127, 11882.
812. Calderon N.; Chen, H.Y.; Scott, K.W. Tetrahedron Lett. 1967, 3327. See Hughes, W.B. Organomet. Chem. Synth. 1972, 1, 341, see pp. 362–368.; Toreki, R.; Schrock, R.R. J. Am. Chem. Soc. 1990, 112, 2448.
813. Banks, R.L.; Bailey, G.C. Ind. Eng. Chem. Prod. Res. Dev., 1964, 3, 170. See also, Banks, R.L. CHEMTECH 1986, 112.
814. Gilbertson, S.R.; Hoge, G.S.; Genov, D.G. J. Org. Chem. 1998, 63, 10077; Maier, M.E.; Bugl, M. Synlett 1998, 1390; Stefinovic, M.; Snieckus, V. J. Org. Chem. 1998, 63, 2808.
815. See Banks, R.L. Fortschr. Chem. Forsch. 1972, 25, 39, pp. 41–46.
816. Schwab, P.; Grubbs, R.H.; Ziller, J.W. J. Am. Chem. Soc. 1996, 118, 100.
817. Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Org. Lett. 1999, 1, 953.
818. Bazan, G.C.; Oskam, J.H.; Cho, H.-N.; Park, L.Y.; Schrock, R.R. J. Am. Chem. Soc. 1991, 113, 6899, and references cited therein.
819. Louie, J.; Grubbs, R.H. Angew. Chem. Int. Ed. 2001, 40, 247.
820. See Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Angew. Chem. Int. Ed. 2005, 44, 4490; Donohoe, T.J.; Fishlock, L.P.; Procopiou, P.A. Chemistry: European J. 2008, 14, 5716; Gradillas, A.; Pérez-Castells, J. Angew. Chem. Int. Ed. 2006, 45, 6086.
821. Schrock, R.R.; Hoveyda, A.H. Angew. Chem. Int. Ed. 2003. 42, 4592; Grela, K.; Kim, M. Eur. J. Org. Chem. 2003, 963; Zhang, W.; Kraft, S.; Moore, J.S. J. Am. Chem. Soc. 2004, 126, 329; Iyer, K.; Rainier, J.D. J. Am. Chem. Soc. 2007, 129, 12604; Vougioukalakis, G.C.; Grubbs, R.H. J. Am. Chem. Soc. 2008, 130, 2234; Matsugi, M.; Curran, D.P. J. Org. Chem. 2005, 70, 1636; Rix, D.; Caijo, F.; Laurent, I.; Boeda, F.; Clavier, H.; Nolan, S.P.; Mauduit, M. J. Org. Chem. 2008, 73, 4225; Katz, T.J. Angew. Chem. Int. Ed. 2005, 44, 3010.
822. Kirkland, T.A.; Lynn, D.M.; Grubbs, R.H. J. Org. Chem. 1998, 63, 9904.
823. See Deiter, S.A.; Martin, S.F. Chem. Rev. 2004, 104, 2199.
824. Takahashi, T.; Kotora, M.; Kasai, K. J. Chem. Soc., Chem. Commun. 1994, 2693.
825. Schneider, M.F.; Junga, H.; Blechert, S. Tetrahedron 1995, 51, 13003.
826. Lee, C.W.; Grubbs, R.H. J. Org. Chem. 2001, 66, 7155.
827. Paquette, L.A.; Méndez-Andino, J. Tetrahedron Lett. 1999, 40, 4301.
828. Brimble, M.A.; Trzoss, M. Tetrahedron 2004, 60, 5613.
829. See Dolman, S.J.; Sattely, E.S.; Hoveyda, A.H.; Schrock, R.R. J. Am. Chem. Soc. 2002, 124, 6991.
830. See Ma, S.; Ni, B.; Liang, Z. J. Org. Chem. 2004, 69, 6305.
831. Yao, Q. Org. Lett. 2002, 4, 427.
832. Demchuk, O.M.; Pietrusiewicz, K.M.; Michrowska, A.; Grela, K. Org. Lett. 2003, 5, 3217.
833. LeFlohic, A.; Meyer, C.; Cossy, J.; Desmurs, J.-R.; Galland, J.-C. Synlett 2003, 667.
834. See Kinderman, S.S.; Van Maarseveen, J.H.; Schoemaker, H.E.; Hiemstra, H.; Rutjes, F.P.J.T. Org. Lett. 2001, 3, 2045.
835. Fürstner, A.; Picquet, M.; Bruneau, C.; Dixneuf, P.H. Chem. Commun. 1998, 1315; Maier, M.E.; Lapeva, T. Synlett 1998, 891; Mori, M.; Sakakibara, N.; Kinoshita, A. J. Org. Chem. 1998, 63, 6082; O'Mahony, D.J.R.; Belanger, D.B.; Livinghouse, T. Synlett 1998, 443.
836. Edwards, S.D.; Lewis, T.; Taylor, R.J.K. Tetrahedron Lett. 1999, 40, 4267.
837. See Rainier, J.D.; Cox, J.M.; Allwein, S.P. Tetrahedron Lett. 2001, 42, 179.
838. Chao, W.; Weinreb, S.M. Org. Lett. 2003, 5, 2505.
839. Schuman, M.; Gouverneur, V. Tetrahedron Lett. 2002, 43, 3513.
840. Kim, S.; Lim, C.J. Angew. Chem. Int. Ed. 2002, 41, 3265.
841. Delgado, M.; Martín, J.D. Tetrahedron Lett. 1997, 38, 6299; Miller, S.J.; Kim, S.-H.; Chen, Z.-R.; Grubbs, R.H. J. Am. Chem. Soc. 1995, 117, 2108.
842. Leconte, M.; Pagano, S.; Mutch, A.; Lefebvre, F.; Basset, J.M. Bull. Soc. Chim. Fr. 1995, 132, 1069.
843. Cefalo, D.R.; Kiely, A.F.; Wuchrer, M.; Jamieson, J.Y.; Schrock, R.R.; Hoveyda, A.H. J. Am. Chem. Soc. 2001, 123, 3139.
844. See Funk, T.W.; Berlin, J.M.; Grubbs, R.H. J. Am. Chem. Soc. 2006, 128, 1840.
845. Sattely, E.S.; Cortez, G.A.; Moebius, D.C.; Schrock, R.R.; Hoveyda, A.H. J. Am. Chem. Soc. 2005, 127, 8526.
846. Gillingham, D.G.; Kataoka, O.; Garber, S.B.; Hoveyda, A.H. J. Am. Chem. Soc. 2004, 126, 12288.
847. Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, Jr., P.J.; Hoveyda, A.H. J. Am. Chem. Soc. 1999, 121, 791.
848. Buijsman, R.C.; van Vuuren, E.; Sterrenburg, J.G. Org. Lett. 2001, 3, 3785. See Clavier, H.; Audic, N.; Mauduit, M.; Guillemin, J.-C. Chem. Commun. 2004, 2282.
849. Fürstner, A.; Ackermann, L.; Beck, K.; Hori, H.; Koch, D.; Langemann, K.; Liebl, M.; Six, C.; Leitner, W. J. Am. Chem. Soc. 2001, 123, 9000.
850. Binder, J.B.; Blank, J.J.; Raines, R.T. Org. Lett. 2007, 9, 4885; Zaman, S.; Curnow, O.J.; Abell, A.D. Austr. J. Chem. 2009, 62, 91; Burtscher, D.; Grela, K. Angew. Chem. Int. Ed. 2009, 48, 442.
851. See Coquerel, Y.; Rodriguez, J. Eur. J. Org. Chem. 2008, 1125. For a solvent-free microwave-induced reaction, see Thanh, G.V.; Loupy, A. Tetrahedron Lett. 2003, 44, 9091.
852. Bargiggia, F.C.; Murray, W.V. J. Org. Chem. 2005, 70, 9636.
853. Yao, Q. Angew. Chem. Int. Ed. 2000, 39, 3896; Schürer, S.C.; Gessler, S.; Buschmann, N.; Blechert, S. Angew. Chem. Int. Ed. 2000, 39, 3898.
854. Hultzsch, K.C.; Jernelius, J.A.; Hoveyda, A.H.; Schrock, R.R. Angew. Chem. Int. Ed. 2002, 41, 589.
855. A recyclable catalyst, see Yao, Q.; Motta, A.R. Tetrahedron Lett. 2004, 45, 2447.
856. Ahn, Y.M.; Yang, K.; Georg, G.I. Org. Lett. 2001, 3, 1411; Cho, J.H.; Kim, B.M. Org. Lett. 2003, 5, 531. See Westhus, M.; Gonthier, E.; Brohm, D.; Breinbauer, R. Tetrahedron Lett. 2004, 45, 3141.
857. Hong, S.H.; Grubbs, R.H. Org. Lett. 2007, 9, 1955.
858. See Chatterjee, A.K.; Choi, T.-L.; Sanders, D.P.; Grubbs, R.H. J. Am. Chem. Soc. 2003,125, 11360.
859. See Schmidt, B. J. Org. Chem. 2004, 69, 7672; Sutton, A.E.; Seigal, B.A.; Finnegan, D.F.; Snapper, M.L. J. Am. Chem. Soc. 2002, 124, 13390.
860. Hong, S.H.; Sanders, D.P.; Lee, C.W.; Grubbs, R.H. J. Am. Chem. Soc. 2005, 127, 17160.
861. See Stewart, I.C.; Douglas, C.J.; Grubbs, R.H. Org. Lett. 2008, 10, 441; Lipshutz, B.H.; Aguinaldo, G.T.; Ghorai, S.; Voigtritter, K. Org. Lett. 2008, 10, 1325.
862. Blanco, O.M.; Castedo, L. Synlett 1999, 557.
863. See Lautens, M.; Maddess, M.L. Org. Lett. 2004, 6, 1883.
864. Rückert, A.; Eisele, D.; Blechert, S. Tetrahedron Lett. 2001, 42, 5245.
865. Choi, T.-L.; Grubbs, R.H. Chem. Commun. 2001, 2648.
866. BouzBouz, S.; Cossy, J. Org. Lett. 2001, 3, 1451; van Otterlo, W.A.L.; Ngidi, E.L.; de Koning, C.D.; Fernandes, M.A. Tetrahedron Lett. 2004, 45, 659.
867. Sundararajan, G.; Prabagaran, N.; Varghese, B. Org. Lett. 2001, 3, 1973.
868. Verbicky, C.A.; Zercher, C.K. Tetrahedron Lett. 2000, 41, 8723.
869. López, F.; Delgado, A.; Rodríguez, J.R.; Castedo, L.; Mascareñas, J.L. J. Am. Chem. Soc. 2004, 126, 10262.
870. See Randl, S.; Connon, S.J.; Blechert, S. Chem. Commun. 2001, 1796; Morgan, J.P.; Morrill, C.; Grubbs, R.H. Org. Lett. 2002, 4, 67.
871. Ahmed, M.; Arnauld, T.; Barrett, A.G.M.; Braddock, D.C.; Flack, K.; Procopiou, P.A. Org. Lett. 2000, 2, 551.
872. Lee, C.W.; Choi, T.-L.; Grubbs, R.H. J. Am. Chem. Soc. 2002, 124, 3224.
873. Basset, J.-M.; Copret, C.; Soulivong, D.; Taoufik, M.; Cazat, J.T. Acc. Chem. Res. 2010, 43, 323; Basset, J.-M.; Copéret, C.; Lefort, L.; Maunders, B.M.; Maury, O.; Le Roux, E.; Saggio, G.; Soignier, S.; Soulivong, D.; Sunley, G.J.; Taoufik, M.; Thivolle-Cazat, J. J. Am. Chem. Soc. 2005, 127, 8604; Basset, J.-M.; Copéret, C.; Soulivong, D.; Taoufik, M.; Thivolle-Cazat, J. Angew. Chem. Int. Ed. 2006, 45, 6082.
874. See Grubbs, R.H.; Miller, S.J.; Fu, G.C. Accts. Chem. Res. 1995, 28, 446.
875. Grela, K. Angew. Chem. Int. Ed. 2008, 47, 5504.
876. Fürstner, A.; Langemann, K. J. Org. Chem. 1996, 61, 3942. Also see, Goldring, W.P.D.; Hodder, A.S.; Weiler, L. Tetrahedron Lett. 1998, 39, 4955; Ghosh, A.K.; Hussain, K.A. Tetrahedron Lett. 1998, 39, 1881.
877. See Kim, M.; Miller, R.L.; Lee, D. J. Am. Chem. Soc. 2005, 127, 12818.
878. Chen, F.-E.; Kuang, Y.-Y.; Dai, H.-F.; Lu, L.; Huo, M. Synthesis 2003, 2629.
879. Wender, P.A.; Sperandio, D. J. Org.Chem. 1998, 63, 4164.
880. Macnaughtan, M.L.; Johnson, M.J.A.; Kampf, J.W. J. Am. Chem. Soc. 2007, 129, 7708.
881. Calderon N.; Ofstead, E.A.; Judy, W.A. J. Polym. Sci. Part A-1 1967, 5, 2209; Wasserman, E.; Ben-Efraim, D.A.; Wolovsky, R. J. Am. Chem. Soc. 1968, 90, 3286; Wolovsky, R.; Nir, Z. Synthesis 1972, 134.
882. Rossi, R.; Diversi, P.; Lucherini, A.; Porri, L. Tetrahedron Lett. 1974, 879; Lal, J.; Smith, R.R. J. Org. Chem. 1975, 40, 775.
883. See Weissman, H.; Plunkett, K.N.; Moore, J.S. Angew. Chem. Int. Ed. 2006, 45, 585. For reviews, see Tamao, K.; Kobayashi, K.; Ito, Y. Synlett 1992, 539; Fürstner, A.; Davies, P.W. Chem. Commun. 2005, 2307.
884. Couteliera, O.; Mortreux, A. Adv. Synth. Catal. 2006, 348, 2038; Mortreux, A.; Petit, F.; Petit, M.; Szymanska-Buza, T. J. Mol. Catal. A: Chemical 1995, 96, 95. However, see McCullough, L.G.; Listemann, M.L.; Schrock, R.R.; Churchill, M.R.; Ziller, J.W. J. Am. Chem. Soc. 1983, 105, 6729.
885. See Mori, M.; Kitamura, T.; Sakakibara, N.; Sato, Y. Org. Lett. 2000, 2, 543; Kitamura, T.; Mori, M. Org. Lett. 2001, 3, 1161.
886. See Gilbertson, S.R.; Hoge, G.S. Tetrahedron Lett. 1998, 39, 2075.
887. Zuercher, W.J.; Scholl, M.; Grubbs, R.H. J. Org. Chem. 1998, 63, 4291.
888. See Kang, B.; Lee, J.M.; Kwak, J.; Lee, Y.S.; Chang, S. J. Org. Chem. 2004, 69, 7661. See Diver, S.T.; Giessert, A.J. Chem. Rev. 2004, 104, 1317.
889. Poulsen, C.S.; Madsen, R. Synthesis 2003, 1. See Lee, H.-Y.; Kim, B.G.; Snapper, M.L. Org. Lett. 2003, 5, 1855; Giessert, A.J.; Brazis, N.J.; Diver, S.T. Org. Lett. 2003, 5, 3819; Kim, M.; Park, S.; Maifeld, S.V.; Lee, D. J. Am. Chem. Soc. 2004, 126, 10242. See also, Kang, B.; Kim, D.-h.; Do, Y.; Chang, S. Org. Lett. 2003, 5, 3041.
890. Hansen, E.C.; Lee, D. Acc. Chem. Res. 2006, 39, 509; Debleds, O.; Campagne, J.-M. J. Am. Chem. Soc. 2008, 130, 1562; Giessert, A.J.; Diver, S.T. J. Org. Chem. 2005, 70, 1046. For a mechanistic study, see Galan, B.R.; Giessert, A.J.; Keister, J.B.; Diver, S.T. J. Am. Chem. Soc. 2005, 127, 5762.
891. See Sanford, M.S.; Ulman, M.; Grubbs, R.H. J. Am. Chem. Soc. 2001, 123, 749; Sanford, M.S.; Love, J.A.; Grubbs, R.H. J. Am. Chem. Soc. 2001, 123, 6543; Cavallo, L. J. Am. Chem. Soc. 2002, 124, 8965; Adlhart, C.; Chen, P. J. Am. Chem. Soc. 2004, 126, 3496.
892. See Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, Wiley, NY, 1988, pp. 244–267; Kingsbury, J.S.; Hoveyda, A.H. J. Am. Chem. Soc. 2005, 127, 4510; Poater, A.; Solans-Monfort, X.; Clot, E.; Copéret, C.; Eisenstein, O. J. Am. Chem. Soc. 2007, 129, 8207; Vyboishchikov, S.F.; Thiel, W. Chemistry: European J. 2005, 11, 3921.
893. See Collman, J.C.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry, 2nd ed., University Science Books, Mill Valley, CA, 1987, pp. 459–520; Lindner, E. Adv. Heterocycl. Chem. 1986, 39, 237. See Romero, P.E.; Piers, W.E. J. Am. Chem. Soc. 2005, 127, 5032; Romero, P.E.; Piers, W.E. J. Am. Chem. Soc. 2007, 129, 1698.
894. See Grubbs, R.H. Prog. Inorg. Chem. 1978, 24, 1; Katz, T.J. Adv. Organomet. Chem. 1977, 16, 283; Calderon N.; Ofstead, E.A.; Judy, W.A. Angew. Chem. Int. Ed. 1976, 15, 401. See also, Kress, J.; Osborn, J.A.; Greene, R.M.E.; Ivin, K.J.; Rooney, J.J. J. Am. Chem. Soc. 1987, 109, 899; Feldman, J.; Davis, W.M.; Schrock, R.R. Organometallics 1989, 8, 2266.
895. Hong, S.H.; Wenzel, A.G.; Salguero, T.T.; Day, M.W.; Grubbs, R.H. J. Am. Chem. Soc. 2007, 129, 7961.
896. Halpern, J. in Wender, I.; Pino, P. Organic Syntheses via Metal Carbonyls, Vol. 2, Wiley, NY, 1977, pp. 705–721; Bishop, III, K.C. Chem. Rev. 1976, 76, 461; Cardin, D.J.; Cetinkaya, B.; Doyle, M.J.; Lappert, M.F. Chem. Soc. Rev. 1973, 2, 99, pp. 132–139; Paquette, L.A. Synthesis 1975, 347; Acc. Chem. Res. 1971, 4, 280.
897. Eaton, P.E.; Chakraborty, U.R. J. Am. Chem. Soc. 1978, 100, 3634.
898. Cassar, L.; Eaton, P.E.; Halpern, J. J. Am. Chem. Soc. 1970, 92, 6336.
899. Sakai, M.; Westberg, H.H.; Yamaguchi, H.; Masamune, S. J. Am. Chem. Soc. 1972, 93, 4611; Paquette, L.A.; Wilson, S.E.; Henzel, R.P. J. Am. Chem. Soc. 1972, 94, 7771.
900. The starting compound here is a derivative of basketane, or 1,8-bishomocubane. For a review of homo-, bishomo-, and trishomocubanes, see Marchand, A.P. Chem. Rev. 1989, 89, 1011.
901. See Paquette, L.A.; Beckley, R.S.; Farnham, W.B. J. Am. Chem. Soc. 1975, 97, 1089.
902. See Sakai, M.; Westberg, H.H.; Yamaguchi, H.; Masamune, S. J. Am. Chem. Soc. 1972, 93, 4611.
903. See Paquette, L.A.; Zon, G. J. Am. Chem. Soc. 1974, 96, 203, 224.
904. Paquette, L.A.; Henzel, R.P.; Wilson, S.E. J. Am. Chem. Soc. 1971, 93, 2335.
905. Gassman, P.G.; Meyer, R.G.; Williams, F.J. Chem. Commun. 1971, 842.
906. Garavelli, M.; Frabboni, B.; Fato, M.; Celani, P.; Bernardi, F.; Robb, M.A.; Olivucci, M. J. Am. Chem. Soc. 1999, 121, 1537.
907. Zimmerman, H.E.; Pincock, J.A. J. Am. Chem. Soc. 1973, 95, 2957.
908. Zimmerman, H.E. Org. Photochem. 1991, 11, 1; Zimmerman, H.E. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 3, Academic Press, NY, 1980, pp. 131–166; Hixson, S.S.; Mariano, P.S.; Zimmerman, H.E. Chem. Rev. 1973, 73, 531. See also, Roth, W.R.; Wildt, H.; Schlemenat, A. Eur. J. Org. Chem.2001, 4081.
909. Zimmerman, H.E.; Hackett, P.; Juers, D.F.; McCall, J.M.; Schröder, B. J. Am. Chem. Soc. 1971, 93, 3653.
910. However, some substrates, generally rigid bicyclic molecules, (e.g., barrelene, which is converted to semi-bullvalene) give the di-π-methane rearrangement only from triplet states.
911. Zimmerman, H.E.; Baum, A.A. J. Am. Chem. Soc. 1971, 93, 3646. See also, Paquette, L.A.; Bay, E.; Ku, A.Y.; Rondan, N.G.; Houk, K.N. J. Org. Chem. 1982, 47, 422.
912. See Zimmerman, H.E.; Boettcher, R.J.; Buehler, N.E.; Keck, G.E. J. Am. Chem. Soc. 1975, 97, 5635. However, see Adam, W.; De Lucchi, O.; Dörr, M. J. Am. Chem. Soc. 1989, 111, 5209.
913. Zimmerman, H.E.; Robbins, J.D.; McKelvey, R.D.; Samuel, C.J.; Sousa, L.R. J. Am. Chem. Soc. 1989, 111, 5209.
914. See Paquette, L.A.; Bay, E. J. Am. Chem. Soc. 1984, 106, 6693; Zimmerman, H.E.; Swafford, R.L. J. Org. Chem. 1984, 49, 3069.
915. See Schuster, D.I. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 3, Academic Press, NY, 1980, pp. 167–279; Houk, K.N. Chem. Rev. 1976, 76, 1; Schaffner, K. Tetrahedron 1976, 32, 641; Dauben, W.G.; Lodder, G.; Ipaktschi, J. Top. Curr. Chem. 1975, 54, 73.
916. For a review, see Demuth, M. Org. Photochem. 1991, 11, 37.
917. See Armesto, D.; Horspool, W.M.; Langa, F.; Ramos, A. J. Chem. Soc. Perkin Trans. 1 1991, 223.
918. See Griffin, G.W.; Chihal, D.M.; Perreten, J.; Bhacca, N.S. J. Org. Chem. 1976, 41, 3931.
919. See Schaffner, K.; Demuth, M. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 3, Academic Press, NY, 1980, pp. 281–348; Zimmerman, H.E. Angew. Chem. Int. Ed. 1969, 8, 1; Kropp, P.J. Org. Photochem.1967, 1, 1; Schaffner, K. Adv. Photochem. 1966, 4, 81; Schultz, A.G.; Lavieri, F.P.; Macielag, M.; Plummer, M. J. Am. Chem. Soc. 1987, 109, 3991, and references cited therein.
920. Schuster, D.I. Acc. Chem. Res. 1978, 11, 65; Zimmerman, H.E.; Pasteris, R.J. J. Org. Chem. 1980, 45, 4864, 4876; Schuster, D.I.; Liu, K. Tetrahedron 1981, 37, 3329.
921. Zimmerman, H.E.; Crumine, D.S.; Döpp, D.; Huyffer, P.S. J. Am. Chem. Soc. 1969, 91, 434.
922. Schaffner, K.; Demuth, M. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 3, Academic Press, NY, 1980, p. 281; Quinkert, G. Angew. Chem. Int. Ed. 1972, 11, 1072; Kropp, P.J. Org. Photochem. 1967, 1, 1.
923. See Carruthers, W. Some Modern Methods of Organic Synthesis 3rd ed., Cambridge University Press, Cambridge, 1986, pp. 263–279.
924. See Stella, L. Angew. Chem. Int. Ed. 1983, 22, 337; Sosnovsky, G.; Rawlinson, D.J. Adv. Free Radical Chem. 1972, 4, 203, see pp. 249–259; Deno, N.C. Methods Free-Radical Chem. 1972, 3, 135, see pp. 136–143.
925. Schmitz, E.; Murawski, D. Chem. Ber. 1966, 99, 1493.
926. Wawzonek, S.; Thelan, P.J. J. Am. Chem. Soc. 1950, 72, 2118.
927. Barton, D.H.R.; Beckwith, A.L.J.; Goosen, A. J. Chem. Soc. 1965, 181; Neale, R.S.; Marcus, N.L.; Schepers, R.G. J. Am. Chem. Soc. 1966, 88, 3051. See Neale, R.S. Synthesis 1971, 1.
928. SeeHesse, R.H. Adv. Free-Radical Chem. 1969, 3, 83; Barton, D.H.R. Pure Appl. Chem. 1968, 16, 1; Saraiva, M.F.; Couri, M.R.C.; Le Hyaric, M.; de Almeida, M.V. Tetrahedron 2009, 65, 3563.
929. Barton, D.H.R.; Beaton, J.M. J. Am. Chem. Soc. 1961, 83, 4083. Also see, Barton, D.H.R.; Beaton, J.M.; Geller, L.E.; Pechet, M.M. J. Am. Chem. Soc. 1960, 82, 2640.
930. See Burke, S.D.; Silks III, L.A.; Strickland, S.M.S. Tetrahedron Lett. 1988, 29, 2761.
931. See Green, M.M.; Boyle, B.A.; Vairamani, M.; Mukhopadhyay, T.; Saunders Jr., W.H.; Bowen, P.; Allinger, N.L. J. Am. Chem. Soc. 1986, 108, 2381; Grossi, L. Chemistry: European J. 2005, 11, 5419.
932. See Nickon, A.; Ferguson, R.; Bosch, A.; Iwadare, T. J. Am. Chem. Soc. 1977, 99, 4518.
933. Cope, A.C.; Fournier, Jr., A.; Simmons Jr., H.E. J. Am. Chem. Soc. 1957, 79, 3905.
934. Schulenberg, J.W.; Archer, S. Org. React. 1965, 14, 1; McCarty, C.G. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 439–447; McCarty, C.G.; Garner, L.A. in Patai, S. The Chemistry of Amidines and Imidates, Wiley, NY, 1975, pp. 189–240. For a review of 1,3-migrations of R in general, see Landis, P.S. Mech. Mol. Migr. 1969, 2, 43.
935. Wheeler, O.H.; Roman, F.; Santiago, M.V.; Quiles, F. Can. J. Chem. 1969, 47, 503.
936. See Wheeler, O.H.; Roman, F.; Rosado, O. J. Org. Chem. 1969, 34, 966; Kimura, M. J. Chem. Soc. Perkin Trans. 2 1987, 205.
937. See Bonnett, R. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 597–662.
938. Landis, P.S. Mech. Mol. Migr. 1969, 2, 43.
939. See Challis, B.C.; Frenkel, A.D. J. Chem. Soc. Perkin Trans. 2 1978, 192.
940. See Yamamoto, J.; Nishigaki, Y.; Umezu, M.; Matsuura, T. Tetrahedron 1980, 36, 3177.
941. See Buncel, E. Mech. Mol. Migr. 1968, 1, 61; Shine, H.J. Aromatic Rearrangements, Elsevier, NY, 1969, pp. 272–284, 357–359; Cox, R.A.; Buncel, E. in Patai, S. The Chemistry of the Hydrazo, Azo, and Azoxy Groups, pt. 2, Wiley, NY, 1975, pp. 808–837.
942. See Shimao, I.; Oae, S. Bull. Chem. Soc. Jpn. 1983, 56, 643.
943. Furin, G.G. Russ. Chem. Rev. 1987, 56, 532; Williams, D.L.H.; Buncel, E. Isot. Org. Chem. 1980, 5, 184; Buncel, E. Acc. Chem. Res. 1975, 8, 132.
944. See Oae, S.; Fukumoto, T.; Yamagami, M. Bull. Chem. Soc. Jpn. 1963, 36, 601.
945. Shemyakin, M.M.; Maimind, V.I.; Vaichunaite, B.K. Chem. Ind. (London) 1958, 755; Bull. Acad. Sci. USSR Div. Chem. Sci. 1960, 808. Also see, Behr, L.C.; Hendley, E.C. J. Org. Chem. 1966, 31, 2715.
946. See Cox, R.A. J. Am. Chem. Soc. 1974, 96, 1059.
947. See Buncel, E.; Keum, S. J. Chem. Soc., Chem. Commun. 1983, 578.
948. Also see Shemyakin, M.M.; Agadzhanyan, Ts.E.; Maimind, V.I.; Kudryavtsev, R.V. Bull. Acad. Sci. USSR Div. Chem. Sci. 1963, 1216; Hendley, E.C.; Duffey, D. J. Org. Chem. 1970, 35, 3579.
949. Cox, R.A.; Dolenko, A.; Buncel, E. J. Chem. Soc. Perkin Trans. 2 1975, 471; Cox, R.A.; Buncel, E. J. Am. Chem. Soc. 1975, 97, 1871.
950. See Shimao, I.; Hashidzume, H. Bull. Chem. Soc. Jpn. 1976, 49, 754.
951. See Shine, H.J.; Subotkowski, W.; Gruszecka, E. Can. J. Chem. 1986, 64, 1108.
952. See Davis, R.L.; Tantillo, D.J. J. Org. Chem. 2010, 75, 1693.
953. Minkin, V.I.; Olekhnovich, L.P.; Zhdanov, Yu.A. Molecular Design of Tautomeric Compounds, D. Reidel Publishing Co., Dordrecht, 1988, pp. 221–246; Minkin, V.I. Sov. Sci. Rev. Sect. B 1985, 7, 51; Reetz, M.T. Adv. Organomet. Chem. 1977, 16, 33. Also see, Mackenzie, K.; Gravaatt, E.C.; Gregory, R.J.; Howard, J.A.K.; Maher, J.P. Tetrahedron Lett. 1992, 33, 5629.
954. See Taylor, G.A. J. Chem. Soc. Perkin Trans. 1 1985, 1181.
955. See Black, T.H.; Hall, J.A.; Sheu, R.G. J. Org. Chem. 1988, 53, 2371; Black, T.H.; Fields, J.D. Synth. Commun. 1988, 18, 125.
956. See Mackenzie, K.; Proctor, G.; Woodnutt, D.J. Tetrahedron 1987, 43, 5981, and cited references.
957. For a review, see Moser, W.H. Tetrahedron 2001, 57, 2065.
958. Brook, A.G. Acc. Chem. Res. 1974, 7, 77; Brook, A.G.; Bassendale, A.R. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 2, Academic Press, NY, 1980, pp. 149–227.
959. Brook, A. G.; Pascoe, J. D. J. Am. Chem. Soc. 197193, 6224.
960. See Linderman, J.J.; Ghannam, A. J. Am. Chem. Soc. 1990, 112, 2392.
961. Wilson, S.R.; Georgiadis, G.M. J. Org. Chem. 1983, 48, 4143.
962. Honda, T.; Mori, M. J. Org. Chem. 1996, 61, 1196.
963. See Smith, III, A.B.; Adams, C. M. Acc. Chem. Res. 2004, 37, 365; Smith, III, A.B.; Kim, D.-S. Org. Lett. 2005, 7, 3247.
964. Sawada, Y.; Sasaki, M.; Takeda, K. Org. Lett. 2004, 6, 2277.
965. Smith, III, A.B.; Pitram, S.M.; Boldi, A.M.; Gaunt, M.J.; Sfouggatakis, C.; Moser, W.H. J. Am. Chem. Soc. 2003, 125, 14435. See Smith, III, A.B.; Xian, M. J. Am. Chem. Soc. 2006, 128, 66.
966. Smith, III, A.B.; Xian, M. J. Am. Chem. Soc. 2006, 128, 66.