March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 19. Oxidations and Reductions
19.B. Reactions
In this chapter, the reactions are classified by the type of bond change occurring to the organic substrate, in conformity with other chapters.9 This means that there is no discussion in any one place of the use of a particular oxidizing or reducing agent (e.g., acid dichromate or LiAlH4; except for a discussion of selectivity of reducing agents Sec. 19.B.ii, A). Some oxidizing or reducing agents are fairly specific in their action, attacking only one or a few types of substrate. Others, like acid dichromate, permanganate, LiAlH4, and catalytic hydrogenation, are much more versatile.10,11
19.B.i. Oxidations11
In some cases, oxidations have been placed in another chapter. The oxidations of an alkene to a diol (Reaction 15-48), and an aromatic compound to a diol (Reaction 15-49), or oxidations to an epoxide (Reaction 15-50) are placed in Chapter 15, for consistency with the concept of addition to a π bond. Diamination of an alkene (Reaction 15-53) and formation of aziridines (Reaction 15-54) are in Chapter 15 for the same reason. Most other oxidations have been placed here. The reactions in this section are classified into groups depending on the type of bond change involved. These groups are Section (A) eliminations of hydrogen, Section (B) oxidations involving cleavage of carbon–carbon bonds, Section (C) reactions involving replacement of hydrogen by oxygen, Section (D) reactions in which oxygen is added to the substrate, and Section (E) oxidative coupling.
A. Eliminations of Hydrogen
19-1 Aromatization of Six-Membered Rings
Hexahydro-terelimination
![]()
Six-membered alicyclic rings can be aromatized in a number of ways.12 Aromatization is accomplished most easily if there are already one or two double bonds in the ring or if the ring is fused to an aromatic ring. The reaction can also be applied to heterocyclic five- and six-membered rings. Many groups may be present on the ring without interference, and even gem-dialkyl substitution does not always prevent the reaction: In such cases, one alkyl group often migrates or is eliminated, but more drastic conditions are usually required for this. In some cases, OH and COOH groups are lost from the ring. Cyclic ketones are converted to phenols. Seven-membered and larger rings are often isomerized to six-membered aromatic rings, although this is not the case for partially hydrogenated azulene systems, which are frequently found in nature; these are converted to azulenes.
There are three types of reagents most frequently used to effect aromatization.
1. Hydrogenation catalysts13 (e.g., Pt, Pd,14 and Ni). Palladium trifluoroacetate also facilitates oxidative aromatization of cyclohexene.15 In this case, the reaction is the reverse of double-bond hydrogenation (15-11 and 15-15), and presumably the mechanism is also the reverse, although not much is known.16 Cyclohexene has been detected as an intermediate in the conversion of cyclohexane to benzene, using Pt.17 The substrate is heated with the catalyst at ~300–350 °C. The reactions can often be carried out under milder conditions if a hydrogen acceptor (e.g., maleic acid, cyclohexene, or benzene) is present to remove hydrogen as it is formed. The acceptor is reduced to the saturated compound. Other transition metals can be used.18 It has been reported that dehydrogenation of 1-methylcyclohexene-1-13C over an alumina catalyst gave toluene with the label partially scrambled throughout the aromatic ring.19 For polycyclic systems, heating with oxygen on activated carbon generates the aromatic compound, as in the conversion of dihydroanthracene to anthracene.20
2. The elements sulfur and selenium, which combine with the hydrogen evolved to give, respectively, H2S and H2Se. Little is known about this mechanism either.21
3. Quinones22 become reduced to the corresponding hydroquinones. Two important quinones often used for aromatizations are chloranil (2,3,5,6-tetrachloro-1,4-benzoquinone) and DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone).23 The latter is more reactive and can be used in cases where the substrate is difficult to dehydrogenate. It is likely that the mechanism involves a transfer of hydride to the quinone oxygen, followed by the transfer of a proton to the phenolate ion.24
Other reagents25 have been used for aromatization of six-membered rings, including atmospheric oxygen, MnO2,26 SeO2, H2SO4, and a Ru catalyst.27 The last-mentioned reagent also dehydrogenates cyclopentanes to cyclopentadienes. In some instances, the hydrogen is not released as H2 or transferred to an external oxidizing agent, but instead serves to reduce another molecule of substrate. This is a disproportionation reaction and can be illustrated by the conversion of cyclohexene to cyclohexane and benzene.
Heteroatom rings, as found in quinoline derivatives, for example, can be generated from amino-ketones with [hydroxy(tosyloxy)iodo]benzene and perchloric acid28 or with NaHSO4–Na2Cr2O7 on wet silica.29Dihydropyridines are converted to pyridines with NaNO2–oxalic acid and wet silica,30 BaMnO4,31 FeCl3–acetic acid,32 or SeO2.33Hantzsch 1,4-dihydropyridines (see Reactions 15-14 and 16-17) are aromatized by treatment with ferric perchlorate in acetic acid.34 Cyclic imines are converted to pyridine derivatives with NCS and then excess sodium methoxide.35 Enamines are aromatized with Sn or Sb compounds.36
Note that hydrogenolysis of cyclohexane leads to n-hexane with hydrogen and an Ir catalyst.37
OS II, 214, 423; III, 310, 358, 729, 807; IV, 536; VI, 731. Also see, OS III, 329.
19-2 Dehydrogenations Yielding Carbon–Carbon Double Bonds
Dihydro-elimination
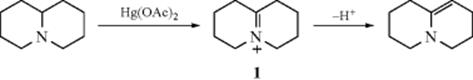
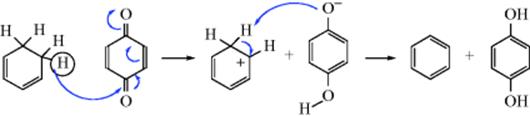
Dehydrogenation of an aliphatic compound to give a double bond in a specific location is not usually a feasible process, although industrially mixtures of alkenes are obtained in this way from mixtures of alkanes (generally by heating with chromia–alumina catalysts). There are, however, some notable exceptions. Heating cyclooctane with an Ir catalyst leads to cyclooctene.38 Treating alkenes that have an allylic hydrogen with CrCl2 converts them to allenes.39 It is not surprising, however, that most of the exceptions generally involve cases where the new double bond can be in conjugation with a double bond or with an unshared pair of electrons already present.40 One example is the synthesis developed by Leonard and Musker,41 in which tertiary amines give enamines (Reaction 10-69) when treated with mercuric acetate42 (see the example above). In this case, the initial product is the iminium ion (1) that loses a proton to give the enamine. Other transition metal catalysts convert amines to enamines, including Co compounds.43Hünig's base (diisopropylethylamine) was converted to the enamine N,N-diisopropyl-N-vinylamine by heating with an Ir catalyst.44
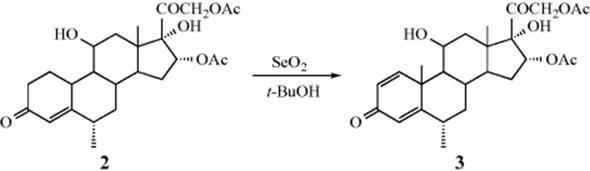
The oxidizing agent SeO2 can in certain cases convert a carbonyl compound to an α,β-unsaturated carbonyl compound by removing H245 (note that this reagent more often gives Reaction 19-17). This reaction has been most often applied in the steroid series, an example being formation of 2 from 3.46 Similarly, SeO2 has been used to dehydrogenate 1,4-diketones47 and 1,2-diarylalkanes. These conversions can also be carried out by certain quinones, most notably DDQ (see Reaction 19-1).24 Molecular oxygen has been used to convert cyclic ketones to the conjugated ketone in the presence of a Pd catalyst.48
Simple aldehydes and ketones have been dehydrogenated (e.g., cyclopentanone → cyclopentenone) by PdCl2,49 by FeCl3,50 and by benzeneseleninic anhydride51 (this reagent also dehydrogenates lactones in a similar manner), among other reagents. In an indirect method of achieving this conversion, the silyl enol ether of a simple ketone is treated with DDQ52 or with triphenylmethyl cation53 (for another indirect method, see Reaction 17-12). Silyl enol ethers give the conjugated ketone upon treatment with ceric ammonium nitrate in DMF54 or with Pd(OAc)2/NaOAc/O2.55
Simple linear alkanes have been converted to alkenes by treatment with certain transition metal compounds.56
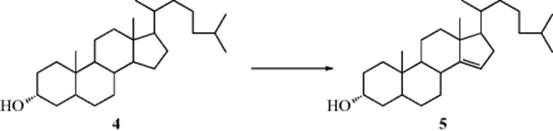
An entirely different approach (remote functionalization) allows specific dehydrogenation, as reported by R. Breslow57 and by J.E. Baldwin et al.58 3α-Cholestanol (4) was converted to 5α-cholest-14-en-3α-ol (5), for example, thus introducing a double bond at a specific site remote from any functional group.59
Certain 1,2-diarylalkenes (ArCH=CHAr′) have been converted to the corresponding alkynes (ArC![]() CAr′) by treatment with t-BuOK in DMF.60 Dihydroindoles are converted to indoles with N,N′,N″-trichloro-1,3,5-triazin-2,4,6-trione and DBU.61
CAr′) by treatment with t-BuOK in DMF.60 Dihydroindoles are converted to indoles with N,N′,N″-trichloro-1,3,5-triazin-2,4,6-trione and DBU.61
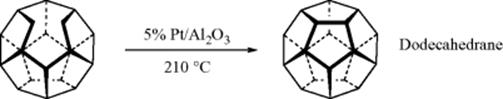
A different kind of dehydrogenation was used in the final step of Paquette's synthesis of dodecahedrane62:
OS V, 428, VII, 4, 473.
19-3 Oxidation or Dehydrogenation of Alcohols to Aldehydes and Ketones
C,O-Dihydro-elimination
![]()
Primary alcohols can be converted to aldehydes and secondary alcohols to ketones in seven main ways:63
1. With Chromium Reagents.64 Secondary alcohols are easily oxidized to ketones by dichromate in acidic media65 at room temperature or slightly above. A solution of chromic and sulfuric acid in water is known as the Jones reagent.66 Secondary alcohols are oxidized to ketones rapidly and in high yield without disturbing any double or triple bonds that may be present (see Reaction 19-10) and without epimerizing an adjacent stereogenic center.67Mixing sodium dichromate with an alcohol, without solvent, provides a method for oxidation when the mixture is shaken.68 Chromium trioxide (CrO3)69 has been used to oxidize primary and alcohols under solvent-free conditions. Chromium trioxide on silica gel, in supercritical CO2, oxidizes alcohols to the corresponding carbonyl.70 For acid-sensitive compounds, trimethylsilyl chromates71 can be used. Chromium trioxide with aq tert-butylhydroperoxide oxidizes benzylic alcohols with microwave irradiation.72 Phase-transfer catalysis is particularly useful,73 especially when the substrates are generally insoluble in water (see Sec. 10.G.v). A catalytic amount of Cr(acac)3 in conjunction with H5IO5 oxidizes benzylic alcohols to aldehydes.74
The Jones reagent can also oxidize primary allylic alcohols to the corresponding aldehydes,75 although overoxidation to the carboxylic acid is a problem.76 Oxidative cleavage of primary alcohols has been observed in the presence of molecular sieves 3 Å.77 One way to mitigate overoxidation is to distil the aldehyde as it is formed, but this is not always possible. Due to these problems, other oxidizing conditions have been used to convert at least some primary alcohols to aldehydes.78 Perhaps the three most commonly used Cr(VI) reagents used for the oxidation of allylic alcohols include79 dipyridine Cr(VI) oxide (Collins' reagent),80 pyridinium chlorochromate (PCC),81 and pyridinium dichromate (PDC).82 The PCC is somewhat acidic, and acid-catalyzed rearrangements have been observed.83
Analogous to the use of pyridine for PCC and PDC, a variety of amines and diamines have been converted to tetraalkylammonium halochromates or dichromates, including N-benzyl 1,4-diazabicyclo[2.2.2]octane ammonium dichromate with microwave irradiation,84 γ-picolinium chlorochromate,85 and quinolinium fluorochromate.86 Benzyltriphenylphosphonium chlorochromate has been used in a similar manner.87 Oxidizing agents have been supported on a polymer,88 including chromic acid,89 as well as poly[vinyl(pyridinium fluorochromate)].90 Triphenylmethylphosphonium dichromate is effective for selective oxidation of benzylic alcohols.91
Studies on the mechanism of oxidation with acid dichromate92 led to the currently accepted mechanism as proposed by Westheimer93 (cf. the first two steps with Sec. 19.A, category 4).
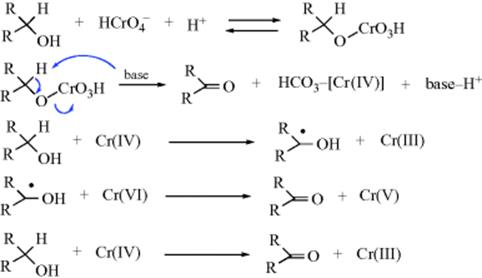
The base in the second step may be water, although it is also possible94 that in some cases no external base is involved and that the proton is transferred directly to one of the CrO3H oxygen atoms in which case the Cr(IV) species produced would be H2CrO3. Part of the evidence for this mechanism was the isotope effect of ~6 found on use of MeCDOHMe, showing that the α hydrogen is removed in the rate-determining step.95 Note that, as in Reaction 19-23 the substrate is oxidized by three different oxidation states of chromium.96
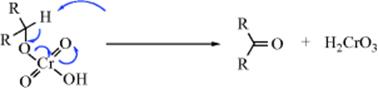
With other oxidizing agents discussed below, mechanisms are less clear.97 It seems certain that some oxidizing agents operate by a hydride-shift mechanism,98 for example, dehydrogenation with triphenylmethyl cation99 and the Oppenauer oxidation, and some by a free radical mechanism (e.g., oxidation with S3O82−100 and with VO2+101). A summary of many proposed mechanisms is given by Littler.102
2. With Manganese and Other Metal Oxidizing Agents. Potassium permanganate (KMnO4) has been used for the oxidation of alcohols.103 Benzylic and allylic alcohols have been selectively oxidized to the aldehydes in the presence of saturated alcohols by the use of potassium permanganate (KMnO4) under phase-transfer conditions.104 Phase-transfer catalysis has also been used with chromic acid,105 and ruthenium tetroxide.106 Ultrasound has been used for KMnO4 oxidations.107 Permanganate supported on a polymer has been used.108
Permanganate109 is an important reagent for the selective oxidation of benzylic alcohols primary and benzylic alcohols, in preference to aliphatic substrates. 110A variation oxidizes alcohols with MnO2/AlCl3.111
An alternative to MnO2 is the oxidation of allylic and benzylic alcohols with Me3NO in the presence of CHDFe(CO)3.112 Similar oxidation occurs with NaBrO3 in aq MeCN113 or K2FeO4 on clay.114 The reaction of AuCl with an anionic ligand leads to oxidation of primary alcohols to aldehydes.115 The Grubbs' catalyst, PhCH=Ru(PCy3)2Cl2, where Cy = cyclohetyl, (see 156 in Reaction 18-37), in the presence of KOH, oxidized alcohols.116
Tetrapropylammonium perruthenate (Pr4N+ RuO4−; also called TPAP; the Ley reagent)117 is an important oxidizing agent that is compatible with the presence of other functionality in the molecule.118 In the presence of molecular oxygen, oxidation of alcohols is catalytic in TPAP.119 This reagent has been bound to a polymer.120 Methods have been developed for recovery of the catalyst and reuse of TPAP.121
Many other oxidizing agents have been employed. Examples include ruthenium tetroxide,122 MeReO3,123 HNO3 with a Yb(OTf)3 catalyst,124 FeBr3–H2O2,125 ceric ammonium nitrate in an ionic liquid,126 a Bi catalyst,127 O2with transition metal catalysts,128 and with RuO2 and a zeolite catalyst.129 Microwave induced oxidation of benzylic alcohols was reported using zeolite-supported ferric nitrate.130
Reagents that can be used specifically to oxidize a secondary OH group in the presence of a primary OH group131 are H2O2–ammonium molybdate,132 or urea–H2O2 with MgBr2,133 while RuCl2(PPh3)3–benzene,134 osmium tetroxide,135 and Br2–Ni(OBz)2136 all oxidize primary OH groups in the presence of a secondary OH group.137 Certain zirconocene complexes can selectively oxidize only one OH group of a diol, even if both are primary.138 α-Hydroxy ketones are oxidized to 1,2-diketones with Bi(NO3)3 and a Cu(OAc)2 catalyst,139 ferric chloride (solid state),140 or O2 and a V catalyst.141 1,2-Diols are oxidized to chiral α-hydroxy-ketones using NBS with a chiral Cu catalyst.142
3. The Oppenauer Oxidation. When a ketone in the presence of an aluminum alkoxide is used as the oxidizing agent (it is reduced to a secondary alcohol), the reaction is known as the Oppenauer oxidation.143 This is the reverse of the Meerwein–Ponndorf–Verley reaction (19-36) and the mechanism is also the reverse. The ketones most commonly used are acetone, butanone, and cyclohexanone. A common base is aluminum tert-butoxide. The chief advantage of the method is its high selectivity. Although the method is most often used for the preparation of ketones, it has also been used for aldehydes. An Ir catalyst144 has been developed for the Oppenauer oxidation, and also a water-soluble Ir catalyst.145 Homogeneous water-soluble complexes catalyze the reaction.146 An uncatalyzed reaction under supercritical conditions was reported.147
4. DMSO Based Reagents. The use of oxalyl chloride and DMSO at low temperature is called the Swern oxidation148 and is widely used. A sulfonium salt is produced in situ, which reacts with the alcohol to generate the key intermediate required for oxidation.149 Maintaining the low-reaction temperature is essential in this reaction, however, since the reagent generated in situ decomposes at temperatures significantly below ambient. Note that Swern oxidation of molecules having alcohol moieties, as well as a disulfide, leads to the ketone without oxidation of the sulfur.150 Sulfoxides other than DMSO can be used in conjunction with oxalyl chloride for the oxidation of alcohols,151 including fluorinated sulfoxides152 and a polymer-bound sulfoxide.153
Similar oxidation of alcohols has been carried out with DMSO and other reagents154 in place of DCC: acetic anhydride,155 SO3–pyridine–triethylamine,156 trifluoroacetic anhydride,157 pivaloyl chloride,158 tosyl chloride,159Ph3P+Br−,160 trimethylamine N-oxide,161 a Mo catalyst and O2,162 and methanesulfonic anhydride.517 Dimethyl sulfoxide in 48% HBr oxidizes benzylic alcohols to aryl aldehydes.163 An alcohol is treated with DMSO, DCC,164and anhydrous phosphoric acid165 in what is called Moffatt oxidation. In this way, a primary alcohol can be converted to the aldehyde with no carboxylic acid being produced. The strong acid conditions are sometimes a problem, and complete removal of the dicyclohexylurea byproduct can be difficult.
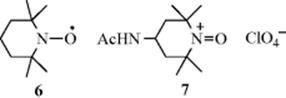
5. TEMPO and Related Reagents. The nitroxyl radical TEMPO (6) has been used in conjunction with coreagents, including mcpba,166 O2 with transition metal catalysts,167 O2 with HBr and tert-butylnitrite,168 CuBr2(bpy)–air (bpy = 2,2′-bipyridyl),169 CuBr·SMe2 in perfluorous solvents,170 bromohydantoins,171 enzymes,172 carbenes,173 NaNO2–HCl,174 NaIO4,175 and H5IO6.176 Silica-supported TEMPO,177 polymer-bound TEMPO,178 and PEG–TEMPO179 have been used. The TEMPO derived ionic liquids,180 or ionic liquid-supported TEMPO181 have been used for the oxidation of alcohols. The TEMPO compound has also been used with a polymer-bound hypervalent iodine reagent.182 A catalytic reaction using 5% TEMPO and 5% CuCl with O2 in an ionic liquid oxidizes benzylic alcohols to the corresponding aldehyde.183 Ion-supported TEMPO oxidation in water is possible.184
Other nitroxyl radical oxidizing agents are known.185 A related oxidizing agent is oxoammonium salt 7 (Bobbitt's reagent), a stable and nonhygroscopic salt that oxidizes primary and secondary alcohols in dichloromethane.186 The mechanism of oxidation for 7 has been examined.187
6. With Hypervalent Iodine Reagents.188 Treatment of 2-iodobenzoic acid with KBrO3 in H2SO4 and heating the resulting product to 100 °C with acetic anhydride and acetic acid189 gives hypervalent iodine reagent (8), the so-called Dess–Martin Periodinane.190 This reagent reacts with alcohols at ambient temperature to give the corresponding aldehyde or ketone.191 The reaction is accelerated by water192 and a water-soluble periodinane [o-iodoxybenzoic acid (9), IBX]193 has been prepared that oxidized allylic alcohols to conjugated aldehydes.194 2-Methyl-2-propanol has been used as a solvent.195 The reagent has an indefinite shelf-life in a sealed container, but hydrolysis occurs upon long-term exposure to atmospheric moisture. A note of CAUTION! The Dess–Martin reagent can be shock sensitive under some conditions and explode ~200 °C.196
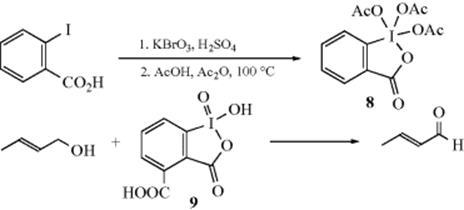
Iodine has been used as a cocatalyst.197 Other hypervalent iodine oxidizing reagents are known,198 including PhI(OAc)2/TEMPO,199 PhI(OAc)2–chromium salen,200 PhI(OAc)2 supported on alumina with microwave irradiation,201 and an ion-supported hypervalent iodine(III) reagent.202 Microwave irradiation of benzylic alcohols with PhI(OH)OTs gave the corresponding aldehyde.203 Hypervalent iodine compounds have been used in ionic liquids.204 Heating benzylic alcohols with o-iodoxybenzoic acid under solvent-free conditions gave the aldehyde.205 2-Iodobenzenesulfonic acid is a very active catalyst for oxidation of alcohols using Oxone.206
7. By Catalytic Dehydrogenation. For the conversion of primary alcohols to aldehydes, dehydrogenation catalysts have the advantage over strong oxidizing agents that further oxidation to the carboxylic acid is prevented. Copper chromite is often used, but other catalysts (e.g., Ag and Cu), have also been employed. Many ketones were prepared in this manner. Catalytic dehydrogenation is more often used industrially than as a laboratory method. However, procedures using Cu(II) complexes,207 Rh complexes,208 Ru complexes,209 Raney nickel,210 and Pd complexes211 (under phase transfer conditions)212 have been reported. Allylic alcohols213 are oxidized to the corresponding saturated aldehyde or ketone by heating with a Rh catalyst, and benzylic alcohols are converted to the aldehyde with a Rh catalyst.214 Propargylic alcohols are oxidized by heating with a V catalyst.215 Secondary alcohols are oxidized with Bi(NO3)3 on Montmorillonite.216 Biooxidation is possible as well via hydrogen transfer.217
8. Miscellaneous Reagents.218 Nitric acid in dichloromethane oxidizes benzylic alcohols to the corresponding ketone.219 Bromine is an effective oxidant, and iodine under photochemical conditions has been used.220 Heating a 1,2-diol with NBS in CCl4 gave the 1,2-diketone.221 Iodine has been used in conjunction with DMSO and hydrazine.222 Enzymatic oxidations have been reported.223 Oxidation of alcohols in water is possible using I2O5.224Dimethyl dioxirane225 oxidizes benzylic alcohols to the corresponding aldehyde,226 and dioxirane reagents are sufficiently mild that an α,β-epoxy alcohol was oxidized to the corresponding ketone, without disturbing the epoxide, using methyl trifluoromethyl dioxirane.227 Hydrogen peroxide with urea oxidizes aryl aldehydes in formic acid.228tert-Butylhydroperoxide with a Cu catalyst gives oxidation in an ionic liquid.229 Potassium monoperoxysulfate in the presence of a chiral ketone oxidizes 1,2-diols to α-hydroxy ketones enantioselectively.230 Potassium monoperoxysulfate also oxidizes secondary alcohols in the presence of O2.231 Air, in the presence of a zeolite oxidizes benzylic alcohols.232 Periodic acid oxidizes aldehydes or ketones in the presence of a PCC catalyst.233 Sodium hypochlorite in acetic acid234 or in water with β-cyclodextrin235 is a useful oxidizing agent. Calcium hypochlorite on moist alumina with microwave irradiation has been used to oxidize benzylic alcohols.236 Hydrogen bromide in aq H2O2 oxidizes secondary alcohols to ketones.237 With ultrasound, DDQ selectively oxidizes a benzylic or allylic hydroxyl group of 1,2-diols with those substituents.238 Photoxidation of alcohols is possible in the presence of a catalytic amount of NBS.239 A mixture of I2–KI–K2CO3–H2O oxidizes alcohols to aldehydes or ketones under anaerobic conditions.240 Similarly KBrO3/ZrClO2·8H2O can be used to oxidize alcohols.241 Oxone oxidizes alcohols, catalyzed by AlCl3.242
Tetrabutylammonium periodate243 and benzyltriphenylphosphonium periodate.244 oxidizes primary alcohols to aldehydes. On the other hand, Fremy's salt (see Reaction 19-4) selectively oxidizes benzylic alcohols and not allylic or saturated ones.245
In a related reaction to the oxidation of alcohols, it is possible to oxidize ethers to aldehydes. Oxidation of trimethylsilyl ethers with O2, a catalytic amount of N-hydroxyphthalimide and a Co catalyst, give an aldehyde.246Microwave irradiation with BiCl2 oxidizes benzylic TMS ethers to the aldehyde.247 Microwave irradiation on zeolite supported ferric nitrate has been used.248O-Tetrahydropyranyl ethers (O-THP) have been oxidized to the aldehyde with ferric nitrate on zeolites,249 and the Pd catalyzed oxidation of allylic esters to conjugated ketones is known.250N-Bromosuccinimide with β-cyclodextrin oxidizes tetrahydropyranyl ethers in water.251
OS I, 87, 211, 241, 340; II, 139, 541; III, 37, 207; IV, 189, 192, 195, 467, 813, 838; V, 242, 310, 324, 692, 852, 866; VI, 218, 220, 373, 644, 1033; VII, 102, 112, 114, 177, 258, 297; VIII, 43, 367, 386; IX, 132, 432. Also see, OS IV, 283; VIII, 363, 501.
19-4 Oxidation of Phenols and Aromatic Amines to Quinones
1/O,6/O-Dihydro-elimination
![]()
Ortho and para diols are easily oxidized to o- and p-quinones, respectively.252 Either or both OH groups can be replaced by NH2 groups to give the same products, although for the preparation of o-quinones only OH groups are normally satisfactory. The reaction has been successfully carried out with other groups para to OH or NH2; halogen, OR, Me, t-Bu, and even H, although yields are poor with the latter. Many oxidizing agents have been used: acid dichromate,253 silver oxide, silver carbonate, lead tetraacetate, HIO4, NBS–H2O–H2SO4,254 dimethyl dioxirane,255 and atmospheric oxygen.256 Oxidation has been done photochemically with O2 and tetraphenylporphine.257 A particularly effective reagent for rings with only one OH or NH2 group is (KSO3)2N–O· (dipotassium nitrosodisulfonate; Fremy's salt), which is a stable free radical.258 A mixture of 4-iodophenoxyacetic acid and Oxone is an effective catalyst for the oxidation of p-alkoxyphenols to p-quinones.259 A supported iron phthalocyanine facilitates the aromatic oxidation of phenols.260
Less is known about the mechanism than is the case for oxidizing simple alcohols in Reaction 19-3, and it seems to vary with the oxidizing agent. For oxidation of catechol with NaIO4, it was found that the reaction conducted in H218O gave unlabeled quinone.261 Therefore the following mechanism262 was proposed:
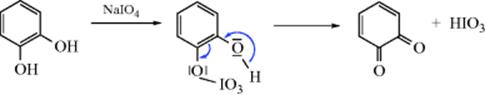
When catechol was oxidized with MnO4− under aprotic conditions, a semiquinone radical ion intermediate was involved.263 For autoxidations264 (i.e., with atmospheric O2) a free radical mechanism is known to operate.265
OS I, 383, 482, 511; II, 175, 254, 430, 553; III, 663, 753; IV, 148; VI, 412, 480, 1010.
19-5 Dehydrogenation of Amines to Nitriles or Imines
1/1/N,2/2/C-Tetrahydro-bielimination
![]()
Primary amines at a primary carbon can be dehydrogenated to nitriles. The reaction has been carried out with a variety of reagents, among others, I2 in aq NH3,266 IBX see 19-3, category 6),267 NaOCl,268 Me3N–O/OsO4,269Ru/Al2O3/O2,270 and CuCl/O2/pyridine.271 Iodine and 1,3-diiodo-5,5-dimethylhydantoin in aq ammonia converted both amines and alcohols to nitriles.272 Dehydrogenation of amines has been done in aq micelles.273
Several methods have been reported for the dehydrogenation of secondary amines to imines.274 Among them275 are treatment with (1) iodosylbenzene (PhIO) alone or in the presence of a Ru complex,276 (2) DMSO and oxalyl chloride,277 and (3) t-BuOOH and a Rh catalyst.278N-Tosyl aziridines are converted to N-tosyl imines when heated with a Pd catalyst.279 An interesting variation treats pyrrolidine with iodobenzene and a Rh catalyst to give 2-phenylpyrroline.280
A reaction that involves dehydrogenation to an imine that then reacts further is the reaction of primary or secondary amines281 with Pd black.282 The imine initially formed by the dehydrogenation reacts with another molecule of the same or a different amine to give an aminal, which loses NH3 or RNH2 to give a secondary or tertiary amine. An example is the reaction between N-methylbenzylamine and butylmethylamine, which produces 95% N-methyl-N-butylbenzylamine.
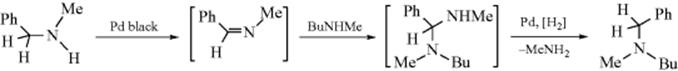
In a related reaction, alkyl azides react with BrF3 to give the corresponding nitrile.283
19-6 Oxidation of Hydrazines, Hydrazones, and Hydroxylamines
1/N,2/N-Dihydro-elimination
![]()
N,N′-Diarylhydrazines (hydrazo compounds) are oxidized to azo compounds by several oxidizing agents, including NaOBr, K3Fe(CN)6 under phase-transfer conditions284 FeCl3,285 MnO2 (this reagent yields cis azobenzenes),286CuCl2, and air and NaOH.287 The reaction is also applicable to N,N′-dialkyl- and N,N′-diacylhydrazines. Hydrazines (both alkyl and aryl) substituted on only one side also give azo compounds,288 but these are unstable and decompose to nitrogen and the hydrocarbon:
![]()
Aniline derivatives are converted to azo compounds by heating with cetyltrimethylammonium dichromate in chloroform.289 When hydrazones are oxidized with HgO, Ag2O, MnO2, PbO4, or certain other oxidizing agents, diazo compounds (R2C=N–NH2 → R2C=N+=N−) are obtained290:
Hydrazones of the form ArCH=NNH2 react with HgO in solvents, (e.g., diglyme or ethanol) to give nitriles (ArCN).291 It is possible to oxidize dimethylhydrazones (R–C=N–NMe2) to the corresponding nitrile (R–C![]() N) with magnesium monoperoxyphthalate (MMPP),292 or with dimethyl dioxirane.293 Oxone on wet alumina also converts hydrazones to nitriles with microwave irradiation.294 Oximes of aromatic aldehydes are converted to aryl nitriles with InCl3295 (ketoximes give a Beckmann rearrangement, Reaction 18-17).
N) with magnesium monoperoxyphthalate (MMPP),292 or with dimethyl dioxirane.293 Oxone on wet alumina also converts hydrazones to nitriles with microwave irradiation.294 Oximes of aromatic aldehydes are converted to aryl nitriles with InCl3295 (ketoximes give a Beckmann rearrangement, Reaction 18-17).
In a related reaction, primary aromatic amines have been oxidized to azo compounds by a variety of oxidizing agents, among them MnO2, lead tetraacetate, O2 and a base, BaMnO4,296 and sodium perborate in acetic acid. tert-Butyl hydroperoxide has been used to oxidize certain primary amines to azoxy compounds.297
Nitrones [C=N+(R)–O−] are generated by the oxidation of N-hydroxyl secondary amines with 5% aq NaOCl.298 Secondary amines (e.g., dibenzylamine) can be converted to the corresponding nitrone by heating with cumyl hydroperoxide in the presence of a titanium catalyst.299
OS II, 496; III, 351, 356, 375, 668; IV, 66, 411; V, 96, 160, 897; VI, 78, 161, 334, 392, 803, 936; VII, 56. Also see, OS V, 258. For oxidation of primary amines, see OS V, 341.
B. Oxidations Involving Cleavage of Carbon–Carbon Bonds300
19-7 Oxidative Cleavage of Glycols and Related Compounds
2/O-De-hydrogen-uncoupling
![]()
1,2-Diols (glycols) are easily cleaved under mild conditions and in good yield with periodic acid or lead tetraacetate.301 The reaction generates 2 molar equivalents of aldehyde, or 2 molar equivalents of ketone, or 1 molar equivalent of each, depending on the groups attached to the two carbons. The yields are so good that alkenes are often converted to diols (Reaction 15-48), and then cleaved with HIO4 or Pb(OAc)4 rather than being cleaved directly with ozone (Reaction 19-9) or dichromate or permanganate (Reaction 19-10). The diol can be generated and cleaved in situ from an alkene to give the carbonyl compounds.302 A number of other oxidizing agents also give the same products, among them303 aq sodium hypochlorite (NaOCl),304 activated MnO2,305 O2 and a Ru catalyst,306 or PCC.307 Permanganate, dichromate, and several other oxidizing agents308 also cleave glycols, giving carboxylic acids rather than aldehydes, but these reagents are seldom used synthetically.
The two reagents (periodic acid and lead tetraacetate) are complementary, since periodic acid is best used in water and lead tetraacetate in organic solvents. Chiral lead carboxylates have been prepared for the oxidative cleavage of 1,2-diols.309 When three or more OH groups are located on adjacent carbons, the middle one (or ones) is converted to formic acid.
Other compounds that contain oxygen atoms or nitrogen atoms on adjacent carbons undergo similar cleavage:
Cyclic 1,2-diamines are cleaved to diketones with dimethyl dioxirane.310 α-Diketones and α-hydroxy ketones are also cleaved by alkaline H2O2.311 Periodic acid (HIO4) has been used to cleave epoxides to aldehydes,312 for example,
![]()
α-Hydroxy and α-keto acids are not cleaved by HIO4 but are cleaved by NaIO4 in methanol in the presence of a crown ether Pb(OAc)4,313 alkaline H2O2, and other reagents. These are oxidative decarboxylations. α-Hydroxy acids give aldehydes or ketones, and α-keto acids give carboxylic acids. Also see, Reaction 19-12 and 19-13.
The mechanism of glycol oxidation with Pb(OAc)4 was proposed by Criegee et al.:314
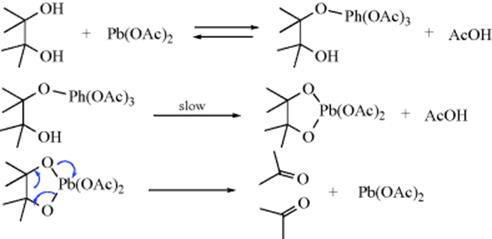
This mechanism is supported by (1) the kinetics are second order (first order in each reactant); (2) added acetic acid retards the reaction (drives the equilibrium to the left); and (3) cis-glycols react much more rapidly than transglycols.315 For periodic acid, the mechanism is similar, with the intermediate (10)316
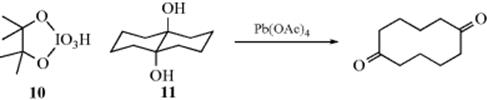
However, the cyclic-intermediate mechanism cannot account for all glycol oxidations, since some glycols that cannot form such an ester (e.g., 11) are nevertheless cleaved by lead tetraacetate (although other glycols that cannot form cyclic esters are not cleaved, by either reagent317). To account for cases like 11, a cyclic transition state has been proposed:315
OS IV, 124; VII, 185; VIII, 396.
19-8 Oxidative Cleavage of Ketones, Aldehydes, and Alcohols
Cycloalkanone oxidative ring opening
![]()
Oxidative cleavage of open-chain ketones or alcohols318 is a preparative procedure that is seldom useful, not because these compounds do not undergo oxidation (they do, except for diaryl ketones), but because the result is generally a hopeless mixture. Aryl methyl ketones (e.g., acetophenone), however, are readily oxidized to aryl carboxylic acids with Re2O7 and 70% aq tert-butyl hydroperoxide.319 Oxygen with a mixture of Mn and Co catalysts give similar oxidative cleavage,320 as do hypervalent iodine compounds.321 Aldehydes, (e.g., PhCH2CHO) are cleaved to benzaldehyde with phosphonium dichromate in refluxing acetonitrile.322 1,3-Diketones (e.g., 1,3-diphenyl-1,3-propanedione) are oxidatively cleaved with aq Oxone to give benzoic acid.323 Cyclic α-chloro ketones are cleaved to give an α,ω-functionalized compound (acetal–ester) when treated with cerium (IV) sulfate tetrahydrate and O2.324
Despite problems with acyclic ketones, the reaction is useful for the conversion of cyclic ketones and the corresponding secondary alcohols to the dicarboxylic acid in good yield. The formation of adipic acid from cyclohexanone (shown above) is an important industrial procedure. Dichromate in acidic media and permanganate are the most common oxidizing agents, although autoxidation (oxidation with atmospheric oxygen) in alkaline solution325 and potassium superoxide under phase-transfer conditions326 have also been used. O-Silyl-ketones have been cleaved to esters using electrolysis in alcohol solvents.327
Cyclic 1,3-diketones, which exist mainly in the mono-enolic form, can be cleaved with sodium periodate with loss of one carbon, for example,328
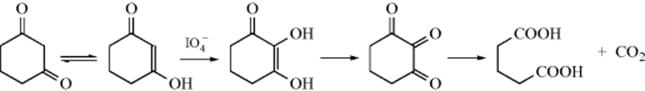
The species actually undergoing the cleavage is the triketone, so this is an example of Reaction 19-7. Cyclic 1,3-diketones are converted to α,ω-diesters with an excess of KHSO5 in methanol.329
OS I, 18; IV, 19; VI, 690. See also, OS VI, 1024.
19-9 Ozonolysis
Oxo-uncoupling
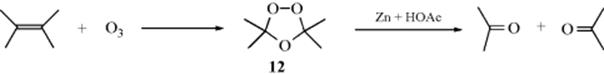
When compounds containing double bonds are treated with ozone, usually at low temperatures, initial formation of a 1,2,3-trioxolane is followed by formation of compounds called 1,2,4-trioxolanes (ozonides, 12). These compounds can be isolated but, because some of them are explosive, they are decomposed with Zn and acetic acid or catalytic hydrogenation. More commonly they are decomposed with DMS330 to give 2 molar equivalents of aldehyde, or 2 molar equivalents of ketone, or 1 molar equivalent of each, depending on the groups attached to the alkene.331 The decomposition of 12 has also been carried out with triethylamine332 and with reducing agents (e.g., trimethyl phosphite333 or thiourea).334 However, ozonides can also be oxidized with oxygen, peroxyacids, or H2O2 to give ketones and/or carboxylic acids. Note that the presence of a hydrogen atom on the C=C unit (C=C–H) leads to differences in oxidation or reduction of 12. In such a system, oxidation leads to the acid, whereas reduction leads to the aldehyde. Note that the presence of a hydrogen atom on the C=C unit (C=C–H) leads to differences in oxidation or reduction of 12. In such a system, oxidation leads to the acid, whereas reduction leads to the aldehyde. It is also possible to reduce 12 with LiAlH4, NaBH4, BH3, or catalytic hydrogenation with excess H2 to give 2 molar equivalents of alcohol.335 Ozonides can be treated with ammonia, hydrogen, and a catalyst to give the corresponding amines,336 or with an alcohol and anhydrous HCl to give the corresponding carboxylic esters.337 Ozonolysis is therefore an important synthetic reaction.338 Ozonolysis can be done in solvent–water mixtures.339
Many alkenes undergo ozonolysis, including cyclic alkenes, where cleavage gives rise to one bifunctional product (an α,ω-difunctional molecule). Alkenes in which the double bond is connected to electron-donating groups react many times faster than those in which it is connected to electron-withdrawing groups.340 Ozonolysis of compounds containing more than one double bond generally leads to cleavage of all the bonds. In some cases, especially when bulky groups are present, conversion of the substrate to an epoxide (15-50) becomes an important side reaction and can be the main reaction.341 Rearrangement is possible in some cases.342 Ozonolysis of triple bonds343 is less common and the reaction proceeds less easily, since ozone is an electrophilic agent344 and prefers double to triple bonds (Sec. 15.B.i). Compounds that contain triple bonds generally give carboxylic acids, although sometimes ozone oxidizes them to α-diketones (Reaction 19-26).
Aromatic compounds are attacked less readily than alkenes, but cleavage is known. Aromatic compounds behave as if the double bonds in the Kekulé structures were really there. Thus benzene gives 3 molar equivalents of glyoxal (HCOCHO), and o-xylene gives a glyoxal/MeCOCHO/MeCOCOMe ratio of 3 : 2 : 1, which shows that in this case cleavage is statistical. With polycyclic aromatic compounds the site of attack depends on the structure of the molecule and on the solvent.345
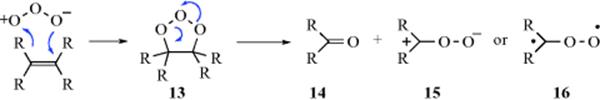
Although a large amount of work has been done on the mechanism of ozonization (formation of 12), not all the details are known. Note that a primary ozonide has been trapped.346 Criegee347 formulated the basic mechanism. The first step of the Criegee mechanism348 is a 1,3-dipolar addition (Reaction 15-58) of ozone to the substrate to give the “initial” or “primary” ozonide, the structure of which has been shown to be the 1,2,3-trioxolane (13) by microwave and other spectral methods.349 However, 13 is highly unstable and cleaves to an aldehyde or ketone (14) and an intermediate350 that Criegee showed as a zwitterion (15), but which may be a diradical (16). This intermediate is usually referred to as a carbonyl oxide.351 The carbonyl oxide, which will be represented as 15, can then undergo various reactions, three of which lead to normal products. One is a recombination with 14, which leads to ozonide 12. The second is a dimerization to the bis(peroxide) 17, and the third a kind of dimerization to 18.352 If the first path is taken (this is normally possible only if 14 is an aldehyde; most ketones do not do this353,354 the product is an ozonide (1,2,4-trioxolane, 12),355 and hydrolysis
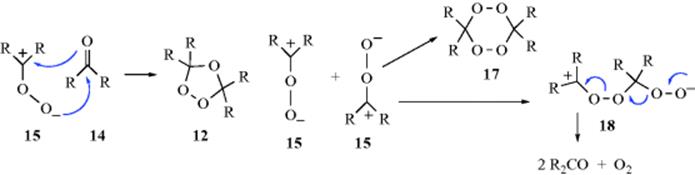
of the ozonide gives the normal products. If 17 is formed, hydrolysis of it gives one of the products, and, of course, 14, which then does not undergo further reaction, is the other. If intermediate 18 is formed, direct decomposition is possible, as shown, to give the normal products and oxygen. In protic solvents, 15 is converted to a hydroperoxide, and these have been isolated [e.g., Me2C(OMe)OOH from Me2C=CMe2 in methanol]. Further evidence for the mechanism is that 17 can be isolated in some cases (e.g., from Me2C=CMe2). But perhaps the most impressive evidence comes from the detection of cross-products. In the Criegee mechanism, the two parts of the original alkene break apart and then recombine to form the ozonide. In the case of an unsymmetrical alkene (RCH=CHR′) there should be three ozonides:
![]()
since there are two different aldehydes (14) and two different species (15). These compounds can recombine in the three ways shown. Actually six ozonides, corresponding to the cis and trans forms of these three, were isolated and characterized for methyl oleate.356 Similar results have been reported for smaller alkenes (e.g., 2-pentene, 4-nonene, and even 2-methyl-2-pentene.357 The last-mentioned case is especially interesting, since it is quite plausible that this compound would cleave in only one way, so that only one ozonide (in cis and trans versions) would be found; but this is not so, and three were found for this case too. However, terminal alkenes give little or no cross-ozonide formation.358 In general, the less alkylated end of the alkene tends to go to 14 and the other to 15. Still other evidence359 for the Criegee mechanism is (1) When Me2C=CMe2 was ozonized in the presence of HCHO, the ozonide (19) could be isolated354; (2) 15 prepared in an entirely different manner (photooxidation of diazo compounds), reacted with aldehydes to give ozonides360; and (3) cis-and trans-alkenes generally give the same ozonide, which would be expected if they cleave first.361 However, this was not true for Me3CCH=CHCMe3, where the cis-alkene gave the cis-ozonide (chiefly), and the trans gave the trans.362
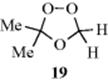
The latter result is not compatible with the Criegee mechanism. Also incompatible with the Criegee mechanism was the finding that the cis/trans ratios of symmetrical (cross) ozonides obtained from cis- and trans-4-methyl-2-pentene were not the same.363 If the Criegee mechanism operated as shown above, the cis/trans ratio for each of the two cross ozonides would have to be identical for the cis- and trans-alkenes, since in this mechanism they are completely cleaved.
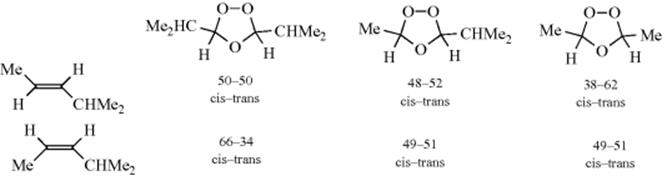
The above stereochemical results have been explained364 on the basis of the Criegee mechanism with the following refinements: (1) The formation of 13 is stereospecific, as expected from a 1,3-dipolar cycloaddition. (2) Once formed, 15 and 14 remain attracted to each other, much like an ion pair. (3) Intermediate 15 exists in syn and anti forms, which are produced in different amounts and can hold their shapes, at least for a time. This is plausible if we remember that a C=O canonical form contributes to the structure of 19. (4) The combination of 15 and 14 is also a 1,3-dipolar cycloaddition, so configuration is retained in this step too.365
Evidence that the basic Criegee mechanism operates even in these cases comes from ![]() labeling experiments, making use of the fact, mentioned above, that mixed ozonides (e.g., 15) can be isolated when an external aldehyde is added. Both the normal and modified Criegee mechanisms predict that if
labeling experiments, making use of the fact, mentioned above, that mixed ozonides (e.g., 15) can be isolated when an external aldehyde is added. Both the normal and modified Criegee mechanisms predict that if ![]() -labeled aldehyde is added to the ozonolysis mixture, the label will appear in the ether oxygen (see the reaction between 15 and 14), and this is what is found.366 There is evidence that the anti-15 couples much more readily than the syn-15.367
-labeled aldehyde is added to the ozonolysis mixture, the label will appear in the ether oxygen (see the reaction between 15 and 14), and this is what is found.366 There is evidence that the anti-15 couples much more readily than the syn-15.367
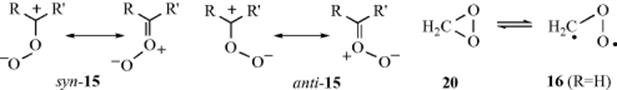
The ozonolysis of ethylene368 in the liquid phase (without a solvent) was shown to take place by the Criegee mechanism.369 This reaction has been used to study the structure of the intermediate 15 or 16. The compound dioxirane (20) was identified in the reaction mixture370 at low temperatures and is probably in equilibrium with the biradical 16 (R = H). Dioxirane has been produced in solution, but it oxidatively cleaves dialkyl ethers (e.g., Et–O–Et) via a chain-radical process,371 so the choice of solvent is important.
Ozonolysis in the gas phase is not generally carried out in the laboratory. However, the reaction is important because it takes place in the atmosphere and contributes to air pollution.372 There is much evidence that the Criegee mechanism operates in the gas phase too, although the products are more complex because of other reactions that also take place.373
OS V, 489, 493; VI, 976; VII, 168; IX, 314. Also see, OS IV, 554. For the preparation of ozone, see OS III, 673.
19-10 Oxidative Cleavage of Double Bonds and Aromatic Rings
Oxo-de-alkylidene-bisubstitution, and so on
![]()
Carbon–carbon double bonds can be cleaved by many oxidizing agents,374 the most common of which are permanganate in neutral or acid media and dichromate in acid media. The products are generally 2 molar equivalents of ketone, 2 molar equivalents of carboxylic acid, or 1 molar equivalent of each, depending on what groups are attached to the alkene. With ordinary solutions of permanganate or dichromate, yields are generally low and the reaction is seldom a useful synthetic method; but high yields can be obtained by oxidizing with KMnO4 dissolved in benzene containing the crown ether dicyclohexano-18-crown-6 (see Sec. 3.C.ii).375 The crown ether coordinates with K+, permitting the KMnO4 to dissolve in benzene. Another reagent frequently used for synthetic purposes is the Lemieux-von Rudloff reagent: HIO4 containing a trace of MnO4−.376 The MnO4− is the actual oxidizing agent, being reduced to the manganate stage, and the purpose of the HIO4 is to reoxidize the manganate back to MnO4−. Another reagent that behaves similarly is NaIO4–ruthenium tetroxide.377 Oxidative cleavage of alkenes is catalyzed by Ru with IO(OH)5.378 Cyclic alkenes are cleaved to α,ω-diketones, keto-acids, or dicarboxylic acids. Cyclic alkenes are cleaved to dialdehydes with KMnO4·CuSO4 in dichloromethane.379 A combination of RuCl3/HIO5 oxidatively cleaves cyclic alkenes to dicarboxylic acids.380
The Barbier–Wieland procedure for decreasing the length of a chain by one carbon involves oxidative cleavage by acid dichromate (NaIO4–RuO4 has also been used), but this is cleavage of a 1,1-diphenyl alkene (21), which generally gives good yields. Addition of a catalytic amount of OsO4 to Jones reagent (Reaction 19-3) leads to good yields of the carboxylic acid from simple alkenes.381 A combination of Oxone and OsO4 in DMF cleaves alkenes to carboxylic acids.382 Cleavage of alkynes is generally rather difficult, but treatment of internal alkynes with an excess of Oxone with a Ru catalyst leads to aliphatic carboxylic acids.383
With certain reagents, the oxidation of double bonds can be stopped at the aldehyde stage, and in these cases the products are the same as in the ozonolysis procedure. Among these reagents are tert-butyl iodoxybenzene,384KMnO4 in THF–H2O,385 and NaIO4–OsO4.386 Enol ethers, [RC(OR′)=CH2] have been cleaved to carboxylic esters [RC(OR′)=O] by atmospheric oxygen.387 Oxidative cleavage of alkenes is catalyzed by a Mn–porphyrin complex.388
The mechanism of oxidation probably involves in most cases the initial formation of a glycol (Reaction 15-29) or cyclic ester,389 and then further oxidation as in Reaction 19-7.390 In line with the electrophilic attack on the alkene, triple bonds are more resistant to oxidation than double bonds. Terminal triple-bond compounds can be cleaved to carboxylic acids with Tl(III)NO3391 or with [bis(trifluoroacetoxy)iodo]pentafluorobenzene [i.e., C6F5I(OCOCF3)2].392
Aromatic rings can be cleaved with strong enough oxidizing agents. An important laboratory reagent for this purpose is RuO4 along with a cooxidant (e.g., NaIO4 or NaOCl and household bleach can be used). Ruthenium tetroxide is an expensive reagent, but the cost can be greatly reduced by the use of an inexpensive cooxidant (e.g., NaOCl), the function of which is to oxidize RuO2 back to ruthenium tetroxide. Examples393 are the oxidation of naphthalene to phthalic acid394 and, even more remarkably, of cyclohexylbenzene to cyclohexanecarboxylic acid395 (note the contrast with Reaction 19-11). The latter conversion was also accomplished with ozone.396 Another reagent that oxidizes aromatic rings is air catalyzed by V2O5. The oxidations of naphthalene to phthalic anhydride and of benzene to maleic anhydride by this reagent are important industrial procedures.397o-Diamines have been oxidized with nickel peroxide, with lead tetraacetate,398 and with O2 catalyzed by CuCl:399
The last-named reagent also cleaves o-dihydroxybenzenes (catechols) to give, in the presence of MeOH, the monomethylated dicarboxylic acids (HO2C–C=C–C=C–CO2Me).400
OS II, 53, 523; III, 39, 234, 449; IV, 136, 484, 824; V, 393; VI, 662, 690; VII, 397; VIII, 377, 490; IX, 530. Also see, OS II, 551.
19-11 Oxidation of Aromatic Side Chains
Oxo,hydroxy-de-dihydro,methyl-tersubstitution
![]()
Alkyl chains on aromatic rings can be oxidized to CO2H groups by many oxidizing agents, including permanganate, nitric acid, and acid dichromate.401 The method is most often applied to the methyl group (CH3 → CO2H), although longer side chains can also be cleaved. Tertiary alkyl groups are resistant to oxidation, and when they are oxidized, ring cleavage usually occurs too.402 It is usually difficult to oxidize an R group on a fused aromatic system without cleaving the ring or oxidizing it to a quinone (Reaction 19-19). However, this has been done (e.g., 2-methylnaphthalene was converted to 2-naphthoic acid) with aq Na2Cr2O7.403 Aryl methyl groups are oxidized to aryl CO2H with NaOCl in acetonitrile,404 or with NBS in aqueous NaOH under photochemical conditions.405 Functional groups can be present anywhere on the side chain and, if in the α position, greatly increase the ease of oxidation. An exception is an α phenyl group. In such cases, the reaction stops at the diaryl ketone stage. Molecules containing aryl groups on different carbons cleave so that each ring gets one carbon atom, as in the cleavage of the 9,10-bond of dihydrophenanthrenes (21 to 22).
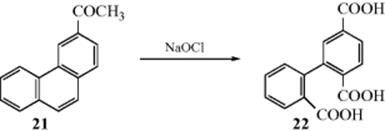
It is possible to oxidize only one alkyl group of a ring that contains more than one. The order of reactivity406 toward most reagents is CH2Ar > CHR2 > CH2R > CH3.407 Groups on the ring susceptible to oxidation (OH, NHR, NH2, etc.) must be protected. The oxidation can be performed with oxygen, in which case it is autoxidation, and the mechanism is like that in Reaction 14-7, with a hydroperoxide intermediate.408 With this procedure it is possible to isolate ketones from ArCH2R, and this is often done.409
The mechanism has been studied for the following closely related reaction: Ar2CH2 + CrO3 → Ar2C=O.410 A deuterium isotope effect of 6.4 was found, indicating that the rate-determining step is either Ar2CH2 → Ar2CH· or Ar2CH2 → Ar2CH+. Either way this explains why tertiary groups are not converted to CO2H and why the reactivity order is CHR2 > CH2R > CH3, as mentioned above. Both free radicals and carbocations exhibit this order of stability (Chapter 5). The two possibilities are examples of categories 2 and 3 in Section 19.A. Just how the radical or the cation goes on to the product is not known.
When the alkyl group is one oxidizable to CO2H (Reaction 19-11), cupric salts are oxidizing agents, and the OH group is found in a position ortho to that occupied by the alkyl group.411 This reaction is used industrially to convert toluene to phenol.
In another kind of reaction, an aromatic aldehyde (ArCHO) or ketone (ArCOR′) is converted to a phenol (ArOH) on treatment with alkaline H2O2,412 but there must be an OH or NH2 group in the ortho or para position. This is called the Dakin reaction.413 The mechanism may be similar to that of the Baeyer–Villiger Reaction (18-19).414 The intermediate 23 has been isolated.415 The reaction has been performed on aromatic aldehydes with an alkoxy group in the ring, and no OH or NH2. In this case, acidic H2O2 was used.416 The Dakin reaction has been done in ionic liquids.417
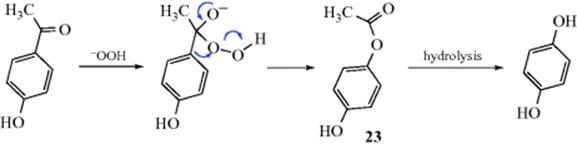
OS I, 159, 385, 392, 543; II, 135, 428; III, 334, 420, 740, 791, 820, 822; V, 617, 810. OS I, 149; III, 759.
19-12 Oxidative Decarboxylation
Acetoxy-de-carboxy-substitution
![]()
Hydro-carboxyl-elimination
![]()
Carboxylic acids can be decarboxylated418 with lead tetraacetate to give a variety of products: an ester (ROAc), the alkane (RH) (see Reaction 12-40), an alkene if α,β hydrogen is present, as well as numerous other products arising from rearrangements, internal cyclizations,419 and reactions with solvent molecules. When R is tertiary, the chief product is usually the alkene. High yields of alkenes can also be obtained when R is primary or secondary using Cu(OAc)2 along with the Pb(OAc)4.420 In the absence of Cu(OAc)2, primary acids give mostly alkanes (though yields are generally low) and secondary acids may give carboxylic esters or alkenes. Other oxidizing agents,421including Co(III), Ag(II), Mn(III), and Ce(IV), have also been used to effect oxidative decarboxylation.422
The mechanism with lead tetraacetate is generally accepted to be of the free radical type.423 First, there is an interchange of ester groups:
![]()
A free radical chain mechanism follows (shown for 24 although 25 and other lead esters can behave similarly)
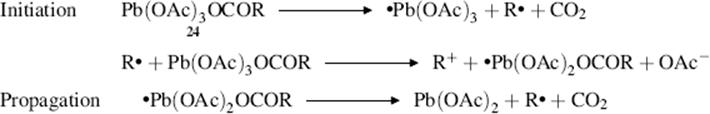
Products can then be formed either from R√ or R+. Primary R√ abstract H from solvent molecules to give RH. The R+ ion can lose H+ to give an alkene, react with HOAc to give the carboxylic ester, react with solvent molecules or with another functional group in the same molecule, or rearrange, thus accounting for the large number of possible products. The radical R√ can also dimerize to give RR. The effect of Cu2+ ions424 is to oxidize the radicals to alkenes, thus producing good yields of alkenes from primary and secondary substrates. The Cu2+ ion has no effect on tertiary radicals, because these are efficiently oxidized to alkenes by lead tetraacetate.
![]()
In another type of oxidative decarboxylation, arylacetic acids can be oxidized to aldehydes with one less carbon (ArCH2COOH → ArCHO) by tetrabutylammonium periodate.425 Simple aliphatic carboxylic acids were converted to nitriles with one less carbon (RCH2COOH → RC![]() N) by treatment with trifluoroacetic anhydride and NaNO2 in F3CCO2H.426
N) by treatment with trifluoroacetic anhydride and NaNO2 in F3CCO2H.426
See also, Reaction 14-37.
19-13 Bis(decarboxylation)
Dicarboxy-elimination
![]()
Compounds containing carboxyl groups on adjacent carbons (succinic acid derivatives) can be bis(decarboxylated) with lead tetraacetate in the presence of O2.417 The reaction is of wide scope. The elimination is stereoselective, but not stereospecific (both meso- and dl-2,3-diphenylsuccinic acid gave trans-stilbene)427; a concerted mechanism is thus unlikely. The following mechanism is compatible with the data:
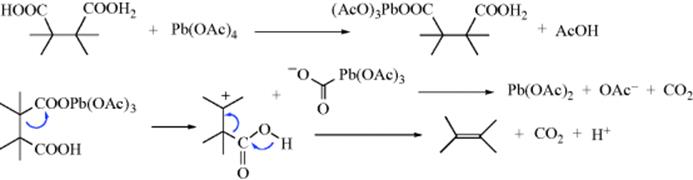
though a free radical mechanism seems to hold in some cases. Bis(decarboxylation) of succinic acid derivatives to give alkenes428 has also been carried out by other methods.429 Compounds containing geminal carboxyl groups (disubstituted malonic acid derivatives) can be bis(decarboxylated) with lead tetraacetate,430gem-diacetates (acylals) being produced, which are easily hydrolyzable to ketones:431
![]()
A related reaction involves α-substituted aryl nitriles having a sufficiently acidic α hydrogen, which can be converted to ketones by oxidation with air under phase-transfer conditions.432 The nitrile is added to NaOH in benzene or DMSO, containing a catalytic amount of triethylbenzylammonium chloride (TEBA).433 This reaction could not be applied to aliphatic nitriles, but an indirect method for achieving this conversion is given in Reaction 19-60.
C. Reactions Involving Replacement of Hydrogen by Heteroatoms
19-14 Hydroxylation at an Aliphatic Carbon
Hydroxylation or Hydroxy-de-hydrogenation
![]()
Compounds containing susceptible C–H bonds can be oxidized to alcohols.434 Nearly always, the C–H bond involved is tertiary, so the product is a tertiary alcohol. This is partly because tertiary C–H bonds are more susceptible to free radical attack than primary and secondary bonds and partly because the reagents involved would oxidize primary and secondary alcohols further. In the best method, the reagent is ozone and the substrate is absorbed on silica gel.435 Yields as high as 99% have been obtained by this method. Other reagents are chromic acid,436 ruthenium tetroxide (RuO4),437 thallium acetate,438 sodium chlorite (NaClO2) with a metalloporphyrin catalyst,439 OsO4,440 and certain peroxybenzoic acids.441 Alkanes and cycloalkanes have been oxidized at secondary positions, to a mixture of alcohols and trifluoroacetates, by 30% aq H2O2 in trifluoroacetic acid.442 This reagent does not oxidize the alcohols further and ketones are not found. As in the case of chlorination with N-haloamines and sulfuric acid (see Reaction 14-1), the ω − 1 position is the most favored. Another reagent443 that oxidizes secondary positions is iodosylbenzene, catalyzed by Fe(III)–porphyrin catalysts.444 Use of an optically active Fe(III)–porphyrin gave modest enantioselective hydroxylation.445
When chromic acid is the reagent, the mechanism is probably as follows: a Cr6+ species abstracts a hydrogen to give R3C√, which is held in a solvent cage near the resulting Cr5+ species. The two species then combine to give R3COCr4+, which is hydrolyzed to the alcohol. This mechanism predicts retention of configuration; this is largely observed.446 The oxidation by permanganate also involves predominant retention of configuration, and a similar mechanism has been proposed.447
Treatment of double-bond compounds with selenium dioxide introduces an OH group into the allylic position (see also, Reaction 19-17).448 This reaction also produces conjugated aldehydes in some cases.449 Allylic rearrangements are common. There is evidence that the mechanism does not involve free radicals, but includes two pericyclic steps (A and B):450
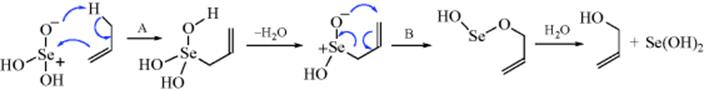
The step marked A is similar to the ene reaction (Reaction 15-23). The step marked B is a [2,3]-sigmatropic rearrangement (see Reaction 18-35). The reaction can also be accomplished with tert-butyl hydroperoxide, if SeO2 is present in catalytic amounts (the Sharpless method).451 The SeO2 is the actual reagent; the peroxide reoxidizes the Se(OH)2.452 This method makes work up easier, but gives significant amounts of side products when the double bond is in a ring.453 Alkynes generally give α,α′ dihydroxylation.454 Allylic hydroxylation455 with selenium dioxide often gives aldehydes, but in the presence of acetic anhydride and oxygen, SeO2 converts alkenes to homoallylic acetates as the major product, C=C–C–C → C=C–C–C–OAc.456
Hydroxylation of unactivated sp3 hybridized bonds is possible using an oxaziridine-mediated, organocatalyzed reaction.457 Ruthenium tetroxide oxidizes alkanes.458 Nanocrystalline cobalt oxide is another catalyst for alkane oxidation.459 The H2O2–NaVO3–H2SO4 system facilitates alkane oxidation in aqueous acetonitrile.460
Benzylic methylene groups are more readily oxidized to benzylic alcohols when compared to simple alkanes. Typical reagents include manganese salen and PhIO461 or peroxides.462 Oxidation to an acetoxy benzyl derivative was accomplished with PhI(OAc)2 in acetic acid with a Pd catalyst,463 and with PhI(OH)OTs in aq DMSO.464 With minimal water, cerium (IV) triflate converts benzylic arenes to benzylic alcohols, although the major product is the ketone when >15% of water is present.465
Allylic benzyloxylation occurs when an alkene is treated with t-BuOOCOPh and a Cu–Na zeolite,466 a Cu catalyst,467 or with a chiral Cu catalyst to give modest enantioselectivity.468 Allylic methylene groups can be converted to ester (–CH–OCOR) derivatives in a similar manner using copper triflate.469 Cupric acetate has been used,470 as well as Cu2O.471 A chiral Lewis acid has been used for an enantioselective allylic CH oxidation to an allylic acyl derivative.472 α-Acetoxylation of allylic alkenes can proceed with allylic rearrangement.473
Hydroxylation can be accomplished using enzymatic systems. In the presence of Bacillus megaterium and oxygen, cyclohexane is converted to cyclohexanol.474 Allylic oxidation to an allylic alcohol was accomplished with cultured cells of Gossypium hirsutum.475 Benzylic arenes are converted to the corresponding α-hydroxy compound by treatment with the enzymes of B. megaterium, with modest enantioselectivity.476 The reaction of tetradecanoic acid with the α-oxidase from Pisum sativum, in the presence of molecular oxygen, gives 2(R)-hydroxytetradecanoic acid with high asymmetric induction.477
Simple alkanes can be converted to esters with dialkyloxiranes. Cyclic alkanes are oxidized to alcohols with dimethyl dioxirane.478 Cyclohexane was converted to cyclohexyl trifluoroacetate with di(trifluoromethyl) dioxirane and trifluoroacetic anhydride479 and also with RuCl3/MeCO3H/CF3CO2H.480 Dimethyl dioxirane converts alkanes to alcohols in some cases.481 Adamantane is converted to adamantyl alcohol with DDQ (see Reaction 19-1, category 3) and triflic acid.482 The mechanism of oxygen insertion into alkanes has been examined.483
It is possible to perform the conversion CH2 → C=O on an alkane, with no functional groups at all, although the most success has been achieved with substrates in which all CH2 groups are equivalent (e.g., unsubstituted cycloalkanes). Hydrogen peroxide and trifluoroacetic acid has also been used for oxidation of alkanes.484 With this method, cyclohexane was converted with 72% efficiency to give 95% cyclohexanone and 5% cyclohexanol.485 The same type of conversion, with lower yields (20–30%), has been achieved with the Gif system.486 There are several variations. One consists of pyridine–acetic acid, with H2O2 as oxidizing agent and tris(picolinato)iron(III) as catalyst.487 Other Gif systems use O2 as oxidizing agent and Zn as a reductant.488 The selectivity of the Gif systems toward alkyl carbons is CH2 > CH ≥ CH3, which is unusual, and shows that a simple free radical mechanism (see Sec. 14.A.iv) is not involved.489 Another reagent that can oxidize the CH2 of an alkane is methyl(trifluoromethyl)dioxirane, but this produces CH–OH more often than C=O (see Reactions 19-14 and 19-15).490 Cyclic alkanes are oxidized to a mixture of the alcohol and the ketone with PhI(OAc)2 and a manganese complex in an ionic liquid.491 Oxidation of cyclic alkanes to cyclic ketones was accomplished using a Ru catalyst.492
OS IV, 23; VI, 43, 946; VII, 263, 277, 282.
19-15 Oxidation of Methylene to OH, O2CR, or OR
Hydroxy (or alkoxy) -de-dihydro-bisubstitution

Methyl or methylene groups α to a carbonyl can be oxidized to give α-hydroxy ketones, aldehydes, or carboxylic acid derivatives. Ketones can be α hydroxylated in good yields, without conversion to the enolates, by treatment with the hypervalent iodine reagents493 o-iodosobenzoic acid.494 Dioxygen (O2) and a chiral phase-transfer catalyst gave enantioselective α hydroxylation of ketones, if the α position was tertiary.495 Dimethyl dioxirane is quite effective for hydroxylation of 1,3-dicarbonyl compounds,496 and O2 with a Mn catalyst also gives hydroxylation.497 Oxygen with a Ce catalyst α-hydroxylates β-keto esters.498 The Pd–C catalyzed α-oxygenation of 1,3-dicarbonyl compounds can be accomplished using O2.499 An engineered Cytochrome P450 BM-3 is effective for the enantioselective α-hydroxylation of esters of benzylic acids.500 The reaction of ketones with Ti(OiPr)4, diethyl tartrate and tert-butylhydroperoxide gave the α-hydroxy ketone with good enantioselectively, albeit in low yield.501 α-Hydroxylation of ketones was reported using H2O2 and 12-tungstophosphosphoric acid–cetylpyridinium chloride as a catalyst.502 Hypervalent iodine(III) sulfonate has been used for the α-hydroxylation of aryl ketones.503
Ketones and carboxylic esters can be α hydroxylated by treatment of their enolate anions (prepared by adding the ketone or ester to LDA) with a Mo peroxide reagent (MoO5–pyridine–HMPA; called MoOPH) in THF–hexane at −70°C.504 The enolate forms of amides and esters505 and the enamine derivatives of ketones506 can similarly be converted to their α-hydroxy derivatives by reaction with molecular oxygen. The MoO5 method can also be applied to certain nitriles.507
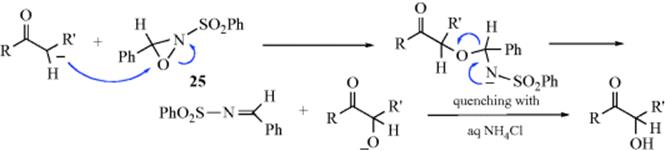
Ketones are converted to α-hydroxy ketones by reaction of the enolate anion with a 2-sulfonyloxaziridine (e.g., 25).508 This is not a free radical process; the mechanism shown is likely. The method is also successful for carboxylic esters509 and N,N-disubstituted amides,510 and can be made enantioselective by the use of a chiral oxaziridine.511 Dimethyldioxirane also oxidizes the enolate anions of ketones to α-hydroxy ketones.512 Titanium enolates are oxidized with tert-butyl hydroperoxide513 or with dimethyldioxirane514 and hydrolyzed with aq ammonium fluoride to give the α-hydroxy ketone. Ketones are converted to the α-oxamino derivative (O=C–CH2– → O=C–CHONHPh) with excellent enantioselectivity using PhN=O and l-proline515 or (S)-proline.516 Aldehydes undergo a similar oxidation.517 α-Lithio sulfones have been hydroxylated with Me3SiOOt-Bu.518
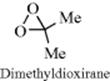
Ketones have been α hydroxylated by conversion to the silyl enol ether, followed by treatment with m-chloroperoxybenzoic acid,166 or with certain other oxidizing agents.519 α-Hydroxyketones can be accomplished from silyl enol ethers with a catalytic amount of MeReO3 and H2O2.520 When the silyl enol ethers are treated with iodosobenzene in the presence of trimethylsilyl trifluoromethyl sulfonate, the product is the α-keto triflate.521 Silyl ketene ethers are converted to α-hydroxy esters with H2O2 and methyl trioxorhenium.522 The α′-position of α,β-unsaturated ketones can be selectively oxidized.523N-Acyl amines are converted to the α-hydroxy derivative with PhIO and a Mn salen catalyst.524 Note that homoallylic-type oxidation occurs when an α,α-dimethyl oxime ether is treated with PhI(OAc)2 and a Pd catalyst in acetic acid/acetic anhydride, converting one of the methyl groups to an acetoxymethyl.525
α-Acetoxylation of ketones with concurrent α-arylation occurs when ketones react with Mn(OAc)3 in benzene.526 α-Acetoxylation of ketones can occur under similar conditions without arylation.527 α-Methyl ketones are converted to the α-acetoxy derivative under the same conditions.528 Iodobenzene with 30% aq H2O2 and acetic anhydride generates α-acetoxy ketones.529 Thallium(III) triflate converts acetophenone to α-formyloxy acetophenone.530Methanesulfonic acid and CuO converts ketones to α-mesyloxy (–OMs) ketones531 and PhI(OH)OTs converts ketones to α-tosyloxy (–OTs) ketones.532 N-Methyl-O-tosylhydroxylamine is another reagent that effects direct α-oxytosylation of ketones and aldehydes.533 α-Acetoxylation of ketones results from in situ generation of hypervalent iodine species in the presence of acetic acid.534
OSCV 7, 277; OSCV 7, 263; OSCV 6, 43
19-16 Oxidation of Methylene to Heteroatom Functional Groups Other Than Oxygen or Carbonyl
Amino (or amido) -de-dihydro-bisubstitution
![]()
α-Amination or amidation of a CH unit is possible in some cases. Cyclic alkanes are converted to the N-alkyl N-tosylamine with PhI=NTs and a Cu complex.535 Benzylic (CH) as in ethylbenzene, is oxidized with PhI(OAc)2 in the presence of TsNH2 and a fluorinated manganese porphyrin to give the corresponding N-tosylamine [PhCHMe(NHTs)].536 Alkenes with an allylic CH react with PhI=NTs and a Ru catalysts to give an allylic N-tosylamine.537 When an α-keto ester reacts with DEAD and a chiral Cu complex, an α-carbamate is formed, RCH(NHCO2Et)C(=O)CO2Et, with modest enantioselectivity.538
Cyclic amines react with Pseudomonas oleovorans GPol to give hydroxy amines; N-benzylpyrrolidine is converted to 3-hydroxy N-benzylpyrrolidine.539 Sphingomonas sp. HXN-200 gives similar results,540 and lactams are converted to the corresponding 3-hydroxy lactam with sphingomonas sp. HXN-200.541 N-Benzyl piperidine is converted to the 4-hydroxy derivative under the same conditions.542 N-Benzyl phthalimide reacts with NBS, NaOAc, and acetic acid to give N-(α-acetoxybenzyl)phthalimide.543
Tetrahydrofuran was converted to the hemiacetal 2-hydroxytetrahydrofuran, which was relatively stable under the conditions used, by electrolysis in water.544 α-Hydroxy ethers are generated by reaction of SO2/O2 and a V catalyst with ethers.545
Similar reactions are possible, in some cases, to produce sulfur containing compounds.
Sulfo-de-dihydro-bisubstitution
![]()
Cyclic alkanes are converted to the corresponding alkylsulfonic acid with SO2/O2 and a V catalyst.546
19-17 Oxidation of Methylene to Carbonyl
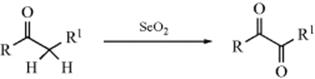
Oxo-de-dihydro-bisubstitution
Methyl or methylene groups α to a carbonyl can be oxidized with selenium dioxide to give, respectively, α-keto aldehydes (see Reaction 19-18) and α-diketones.547 The reaction can also be carried out α to an aromatic ring or to a double bond, although in the latter case, hydroxylation (see 19-14) is the more common result. Selenium dioxide, (SeO2) is often used, but the reaction has also been carried out with other oxidizing agents,548 including hypervalent iodine compounds.549 Sodium nitrite/HCl oxidizes cyclic ketones to the diketone.550 Substrates most easily oxidized contain two aryl groups on CH2, and these substrates can be oxidized with many oxidizing agents (see Reaction 19-11). The benzylic position of arenes have been oxidized to alkyl aryl ketones with several oxidizing agents, including the Jones reagent,551 CrO3 on silica,552 PCC,553 DDQ,554 KMnO4 supported on MnO2,555 KMnO4/CuSO4neat556 or with ultrasound,557 manganese salen/PhIO,558 tert-butylhydroperoxide and a Ru catalyst,559 or H2O2 with a Cu catalyst.560 The combination of O2 and mcpba oxidizes benzylic arenes to aryl ketones.561 The combination of HBr and H2O2 gives a similar oxidation.562 Methyl ketones are oxidized to the α-keto ester in a two-step procedure using a fluorous selenic acid with an iodoxy benzene, followed by treatment with sodium metabisulfite (Na2S2O5).563
Alkenes of the form C=C–CH2 (an allylic position) have been oxidized to α,β-unsaturated ketones564 by sodium dichromate in HOAc-Ac2O, by t-BuOOH and Cr compounds,565 t-BuOOH and a Pd566 or Rh567 catalyst. Thallium(III) nitrate in aq acetic acid converts allylic alkenes to the corresponding saturated ketone, even in the presence of a primary alcohol elsewhere in the molecule.568 The propargylic position of internal alkynes are oxidized to give propargylic ketones with an iron catalyst,569 with a dirhodium catalyst in water,570 or with O2/t-BuOOH in the presence of a Cu catalyst.571 Chloramine-T (see Reaction 15-54), O2, and an Fe catalyst give selective oxidation of hydrocarbons to ketones.572
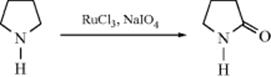
Cyclic amines are oxidized to lactams using a mixture of RuCl3 and NaIO4.573 Lactams are also formed using KMnO4 with benzyltriethylammonium chloride.574 Tertiary amines are converted to amides575 and cyclic tertiary amines can be converted to lactams by oxidation with a Hg(II)–EDTA (EDTA = ethylenediaminetetraacetic acid) complex in basic solution.576 Lactams, which need not be N-substituted, can be converted to cyclic imides by oxidation with a hydroperoxide or peroxyacid and an Mn(II) or Mn(III) salt.577 Lactams are oxidized to cyclic imides with oxygen and Co(OAc)2 in the presence N-hydroxysuccinimide.578
Ethers in which at least one group is a primary alkyl can be oxidized to the corresponding carboxylic esters in high yields with ruthenium tetroxide.579 Molecular oxygen with a binuclear Cu(II) complex580 or PdCl2/CuCl2/CO581also converts ethers to esters. Cyclic ethers are oxidized to lactones.582 Cyclic ethers are oxidized to lactones with CrO3/Me3SiONO2.583 Lactones are also formed from cyclic ethers with NaBrO3–KHSO4 in water.584 The reaction has also been accomplished with CrO3 in H2SO4,585 and with benzyltriethylammonium permanganate.586
Two mechanisms have been suggested for the reaction with SeO2. One of these involves a selenate ester of the enol:587
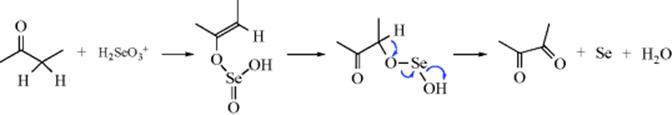
In the other proposed mechanism,588 the principal intermediate is α,β-ketoseleninic acid (O=C–CH–SeO2H) and a selenate ester is not involved.
Oxidation of CH2 to C=O groups is possible even if they are not near any functional groups, indirectly, by the remote oxidation method of Breslow57 (see Reaction 19-2). One of the CH2 groups of n-hexadecanol monosuccinate [CH3(CH2)14CH2OCOCH2CH2CO2H] was oxidized to a C=O group to give a mixture of it and benzophenone-4-carboxylic acid [p-PhCOC6H4CO2H] in CCl4.589 Other remote oxidations590 also have been reported. Among these are conversion of aryl ketones [ArCO(CH2)3R] to 1,4-diketones [ArCO(CH2)4COR] by photoirradiation in the presence of such oxidizing agents as K2Cr2O7 or KMnO4,591 and conversion of alkyl ketones [RCO(CH2)3R′] to 1,3- and 1,4-diketones with Na2S2O8 and FeSO4.592 2-Octanol was oxidized to give 2-propyl-5-methyl γ-butyrolactone with lead tetraacetate in a CO atmosphere.593
OS I, 266; II, 509; III, 1, 420, 438; IV, 189, 229, 579; VI, 48; IX, 396. Also see, OS IV, 23.
19-18 Oxidation of Arylmethanes to Aldehydes
Oxo-de-dihydro-bisubstitution
![]()
Methyl groups on an aromatic ring can be oxidized to an aldehyde by several oxidizing agents. The reaction is a special case of 19-17. When the reagent is chromyl chloride (CrO2Cl2), the reaction is called the Étard reaction594and the yields are high.595 Another oxidizing agent is a mixture of CrO3 and Ac2O, where the reaction stops at the aldehyde stage because the initial product is ArCH(OAc)2 (an acylal), which is resistant to further oxidation. Hydrolysis of the acylal gives the aldehyde.
Among other oxidizing agents596 that have been used to accomplish the conversion of ArCH3 to ArCHO are ceric ammonium nitrate,597 PCC,598 hypervalent iodoso compounds (see Reaction 19-3),599 Bi-t-BuOOH,600 and urea–H2O2 with microwave irradiation.601 Oxidative of benzylic positions to the corresponding carbonyl has been reported using two heterogeneous catalysts.602 Oxidation of ArCH3 to carboxylic acids is considered at Reaction 19-11.
Conversion of ArCH3 to ArCHO can also be achieved indirectly by bromination to give ArCHBr2 (14-1), followed by hydrolysis (Reaction 10-2).
The mechanism of the Étard reaction is not completely known.603 An insoluble complex is formed on addition of the reagents, which is hydrolyzed to the aldehyde. The complex is probably a kind of acylal, but the identity of the structure is not fully settled, although many proposals have been made as to its structure and as to how it is hydrolyzed. It is known that ArCH2Cl is not an intermediate (see Reaction 19-20), since it reacts only very slowly with chromyl chloride. Magnetic susceptibility measurements604 indicate that the complex from toluene is 26, a structure first proposed by Étard. According to this proposal, the reaction stops after only two hydrogen atoms have been replaced because of the insolubility of 26. There is a disagreement on how 26 is formed, assuming that the complex has this structure. Both an ionic605 and a free radical606 process have been proposed. An entirely different structure for the complex was proposed by Nenitzescu et al.607 On the basis of ESR studies, they proposed that the complex is PhCH2OCrCl2OCrOCl2OH, which is isomeric with 26. However, this view has been challenged by Wiberg and Eisenthal,606 who interpret the ESR result as being in accord with 26. Still another proposal is that the complex is composed of benzaldehyde coordinated with reduced chromyl chloride.608
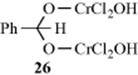
OS II, 441; III, 641; IV, 31, 713.
19-19 Oxidation of Aromatic Hydrocarbons to Quinones
Arene-quinone transformation
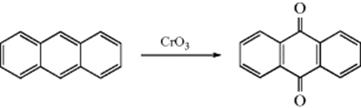
Condensed aromatic systems (including naphthalenes) can be directly oxidized to quinones by various oxidizing agents.609 Yields are generally not high, although good yields have been reported with ceric ammonium sulfate.610Benzene cannot be so oxidized by strong oxidizing agents, but can be electrolytically oxidized to benzoquinone.611 Naphthalene derivatives, however, are oxidized to naphthoquinones with H5IO6 and CrO3.612 1,4-Dimethoxy aromatic compounds are oxidized to p-quinones with an excess of CoF3 in water–dioxane.613
OS IV, 698, 757. Also see, OS II, 554.
19-20 Oxidation of Primary Halides and Esters of Primary Alcohols to Aldehydes614
Oxo-de-hydro,halo-bisubstitution
![]()
Primary alkyl halides (chlorides, bromides, and iodides) can be oxidized to aldehydes easily and in good yields with DMSO,615 in what has been called the Kornblum reaction. In Kornblum's original work, the reaction of α-halo ketones with DMSO at elevated temperatures gave good yields of the corresponding glyoxal (an α-keto-aldehyde).616 If the glyoxal could be removed from the reaction medium by distillation as it was formed, the reaction was very efficient. In many cases, it was difficult to isolate high-boiling glyoxals from DMSO. Primary and secondary617 alkyl iodides or tosylates618 can be converted to aldehydes or ketones, although they are much less reactive than α-halo ketones. Primary chlorides with DMSO, NaBr, and ZnO give the corresponding aldehyde when heated to 140 °C.619 Benzylic halides are oxidized to aryl aldehydes with MnO2620 or with NaIO4–LiBr.621 Hydrogen peroxide in ethanol oxidizes organic halides to carbonyl compounds.622 Pyridine N-oxide in the presence of silver oxide oxidizes benzylic and allylic halides.623
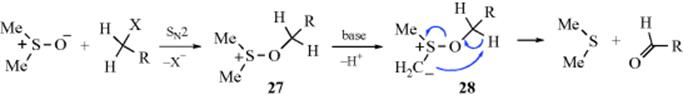
The mechanism of these DMSO oxidations is probably that shown with 27 and 28,624 although in some cases the base abstracts a proton directly from the carbon being oxidized, in which case the ylid (28) is not an intermediate. Alkoxysulfonium salts (27) have been isolated.625 This mechanism predicts that secondary compounds should be oxidizable to ketones, and this is the case. In a related procedure for the oxidation of alcohols, the intermediate 27626is formed without the use of DMSO by treating the substrate with a complex generated from chlorine or NCS and DMS.627 Also see the Swern oxidation in Reaction 19-3.
Another way to oxidize primary alkyl halides to aldehydes is by the use of hexamethylenetetramine followed by water. However, this reaction, called the Sommelet reaction,628 is limited to benzylic halides. The reaction is seldom useful when the R in RCH2Cl is alkyl. The first part of the reaction is conversion to the amine (ArCH2NH2), which can be isolated. Reaction of the amine with excess hexamethylenetetramine gives the aldehyde. It is this last step that is the actual Sommelet reaction, although the entire process can be conducted without isolation of intermediates. Once the amine is formed, it is converted to an imine (ArCH2N=CH2) with formaldehyde liberated from the reagent. The key step then follows: transfer of hydrogen from another mole of the arylamine to the imine. This last imine is then hydrolyzed by water to the aldehyde. Alternatively, the benzylamine may transfer hydrogen directly to hexamethylenetetramine.
Pyridine followed by p-nitrosodimethylaniline and then water converts benzylic halides to aldehydes, and is called the Kröhnke reaction. Primary halides and tosylates have been oxidized to aldehydes by trimethylamine N-oxide,629 and by pyridine N-oxide with microwave irradiation.630
Epoxides631 have been used to give α-hydroxy ketones or aldehydes.632
OS II, 336: III, 811; IV, 690, 918, 932; V, 242, 668, 825, 852, 872. Also see, OS V, 689; VI, 218.
19-21 Oxidation of Amines or Nitro Compounds to Aldehydes, Ketones, or Dihalides
Oxo-de-hydro,amino-bisubstitution (overall transformation)
![]()
Primary aliphatic amines can be oxidized to aldehydes or ketones. using silver compounds as shown.633 Other reagents have been used,634 including N-bromoacetamide635 (for benzylic amines), or aq NaOCl with phase-transfer catalysts.636 Several indirect methods for achieving the conversion RR′CHNH2 → RR′C=O (R′ = alkyl, aryl, or H) have been reported.637
Primary, secondary, and tertiary aliphatic amines have been cleaved to give aldehydes, ketones, or carboxylic acids with aq bromine638 and with neutral permanganate.639 The other product of this reaction is the amine with one less alkyl group. Reaction of a primary amine with benzoyl peroxide/CsCO3 and subsequent heating of the hydroxylamine product gives the ketone.640 In a different type of procedure, primary alkyl primary amines can be converted to gem-dihalides [RCH2NH2 → RCHX2 (X = Br or Cl)] by treatment with an alkyl nitrite and the anhydrous copper(I) halide.641
Primary and secondary aliphatic nitro compounds have been oxidized to aldehydes and ketones, respectively (RR′CHNO2 → RR′C=O), with sodium chlorite under phase-transfer conditions,642 tetrapropylammonium perruthenate (TPAP),643 as well as with other reagents.644
19-22 Oxidation of Primary Alcohols to Carboxylic Acids or Carboxylic Esters
Oxo-de-dihydro-bisubstitution
![]()
Primary alcohols can be oxidized to carboxylic acids by many strong oxidizing agents including chromic acid, permanganate,645 nitric acid,646 or H5IO6/CrO3.647 The reaction can be looked on as a combination of 19-3 and 19-23. Aliphatic primary alcohols are converted to the carboxylic acid with 30% aq H2O2, tetrabutylammonium hydrogen sulfate and a W catalyst with microwave irradiation.648 Benzylic alcohols are oxidized to benzoic acid derivatives by treatment first with TEMPO649 (Sec. 5.C.i), and then NaClO2.650 Oxidation with 5% aq NaOCl and a Ni catalyst oxidizes primary alcohols to the corresponding acid.651 Similar oxidation to the acid occurred with NaIO4/RuCl3 in aq acetonitrile,652 or 30% aq H2O2 and a Co salen catalyst.653 Oxammonium salts and NaClO2 oxidize alcohols to carboxylic acids.654
When acidic conditions are used, a considerable amount of carboxylic ester (RCOOCH2R) is often isolated, although this is probably not formed by a combination of the acid with unreacted alcohol, but by a combination of intermediate aldehyde with unreacted alcohol to give an acetal or hemiacetal, which is oxidized to the ester.655 A mixture of Oxone and NaCl converts alcohols to symmetrical esters.656 Aliphatic alcohols are converted to a symmetrical ester (RCH2OH → RCOOCH2R) by oxidation with PCC on aluminum without solvent.657 Hydrogen with a Ru–CO complex converts primary alcohols (ROH) to an ester (RCO2R).658 Iodine has been used to convert alcohols to esters.659 Hydrogen transfer with a Ru catalyst has been used to convert primary alcohols to methyl esters.660 Oxone in aq methanol also converts aryl aldehydes to the corresponding ester.661 Allylic alcohols are converted to conjugated esters with MnO2 and NaCN in methanol–acetic acid.662 Primary alcohols are oxidized to the methyl ester with trichloroisocyanuric acid in methanol.663 This reagent also converts diols to lactones. Lactones can be prepared by oxidizing diols in which at least one OH is primary,664 and addition of a chiral additive (e.g., sparteine) leads to lactones with high asymmetric induction.665
Primary alcohols (RCH2OH) can be directly oxidized to acyl fluorides (RCOF) with cesium fluoroxysulfate.666 2-(3-Hydroxypropyl)aniline was oxidized to an acyl derivative that cyclized to give a lactam when heated with a Rh catalyst.667
OS I, 138, 168; IV, 499, 677; V, 580; VII, 406; IX, 462; 81, 195. Also see, OS III, 745.
19-23 Oxidation of Aldehydes to Carboxylic Acids, Carboxylic Esters, and Related Compounds
Hydroxylation or Hydroxy-de-hydrogenation
![]()
Oxidation of aldehydes to carboxylic acids is quite common668 and has been carried out with many oxidizing agents, including permanganate in acid, basic, or neutral solution,669 chromic acid,670 bromine, and Oxone.671 Silver oxide is a fairly specific oxidizing agent for aldehydes and does not readily attack other groups. Benedict's and Fehling's solutions oxidize aldehydes,672 and there is a test for aldehydes that depends on this reaction, but the method is seldom used for preparative purposes and gives very poor results with aromatic aldehydes. α,β-Unsaturated aldehydes can be oxidized by sodium chlorite without disturbing the double bond.673 Aldehydes are also oxidized to carboxylic acids by atmospheric oxygen, but the actual direct oxidation product in this case is the peroxy acid (RCO3H),674 which with another molecule of aldehyde disproportionates to give two molecules of acid (see Reaction 14-7).675 The air oxidation of aldehydes to carboxylic acids is mediated by a mixture of Pd/C–NaBH4 and KOH.676 An aldehyde can be converted to the carboxylic acid by treatment with 30% H2O2 and methyl(trioctyl)ammonium hydrogen sulfate at 90 °C.677 Aryl aldehydes are similarly oxidized by a mixture of H2O2 and selenium dioxide (SeO2).678 Polymer-bound hypervalent iodine + TEMPO oxidizes aldehydes to acids.679 Hydrogen peroxide oxidizes aldehydes to carboxylic acids in the presence of a AgNO3 catalyst680 or a Pd catalyst.681
Aryl aldehydes are converted to the corresponding aryl carboxylic ester with H2O2 and a V2O5 catalyst682 or a titanosilicate683 in an alcohol solvent. Esterification of aldehydes with alcohols uses an Ir catalyst.684 The reaction of aldehydes with aq alcohols, in the presence of iodine and NaNO2, gives an ester.685 Organoboronic acids and molecular oxygen convert aldehydes to an ester using a Pd catalyst.686 N-heterocyclic carbenes catalyze oxidation of aldehydes to the corresponding ester.687 Aldehydes (RCHO) can be directly converted to carboxylic esters (RCOOR′) by treatment with Br2 in the presence of an alcohol.688
Aldehydes react with amines, mediated by La catalysts, to give amides.689
Mechanisms of aldehyde oxidation690 are not firmly established, but there are at least two main types: a free radical mechanism and an ionic one. In the free radical process, the aldehyde hydrogen is abstracted to leave an acyl radical, which obtains OH from the oxidizing agent. In the ionic process, the first step is addition of a species −OZ to the carbonyl bond to give 29 in alkaline solution and 30 in acid or neutral solution. The aldehyde hydrogen of 29or 30 is then lost as a proton to a base, while Z leaves with its electron pair.
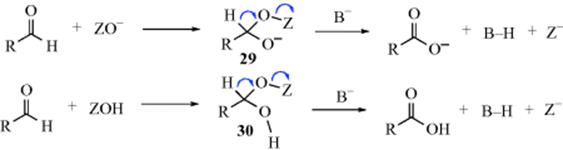
For oxidation with acid dichromate the picture seems to be quite complex, with several processes of both types going on:691
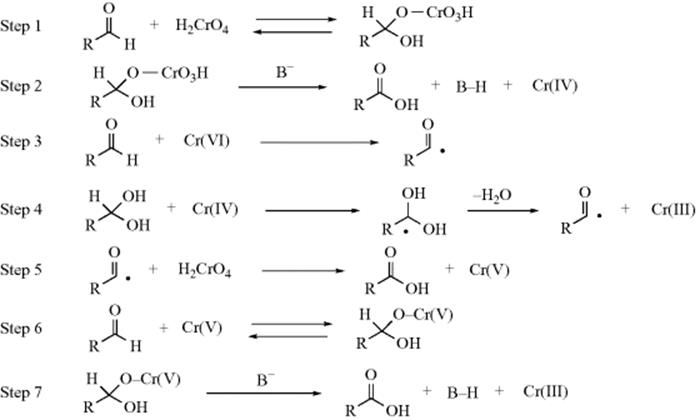
Steps 1 and 2 constitute an oxidation by the ionic pathway by Cr(VI), and steps 6 and 7 a similar oxidation by Cr(V), which is produced by an electron-transfer process. Either Cr(VI) (step 3) or Cr(IV) (step 4) [Cr(IV) is produced in step 2] may abstract a hydrogen and the resulting acyl radical is converted to carboxylic acid in step 5. Thus, Cr in three oxidation states is instrumental in oxidizing aldehydes. Still another possible process has been proposed in which the chromic acid ester decomposes as follows:692
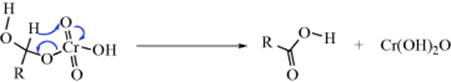
The mechanism with permanganate is less well known, but an ionic mechanism has been proposed693 for neutral and acid permanganate, similar to steps 1 and 2 for dichromate:
![]()
For alkaline permanganate, the following mechanism has been proposed:694
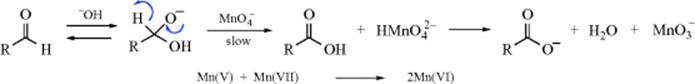
OS I, 166; II, 302, 315, 538; III, 745; IV, 302, 493, 499, 919, 972, 974.
The conversion of thioketones to sulfines (R2C=S=O) is difficult to categorize into the sections available, and it placed after oxidation of ketones and aldehydes. The reaction of a thioketone with H2O2 and a catalytic amount of MTO (methyl trioxorhenium) gives the sulfine.695
19-24 Oxidation of Carboxylic Acids to Peroxy Acids
Peroxy-de-hydroxy-substitution
![]()
The oxidation of carboxylic acids with H2O2 and an acid catalyst is the best general method for the preparation of peroxy acids.696 A mixture of Me2C(OMe)OOH and DCC has also been used.697 Concentrated H2SO4 is a common catalyst for aliphatic R. The reaction is in equilibrium and is driven to the right by removal of water or by the use of excess reagents. For aromatic R, the best catalyst is methanesulfonic acid, which is also used as the solvent.
D. Reactions in which Oxygen is Added to the Substrate
19-25 Oxidation of Alkenes to Aldehydes and Ketones
1/Oxo-(1/→2/hydro)-migro-attachment
![]()
Monosubstituted and 1,2-disubstituted alkenes can be oxidized to aldehydes and ketones by PdCl2, where the PdCl2 is reduced to Pd.698 Similar salts of noble metals also work, but 1,1-disubstituted alkenes generally give poor results. The reaction is used industrially to prepare acetaldehyde from ethylene (the Wacker process),699 but it is also suitable for laboratory preparations. The reagent is expensive, so the reaction is usually carried out with a cooxidant, often CuCl2, whose function is to reoxidize the Pd to Pd(II). The CuCl2 is reduced to Cu(I), which itself is reoxidized to Cu(II) by air, so that atmospheric oxygen is the only oxidizing agent actually used up. Many other cooxidants have been tried, among them O3, Fe3+, and PbO2. Terminal alkenes are oxidized to methyl ketones with O2 and a Pd catalyst.700 The principal product is an aldehyde only from ethylene: With other alkenes Markovnikov's rule is followed, and ketones are formed predominantly.
The generally accepted mechanism shown below involves π complexes of Pd.701 This mechanism accounts for the fact, established by deuterium labeling, that the four hydrogen atoms of the acetaldehyde all come from the original ethylene and none from the solvent.
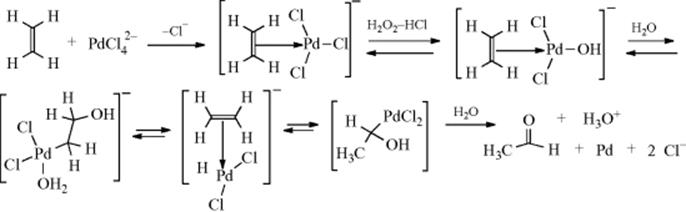
Similar reactions have been carried out with other oxidizing agents. An example involving migration of an alkyl group instead of hydrogen is oxidation of Me2C=CMe2 with peroxytrifluoroacetic acid–boron trifluoride to give Me3COMe (pinacolone).702 This reaction consists of epoxidation (15-50) followed by pinacol rearrangement of the epoxide (18-2). A migration is also involved in the conversion of ArCH=CHCH3 to ArCH(CH3)CHO by treatment with I2–Ag2O in aq dioxane.703
Other reagents used have been Pb(OAc)4–F3CCO2H704 (e.g., PhCH=CH2 → PhCH2CHO), H2O2 and a Pd catalyst,705 H2O–PdCl2–polyethylene glycol.706 Terminal alkenes react with ceric ammonium nitrate in methanol to give α-methoxy ketones.707
Alkenes have also been converted to more highly oxidized products. Examples are (1) Treatment with KMnO4 in aq acetone containing acetic acid gives α-hydroxy ketones.708 (2) 1,2-Disubstituted and trisubstituted alkenes give α-chloro ketones when oxidized with chromyl chloride in acetone: RCH=CR1R2 → RCOCClR1R2.709 (3) α-Iodo ketones can be prepared by treating alkenes with bis(sym-collidine)iodine(I) tetrafluoroborate.710 (4) Potassium permanganate in acetic anhydride oxidizes large-ring cycloalkenes to 1,2-diketones.711
Enol ethers are oxidized to carboxylic esters (RCH=CHOR′ → RCH2COOR′) with PCC712 and enamines to α-amino ketones with N-sulfonyloxaziridines.713 Enamines (R1R4C=CR2NR23, R4 ≠ H) do not give these products, but lose the amino group to give α-hydroxy ketones [R1R4C(OH)COR2].713 Carboxylic acids can be prepared from terminal alkynes by conversion of the alkyne to its phenylthio ether (RC![]() CSPh) and treatment of this with HgSO4 in HOAc–H2SO4.714 Aza-Wacker reactions are known.715
CSPh) and treatment of this with HgSO4 in HOAc–H2SO4.714 Aza-Wacker reactions are known.715
OS VI, 1028; VII, 137; VIII, 208.
19-26 The Oxidation of Alkynes to α-Diketones
Dioxo-biaddition

Internal alkynes have been oxidized716 to α-diketones by several oxidizing agents,717 including neutral KMnO4,718 bis(trifluoroacetoxy)iodobenzene,719 NaIO4–RuO2,720 MeReO3/H2O2,721 or oxygen and a mixture of Pd and Cu catalysts.722 A Ru complex with a small amount of trifluoroacetic acid converts internal alkynes to the α-diketone.723 Ozone generally oxidizes triple-bond compounds to carboxylic acids (Reaction 19-9), but α-diketones are sometimes obtained.724 Selenium dioxide (SeO2) with a small amount of H2SO4 oxidizes alkynes to α-diketones, as well as arylacetylenes to α-keto acids (ArC![]() CH → ArCOCO2H).725 A mixture of formic acid, methanesulfonic acid and DMSO with an HBr catalyst converts alkynes to α-diketones.726
CH → ArCOCO2H).725 A mixture of formic acid, methanesulfonic acid and DMSO with an HBr catalyst converts alkynes to α-diketones.726
19-27 Oxidation of Amines to Nitroso Compounds and Hydroxylamines and Related
N-Oxo-de-dihydro-bisubstitution
![]()
Primary aromatic amines can be oxidized727 to nitroso compounds. Most often the conversion is accomplished by Caro's acid (H2SO5) or with H2O2 in HOAc.728 Other reagents used for this oxidation are sodium perborate729H2O2 with a Ti complex,730 HOF generated in situ,731 and Na2WO4/H2O2.732 Hydroxylamines, which are probably intermediates in most cases, can sometimes be isolated, but under the reaction conditions they are generally oxidized to the nitroso compounds. Primary aliphatic amines can be oxidized in this manner, but the nitroso compound is stable only if there is no α hydrogen. If there is an α hydrogen, the compound tautomerizes to the oxime.733
The mechanism with H2SO5 has been postulated to be an example of category 5 (Sec. 19.A).734
Secondary amines (R2NH) are oxidized to hydroxylamines (R2NHOH), which are resistant to further oxidation, by dimethyldioxirane735 and by benzoyl peroxide and Na2HPO4.736 Oxone on silica also oxidizes secondary alcohols to the hydroxylamine.737 Hydroxylamines are formed when secondary amines react with the enzyme cyclohexanone monooxygenase.738 Carbamates (e.g., N-Boc amines) are converted to the N-hydroxy compound with bis(trifluoromethyl)dioxirane.739 Dialkylamines are oxidized to the N-nitroso compound with N2O2 on poly(vinylpyrrolidinone).740
OS III, 334; VIII, 93; 80, 207.
19-28 Oxidation of Primary Amines, Oximes, Azides, Isocyanates, or Nitroso Compounds to Nitro Compounds
![]()
Tertiary alkyl primary amines can be oxidized to nitro compounds in excellent yields with KMnO4.741 This type of nitro compound is not easily prepared in other ways. All classes of primary amine (including primary, secondary, and tertiary alkyl, as well as aryl) are oxidized to nitro compounds in high yields with dimethyldioxirane.742 Other reagents that oxidize various types of primary amines to nitro compounds are dry ozone,743 various peroxyacids,744MeReO3/H2O2,745 Oxone,746tert-butyl hydroperoxide in the presence of certain Mo and V compounds,747 and sodium perborate.748 An aqueous solution of fluorine oxidizes amino esters to α-nitro esters.749
Dimethyldioxirane in wet acetone oxidizes isocyanates to nitro compounds (RNCO → RNO2).750 Oximes can be oxidized to nitro compounds with peroxytrifluoroacetic acid, or sodium perborate,751 among other ways.741Secondary hydroxylamines are also oxidized to nitrones with MnO2 in dichloromethane.752 Primary and secondary alkyl azides have been converted to nitro compounds by treatment with Ph3P followed by ozone.753 An aqueous solution of fluorine also oxidizes azides to the corresponding nitro compound.754 Aromatic nitroso compounds are easily oxidized to nitro compounds by many oxidizing agents.755
OS III, 334; V, 367, 845; VI, 803; 81, 204.
19-29 Oxidation of Tertiary Amines to Amine Oxides
N-Oxygen-attachment
![]()
Tertiary amines can be converted to amine oxides by oxidation. Hydrogen peroxide is often used, but peroxyacids are also important reagents for this purpose. Pyridine and its derivatives are oxidized by peroxyacids756 rather than hydrogen peroxide. Note, however, that urea–H2O2 in formic acid does indeed oxidize pyridine.757 Oxidation with Caro's acid has been shown to proceed in this manner:758
This mechanism is the same as that of Reaction 19-27; the products differ only because tertiary amine oxides cannot be further oxidized. The mechanism with other peroxyacids is probably the same. A green procedure for oxidation of tertiary amines has been developed, using a Mg–Al complex with aq H2O2.759 Nitrones can be prepared using a ball-mill.760
An alternative oxidation using O2 and a RuCl3 catalyst converted pyridine to pyridine N-oxide.761 Bromamine-T and RuCl3 in aq acetonitrile also oxidizes pyridine to the N-oxide.762 Tertiary amines are oxidized to the N-oxide with O2 and Fe2O3 in the presence of an aliphatic aldehyde.763 Oxygen and a Co–Schiff base complex also oxidizes tertiary amines, including pyridine.764
Analogous to the oxidation of tertiary amines, tertiary phosphines are oxidized to phosphine oxides (R3P=O). Triphenylphosphine is converted to triphenylphosphine oxide with N2O at 100 °C, for example. Triphenylphosphine is also oxidized with PhIO on Montmorillonite K-10.765 tert-Butylhydroperoxide oxides Ph3P → BH3 to Ph3P=O.766 P-Stereogenic phosphine oxides have been prepared.767
OS IV, 612, 704, 828; VI, 342, 501; VIII, 87.
19-30 Oxidation of Thiols and Other Sulfur Compounds to Sulfonic Acids
Thiol-sulfonic acid oxidation
![]()
Thiols, sulfoxides, sulfones, disulfides,768 and other sulfur compounds can be oxidized to sulfonic acids with various oxidizing agents, but for synthetic purposes the reaction is most important for thiols.769 Among oxidizing agents used are boiling nitric acid, barium permanganate, and dimethyl dioxirane.770 Autoxidation (oxidation by atmospheric oxygen) can be accomplished in basic solution.771 Oxidation of thiols with chlorine and water gives sulfonyl chlorides directly.772 Thiols can also be oxidized to disulfides (Reaction 19-34).
OS II, 471; III, 226. Also see, OS V, 1070.
19-31 Oxidation of Thioethers to Sulfoxides and Sulfones
S-Oxygen-attachment
![]()
Thioethers can be oxidized to sulfoxides by 1 equiv of 30% H2O2 or by many other oxidizing agents,773 including NaIO4,774 H2O2, and a Sc(OTf)3 catalyst,775 HIO3/wet SiO2,776 dioxiranes,777 MeReO3/H2O2,778 O2 and a ceric ammonium nitrate catalyst,779 KO2/Me3SiCl,780 hexamethylene triamine–Br2 with CHCl3–H2O,781 H5IO6/FeCl3,782 hypervalent iodine compounds,783 and peroxyacids.784 Sulfoxides can be further oxidized to sulfones by another equivalent of H2O2, KMnO4, sodium perborate, or a number of other agents. If enough oxidizing agent is present, thioethers can be directly converted to sulfones without isolation of the sulfoxides.785 Thioethers can be oxidized directly to the sulfone by treatment with TPAP,786 H2O2787 and an Fe catalyst,788 a Zr catalyst,789 a Ta catalyst,790 a V catalyst,791 an Au catalyst,792 a Mo catalyst,793 a flavin-ionic liquid catalyst,794 urea–H2O2,795 peroxy monosulfate and a Mn catalyst,796 or nitric acid with P2O5 on silica gel.797 These reactions give high yields, and many functional groups do not interfere.798
As with tertiary amines (Reaction 19-29), racemic thioethers can be kinetically resolved by oxidation to sulfoxides with an optically active reagent, and this has often been done.799 In addition, the use of chiral additives in conjunction with various oxidizing agents leads to chiral nonracemic sulfoxide with good-to-excellent enantioselectivity.800 Asymmetric oxidation using bacterial monooxygenases is known,801 and horseradish peroxidase gives modest enantioselectivity.802 Chiral sulfur reagents are also known.803 It is possible to oxidize a thioether to a sulfoxide in the presence of an alcohol moiety using MnO2/HCl.804 N-Sulfonyloxaziridines can be used to oxidize sulfides to sulfoxides.805 Selenides (R2Se) can be oxidized to selenoxides and selenones.806 Alkyl disulfides give oxidation of one sulfur to give a RS–S(=O)R compound with good enantioselectivity when using aq H2O2, a catalytic amount of a V catalyst, and a chiral Schiff base ligand.807
When the oxidizing agent is a peroxide, the mechanism808 of oxidation to the sulfoxide is similar to that of Reaction 19-29.809
The second oxidation, which is normally slower than the first, which is why sulfoxides are so easily isolable, has the same mechanism in neutral or acid solution, but in basic solution it has been shown that the conjugate base of the peroxy compound (R′OO−) also attacks the SO group as a nucleophile:810
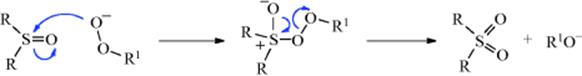
There are some reagents that oxidize sulfoxides in preference to sulfides (e.g., NaMnO4).811
OS V, 791; VI, 403, 404, 482; VII, 453, 491; VIII, 464, 543; IX, 63; 80, 190. Also see, OS V, 723; VI, 23.
E. Oxidative Coupling
19-32 Coupling Involving Carbanions
De-hydro,chloro-coupling
![]()
Alkyl halides with an electron-withdrawing group on the halogen-bearing carbon can be dimerized to alkenes by treatment with bases. The Z group may be nitro, aryl, and so on. It is likely that in most cases the mechanism812involves nucleophilic substitution followed by elimination813 (illustrated for benzyl chloride):
![]()
α,α-Dibromotoluenes (ArCHBr2) give tolanes (ArC![]() CAr) by debromination of the intermediates ArCBr=CBrAr.814 In a related reaction, diarylmethane dihalides (Ar2CX2) have been dimerized to tetraaryl alkenes (Ar2C=CAr2) with Cu,815 and with iron(II) oxalate dihydrate.816
CAr) by debromination of the intermediates ArCBr=CBrAr.814 In a related reaction, diarylmethane dihalides (Ar2CX2) have been dimerized to tetraaryl alkenes (Ar2C=CAr2) with Cu,815 and with iron(II) oxalate dihydrate.816
A somewhat different type of coupling is observed when salts of β-keto esters, arylacetonitriles (ArCH2CN), and other compounds of the form ZCH2Z′ are treated with an oxidizing agent (e.g., iodine,817 or Cu(II) salts.]818Arylmethanesulfonyl chlorides (ArCH2SO2Cl) couple to give ArCH=CHAr when treated with Et3N.819
OS II, 273; IV, 372, 869, 914; VIII, 298. Also see, OS I, 46; IV, 877.
19-33 Dimerization of Silyl Enol Ethers or of Lithium Enolates
3/O-De-trimethylsilyl-1/C-coupling
Silyl enol ethers can be dimerized to symmetrical 1,4-diketones by treatment with Ag2O in DMSO or certain other polar aprotic solvents.820 The reaction has been performed with R2, R3 = hydrogen or alkyl, although best yields are obtained when R2 = R3 = H. In certain cases, unsymmetrical 1,4-diketones have been prepared by using a mixture of two silyl enol ethers. Other reagents that have been used to achieve either symmetrical or cross-coupled products are iodosobenzene–BF3–OEt2,821 ceric ammonium nitrate,822 and lead tetraacetate.823 If R1 = OR (in which case the substrate is a ketene silyl acetal), dimerization with TiCl4 leads to a dialkyl succinate (31, R1 = OR).824
In a similar reaction, lithium enolates [RC(OLi)=CH2] were dimerized to 1,4-diketones (RCOCH2CH2COR) with CuCl2, FeCl3, or copper(II) triflate, in a nonprotic solvent.825
OS VIII, 467.
19-34 Oxidation of Thiols to Disulfides
S-De-hydrogen-coupling
![]()
Thiols are easily oxidized to disulfides.826 Hydrogen peroxide is the most common reagent,827 but many oxidizing agents give the reaction, among them Br2 on hydrated silica,828 sodium perborate,829 SmI2,830 PPh3 with a Rh catalyst,831 cetyltrimethylammonium dichromate,832 and NO. Hydrogen peroxide (30%) in hexafluoro-2-propanol converts thiols to disulfides,833 and solventless reactions on MnO2,834 PCC (Reaction 19-3, category 1)835 or SO2Cl2836 are also effective. A potassium phosphate catalyst has been used for the oxidative coupling of thiols to disulfides.837 Strong oxidizing agents may give Reaction 19-26. Even oxygen in the air oxidizes thiols on standing, if a small amount of base is present. The reaction is reversible (see Reaction 19-75), and the interconversion between cysteine and cystine is an important one in biochemistry.
The mechanism has been studied for several oxidizing agents and varies with the agent.838 For oxygen it is839
With respect to the sulfur, this mechanism is similar to that of Reaction 14-16, involving as it does loss of a proton, oxidation to a free radical, and radical coupling.
Unsymmetrical disulfides can be prepared840 by treatment of a thiol RSH with diethyl azodicarboxylate (EtOOCN=NCOOEt) to give an adduct, to which another thiol (R′SH) is then added, producing the disulfide (RSSR′).841
OS III, 86, 116.
19.B.ii. Reductions
For the most part, reductions have been grouped into this chapter, with a few notable exceptions. Catalytic hydrogenation of alkenes and alkynes in Reactions 15-11 and 15-12, hydrogenation of aromatic rings in Reaction 15-13, and reductive cleavage of cyclopropanes in Reaction 15-15 were placed in Chapter 15 to coincide with addition reactions, and protonolysis of alkyl boranes in Reaction 15-16 was placed there also for continuity. In general, reductions of functional groups encompass a variety of reaction types. The reactions in this section are classified into groups depending on the type of bond change involved. These groups are (1) attack at carbon (C–O and C=O), (2) attack at non-carbonyl multiple bonds to heteroatoms, (3) reactions in which a heteroatom is removed from the substrate, (4) reduction with cleavage, (5) reductive coupling, and (6) reactions in which an organic substrate is both oxidized and reduced. Most of the reagents in this section are metal hydrides, metals with an acid or a protic solvent, hydrogen gas with a catalyst, and so on. Other reducing agents are available, and will be introduced in the appropriate section. Note that plants can be used as reducing agents.842
A. Selectivity843
It is often necessary to reduce one group in a molecule without affecting another reducible group (this is called chemoselectivity), and reducing agents are available that will do this. The most common broad-spectrum reducing agents are the metal hydrides844 or hydrogen (with a catalyst).845 Many different metal hydride systems and hydrogenation catalysts have been investigated in order to find conditions under which a given group will be reduced chemoselectively. The ease of reduction for various functional groups toward catalytic hydrogenation is acyl halides > alkyl nitro compounds > alkynes > aldehydes > alkenes > ketones > benzylic ethers > nitriles > esters > amides.846 Futher, Table 19.2 and 19.4 list the reactivity of various functional groups with LiAlH4, and BH3, respectively.846 Table 19.5 shows which groups can be reduced by catalytic hydrogenation and various metal hydrides.847Of course, the tables cannot be exact, because the nature of R and the reaction conditions obviously affect reactivity. Nevertheless, the tables do give a fairly good indication of which reagents reduce which groups.848 Lithium aluminum hydride is very powerful and unselective reagent.849 Other metal hydrides are generally used when chemoselectivity is required. As will be seen in Reaction 19-36, less reactive (and more selective) reagents have been prepared by replacing some of the hydrogen atoms of LiAlH4 with alkoxy groups.850 Most of the metal hydrides are nucleophilic reagents and attack the carbon atom of a carbon-hetero single or multiple bond. However, BH3857,858and AlH3859 are electrophiles (Lewis acids) and attack the heteroatom. This accounts for the different patterns of selectivity in the tables.
Table 19.2 The Ease of Reduction of Various Functional Groups Toward Catalytic Hydrogenation846
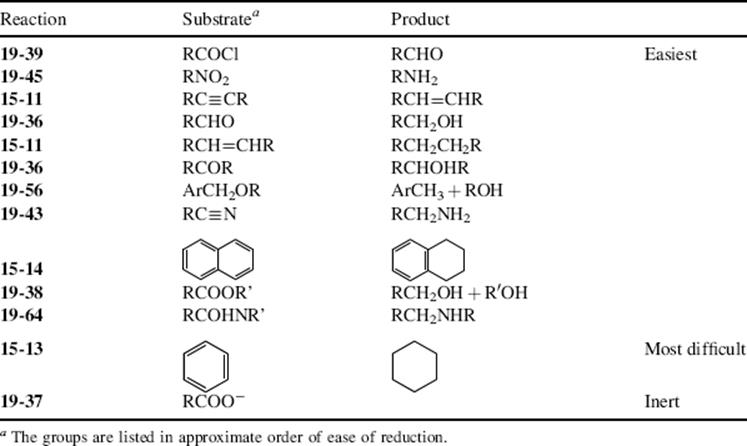
Table 19.3 The Ease of Reduction of Various Functional Groups with LiAlH4 in Ethera
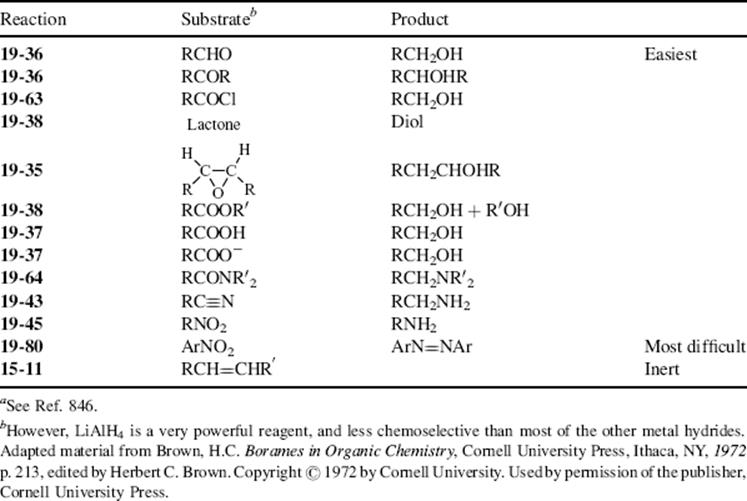
Table 19.4 The Ease of Reduction of Various Functional Groups With Boranea
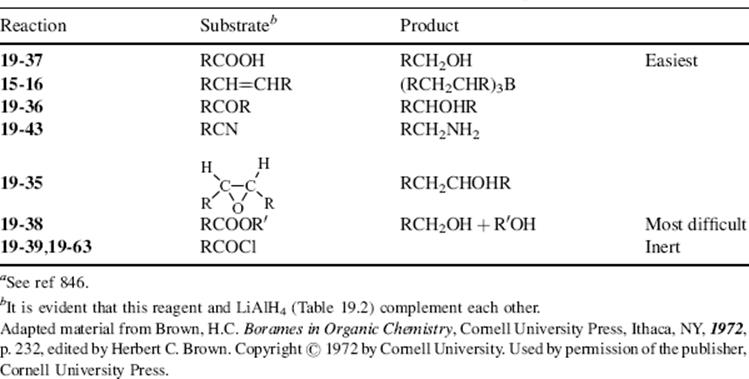
Table 19.5 Reactivity of Various Functional Groups With Some Metal Hydrides and Toward Catalytic Hydrogenationa
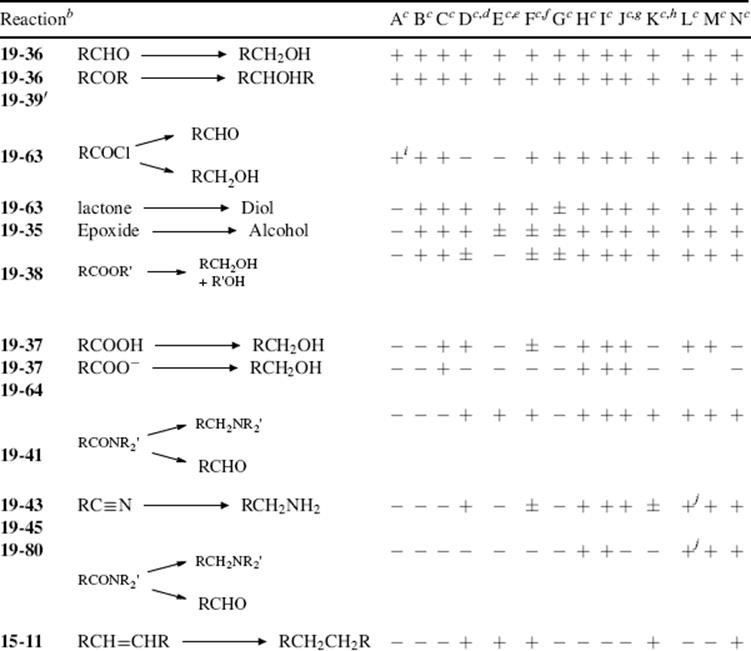
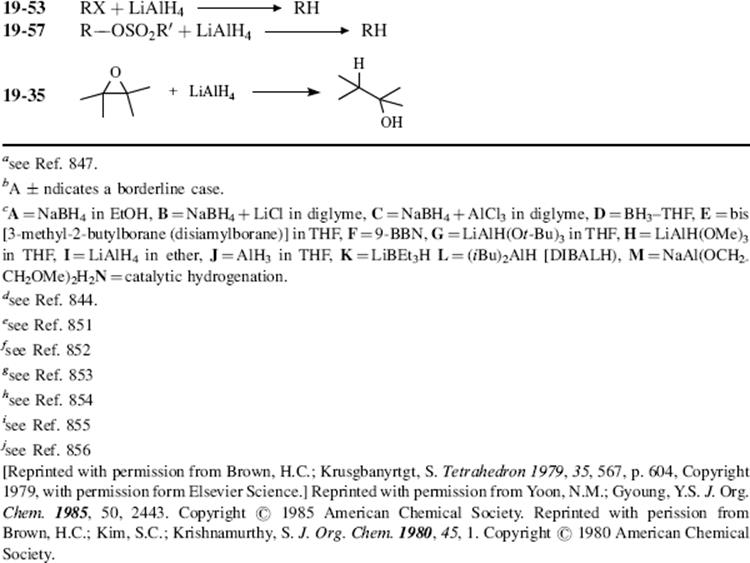
B. Attack at Carbon (C–O And C=O)
19-35 Reduction of Epoxides
(3) OC-seco-Hydro-de-alkoxylation
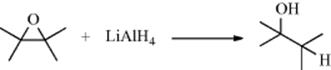
Reduction of epoxides is a special case of Reaction 19-56 and is easily carried out.860 A common reagent is LiAlH4,861 which reacts by the SN2-type mechanism, giving inversion of configuration. An epoxide on a substituted cyclohexane ring cleaves in such a direction as to give an axial alcohol. As expected for an SN2 mechanism, the hydrogen atom is usually delivered to the less substituted carbon. The reaction has also been carried out with other reagents (e.g., sodium amalgam in EtOH, Li in ethylenediamine,862 and by catalytic hydrogenolysis).863 Chemoselective and regioselective ring opening of allylic epoxides and of epoxy ketones and esters has been achieved with SmI2,864 HCOOH–NEt3 and a Pd catalyst,865 and sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al; also known as Vitride).866 Highly hindered epoxides can be conveniently reduced, without rearrangement, with lithium triethylborohydride (called Super Hydride).867 For certain substrates, the epoxide ring can be opened the other way by reduction with NaBH4–ZrCl4,868 Pd/C and HCOONH4,869 or with BH3 in THF.870
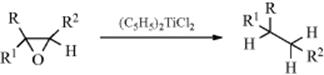
Epoxy ketones are selectively reduced with lithium naphthalenide871 or Cp2TiCl in THF/MeOH872 to the β-hydroxyketone. Other reduction methods can lead to the epoxy alcohol. Reduction of epoxy amides with SmI2 in methanol gave the α-hydroxyamide.873
Epi-sulfides can be reduced to give the alkene using Bu3SnH in the presence of BEt3.874
The usual product of epoxide reductions is the alcohol, but epoxides are reduced all the way to the alkane by titanocene dichloride875 and by Et3SiH–BH3.876
19-36 Reduction of Aldehydes and Ketones to Alcohols877
C,O-Dihydro-addition
![]()
Aldehydes can be reduced to primary alcohols, and ketones to secondary alcohols, by a number of reducing agents.878 Among the most common are the metal hydrides (e.g., LiAlH4, NaBH4, and related compounds).879 These reagents have two main advantages over many other reducing agents: They do not reduce carbon–carbon double or triple bonds (with the exception of propargylic alcohols),880 and with LiAlH4 all four hydrogen atoms are theoretically usable for reduction. Methods are available for titering hydride reagents.881 The scope of these reagents with ketones is similar to that with aldehydes. Lithium aluminum hydride reduces even sterically hindered ketones.
The reaction is broad and general. Lithium aluminum hydride easily reduces aliphatic, aromatic, alicyclic, and heterocyclic aldehydes or ketones, containing double or triple bonds and/or nonreducible groups (e.g., NR3, OH, OR, and F). If the molecule contains a group reducible by LiAlH4 (e.g., NO2, CN, COOR), then that group is usually reduced. Since LiAlH4 reacts readily with water and alcohols, protic solvents must be excluded. Despite limited solubility, common solvents are ether and THF.
The compound NaBH4 (sodium borohydride) has a similar scope, but is less reactive, so it is more selective and so may be used with NO2, Cl, COOR, CN, and so on in the molecule. Another advantage of NaBH4 is that it can be used in water or alcoholic solvents,and so reduces compounds, (e.g., sugars) that are not soluble in ethers.882 Other solvents can be used with some modification of the borohydride. For example, butyltriphenylphosphonium borohydride reduces aldehydes to alcohols in dichloromethane.883 Reduction with solid acid activated NaBH4 has been reported.884 A polymer-bound phase-transfer material with NaBH4 in wet THF has also been used.885 Sodium borohydride on alumina, under microwave irradiation, is an effective reagent.886 Sodium borohydride has been used on silica gel.887
Red-Al was prepared by Vit in 1967,888 and its reducing power is close to that of LiAlH4. In addition, it is stable to dry air (it does not ignite in even moist air or oxygen), and is thermally stable up to 200 °C. The greatest practical utility of Red-Al is its solubility in aromatic hydrocarbon and ether solvents, which allows it to be conveniently used for applications that require inverse addition of hydrides. Red-Al essentially reacts the same as LiAlH4, reducing aldehydes, ketones889 and acid derivatives to alcohols.890 Reduction of conjugated carbonyls gives primarily 1,2-reduction to an allylic alcohol.891 Other functional groups can be reduced.892
The C=C units in compounds that contain double bonds are generally not affected by metallic hydrides, and the C=C unit may be isolated or conjugated, but double bonds that are conjugated with the C=O group may or may not be reduced, depending on the substrate, reagent, and reaction conditions.893 Some reagents that reduce only the C=O bonds of α,β-unsaturated aldehydes and ketones are AlH3,894 NaBH4, or LiAlH4 in the presence of lanthanide salts,895 Co complexes,896 NaBH4–LiClO4,897 Ni compounds,898 NaBH3(OAc),899 Zn(BH4)2900 on Y-zeolite,901 and Et3SiH.902 Also, both LiAlH4903 and NaBH4904 predominantly reduce only the C=O bonds of C=C–C=O systems in most cases, although substantial amounts of fully saturated alcohols have been found in some cases903 (Reaction 15-14). For some reagents that reduce only the C=C bonds of conjugated aldehydes and ketones, see Reaction 15-11. A mixture of InCl3 and NaBH4 reduced both the C=C and C=O units of conjugated ketones.905
When a functional group is selectively attacked in the presence of a different functional group, the reaction is said to be chemoselective.906 A number of reagents have been found to reduce aldehydes much faster than ketones. Among these907 are sodium triacetoxyborohydride908 (NaBH4–HCO2H),909 zinc borohydride in THF,910 a complex of LiAlH4 and N-methyl-2-pyrrolidinone (of particular interest since it is stable in air and to heating),911 and Raney nickel.912
Ketones can be chemoselectively reduced in the presence of aldehydes with NaBH4 in aq EtOH at −15 °C in the presence of cerium trichloride (CeCl3).913 The reagent lithium n-dihydropyridylaluminum hydride reduces diaryl ketones much better than dialkyl or alkyl aryl ketones.914 Most other hydrides reduce diaryl ketones more slowly than other types of ketones. Saturated ketones can be reduced in the presence of α,β-unsaturated ketones with NaBH4–50% MeOH–CH2Cl2 at −78 °C,915 and with zinc borohydride.916
In general, NaBH4 reduces carbonyl compounds in the order aldehydes > α,β-unsaturated aldehydes > ketones > α,β-unsaturated ketones, and a carbonyl group of one type can be selectively reduced in the presence of a carbonyl group of a less reactive type.917 A number of reagents will preferentially reduce the less sterically hindered of two carbonyl compounds, but by the use of Dibal-H in the presence of the Lewis acid methylaluminum bis(2,16-di-tert-butyl-4-methylphenoxide), it was possible selectively to reduce the more hindered of a mixture of two ketones.918 It is obvious that reagents can often be found to reduce one kind of carbonyl function in the presence of another.919For a discussion of selectivity in reduction reactions (see Sec. 19.B.ii-A). A syn-selective reduction of β-hydroxy ketones was achieved using (iPrO)2TiBH4.920 Quinones are reduced to hydroquinones by LiAlH4, SnCl2–HCl, or sodium hydrosulfite (Na2S2O4), as well as by other reducing agents.
The reagent lithium tri-sec-butylborohydride [LiBH(sec-Bu)3, L-Selectride] reduces cyclic and bicyclic ketones in a highly stereoselective manner.921 For example, 2-methylcyclohexanone gave cis-2-methylcyclohexanol with an isomeric purity > 99%. Both L-Selectride and the potassium salt (K-Selectride) reduce carbonyls in cyclic and acyclic molecules with high diastereoselectivity.922 The more usual reagents (e.g., LiAlH4 and NaBH4) reduce relatively unhindered cyclic ketones either with little or no stereoselectivity923 or give predominant formation of the more stable isomer (axial attack).924 Reduction of cyclohexanone derivatives with the very hindered LiAlH(Cet2CMe3)3 gave primarily the cis-alcohol.925 Cyclohexanones that have a large degree of steric hindrance near the carbonyl group usually give predominant formation of the less stable alcohol, even with LiAlH4 and NaBH4.
Other reagents reduce aldehydes and ketones to alcohols,926 including the following:
1. Hydrogen and a Catalyst.927 Common heterogeneous catalysts for carbonyls are Pt and Ru,928 and homogeneous catalysts are commonly used,929 especially for asymmetric hydrogenation (see A below). Before the discovery of the metal hydrides, this was one of the most common ways of effecting this reduction, but it suffers from the fact that C=C, C![]() C, C=N and C
C, C=N and C![]() N bonds are often more susceptible to attack than C=O bonds.930 For aromatic aldehydes and ketones, reduction to the hydrocarbon (Reaction 19-61) is a side reaction via hydrogenolysis of the initially produced alcohol (Reaction 19-54). The mechanism of catalytic hydrogenation of aldehydes and ketones is probably similar to that of reaction 15–11.931
N bonds are often more susceptible to attack than C=O bonds.930 For aromatic aldehydes and ketones, reduction to the hydrocarbon (Reaction 19-61) is a side reaction via hydrogenolysis of the initially produced alcohol (Reaction 19-54). The mechanism of catalytic hydrogenation of aldehydes and ketones is probably similar to that of reaction 15–11.931
2. Sodium in Ethanol.932 This is called the Bouveault–Blanc procedure and was more popular for the reduction of carboxylic esters (Reaction 19-38) than of aldehydes or ketones before the discovery of LiAlH4.
For the reaction with sodium in ethanol the following mechanism933 has been suggested:934
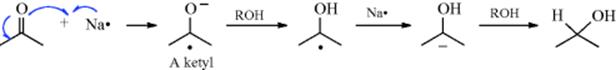
The ketyl intermediate can be isolated.935 Lithium is often a superior metal for alkali metal reductions.936
3. Other Metal Reductions. A single carbonyl group of an α-diketone can be reduced to give an α-hydroxy ketone, by heating with Zn powder in aq DMF937 or Zn in methanol in the presence of benzyltriethylammonium chloride.938 Aluminum and NaOH in aq methanol reduces ketones.939 β-Hydroxy ketones are reduced with good antiselectivity using an excess of SmI2 in water,940 and other ketones or aldehydes are reduced with SmI2941 in aq THF,942 or in alcohols.943 Other metals can be used, including FeCl3/Zn in aq DMF944 or Mg in alcohols.945 1,2-Diketones were reduced to the α-hydroxy ketone with TiI4 in acetonitrile, followed by hydrolysis.946 Ammonia and aq TiCl3 in methanol reduces ketones.947
4. Isopropyl Alcohol and Aluminum Isopropoxide. This is called the Meerwein–Ponndorf–Verley reduction.948 It is reversible, and the reverse reaction is known as the Oppenauer oxidation (see 19-3):
![]()
The equilibrium is shifted by removal of the acetone by distillation. There is a report of the reduction of benzaldehyde to benzyl alcohol by heating with 2-propanol at 225 °C for 1 day.949 The reaction usually takes place under very mild conditions and is highly specific for aldehydes and ketones, so that C=C bonds (including those conjugated with the C=O bonds) and many other functional groups can be present without themselves being reduced.950This includes acetals, so that one of two carbonyl groups in a molecule can be specifically reduced if the other is first converted to an acetal. β-Keto esters, β-diketones, and other ketones and aldehydes with a relatively high enol content do not give this reaction. Zeolites have been used as a medium for this reduction.951 This reduction can be done catalytically952 and an aluminum-free, Zr-zeolite catalyst has been developed.953 Microwave irradiation of a ketone with 2-propanol, KOH, and activated alumina gives good yields of the alcohol.954 When the carbonyl substrate has a stereogenic center, the reaction proceeds with good diastereoselectivity.955 The use of chiral metal complexes leads to chiral hydrogen transfer.956 A combination of 2-propanol with BINOL and AlMe3 leads to reduction of α-chloroketones to the chlorohydrin with good enantioselectivity.957
The Meerwein–Ponndorf–Verley reaction usually958 involves a cyclic transition state:959
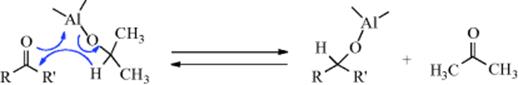
but in some cases 2 molar equivalents of aluminum alkoxide are involved: one attacking the carbon and the other the oxygen, a conclusion that stems from the finding that in these cases the reaction was 1.5 order in alkoxide.960The alcohol solvent acts as a hydrogen donor in this reaction.961 Although, for simplicity, the alkoxide is shown as a monomer, it actually exists as trimers and tetramers, which are the actual reactive species.962 Note that supercritical 2-propanol has been used for reduction of ketones, without the need for a catalyst.963
5. Boranes. Borane (BH3) and substituted boranes reduce aldehydes and ketones in a manner similar to their addition to C=C bonds (Reaction 15-16).964 That is, the boron adds to the oxygen and the hydrogen to the carbon:965
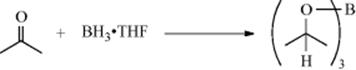
The resulting borate is then hydrolyzed to the alcohol. A variety of alkylboranes can be used for reduction.966 Both 9-BBN967 (Reaction 15-16) and BH3–Me2S968 reduce only the C=O group of conjugated aldehydes and ketones.969 Tributylborane in ionic solvents reduces aldehydes to alcohols.970 Enantioselective borane reductions lead to chiral alcohols.971 Spiroborate esters have been used for enantioselective reduction,972 and chiral boronic esters have also been used.973
Alane (AlH3) derivatives can also be used, including diisobutylaluminum hydride.974
6. Tin Hydrides. Tributyltin hydride reduces aldehydes to primary alcohols by simply heating in methanol.975 A mixture of Bu3SnH and phenylboronic acid (Reaction 12-28) reduces aldehydes in dichloromethane.976 Reduction of ketones was achieved with Bu2SnH2 and a Pd catalyst.977 Using triaryltin hydrides with BF3·OEt2, where aryl is 2,6-diphenylbenzyl, selective reduction of aliphatic aldehydes in the presence of a conjugated aldehyde was achieved.978 Tris(trimethylsilyl)methane has been used as a tin-free radical reducing agent.979
7. Cannizzaro Reaction. In the Cannizzaro reaction (see 19-81), aldehydes without an α hydrogen are reduced to alcohols.
8. Silanes. In the presence of bases, certain silanes can selectively reduce carbonyls.980 Transition metal complexes also catalyze hydrosilylation of ketones.981 Controlling temperature and solvent leads to different ratios of synand anti products.982 Silanes reduce ketones in the presence of BF3·OEt2.983 Ketones are reduced with Cl3SiH in the presence of pyrrolidine carbaldehyde984 or under photochemical conditions.985 Polymethylhydrosiloxane with tetrabutylammonium fluoride reduces α-amino ketones to give the syn amino alcohol.986
9. Ammonium Formates. Sodium formate and trialkylammonium formates can be used to reduce aldehydes and ketones to the corresponding alcohol. Decanal was reduced to decanol, for example, using sodium formate in N-methyl-2-pyrrolidinone as a solvent.987 A mixture of formic acid and ethyl magnesium bromide was used to reduce decanal to decanol in 70% yield.988 Transfer hydrogenation also occurs with formic acid–triethylamine and a Ru catalyst989 in water.990
10. Enzymatic Reductions. Successful asymmetric reductions (see Section A) have been achieved with biologically derived reducing agents991 (e.g., baker's yeast),992 enzymes from other organisms,993 or with other biocatalysts.994 Ionic liquids have been used in conjugation with enzymatic reduction,995 and enzymatic reduction has been done in supercritical CO2.996
With most reagents there is an initial attack on the carbon of the carbonyl group by a hydride equivalent (H−) although with BH3997 the initial attack is on the oxygen. Detailed mechanisms are not known in most cases.998 With LiAlH4 or NaBH4, the attacking species is the AlH4− (or BH4−) ion, which, in effect, transfers H− to the carbon. The following mechanism has been proposed for LiAlH4:999
Evidence that the cation plays an essential role, at least in some cases, is that when the Li+ was effectively removed from LiAlH4 (by the addition of a crown ether), the reaction did not take place.1000 Complex 32 must be hydrolyzed to the alcohol. For NaBH4, the Na+ does not seem to participate in the transition state, but kinetic evidence shows that an OR group from the solvent does participate and remains attached to B:1001
Free H− cannot be the attacking entity in most reductions with boron or aluminum hydrides because the reactions are frequently sensitive to the size of the MH4− [or MRmHn− or M(OR)mHn−etc.].
The question of whether the initial complex in the LiAlH4 reduction (32, or H–C–OAl− H3 = 33) can reduce another carbonyl to give (H–C–O)2Al− H2, and so on has been controversial. It has been shown1002 that this is probably not the case but that, more likely, 33 disproportionates to (H–C–O)4Al− and AlH4−, which is the only attacking species. Disproportionation has also been reported in the NaBH4 reaction.1003
Aluminate (33) is essentially LiAlH4 with one of the hydrogen atoms replaced by an alkoxy group (i.e., LiAlH3OR). The fact that 33 and other alkoxy derivatives of LiAlH4 are less reactive than LiAlH4 itself has led to the use of such compounds as reducing agents that are less reactive and more selective than LiAlH4.1004 An example is LiAlH(O-t-Bu)3 (Reactions 19-39–19-41; see also, Table 19.5). As an example of chemoselectivity in this reaction it may be mentioned that LiAlH(Ot-Bu)3 has been used to reduce only the keto group in a molecule containing both keto and carboxylic ester groups.1005 However, the use of such reagents is sometimes complicated by the disproportionation mentioned above, which may cause LiAlH4 to be the active species, even if the reagent is an alkoxy derivative. Another highly selective reagent (reducing aldehydes and ketones, but not other functional groups), which does not disproportionate, is potassium triisopropoxyborohydride.1006
For other reduction reactions of aldehydes and ketones, see 19-61, 19-76, and 19-81.1007
A. Asymmetric Reduction
Unsymmetrical ketones are prochiral (Sec. 4.M); that is, reduction creates a new stereogenic center:
![]()
The relative effectiveness of various reagents for reduction of eight other types of ketone was determined, using several different reducing agents. The ketones examined included heterocyclic, aralkyl, β-keto esters, β-keto acids,1008and so on.991 Much effort has been put into finding optically active reducing agents that will produce one enantiomer of the alcohol enantioselectively.1009 Each reagent tends to show a specificity for certain types of ketones.1010Good enantioselectivity is usually obtained with the proper reagent.1011 Substituents that are remote to the carbonyl group can play a role in facial selectivity of the reduction.1012 Asymmetric reduction has been accomplished using bio-reagents (e.g., enzymes, see item 10 above).
Asymmetric reduction with very high enantioselectivity has also been achieved with achiral reducing agents and optically active catalysts.1013 Homogeneous catalytic asymmetric hydrogenation leads to reduction of substrates with high enantioselectivity.1014 A typical chiral ligand is 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl-ruthenium acetate (BINAP, 34), used with a metal catalysts [e.g., Ru(OAc)2],1015 β-Keto esters are reduced enantioselectively, for example.1016 A variety of chiral additives and/or ligands have been used with catalytic hydrogenation reactions. Many functional groups can be tolerated.1017 Asymmetric catalytic hydrogenation has been done in ionic liquids.1018
A second approach is reduction with BH3–THF or catecholborane,1019 using an oxazaborolidine (35, R = H, Me, or n-Bu; Ar = Ph or β-naphthyl)1020 or other chiral compounds1021 as a catalyst. Both a polymer-bound oxazaborolidine1022 and a dendritic chiral catalyst have been used in conjunction with borane,1023 as well as other chiral additives can be used.1024 Chiral sulfonamides have been used as additives.1025
Lithium aluminum hydride in combination with a chiral diol1026 or other chiral ligands1027 leads to enantioselective reduction, often in the presence of a transition metal complex.1028 Chiral additives have also been used with NaBH4.1029 Examples include LiBH4/NiCl2 and a chiral amino alcohol,1030 NaBH4 with chiral Lewis acid complexes,1031 or NaBH4/Me3SiCl and a chiral ligand.1032 A mixture of NaBH4 and Me3SiCl with a catalytic amount of a chiral, polymer-bound sulfonamide leads to asymmetric reduction.1033
Enantioselective reduction is possible with the other methods mentioned above. Transition metal catalyzed asymmetric transfer hydrogenation is effective for the preparation of chiral alcohols.1034 Reduction with silanes and transition metal catalysts (e.g., Ru compounds) is also very effective.1035 Chiral Ru catalysts have been used with triethylammonium formate for the enantioselective reduction.1036 The transition metal catalyzed hydrosilylation of ketones gives chiral alcohols in the presence of suitable chiral ligands.1037 Enantioselective reduction was observed with PhSiH3 and Cu compounds with a chiral ligand,1038 with a mixture of Ru and Ag catalysts,1039 or with Mn(dpm)3 and oxygen (dpm = diphenylmethylene).1040 Enantioselective hydrosilylation is possible using chiral organocatalysts.1041 A chiral Sm complex has been used in conjunction with 2-propanol.1042 Chiral mercapto alcohols have also been used for asymmetric reduction.1043
Enantioselective reduction is usually not possible for aldehydes,1044 since the products are primary alcohols in which the reduced carbon is not chiral. Instead deuterated aldehydes (RCDO) give a chiral product, and these have been reduced enantioselectively with B-(3-pinanyl)-9-borabicyclo[3.3.1]nonane (Alpine-Borane) with almost complete optical purity.1045 Other chiral boranes can be used to reduce aldehydes or ketones.1046
In the above cases, an optically active reducing agent or catalyst interacts with a prochiral substrate. Asymmetric reduction of ketones has also been achieved with an achiral reducing agent, if the ketone is complexed to an optically active transition metal Lewis acid.1047
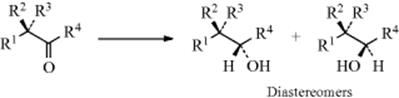
There are other stereochemical aspects to the reduction of aldehydes and ketones. If there is a stereogenic center α to the carbonyl group,1048 even an achiral reducing agent can give more of one diastereomer than of the other (a diastereoselective reduction). Such reductions have been carried out with considerable success.1049 In most such cases, Cram's rule (Sec. 4.H., category 1) is followed, but exceptions are known.1050
OS I, 90, 304, 554; II, 317, 545, 598; III, 286; IV, 15, 25, 216, 660; V, 175, 294, 595, 692; VI, 215, 769, 887; VII, 129, 215, 241, 402, 417; VIII, 302, 312, 326, 527; IX, 58, 362, 676.
19-37 Reduction of Carboxylic Acids to Alcohols
Dihydro-de-oxo-bisubstitution
![]()
Carboxylic acids are easily reduced to primary alcohols by LiAlH4.1051 The conditions are particularly mild, the reduction proceeding quite well at room temperature. Other hydrides have also been used,1052 but not NaBH4 (see Table 19.5).1053 A combination of NaBH4 and an arylboronic acid (Reaction 12-28) is also effective.1054 A mixture of NaBH4, Me2SO4, and B(OMe)3 is effective for the reduction of hydroxyl-substituted aromatic carboxylic acids.1055 Benzyltriethylammonium borohydride is dichloromethane reduces carboxylic acids to the alcohol.1056 Catalytic hydrogenation is generally ineffective.1057
Borane (BH3) is particularly good for carboxyl groups (Table 19.4) and permits selective reduction of them in the presence of many other groups (although the reaction with double bonds takes place at about the same rate in ether solvents).1058 For many years, borane was the reagent of choice for this reduction. Borane also reduces carboxylic acid salts.1059 Aluminum hydride reduces COOH groups without affecting carbon–halogen bonds in the same molecule. The reduction has also been carried out with SmI2 in basic media1060 or aq H3PO4,1061 or simply with SmI2 in water.1062 A mixture of NaBH4 and I2 has been used to reduced amino acids to amino alcohols.1063
OS III, 60; VII, 221; 530; VIII, 26, 434, 528.
19-38 Reduction of Carboxylic Esters to Alcohols
Dihydro,hydroxy-de-oxo,alkoxy-tersubstitution
![]()
Lithium aluminum hydride reduces carboxylic esters to give two different alcohols, as shown.1064 The reaction is of wide scope and has been used to reduce many esters. Where the interest is in obtaining R′OH, this is a method that is often a working equivalent of “hydrolyzing” esters. Reduction of lactones yields diols.1065 Among the reagents used for this reduction1066 are Dibal-H, lithium triethylborohydride, LiAlH(Ot-Bu)3,1067 and BH3–SMe2 in refluxing THF.1068 Although NaBH4 reduces phenolic esters, especially those containing electron-withdrawing groups,1069 its reaction with other esters is usually so slow that it is not the reagent of choice, but there are many exceptions.1070) However, it is generally possible to reduce an aldehyde or ketone without reducing an ester function in the same molecule. Note that NaBH4 reduces esters in the presence of certain compounds (see Table 19.5).1071Note that NaBH4 in DMF–MeOH reduces aryl carboxylic esters to benzylic alcohols,1072 and NaBH4–LiCl with microwave irradiation also reduces esters to primary alcohols.1073
Carboxylic esters can also be reduced to alcohols by hydrogenation over copper chromite catalysts,1074 although high pressures and temperatures are required. Ester functions generally survive low-pressure catalytic hydrogenations, but homogeneous catalytic hydrogenation procedures have been developed.1075 Before the discovery of LiAlH4, the most common way of carrying out the reaction was with sodium in ethanol, a method known as the Bouveault–Blanc procedure.1076 This procedure is still sometimes used where selectivity is necessary (see also, Reactions 19-62, 19-65, and 19-59).
Silanes (e.g., Ph2SiH2), with a catalytic amount of triphenylphosphine and a Rh catalyst reduced esters to primary alcohols.1077 Aliphatic silanes (e.g., EtMe2SiH) also reduced esters with a Ru catalyst.1078
OS II, 154, 325, 372, 468; III, 671; IV, 834; VI, 781; VII, 356; VIII, 155; IX, 251.
19-39 Reduction of Acyl Halides
Hydro-de-halogenation or Dehalogenation
![]()
Acyl halides can be reduced to aldehydes1079 by treatment with lithium tri-tert-butoxyaluminum hydride in diglyme at −78 °C.1080 The R group may be alkyl or aryl and may contain many types of substituents, including NO2, CN, and EtOOC groups. The reaction stops at the aldehyde stage because steric hindrance prevents further reduction with this reagent. Acyl halides can also be reduced to aldehydes by hydrogenolysis with Pd on barium sulfate as catalyst in what is called the Rosenmund reduction.1081 A convenient hydrogenolysis procedure involves Pd on charcoal as the catalyst, with ethyldiisopropylamine as acceptor of the liberated HCl and acetone as the solvent.1082 The reduction of acyl halides to aldehydes has also been carried out1083 with Bu3SnH,1084 an InCl3 catalyzed reaction using Bu3SnH,1085 NaBH4 in a mixture of DMF and THF,1086 with Sm–PBu3,1087 and with formic acid/NH4OH.1088Polymethylhydrosiloxane (PMHS) reduces acid chlorides to aldehydes in the presence of a Pd catalyst.1089 In some of these cases, the mechanisms are free radical.
There are several indirect methods for the conversion of acyl halides to aldehydes, most of them involving prior conversion of the halides to certain types of amides (see Reaction 19-41). There is also a method in which the COOH group is replaced by a completely different CHO group (Reaction 16-87).
OS III, 551, 627; VI, 529, 1007. Also see, OS III, 818; VI, 312.
19-40 Reduction of Carboxylic Acids, Esters, and Anhydrides to Aldehydes1090
Hydro-de-hydroxylation or Dehydroxylation (overall transformation)
![]()
With most reducing agents, reduction of carboxylic acids generally gives the primary alcohol (Reaction 19-37) and the isolation of aldehydes is not feasible. However, simple straight-chain carboxylic acids have been reduced to aldehydes1091 by treatment with Li in MeNH2 or NH3 followed by hydrolysis of the resulting imine,1092 with
![]()
with thexylchloro(or bromo)borane–SMe21093 (see Reaction 15-16 for the thexyl group), Me2N=CHCl+ Cl− in pyridine,1094 and with diaminoaluminum hydrides.1095 Benzoic acid derivatives were reduced to benzaldehyde derivatives with NaH2PO2 and a diacylperoxide and Pd catalyst.1096 Caproic and isovaleric acids have been reduced to aldehydes in 50% yields or better with Dibal-H (i-Bu2AlH) at −75 to −70 °C.1097
Carboxylic esters have been reduced to aldehydes with Dibal-H at −70 °C, with diaminoaluminum hydrides,1098 and for phenolic esters with LiAlH(Ot-Bu)3 at 0 °C.1099 Pretreatment of the acid with Me3SiCl followed by reduction with Dibal-H also gives the aldehyde.1100 Aldehydes have also been prepared by reducing ethyl thiol esters (RCOSEt) with Et3SiH and a Pd–C catalyst.1101 Thioesters have been reduced to the aldehyde with Li metal in THF at −78 °C, followed by quenching with methanol.1102
Anhydrides, both aliphatic and aromatic, as well as mixed anhydrides of carboxylic and carbonic acids, have been reduced to aldehydes in moderate yields with disodium tetracarbonylferrate [Na2Fe(CO)4].1103 Heating a carboxylic acid, presumably to form the anhydride, and then reaction with Na/EtOH leads to the aldehyde.1104
Acid chlorides are reduced to aldehydes with Bu3SnH and a Ni catalyst.1105
Also see, Reactions 19-62 and 19-38.
OS VI, 312; VIII, 241, 498.
19-41 Reduction of Amides to Aldehydes
Hydro-de-dialkylamino-substitution
![]()
N,N-Disubstituted amides can be reduced to amines with LiAlH4 (see Reaction 19-64), but reduction to an aldehyde is possible.1106 Keeping the amide in excess gives the aldehyde rather than the amine. Sometimes it is not possible to prevent further reduction and primary alcohols are obtained instead. Other reagents1107 that give good yields of aldehydes are Dibal-H,1108 LiAlH(Ot-Bu)3, diaminoaluminum hydrides,1109 disiamylborane (see Reaction 15-16 for the disiamyl group),1110 and Cp2Zr(H)Cl.1111
Aldehydes have been prepared from carboxylic acids or acyl halides by first converting them to certain types of amides that are easily reducible. There are several examples:1112
1. Reissert Compounds.1113 Compounds, such as 36, are prepared from the acyl halide by treatment with quinoline and cyanide ion. Treatment of 36 with sulfuric acid gives the corresponding aldehyde.
2. Acyl Sulfonylhydrazides. Compounds, such as 37, are cleaved with base to give aldehydes. This reaction is known as the McFadyen–Stevens reduction and is applicable only to aromatic aldehydes or aliphatic aldehydes with no α hydrogen.1114 Acyl imide (RCON=NH, see reaction 19-67) has been proposed as an intermediate in this reaction.1115
3. Acyl Imidazoles. Compounds 381116 can be reduced to aldehydes with LiAlH4.
4. Weinreb Amides. A N-Methoxy-N-methyl amide (e.g., 39) is referred to as a Weinreb amide.1117 Reduction with and excess of LiAlH4 or Dibal-H leads to the corresponding aldehyde, as does reaction with Cp2ZrHCl.1118
5. See also, the Sonn–Müller Method (Reaction 19-44).
OS VIII, 68. See OS IV, 641, VI, 115 for the preparation of Reissert compounds.
C. Attack at Non-Carbonyl Multiple Bonded Heteroatoms
19-42 Reduction of the Carbon–Nitrogen Double Bond (C=N)
C,N-Dihydro-addition
![]()
Imines and Schiff bases,1119 hydrazones,1120 and other C=N compounds can be reduced with LiAlH4, NaBH4,1121 Na–EtOH, hydrogen and a catalyst, as well as with other reducing agents.1122 Metal-free catalytic hydrogenation is known.1123 Transfer hydrogenation of imines leads to amines.1124 A mixture of Sm/I21125 or In/NH4Cl1126 also reduces imines. Reduction with Bu2SnClH in HMPA has been shown to be chemoselective for imines.1127 Iminium salts are reduced by LiAlH4 to the corresponding amine, although here there is no “addition” to the nitrogen:1128 Silanes1129 with a triarylborane catalyst reduces N-sulfonyl imines1130 as does TiI4.1131 Imines are reduced with samarium bromide in HMPA,1132 2-propanol with a Ru catalyst,1133 and with triethylammonium formate with microwave irradiation.1134 Oximes are reduced with hydrogen gas and a catalytic amount of 48% HBr.1135
Oximes are generally reduced to amines (Reaction 19-48),1136 but simple reduction to give hydroxylamines can be accomplished with borane1137 or sodium cyanoborohydride.1138 Oxime O-ethers are reduced with Bu3SnH and BF3·OEt2.1139 Diazo compounds (ArN=NAr) are reductively cleaved to aniline derivatives with Zn and ammonium formate in methanol.1140
Reduction of imines has been carried out enantioselectively.1141 Catalytic hydrogenation1142 with a chiral Ir1143 Re,1144 Rh,1145 or Pd1146 catalyst is effective. Catalytic hydrogenation of iminium salts with a chiral Ru catalyst gives the amine.1147 Enantioselective reduction of imines is possible using a mixture of Escherichia coli whole cells and H3N·BH3.1148Hantzsch ester (see Reactions 15-14 and 16-17) reduction of imine-esters, in the presence of a chiral phosphoric acid derivative, leads to chiral amino esters.1149 In a related reaction, enamines were reduced by hydrogenation over a chiral Rh catalyst.1150
Hydrogenation of oximes with Pd/C and a Ni complex gives the imine, and in the presence of a lipase and ethyl acetate the final product was an acetamide, formed with high enantioselectivity.1151 Catalytic-transfer hydrogenation of imines leads with a chiral catalyst to chiral amines.1152 Conjugated N-sulfonyl imines are reduced to the conjugated sulfonamide with good enantioselectivity using a chiral rhodium catalyst in the presence of LiF and PhSnMe3.1153 Phosphinyl imines, R2C=N–P(=O)Ar2, are reduced with high enantioselectivity using a chiral Cu catalyst.1154 Silanes (e.g., PhSiH3) can be used for the reduction of imines, and in the presence of a chiral Ti catalyst the resulting amine was formed with excellent enantioselectivity.1155 The enantioselective reduction of aromatic imines is possible using trichlorosilane.1156 Enzymatic reduction of imines leads to chiral amines.1157
Oxime ethers are reduced with borane and a chiral spiroborate ester catalyst.1158
Isocyanates have been catalytically hydrogenated to N-substituted formamides: RNCO → R–NH–CHO.1159 Isothiocyanates were reduced to thioformamides with SmI2 in HMPA/t-BuOH.1160
OS III, 328, 827; VI, 905; VIII, 110, 568. Also see, OS IV, 283.
19-43 The Reduction of Nitriles to Amines
CC,NN-Tetrahydro-biaddition
![]()
Nitriles can be reduced to primary amines with many reducing agents,1161 including LiAlH4, and BH3·SMe2.1162 The reagent NaBH4 does not generally reduce nitriles except in alcoholic solvents with a catalyst (e.g., CoCl2,1163NiCl2,1164 or Raney nickel).1165 Lithium dimethylaminoborohydride (LiBH3NMe2) reduces aryl nitriles to the corresponding benzylamines.1166
The reduction of nitriles is of wide scope and has been applied to many nitriles. Catalytic hydrogenation converts nitriles to primary amines,1167 but secondary amines [(RCH2)2NH] are often side products.1168 These can be avoided by adding a compound (e.g., acetic anhydride), which removes the primary amine as soon as it is formed,1169 or by the use of excess ammonia to drive the equilibria backward.1170 Sponge nickel1171 or nickel on silica gel1172have been used for the catalytic hydrogenation of aryl nitriles to amines.
Attempts to stop with the addition with only 1 equiv of hydrogen, have failed; that is, to convert the nitrile to an imine, except where the imine is subsequently hydrolyzed (Reaction 19-44).
N-Alkylnitrilium ions are reduced to secondary amines by NaBH4.1173
![]()
Since nitrilium salts can be prepared by treatment of nitriles with trialkyloxonium salts (see Reaction 16-8), this is a method for the conversion of nitriles to secondary amines.
Note that the related compounds, the isonitriles (R–N+![]() CO−, also called isocyanides) have been reduced to N-methylamines with LiAlH4, as well as with other reducing agents.
CO−, also called isocyanides) have been reduced to N-methylamines with LiAlH4, as well as with other reducing agents.
OS III, 229, 358, 720; VI, 223.
19-44 The Reduction of Nitriles to Aldehydes
Hydro,oxy-de-nitrilo-tersubstitution
![]()
There are two principal methods for the reduction of nitriles to aldehydes.1174 In one of these, known as the Stephen reduction, the nitrile is treated with HCl to form an iminium salt (40). Subsequent reduction of (40) with anhydrous SnCl2 gives RCH=NH, which precipitates as a complex with SnCl4 and is then hydrolyzed (Reaction 16-2) to the aldehyde. The Stephen reduction is most successful when R is aromatic, but it can be done for aliphatic R up to about six carbons.1175 It is also possible to prepare 40 in a different way, by treating ArCONHPh with PCl5, which can then be converted to the aldehyde. This is known as the Sonn–Müller method. Aqueous formic acid in the presence of PtO2, followed by treatment with aqueous acid converts aryl nitriles to aryl aldehydes.1176
The other way of reducing nitriles to aldehydes involves using a metal hydride reducing agent to add 1 molar equivalent of hydrogen and subsequent hydrolysis, in situ, of the resulting imine (which is undoubtedly coordinated to the metal). This reaction has been carried out with LiAlH4, LiAlH(OEt)3,1177 LiAlH(NR2)3,1178 and Dibal-H.1179 The metal hydride method is useful for aliphatic and aromatic nitriles.
OS III, 626, 818; VI, 631.
19-45 Reduction of Nitro Compounds to Amines
![]()
Both aliphatic1180 and aromatic nitro compounds can be reduced to amines, although the reaction has been applied much more often to aromatic nitro compounds, owing to their greater availability. Many reducing agents have been used to reduce aromatic nitro compounds, the most common being Zn, Sn, or Fe (or sometimes other metals) and acid, and catalytic hydrogenation.1181 Chemoselective catalytic hydrogenation of nitro compounds is possible.1182 Transfer hydrogenation is used to reduce nitro compounds.1183 Indium metal in aq ethanol with ammonium chloride1184 or with water in aq THF1185 also reduces aromatic nitro compounds to the corresponding aniline derivative. Indium metal in methanol, with acetic anhydride and acetic acid, converts aromatic nitro compounds to the acetanilide.1186 Both samarium metal in methanol with ultrasound,1187 and a mixture of SmI2–water and an amine reduce nitro compounds.1188 Alternative reduction methods use ultrasound with Al(Hg) in aq THF1189 or with stannous chloride in an ionic liquid.1190 Some other reagents used1191 were Et3SiH/RhCl(PPh)3,1192 AlH3–AlCl3, formic acid and Pd–C,1193 or formic acid with Raney nickel in methanol.1194 The reaction with sulfides or polysulfides is called the Zinin reduction.1195 Amines are also the products when nitro compounds, both alkyl and aryl, are reduced with HCOONH4–Pd–C.1196 Many other functional groups (e.g., COOH, COOR, CN, amide) are not affected by this reagent (although ketones are reduced, see Reaction 19-33). With optically active alkyl substrates this method gives retention of configuration.1197
Lithium aluminum hydride reduces aliphatic nitro compounds to amines, but with aromatic nitro compounds the products with this reagent are azo compounds (Reaction 19-80). Most metal hydrides, including NaBH4 and BH3, do not reduce nitro groups at all, although both aliphatic and aromatic nitro compounds have been reduced to amines with NaBH4 and various catalysts (e.g., NiCl2 or CoCl21198 and ZrCl4).1199 Borohydride exchange resin (BER) in the presence of Ni(OAc)2, however, gives the amine.1200 Treatment of aromatic nitro compounds with NaBH4 alone has resulted in reduction of the ring to a cyclohexane ring with the nitro group still intact1201 or in cleavage of the nitro group from the ring.1202 With (NH4)2S or other sulfides or polysulfides it is often possible to reduce just one of two or three nitro groups on an aromatic ring or on two different rings in one molecule.1203 Bakers yeast reduces aromatic nitro compounds to aniline derivatives.1204 A combination of NaH2PO2/FeSO4 with microwave irradiation reduces aromatic nitro compounds to aniline derivatives.1205 Hydrazine on alumina, with FeCl3 and microwave irradiation, accomplishes this reduction.1206 Hydrazine–formic acid with Raney nickel in methanol reduces aromatic nitro compounds.1207 Heating aromatic nitro compounds with 57% HI reduces the nitro group to the amino group.1208
With some reducing agents, especially with aromatic nitro compounds, the reduction can be stopped at an intermediate stage, and hydroxylamines (Reaction 19-46), hydrazobenzenes, azobenzenes (Reaction 19-80), and azoxybenzenes (Reaction 19-79) can be obtained in this manner. However, nitroso compounds, which are often postulated as intermediates, are too reactive to be isolated, if indeed they are intermediates. Reduction by metals in mineral acids cannot be stopped, but always produces the amine.
The mechanisms of these reductions have not been much studied, although it is usually presumed that, at least with some reducing agents, nitroso compounds and hydroxylamines are intermediates. Both of these types of compounds give amines when exposed to most of these reducing agents (Reaction 19-47), and hydroxylamines can be isolated (Reaction 19-46). With metals and acid the following path has been suggested1209:
Certain aromatic nitroso compounds (Ar–NO) can be obtained in good yields by irradiation of the corresponding nitro compounds in 0.1 M aq KCN with UV light.1210 The reaction has also been performed electrochemically.1211When nitro compounds are treated with most reducing agents, nitroso compounds are either not formed or react further under the reaction conditions and cannot be isolated.
Reductive alkylation of aromatic nitro compounds is possible. The reaction of nitrobenzene with allylic or benzyl halides in the presence of an excess of tin metal in methanol, leads to the N,N-diallyl or dibenzyl aniline.1212 A similar reaction occurs with nitrobenzene, allyl bromide, and In metal in aq acetonitrile.1213
OS I, 52, 240, 455, 485; II, 130, 160, 175, 254, 447, 471, 501, 617; III, 56, 59, 63, 69, 73, 82, 86, 239, 242, 453; IV, 31, 357; V, 30, 346, 552, 567, 829, 1067, 1130; 81, 188.
19-46 Reduction of Nitro Compounds to Hydroxylamines
![]()
When aromatic nitro compounds are reduced with zinc and water under neutral conditions,1214 hydroxylamines are formed. Among other reagents used for this purpose have been SmI2,1215 N2H4–Rh–C,1216 and KBH4/BiCl3.1217Borane in THF reduces aliphatic nitro enolate anions to hydroxylamines1218:
![]()
Nitro compounds have been reduced electrochemically, to hydroxylamines, as well as to other products.1219
OS I, 445; III, 668; IV, 148; VI, 803; VIII, 16.
19-47 Reduction of Nitroso Compounds and Hydroxylamines to Amines
N-Dihydro-de-oxo-bisubstitution
![]()
N-Hydro-de-hydroxylation or N-Dehydroxylation
![]()
Nitroso compounds and hydroxylamines can be reduced to amines by the same reagents that reduce nitro compounds (Reaction 19-45). Reaction with CuCl, and then phenylboronic acid (Reaction 12-28), also reduces nitroso compounds to the amine.1220 A hydroxylamine can be reduced to the amine with CS2 in acetonitrile.1221 Indium metal in EtOH/aq NH4Cl reduces hydroxylamines to the amine.1222 N-Nitroso compounds are similarly reduced to hydrazines (R2N–NO → R2N–NH2).1223
OS I, 511; II, 33, 202, 211, 418; III, 91; IV, 247. See also, OS VIII, 93.
19-48 Reduction of Oximes to Primary Amines or Aziridines
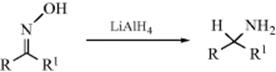
Both aldoximes and ketoximes can be reduced to primary amines with LiAlH4. The reaction is slower than similar reduction of ketones, so that, for example, PhCOCH=NOH gave 34% PhCHOHCH=NOH.1224 Among other reducing agents that give this reduction1225 are zinc and acetic acid, BH3,1226 NaBH3CN–TiCl3,1227 PMHS with Pd-C,1228 and sodium and an alcohol.1229 Catalytic hydrogenation is also effective.1230 Reduction of oximes with In metal in acetic anhydride/acetic acid–THF leads to the acetamide.1231
The reduction has been performed enantioselectively with Baker's yeast1232 and with Ph2SiH2 and an optically active Rh complex catalyst.1233 Oxime O-ethers are reduced to the amine with modest enantioselectivity using a chiral boron compound.1234
When the reducing agent is Dibal-H, the product is a secondary amine, arising from a rearrangement1235:
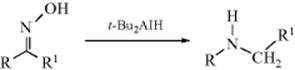
With certain oximes (e.g., those of the type ArCH2CR=NOH), treatment with LiAlH4 gives aziridines,1236 for example,
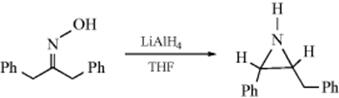
Hydrazones, arylhydrazones, and semicarbazones can also be reduced to amines with various reducing agents, including Zn–HCl and H2 and Raney nickel.
Oximes have been reduced in a different way, to give imines (RR′C=NOH → RR′C=NH), which are generally unstable, but which can be trapped to give useful products. Among reagents used for this purpose have been Bu3P–SPh21237 and Ru3(CO)12.1238 Oximes can also be reduced to hydroxylamines (Reaction 19-42). Nitrones have been reduced to imines using AlCl3·6 H2O/KI followed by Na2S2O3–H2O.1239.
OS II, 318; III, 513; V, 32, 83, 373, 376.
19-49 Reduction of Aliphatic Nitro Compounds to Oximes or Nitriles
![]()
Nitro compounds that contain an α hydrogen can be reduced to oximes with zinc dust in acetic acid1240 or with other reagents, among them CS2–NEt3,1241 CrCl2,1242 and (for α-nitro sulfones) NaNO2.1243 α-Nitro alkenes have been converted to oximes (–C=C–NO2 → –CH–C=NOH) with sodium hypophosphite, In with aq. NH4Cl/MeOH,1244 and with Pb–HOAc–DMF, as well as with certain other reagents.1245
![]()
Primary aliphatic nitro compounds can be reduced to aliphatic nitriles with t-BuN![]() C/BuN=C=O.1246 Secondary compounds give mostly ketones (e.g., nitrocyclohexane gave 45% cyclohexanone, 30% cyclohexanone oxime, and 19% N-cyclohexylhydroxylamine). Tertiary aliphatic nitro compounds do not react with this reagent (see also, Reaction 19-45).
C/BuN=C=O.1246 Secondary compounds give mostly ketones (e.g., nitrocyclohexane gave 45% cyclohexanone, 30% cyclohexanone oxime, and 19% N-cyclohexylhydroxylamine). Tertiary aliphatic nitro compounds do not react with this reagent (see also, Reaction 19-45).
OS IV, 932.
19-50 Reduction of Azides to Primary Amines
N-Dihydro-de-diazo-bisubstitution
![]()
Azides are easily reduced to primary amines by LiAlH4, as well as by a number of other reducing agents,1247 including NaBH4, NaBH4/LiCl,1248 NaBH4/CoCl2/H2O,1249 NaBH4/ZrCl4,1250 H2 and a catalyst, Mg or Ca in MeOH,1251Sm/NiCl2,1252 Sm/I2,1253 CeCl3,1254 Zn/NH4Cl/aq EtOH,1255 baker's yeast,1256 and In metal in EtOH.1257 Triethylsilane has been used for the radical reduction of azides to amines.1258
Reaction with PPh3 leads to a phosphazide (Ph3P=N–N=N–R), which loses nitrogen in what is called the Staudinger reaction1259: a method to prepare phosphazo compounds, but in this case leads to reduction. Alkylation is possible, and the reaction of an alkyl azide with PMe3, and then an excess of iodomethane, leads to the N-methylated amine.1260 The reaction is diastereoselective.1261 Chiral N-heterocyclic carbenes catalyze the Staudinger reactionof ketenes with imines to form β-lactam derivatives.1262 This reaction, combined with RX → RN3 (10-43), is an important way of converting alkyl halides (RX) to primary amines (RNH2); in some cases the two procedures have been combined into one laboratory step.1263 Sulfonyl azides (RSO2N3) have been reduced to sulfonamides (RSO2NH2) by irradiation in isopropyl alcohol1264 and with NaH.1265
OS V, 586; VII, 433.
19-51 Reduction of Miscellaneous Nitrogen Compounds
Isocyanate-methylamine transformation
![]()
Isothiocyanate-methylamine transformation
![]()
N,N-Dihydro-addition
![]()
Diazonium-arylhydrazone reduction
![]()
N-Hydro-de-nitroso-substitution
![]()
Isocyanates and isothiocyanates are reduced to methylamines on treatment with LiAlH4. Azo compounds are not usually reduced by LiAlH4,1266 (indeed these are the products from LiAlH4 reduction of nitro compounds, Reaction 19-80), but they can be reduced to hydrazo compounds by catalytic hydrogenation or with diimide1267 (see Reaction 15-11). Diazonium salts are reduced to hydrazines by sodium sulfite. This reaction probably has a nucleophilic mechanism.1268 The initial product is a salt of hydrazinesulfonic acid, which is converted to the hydrazine by acid treatment. Diazonium salts can also be reduced to arenes (Reaction 19-69). N-Nitrosoamines can be denitrosated to secondary amines by a number of reducing agents, including H2 and a catalyst,1269 BF3–THF–NaHCO3,1270 and NaBH4–TiCl4,1271 as well as by hydrolysis.1272
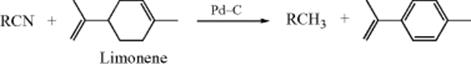
A cyano group can be reduced to a methyl group by treatment with a terpene (e.g., limonene), which acts as reducing agent in the presence of Pd–charcoal.1273 Hydrogen gas (H2) is also effective,1274 although higher temperatures are required. The R group may be alkyl or aryl.
Aryl nitro compounds are reduced to diaryl hydrazines with Al–KOH in methanol.1275
OS I, 442; III, 475. Also see, OS V, 43.
D. Reactions in which a Heteroatom Is Removed from the Substrate
19-52 Reduction of Silanes to Methylene Compounds
Si-Hydrogen-uncoupling
![]()
In certain cases, the C–Si bond of silanes can be converted to C–H. α-Silyl esters are reduced to esters with mercuric acetate and tetrabutylammonium fluoride, for example.1276
19-53 Reduction of Alkyl Halides
Hydro-de-halogenation or Dehalogenation
![]()
This type of reduction can be accomplished with many reducing agents.1277 A powerful but highly useful reagent (LiAlH4)1278 reduces almost all types of alkyl halide, including vinylic, bridgehead, and cyclopropyl halides.1279Reduction with lithium aluminum deuteride serves to introduce deuterium into organic compounds. An even more powerful reducing agent, lithium triethylborohydride (LiEt3BH; Super hydride), rapidly reduces primary, secondary, allylic, benzylic, and neopentyl halides, but not tertiary (these give elimination) or aryl halides.1280 A complex formed from lithium trimethoxyaluminum hydride [LiAlH(OMe)3] and CuI is another powerful reagent, which reduces primary, secondary, tertiary, allylic, vinylic, aryl, and neopentyl halides.1281 Sodium borohydride (NaBH4) is a milder reducing agent that reduces primary, secondary, and some tertiary1282 halides in good yield, in a dipolar aprotic solvent (e.g., Me2SO, DMF, or sulfolane)1283 at room temperature or above without affecting other functional groups that would be reduced by LiAlH4 (e.g., CO2H, CO2R, CN).1284 A mixture of NaBH4 and InCl3 efficiently reduces secondary bromides.1285 Borohydride exchange resin is also an effective reducing agent in the presence of metal catalysts [e.g., Ni(OAc)2].1286
Other reducing agents1287 include Zn (with acid or base), SnCl2, and Et3SiH in the presence of an AlCl3,1288 and also an Ir1289 or In1290 catalyst. Diethyl phosphonate–Et3N,1291 phosphorus tris(dimethylamide) [(Me2N)3P],1292 and organotin hydrides (RnSnH4-n)1293 (chiefly Bu3SnH) usually used in conjunction with a radical-initiator (e.g., AIBN),1294 or with transition metal salts (e.g., InCl3).1295 A water-soluble organotin hydride has been developed [(MeOCH2CH2OCH2CH2CH2)3SnH], which reduces alkyl halides.1296 Raney nickel in 2-propanol reduces primary iodides in the presence of a lactone moiety.1297 Aluminum amalgam efficiently reduced an iodohydrin to the alcohol.1298
Reduction, especially of bromides and iodides, can also be effected by catalytic hydrogenation.1299 Raney nickel by itself can reduce alkyl halides.1300 Homogeneous, chiral transition metal complexes can be used for the asymmetric hydrogenation of halides.1301
Alkali metals (e.g., Li1302 or Na1303 in t-BuOH or THF) are good reducing agents for the removal of all halogen atoms in a polyhalo compound (including vinylic, allylic, geminal, and even bridgehead halogens). Zinc and ammonium chloride in alcohol facilitates dehalogenation with microwave irradiation.1304 Nickel boride facilitates debromination.1305 Propargylic halides can often be reduced with allylic rearrangement to give allenes.1306
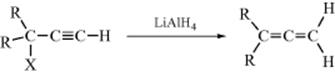
The choice of a reducing agent usually depends on what other functional groups are present since each reducing agent reduces certain groups and not others. This type of selectivity is called chemoselectivity. A chemoselective reagent is one that reacts with one functional group (e.g., halide), but not another (e.g., C=O). For example, there are several reagents that reduce only the halogen of α-halo ketones, leaving the carbonyl group intact.1307 Among them are decaborane with 10% Pd/C,1308 Bi in aq THF1309 or In metal in water,1310 and i-Bu2AlH–SnCl2.1311 Ionic liquids promote the selective debromination of α-bromo ketones.1312 Debromination is also induced by indium metal in a carboxylic acid.1313 In a similar chemoselective reaction, the halogen in α-haloimines has been reduced with SnCl2/MeOH without reducing the C=N bond.1314
Tertiary alkyl, benzylic, and allylic halides are reduced by NaBH3CN–SnCl21315, but do not react with primary or secondary alkyl or aryl halides. Sodium cyanoborohydride (NaBH3CN) in HMPA is another highly selective reagent, in this case for primary and secondary iodo and bromo groups.1316 Most of the reducing agents mentioned reduce chlorides, bromides, and iodides, but organotin hydrides also reduce fluorides.1317 See Section 19.B.ii-A for a discussion of selectivity in reduction reactions.
Alkyl halides, including fluorides and polyhalides, can be reduced with Mg and a secondary or tertiary alcohol (most often 2-propanol).1318 This is actually an example of the occurrence in one step of the sequence:
![]()
More often the process is carried out in two separate steps (Reactions 12-36 and 12-22).
Vinyl halides can be reduced to the corresponding alkene is some cases.1319 When vinyl dibromides (e.g., RCH=CBr2) are treated with (MeO)2P(=O)H and triethylamine, for example, the product is the vinyl bromide (RCH=HBr).1320 Indium metal in ethanol accomplishes the same transformation.1321 Similar reduction occurs when vinyl diiodides are treated with Zn–Cu in acetic acid.1322
With LiAlH4 and most other metallic hydrides, the mechanism usually consists of simple nucleophilic substitution with attack by hydride ion equivalents. The mechanism is SN2 rather than SN1, since primary halides react better than secondary or tertiary (tertiary generally give alkenes or do not react at all) and since Walden inversion has been demonstrated. However, rearrangements found in the reduction of bicyclic tosylates with LiAlH4 indicate that the SN1 mechanism can take place.1323 There is evidence that LiAlH4 and other metal hydrides can also reduce halides by an SET mechanism,1324 especially those (e.g., vinylic,1325 cyclopropyl,1326 or bridgehead halides) that are resistant to nucleophilic substitution. Reduction of halides by NaBH4 in 80% aq diglyme1327 and by BH3 in nitromethane1328 takes place by an SN1 mechanism. It is known that NaBH4 in sulfolane reduces tertiary halides possessing a β hydrogen by an elimination–addition mechanism.1329
The mechanism for reduction of alkyl halides is not always nucleophilic substitution. For example, reductions with organotin hydrides generally1330 take place by free radical mechanisms,1331 as do those with Fe(CO)5.
OS I, 357, 358, 548; II, 320, 393; V, 424; VI, 142, 376, 731; VIII, 82. See also, OS VIII, 583.
19-54 Reduction of Alcohols1332
Hydro-de-hydroxylation or Dehydroxylation
![]()
The hydroxyl groups of most alcohols can seldom be cleaved by catalytic hydrogenation and alcohols are often used as solvents for hydrogenation of other compounds. However, benzyl-type alcohols undergo the reaction readily and have often been reduced.1333 Diaryl and triarylcarbinols are similarly easy to reduce with LiAlH4–AlCl3,1334 with NaBH4 in F3CCOOH,1335 and with iodine, water, and red phosphorus (OS I, 224). Other reagents have been used,1336 including Me3SiCl–NaI,1337 Et3SiH–BF3,1338 SmI2–THF–HMPA,1339 and Sn/HCl. The reduction of secondary alcohols was accomplished using Ph2SiClH and InCl3.1340 1,3-Diols are especially susceptible to hydrogenolysis. Tertiary alcohols can be reduced by catalytic hydrogenolysis when the catalyst is Raney nickel.1341 Allylic alcohols (and ethers and acetates) can be reduced (often with accompanying allylic rearrangement) with Zn amalgam and HCl, as well as with certain other reagents.1342 Reagents that reduce the OH group of α–hydroxy ketones without affecting the C=O group include red phosphorus–iodine,1343 and Me3SiI.1344
Alcohols can also be reduced indirectly by conversion to a sulfonate and reduction of that compound (Reaction 19-57). The two reactions can be carried out without isolation of the sulfonate if the alcohol is treated with pyridine–SO3 in THF, followed by LiAlH4.1345 Another indirect reduction that can be done in one step involves treatment of the alcohol (primary, secondary, or benzylic) with NaI, Zn, and Me3SiCl.1346 In this case, the alcohol is first converted to the iodide, which is reduced. For other indirect reductions of OH, see Reaction 19-59.
The mechanisms of most alcohol reductions are obscure.1347 Hydrogenolysis of benzyl alcohols can give inversion or retention of configuration, depending on the catalyst.1348 The mechanism of electroreduction of allylic alcohols in acidic aqueous media has been examined.1349
OS I, 224; IV, 25, 218, 482; V, 339; VI, 769.
19-55 Reduction of Phenolic and Other Hydroxyaryl Compounds
Hydro-de-hydroxylation or Dehydroxylation, and so on
![]()
Oxygenated compounds (e.g., phenols, phenolic esters, and ethers) can be reduced.1350 Phenols can be reduced by distillation over zinc dust or with HI and red phosphorus, but these methods are quite poor and are seldom feasible. Catalytic hydrogenation has also been used, but the corresponding cyclohexanol (see Reaction 15-13) is a side product.1351
Much better results have been obtained by conversion of phenols to certain esters or ethers and reduction of the latter:
![]()
Ref. 1352
![]()
Ref. 1353
Ref. 1354
With a Pd–C catalyst, phenol derivatives are deoxygenated using Mg and MeO in the presence of ammonium acetate.1355 Palladium-on-carbon also mediated hydrodexoygenation of phenol derivatives in the presence of diethylamine.1356
OS VI, 150. See also, OS VII, 476.
19-56 Replacement of Alkoxyl by Hydrogen
Hydro-de-alkoxylation or Dealkoxylation
![]()
Simple ethers are not normally cleaved by reducing agents, although such cleavage has sometimes been reported1357 [e.g., THF treated with LiAlH4–AlCl31358 or with a mixture of LiAlH(Ot-Bu)3 and Et3B1359 gave 1-butanol; the latter reagent also cleaves methyl alkyl ethers].1360 Certain types of ethers can be cleaved quite well by reducing agents.1361 Among these are allyl aryl,1362 vinyl aryl,1363 benzylic ethers,1333,1364 and anisole1365 (for epoxides, see Reaction 19-35). 7-Oxobicyclo[2.2.1]heptanes can be reductively cleaved with Dibal-H and nickel catalysts.1366 α-Methoxy ketones are demethoxylated (O=C–COMe → O=C–CH) with SmI2.1367
![]()
Acetals and ketals are resistant to LiAlH4 and similar hydrides, and carbonyl groups are often converted to acetals or ketals for protection (Reaction 16-5). However, a combination of LiAlH4 and AlCl31368 does reduce acetals and ketals, removing one group, as shown above.1369 The actual reducing agents in this case are primarily chloroaluminum hydride (AlH2Cl) and dichloroaluminum hydride (AlHCl2), which are formed from the reagents.1370 This conversion can also be accomplished with Dibal-H,1371 as well as with other reagents.1372 Ortho esters are easily reduced to acetals by LiAlH4 alone, offering a route to aldehydes, which are easily prepared by hydrolysis of the acetals (Reaction 10-6). Mixed ketals [R(OMe)OR′] can be demethoxylated (to give RHOR′) with Bn3SnCl/NaCHBH3 in the presence of AIBN.1373
OS III, 693; IV, 798; V, 303. Also see, OS III, 742; VII, 386.
19-57 Reduction of Tosylates and Similar Compounds
Hydro-de-sulfonyloxy-substitution
![]()
Tosylates and other sulfonates can be reduced1374 with LiAlH4,1375 with NaBH4 in a dipolar aprotic solvent,1376 with LiEt3BH, with i-Bu2AlH (Dibal-H),1377 or with Bu3SnH–NaI.1378 The Ni catalyzed reduction of aryl tosylates proceeds in the presence of borane hydrides.1379 The scope of the reaction seems to be similar to that of 19-53. When the reagent is LiAlH4, alkyl tosylates are reduced more rapidly than iodides or bromides if the solvent is Et2O, but the order is reversed in diglyme.1380 The reactivity difference is great enough so that a tosylate function can be reduced in the presence of a halide and vice versa.
OS VI, 376, 762; VIII, 126. See also, OS VII, 66.
19-58 Hydrogenolysis of esters (Barton–McCombie Reaction)
Hydro-de-thioacetoxylation
![]()
Alcohols can readily be converted to carbonate and thiocarbonate derivatives. Under radical conditions,1381 using AIBN (Sec. 14.A.i) and Bu3SnH, the carbonate or thiocarbonate unit is reduced and replaced with hydrogen. The overall process is reduction of the ROH unit to RH and is called the Barton–McCombie reaction.1382 Both PhSiH3/AIBN1383 and PhSiH2–BEt3·O2 can be used.1384 This reaction can be catalytic in Bu3SnH.1385 Variations include reduction of ROCSNHPh derivatives using Ph3SiH/BEt3.1386 Another variation used water as the hydrogen atom source when BMe3 was used.1387 Tetrabutylammonium peroxydisulfate and formate ion has been used.1388
19-59 Reductive Cleavage of Carboxylic Esters
Hydro-de-acyloxylation or Deacyloxylation
![]()
The alkyl group (R) of certain carboxylic esters can be reduced to RH1389 by treatment with lithium in ethylamine.1390 The reaction is successful when R is a tertiary or a sterically hindered secondary alkyl group. A free radical mechanism is likely.1391 Similar reduction, also by a free radical mechanism, has been reported with sodium in HMPA–t-BuOH.1392 In the latter case, tertiary R groups give high yields of RH, but primary and secondary R are converted to a mixture of RH and ROH. Both of these methods provide an indirect method of accomplishing Reaction 19-54 for tertiary R.1393 The same thing can be done for primary and secondary R by treating alkyl chloroformates (ROCOCl) with tri-n-propylsilane in the presence of tert-butylperoxide1394 and by treating thiono ethers [ROC(=S)W, where W can be OAr or other groups] with Ph2SiH21395 or Ph3SiH1396 and a free radical initiator. Allylic acetates can be reduced with NaBH4 and a Pd complex,1397 and with SmI2–Pd(0).1398 For other carboxylic ester reductions, see Reactions 19-62, 19-38, and 19-65.
Note that acid chlorides can be reduced (R–COCl → R–H) using (Me3Si)3SiH/AIBN.1399
OS VII, 139.
19-60 Reduction of Hydroperoxides and Peroxides
![]()
Hydroperoxides can be reduced to alcohols with LiAlH4 or Ph3P1400 or by catalytic hydrogenation. This functional group is very susceptible to catalytic hydrogenation, as shown by the fact that a double bond may be present in the same molecule without being reduced.1401
![]()
The reaction is an important step in a method for the oxidative decyanation of nitriles containing an α hydrogen.1402 The nitrile is first converted to the α-hydroperoxy nitrile by treatment with base at −78 °C followed by O2. The hydroperoxy nitrile is then reduced to the cyanohydrin, which is cleaved (the reverse of Reaction 16-52) to the corresponding ketone. The method is not successful for the preparation of aldehydes (R′ = H).
Peroxides are cleaved to 2 molar equivalents of alcohols by LiAlH4, Mg/MeOH,1403 or by catalytic hydrogenation. Peroxides can be reduced to ethers with P(OEt)3.1404 In a similar reaction, disulfides (RSSR′) can be converted to sulfides RSR′ by treatment with tris(diethylamino)phosphine [(Et2N)3P].1405
OS VI, 130.
19-61 Reduction of Carbonyl to Methylene in Aldehydes and Ketones
Dihydro-de-oxo-bisubstitution
![]()
There are various ways of reducing the C=O group of aldehydes and ketones to CH2.1406 Two old but still popular methods are the Clemmensen reduction1407 and the Wolff–Kishner reduction. The Clemmensen reduction consists of heating the aldehyde or ketone with zinc amalgam and aq HCl.1408 Ketones are reduced more often than aldehydes. In the Wolff–Kishner reduction,1409 the aldehyde or ketone is heated with hydrazine hydrate and a base (usually NaOH or KOH). The Huang–Minlon modification1410 of the Wolff–Kishner reduction, in which the reaction is carried out in refluxing diethylene glycol, has completely replaced the original procedure. A microwave-assisted Huang–Minlon procedure has been reported.1411 The reaction can be carried out under more moderate conditions (room temperature) in DMSO with potassium tert-butoxide as base.1412 A new modification of the reduction treats a ketone with hydrazine in toluene with microwave irradiation, and subsequent reaction with KOH with microwave irradiation completes the Wolff–Kishner reduction.1413 The Wolff–Kishner reduction can also be applied to the semicarbazones of aldehydes or ketones.
The Clemmensen reduction is usually easier to perform, but it fails for acid-sensitive and high-molecular-weight substrates. For these cases, the Wolff–Kishner reduction is quite useful. For high-molecular-weight substrates, a modified Clemmensen reduction, using activated zinc and gaseous HCl in an organic solvent (e.g., as ether or acetic anhydride) has proved successful.1414 The Clemmensen and Wolff–Kishner reductions are complementary, since the former uses acidic and the latter basic conditions.
Both methods are fairly specific for aldehydes and ketones and can be carried out with many other functional groups present. However, certain types of aldehydes and ketones do not give normal reduction products. Under Clemmensen conditions,1415 α-hydroxy ketones give either ketones (hydrogenolysis of the OH, Reaction 19-54) or alkenes, and 1,3-diones usually undergo rearrangement (e.g., MeCOCH2COMe → MeCOCHMe2).1416 Neither method is suitable for α,β-unsaturated ketones, which give pyrazolines1417 under Wolff–Kishner conditions. Under Clemmensen conditions, both groups of these molecules may be reduced or if only one group is reduced, it is the C=C bond.1418 Sterically hindered ketones are resistant to both the Clemmensen and Huang–Minlon procedures, but can be reduced by vigorous treatment with anhydrous hydrazine.1419 In the Clemmensen reduction, pinacols (Reaction 19-76) are often side products.
Other reagents have also been used to reduce the C=O of aldehydes and ketones to CH2.1420 Among these are Me3SiCl followed by Et3SiH/TiCl4,1421 Ni(OAc)2 on borohydride exchange resin,1422 Et3SiH on pyridinium poly(hydrogen fluoride) (PPHF),1423 and, for aryl ketones (ArCOR and ArCOAr), NaBH3CN in THF–aq HCl,1424 Ni–Al in H2O,1425 HCOONH4–Pd–C,1426 or trialkylsilanes in F3CCOOH.1427 Hydrogenation with a heterogeneous Cu–silica catalyst has been used.1428 Silanes (e.g., Et3SiH) and a triarylborane catalyst reduce aliphatic aldehydes to methyl, –CHO → –CH3.1429 Zinc oxide/triethylsilane has been used,1430 and also titanocene dichloride [(C5H5)2TiCl2].1431 Most of these reagents also reduce aryl aldehydes (ArCHO) to methylbenzenes (ArCH3).1432 One carbonyl group of 1,2-diketones can be selectively reduced by H2S with an amine catalyst1433 or by HI in refluxing acetic acid.1434 One carbonyl group of quinones (e.g., 41), can be reduced with Cu and H2SO4 or with Sn and HCl.1435 One carbonyl group of 1,3-diketones was selectively reduced by catalytic hydrogenolysis.1436 Simply heating a ketone in supercritical 2-propanol reduces the ketone to the methylene compound.1437
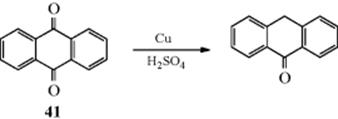
An indirect method of accomplishing the reaction is reduction of tosylhydrazones (R2C=N–NHTs) to R2CH2 with NaBH4, BH3, catecholborane, bis(benzyloxy)borane, or NaBH3CN. The reduction of α,β-unsaturated tosylhydrazones with NaBH3CN, with NaBH4–HOAc, or with catecholborane proceeds with migration of the double bond to the position formerly occupied by the carbonyl carbon, even if this removes the double bond from conjugation with an aromatic ring,1438 for example,
A cyclic mechanism is apparently involved:
Another indirect method is conversion of the aldehyde or ketone to a dithioacetal or ketal, and desulfurization using Raney nickel or another reagent (Reaction 14-27).
The first step in the mechanism1439 of the Wolff–Kishner reduction consists of formation of the hydrazone (16-14). It is this species that undergoes reduction in the presence of base, most likely in the following manner:
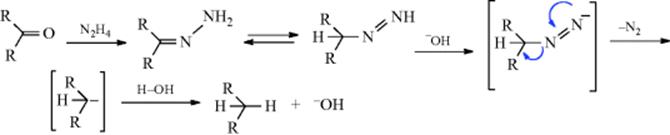
Not much is known about the mechanism of the Clemmensen reduction. Several mechanisms have been proposed,1440 including one going through a zinc–carbene intermediate.1441 One thing reasonably certain is that the corresponding alcohol is not an intermediate, since alcohols prepared in other ways fail to give the reaction. Note that the alcohol is not an intermediate in the Wolff–Kishner reduction either.
It is interesting to see that amines can be deaminated to give the corresponding methylene compounds with low-valent titanium (TiCl3/Li/THF).1442
OS I, 60; II, 62, 499; III, 410, 444, 513, 786; IV, 203, 510; V, 533, 747; VI, 62, 293, 919; VII, 393. Also see, OS IV, 218; VII, 18.
19-62 Reduction of Carboxylic Esters to Ethers
Dihydro-de-oxo-bisubstitution
![]()
Carboxylic esters and lactones have been reduced to ethers, although 2 molar equivalents of alcohol are more commonly obtained (Reaction 19-38). Reduction to ethers has been accomplished with a reagent prepared from BF3–etherate and either LiAlH4, LiBH4, or NaBH4,1443 with trichlorosilane and UV light,1444 and with catalytic hydrogenation. The reaction with the BF3 reagent apparently succeeds with secondary R′, but not with primary R′, which give 19-38. Acyloxy groups are reduced by cleavage of the C–C=O bond, R(Ar)COO–C → C–H) with an excess of Ph2SiH2 and di-tert-butyl peroxide.1445 Esters are reduced to ethers using Et3SiH and TiCl4,1446 BF3,1447 In(III) compounds,1448 or FeCl3.1449 Lactones are converted to cyclic ethers1450 by treatment with Cp2TiCl2 followed by Et3SiH on Amberlyst 15.1451
Thiono esters (RCSOR′) can be reduced to ethers (RCH2OR′) with Raney nickel (Reaction 14-27).1452 Reaction of thio esters (e.g., C–OC(=O)Ph) with Ph2SiH2 and Ph3SnH with BEt3, followed by AIBN (Sec. 14.A.i) leads to reduction of the C=S unit to give an ether.1453 Since the thiono esters can be prepared from carboxylic esters (Reaction 16-11), this provides an indirect method for the conversion of carboxylic esters to ethers. Thiol esters (RCOSR′) have been reduced to thioethers (RCH2SR′).1454
See also, Reactions 19-65 and 19-59.
19-63 Reduction of Cyclic Anhydrides to Lactones and Acid Derivatives to Alcohols
Dihydro-de-oxo-bisubstitution
Cyclic anhydrides are reduced with Zn–HOAc to give lactones, and also with hydrogen and Pt or RuCl2(Ph3P)3,1455 with NaBH4.1456 With cyclic anhydrides the reaction with LiAlH4 can be controlled to give either diols or lactones,1457 although diols are the more usual product. A BINOL–LiAlH4–EtOH complex, however, gives smooth reduction to the lactone.1458 With some reagents the reaction can be accomplished regioselectively; that is, only a specific one of the two C=O groups of an unsymmetrical anhydride is reduced.1459 Open-chain anhydrides either are not reduced at all (e.g., with LiAlH4 or NaBH4) or give 2 molar equivalents of alcohol. The NaBH4 in THF, with dropwise addition of methanol, reduces open-chain anhydrides to 1 equiv of primary alcohol and 1 equiv of carboxylic acid.1460
Acyl halides are reduced1461 to alcohols by LiAlH4 or NaBH4, as well as by other metal hydrides (Table 19.4), but not by borane.
In general, reduction of amides to alcohols is difficult. More commonly the amide is reduced to an amine. An exception uses LiH2NBH3 to give the alcohol.1462 Reduction with sodium metal in propanol also gives the alcohol.1463Acyl imidazoles are also reduced to the corresponding alcohol with NaBH4 in aq HCl.1464
There are no Organic Syntheses references, but see OS II, 526, for a related reaction. See OS VI, 482 for reduction to alcohols and OS IV, 271 for reduction of acyl halides.
19-64 Reduction of Amides to Amines
Dihydro-deoxo-bisubstitution
![]()
A useful reagent is LiAlH4, although the reaction is more difficult than the reduction of most other functional groups. Other groups are often reduced without disturbing an amide function. Although NaBH4 by itself does not reduce amides, it does so in the presence of certain other reagents1465 including iodine.1466 Lithium borohydride reduces acetamides.1467 Substituted amides can be reduced with these powerful reagents, and secondary amides are reduced to secondary amine and tertiary amides to tertiary amines. Borane1468 and sodium in 1-propanol1469 are good reducing agents for all three types of amides. Lithium triethylborohydride produces the alcohol with most N,N-disubstituted amides, but not with unsubstituted or N-substituted amides.1470 Sodium (dimethylamino)borohydride reduces unsubstituted and disubstituted, but not monosubstituted amides.1471
Amides can be reduced1472 to amines by catalytic hydrogenation,1473 but high temperatures and pressures are usually required. Another reagent that reduces disubstituted amides to amines is trichlorosilane.1474 Other silanes (e.g., Et3SiH) in the presence of a Re1475 Pt,1476 In,1477 Zn,1478 or Ru,1479 catalyst, reduce amides to amines. Electrolytic reduction of carbamates to give an amine are possible.1480
Hantzsch esters (see Reactions 15-14 and 16-17) have been used for metal-free reduction of amides to amines.1481
With some RCONR2, LiAlH4 causes cleavage, and the aldehyde (Reaction 10-41) or alcohol is obtained. Lactams are reduced to cyclic amines in high yields with LiAlH4, although cleavage sometimes occurs here too. A mixture of LiBHEt3/Et3SiH is also effective.1482 Lactams are also reduced to cyclic amines with 9-BBN1483 (Reaction 15-16) or LiBH3NMe2.1484
Imides are generally reduced on both sides,1485 although it is sometimes possible to stop with just one. Both cyclic and acyclic imides have been reduced in this manner, although with acyclic imides cleavage is often obtained.1486
Note that imides can be reduced to hydroxy lactams using different reagents, including NaBH4.1487
OS IV, 339, 354, 564; VI, 382; VII, 41.
19-65 Reduction of Carboxylic Acids and Esters to Alkanes
Trihydro-de-alkoxy,oxo-tersubstitution, and so on
![]()
The reagent titanocene dichloride reduces carboxylic esters in a different manner from that of Reactions 19-59, 19-62, or 19-38. The products are the alkane RCH3 and the alcohol R′OH. The mechanism probably involves an alkene intermediate. Aromatic acids can be reduced to methylbenzenes by a procedure involving refluxing first with trichlorosilane in MeCN, then with tripropylamine added, and finally with KOH and MeOH (after removal of the MeCN).1488 The following sequence has been suggested1488:
![]()
Esters of aromatic acids are not reduced by this procedure, so an aromatic COOH group can be reduced in the presence of a COOR′ group.1489 However, it is also possible to reduce aromatic ester groups, by a variation of the trichlorosilane procedure.1490 Both o- and p-hydroxybenzoic acids and their esters have been reduced to cresols (HOC6H4CH3) with sodium bis(2-methoxyethoxy)aluminum hydride [NaAlH2(OC2H4OMe)2, Red-Al].1491 Heating a 2-pyridylbenzyl ester with ammonium formate and a Ru catalyst leads to reduction of the CH3COO unit to the alkane.1492
Carboxylic acids can also be converted to alkanes, indirectly,1493 by reduction of the corresponding tosylhydrazides (RCONHNH2) with LiAlH4 or borane.1494
OS VI, 747.
19-66 Hydrogenolysis of Nitriles
Hydro-de-cyanation
![]()
This transformation is not common, but given the proliferation of nitriles in organic chemistry, it is potentially quite useful. In the presence of mercuric compounds, tertiary nitriles can be reduced to the hydrocarbon with sodium cyanoborohydride.1495 gem-Dinitriles can be reduced to the corresponding mono-nitrile with SmI2.1496 Hydrosilanes facilitate reductive cleavage of nitriles in the presence of a Rh catalyst.1497
19-67 Reduction of the C–N Bond
Hydro-de-amination or Deamination
![]()
Benzylic amines are particularly susceptible to hydrogenolysis by catalytic hydrogenation1498 or dissolving metal reduction.1499 Note that the Wolff–Kishner reduction in Reaction 19-61 involved formation of a hydrazone and deprotonation by base that led to loss of nitrogen and reduction. Ceric ammonium nitrate in aq acetonitrile has also been shown to reductively cleave the N-benzyl group.1500 Primary amines have been reduced to RH with hydroxylamine-O-sulfonic acid and aq NaOH to give the hydrocarbon, nitrogen gas, and the sulfate anion.1501 It is postulated that R–N=N–H is an intermediate that decomposes to the carbocation. An indirect means of achieving the same result is the conversion of the primary amine to the sulfonamide (RNHSO2R′) (Reaction 16-102) and subsequent treatment with NH2OSO2OH1502 or NaOH, and then NH2Cl.1503 Tosylaziridines derived from terminal alkenes are reduced to the corresponding primary tosylamine with polymethylhydrosiloxane/Pd-C.1504 Aziridines can be reduced in the same way as epoxides (Reaction 19-35).
Other indirect methods involve reduction of N,N-ditosylates (Sec. 10.G.iii) with NaBH4 in HMPA1505 and modifications of the Katritzky pyrylium–pyridinium method.1506 Allylic and benzylic amines1333 can be reduced by catalytic hydrogenolysis. Aziridines can be reductively opened with SmI21507 or with Bu3SnH and AIBN.1508 The C–N bond of enamines is reductively cleaved to give an alkene with alane (AlH3)1509:
and with 9-BBN (Reaction 15-16) or borane methyl sulfide (BMS).1510 Since enamines can be prepared from ketones (Reaction 16-13), this is a way of converting ketones to alkenes. In the latter case, BMS gives retention of configuration [an (E)-isomer gives the (E)-product], while 9-BBN gives the other isomer.1510 Diazo ketones are reduced to methyl ketones by HI: RCOCHN2 + HI → RCOCH3.1511
Quaternary ammonium salts can be cleaved with LiAlH4, R4N+ + LiAlH4 → R3N + R−, as can quaternary phosphonium salts (R4P+). Other reducing agents have also been used (e.g., lithium triethylborohydride, which preferentially cleaves methyl groups,1512 and Na in liquid ammonia). When quaternary salts are reduced with Na(Hg) in water, the reaction is known as the Emde reduction. However, this reagent is not applicable to the cleavage of ammonium salts with four saturated alkyl groups.
Nitro compounds (RNO2) can be reduced to RH1513 by sodium methylmercaptide (CH3SNa) in an aprotic solvent1514 or by Bu3SnH.1515 Both reactions have free radical mechanisms.1516 Tertiary nitro compounds can be reduced to RH by NaHTe.1517 The nitro group of aromatic nitro compounds has been removed with sodium borohydride.1518 Reduction of the C–N bond on aromatic amines with Li metal in THF generates the aryl compounds.1519 Sodium nitrite, sodium bisulfite in EtOH/water/acetic acid does a similar reduction.1520 Conversion of the aniline derivative to the methanesulfonamide and subsequent treatment with NaH and NH2Cl gives the same result.1521 The Bu3SnH reagent also reduces isocyanides (RNC, prepared from RNH2 by formylation followed by Reaction 17-31) to RH,1522 a reaction that can also be accomplished with Li or Na in liquid NH3,1523 or with K and a crown ether in toluene.1524 α-Nitro ketones can be reduced to ketones with Na2S2O4–Et3SiH in HMPA–H2O.1525
OS III, 148; IV, 508; VIII, 152.
19-68 Reduction of Amine Oxides and Azoxy Compounds
N-Oxygen-detachment
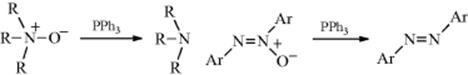
Amine oxides1526 and azoxy compounds (both alkyl and aryl)1527 can be reduced practically quantitatively with triphenylphosphine.1528 Other reducing agents have also been used, including LiAlH4, NaBH4/LiCl,1529 H2–Ni, PCl3, Ga/H2O,1530 or In/TiCl4.1531 Indium metal with aq ammonium chloride in methanol gives good yields of pyridine from pyridine N-oxide.1532 Similar results are obtained using ammonium formate and Raney nickel1533 or zinc.1534Sodium in ethanol, in a sealed tube, reduces pyridine N-oxide to pyridine.1535 Similar reduction was accomplished with Mo(CO)6 in ethanol.1536 Indium (III) chloride has been used for the reduction of quinoline N-oxide to quinoline.1537 Nitrile oxides1538 (R–C![]() N+–O−) can be reduced to nitriles with trialkylphosphines,1539 and isocyanates (RNCO) to isocyanides (RNC) with Cl3SiH–Et3N.1540
N+–O−) can be reduced to nitriles with trialkylphosphines,1539 and isocyanates (RNCO) to isocyanides (RNC) with Cl3SiH–Et3N.1540
Analogous to amino N-oxides, phosphine oxides (R3P=O) are reduced to phosphines (R3P). Treatment of a phosphine oxide with MeOTf followed by reduced with LiAlH41541 or Dibal-H1542 gives the phosphine. Chiral phosphine oxides are reduced to the phosphine with excellent enantioselectivity using PPh3 and Cl3SiH.1543
OS IV, 166. See also, OS VIII, 57.
19-69 Replacement of the Diazonium Group by Hydrogen
Dediazoniation or Hydro-de-diazoniation
![]()
Reduction of a diazonium group (dediazoniation) provides an indirect method for the removal of an amino group from an aromatic ring.1544 A common method uses hypophosphorous acid (H3PO2), although many other reducing agents1545 have been used, including HMPA,1546 thiophenol,1547 and sodium stannite (Na2SnO2). Ethanol was the earliest reagent used, and it frequently gives good yields, but ethers (ArOEt) are often side products. When H3PO2 is used, 5–15 molar equivalents of this reagent are required per molar equivalent of substrate. Diazonium salts can be reduced in nonaqueous media by several methods, including treatment with Bu3SnH or Et3SiH in ethers or MeCN1548 and by isolation as the BF4− salt and reduction of this with NaBH4 in DMF.1549 Aromatic amines can be deaminated (ArNH2 → ArH) in one laboratory step by treatment with an alkyl nitrite in DMF1550 or boiling THF.1551The corresponding diazonium salt is an intermediate.
Not many investigations of the mechanism have been carried out. It is generally assumed that the reaction of diazonium salts with ethanol to produce ethers takes place by an ionic (SN1) mechanism while the reduction to ArH proceeds by a free radical process.1552 The reduction with H3PO2 is also believed to have a free radical mechanism.1553 In the reduction with NaBH4, an aryldiazene intermediate (ArN=NH) has been demonstrated,1554 arising from nucleophilic attack by BH4− on the β nitrogen. Such diazenes can be obtained as moderately stable (half-life of several hours) species in solution.1555 It is not entirely clear how the aryldiazene decomposes, but there are indications that either the aryl radical (AR·) or the corresponding anion (Ar−) may be involved.1556
The dediazoniation reaction is used for functionalization of aromatic rings, to remove an amino group after it has been used to direct one or more other groups to ortho and para positions. For example, the compound 1,3,5-tribromobenzene cannot be prepared by direct bromination of benzene because the bromo group is ortho–para directing; however, this compound is easily prepared by the following sequence:
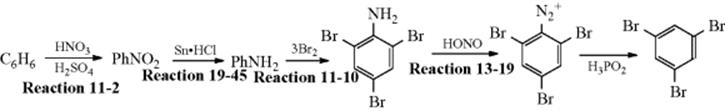
Many other compounds that would otherwise be difficult to prepare are easily synthesized with the aid of the dediazoniation reaction.
Unwanted dediazoniation can be suppressed by using hexasulfonated calix[6]arenes (see Sec. 3.C.ii).1557
OS I, 133, 415; II, 353, 592; III, 295; IV, 947; VI, 334.
19-70 Desulfurization
Hydro-de-thio-substitution, and so on
![]()
Thiols and thioethers,1558 both alkyl and aryl, can be desulfurized by hydrogenolysis with Raney nickel.1559 The hydrogen is usually not applied externally, since Raney nickel typically contains enough hydrogen for the reaction. Other sulfur compounds can be similarly desulfurized, including disulfides, thiono esters,1560 thioamides, sulfoxides, and thioacetals.1561 Reduction of thioacetals is an indirect way of accomplishing reduction of a carbonyl to a methylene group (see Reaction 19-61), and it can also give the alkene if a hydrogen atom is present.1562 In most of the examples given, R can also be aryl. Other reagents1563 have also been used.1564 Reductive cleavage of sulfones and sulfonamides occurs with organobases [e.g., bis(imidazolylidenes)].1565
Lithium aluminum hydride reduces most sulfur compounds with cleavage of the C–S bond, including thiols.1566 Thioesters can be reduced with Ni2B (from NiBr2/NaBH4).1567 β-Ketosulfones are reduced with TiCl4–Zn1568 or TiCl4–Sm.1569
An important special case of RSR reduction is desulfurization of thiophene derivatives. This proceeds with concomitant reduction of the double bonds. Many compounds have been made by alkylation of thiophene to 42, followed by reduction to give the alkane.
Thiophenes can also be desulfurized to alkenes (RCH2CH=CHCH2R′ from 42) with a nickel boride catalyst prepared from NiCl2 and NaBH4 in CH3OH.1570 Only one SR group of a dithioacetal is reduced by treatment with borane–pyridine in trifluoroacetic acid or in CH2Cl2 in the presence of AlCl3.1571 Phenyl selenides (RSePh) can be reduced to RH with Ph3SnH1572 and with nickel boride.1573 Cleavage of the C–Se bond can also be achieved with SmI2.1574
The exact mechanism of the Raney nickel reactions is still in doubt, although they are probably of the free radical type.1575 It has been shown that reduction of thiophene proceeds through butadiene and butene, not through 1-butanethiol or other sulfur compounds; that is the sulfur is removed before the double bonds are reduced. This was demonstrated by isolation of the alkenes and the failure to isolate any potential sulfur-containing intermediates.1576
OS IV, 638; V, 419; VI, 109, 581, 601. See also, OS VII, 124, 476.
19-71 Reduction of Sulfonyl Halides and Sulfonic Acids to Thiols or Disulfides
![]()
Thiols can be prepared by the reduction of sulfonyl halides1577 with LiAlH4. Usually, the reaction is carried out on aromatic sulfonyl chlorides. Zinc and acetic acid, and HI, also give the reduction. Sulfonic acids have been reduced to thiols with a mixture of triphenylphosphine and either I2 or a diaryl disulfide.1578 For the reduction of sulfonyl chlorides to sulfinic acids, see Reaction 16-104.
Disulfides (RSSR) can also be produced.1579 Other sulfonic acid derivatives can be converted to disulfides. Esters (e.g., PhSAc) are converted to disulfides (PhS-SPh) with Clayan and microwave irradiation.1580 Thiobenzoate derivatives (PhSBz) are similarly converted to PhS–SPh with SmI2.1581 In a similar manner, RS–SO3Na is converted to RS–SR when heated with Sm metal in water.1582
OS I, 504; IV, 695; V, 843.
19-72 Reduction of Sulfoxides and Sulfones
S-Oxygen-detachment
Sulfoxides can be reduced to sulfides by many reagents,1583 including LiAlH4, HI, Bu3SnH,1584 H2–Pd–C,1585 NaBH4–NiCl2,1586 NaBH4/I2,1587 catecholborane,1588 BH3 with a Mo catalyst,1589 a Mo/In system,1590 Ti compounds,1591 and Sm/methanolic NH4Cl with ultrasound.1592 Sulfoxides are deoxygenated by treatment with 2,4-diphenyl-1,3-diselenadiphosphetane-2,4-diselenide.1593 Sulfones, however, are usually more difficult to reduce, but they have been reduced to sulfides with Dibal-H.1594 A less general reagent is LiAlH4, which reduces some sulfones to sulfides, but not others.1595 Both sulfoxides and sulfones can be reduced by heating with sulfur, which is oxidized to SO2, although the reaction with sulfoxides proceeds at a lower temperature. It has been shown by using substrate labeled with 35S that sulfoxides simply give up the oxygen to the sulfur, but that the reaction with sulfones is more complex, since ~75% of the original radioactivity of the sulfone is lost.1596 This indicates that most of the sulfur in the sulfide product comes in this case from the reagent. There is no direct general method for the reduction of sulfones to sulfoxides, but an indirect method has been reported.1597 Selenoxides can be reduced to selenides with a number of reagents.1598
OS IX, 446
E. Reduction with Cleavage
19-73 de-Alkylation of Amines and Amides
![]()
Certain amines can be dealkylated, usually under reductive conditions. Both N-allyl amines and N,N-dialkyl allyl amines, are converted to the corresponding amine, R2N–H, with Dibal-H/NiCl2dppp[dppp = 1, 3-bis(diphenylphosphino)propane],1599 and with Pd(dba)2dppb[dppb = 1, 4-bis(diphenylphosphino)butane].1600 A mixture of TiCl3 and Li converts N-benzylamines to the amine.1601 In the case of N,N-dimethyl amines, RuCl3 and H2O2demethylate the amine (ArNMe2 → ArNHMe).1602 Tribenzylamines are dealkylated to give the dibenzylamine with ceric ammonium nitrate in aq acetonitrile.1603 N-Benzyl indoles are cleaved to indoles with O2, DMSO/KO-t-Bu1604or with tetrabutylammonium fluoride.1605 N-Demethylation of alkaloids was reported using a Fe mediated two-step procedure.1606 N-Deallylation and debenzylation of amines occurs with alkali metals on silica.1607
The process is not limited to amines. Amides can also be dealkylated. N-Benzyl amides are debenzylated in the presence of NBS and AIBN.1608 Scandium triflate removes N-tert-butyl from amides.1609
N-Alkyl sulfonamides are dealkylated with PhI(OAc)2 and I2 with ultrasound to give a primary sulfonamide.1610 Similar results are obtained with H5IO6 and a Cr catalyst.1611 tert-Butyl sulfonamides are cleaved to the primary sulfonamide with BCl3.1612
19-74 Reduction of Azo, Azoxy, and Hydrazo Compounds to Amines
Azo, azoxy, and hydrazo compounds can all be reduced to amines.1613 Metals (notably Zn) and acids, and Na2S2O4, are frequently used as reducing agents, and Bu3SnH with a Cu catalyst has been used.1614 Borane reduces azo compounds to amines, although it does not reduce nitro compounds.1615 Lithium aluminum hydride does not reduce hydrazo compounds or azo compounds, although with the latter, hydrazo compounds are sometimes isolated. With azoxy compounds, LiAlH4 gives only azo compounds (Reaction 19-68). Note that azo compounds are reduced to the hydrazine by reaction with hydrazine hydrate in ethanol,1616 or with Fe powder in ammonium chloride.1617
OS I, 49; II, 35, 39; III, 360; X, 327. Also see, OS II, 290.
19-75 Reduction of Disulfides to Thiols
S-Hydrogen-uncoupling
![]()
Disulfides can be reduced to thiols by mild reducing agents1618 (e.g., Zn and dilute acid, In and NH4Cl/EtOH,1619 or Ph3P and H2O).1620 The reaction can also be accomplished simply by heating with alkali.1621 Among other reagents used have been LiAlH4, NaBH4/ZrCl4,1622 Mg/MeOH,1623 and hydrazine or substituted hydrazines.1624
Aryl diselenides are similarly cleaved to selenols (ArSeH) with Cp2TiH followed by Ph2I+X−.1625
OS II, 580. Also see, OS IV, 295.
F. Reductive Coupling
19-76 Bimolecular Reduction of Aldehydes and Ketones to 1,2-Diols and Imines to 1,2-Diamines
2/ O-Hydrogen-coupling and 2/ N-Hydrogen-coupling
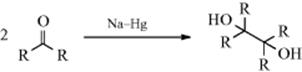
1,2-Diols (pinacols) can be synthesized by reduction of aldehydes and ketones with active metals (e.g., Na, Mg, or Al).1626 Aromatic ketones give better yields than aliphatic ones. The use of a Mg–MgI2 mixture has been called the Gomberg–Bachmann pinacol synthesis.1627 As with a number of other reactions involving Na, there is a direct electron transfer that converts the ketone or aldehyde to a ketyl, which dimerizes.
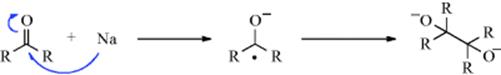
Other reagents have been used,1628 including Sm,1629 SmI2,1630 Pr,1631 Yb,1632 In with ultrasound,1633 InCl3 catalyst with Mg,1634 InCl3/Al in H2O,1635 Al/TiCl3,1636 VCl3/Zn in H2O,1637 activated Mn,1638 Zn,1639 and a low-valent Ti reagent1640 (see Reaction 19-76). Unsymmetrical coupling between two different ketones has been accomplished using TiCl3 in aqueous solution,1641 and coupling of two different aldehydes has been achieved by the use of a V complex.1642 Two aldehydes have also been coupled using Mg in water.1643 Coupling leads to a mixture of syn- and anti-diols. “Syn-selective” reagents are Cp2TiCl2/Mn,1644 TiCl4/Bu4I,1645 TiI4,1646 and NbCl3.1647 “Anti-selective” coupling reactions are also known: Ti–salen,1648 Mg with a NiCl2 catalyst,1649 and Sm/SmCl3.1650 Aryl aldehydes are coupled to give the bis(trimethylsilyl) ether using Mn, Me3SiCl and Cp2TiCl2.1651
Stereoselective pinacol coupling reactions are well known.1652 Chiral additives with pinacol couplings lead to formation of a diol with moderate to good enantioselectivity.1653 A crossed-pinacol coupling was reported using Et2Zn and with a BINOL catalyst gave good enantioselectivity.1654 Enantioselective coupling was reported using a chiral salen–Mo complex.1655 A combination of Mg and Me3SiCl was also used to effect a crossed-pinacol.1656 Chiral metal complexes in conjunction with a metal leads to diol formation with good enantioselectivity.1657
Intramolecular pinacol coupling reactions are known, giving cyclic 1,2-diols.1658 Dialdehydes have been cyclized by reaction with TiCl3 to give cyclic 1,2-diols in good yield.1659
A variation of the pinacol coupling treats acyl nitriles with In metal and ultrasound to give a 1,2-diketone.1660 Another variation couples acetals to give 1,2-diols.1661
The photochemical dimerization of ketones to 1,2-diols is one of the most common photochemical reactions.1662 The substrate, which is usually a diaryl or aryl alkyl ketone, is irradiated with UV light in the presence of a hydrogen donor (e.g., isopropyl alcohol, toluene, or an amine).1663 In the case of benzophenone, irradiated in the presence of 2-propanol, the ketone molecule initially undergoes n → π∗ excitation, and the singlet species thus formed crosses to the T1 state with a very high efficiency.
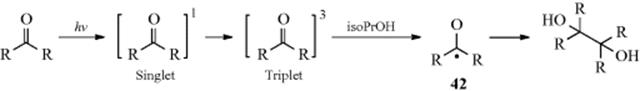
The T1 species abstracts hydrogen from the alcohol (Sec. 7.A.vii, category 4) and then dimerizes. The iPrO√ radical, which is formed by this process, reacts by atom transfer of H√ to another molecule of ground-state benzophenone, producing acetone and another molecule of 51. This mechanism1664 predicts that the quantum yield for the disappearance of benzophenone should be 2, since each quantum of light results in the conversion of 2 equiv of benzophenone to 42. Under favorable experimental conditions, the observed quantum yield does approach 2. Benzophenone abstracts hydrogen with very high efficiency. Other aromatic ketones are dimerized with lower quantum yields, and some (e.g., p-aminobenzophenone, o-methylacetophenone) cannot be dimerized at all in 2-propanol (although p-aminobenzophenone, e.g., can be dimerized in cyclohexane1665). The reaction has also been carried out electrochemically.1666
A coupling reaction similar to pinacol coupling has been used with imines, which dimerize to give 1,2-diamines. A number of reagents have been used, including treatment with TiCl4–Mg,1667 In/aq EtOH,1668 Zn/aq NaOH,1669 or SmI2.1670
![]()
When electroreduction was used, it was possible to obtain cross-products by coupling a ketone to an O-methyl oxime1671: O-Methyl oxime ethers are coupled to give 1,2-diamines using Zn and TiCl4.1672 Aldehydes are converted to 1,2-diamines by treatment with TMS2NH, NaH, and Li metal in 5 M LiClO4 in ether, with sonication.1673 Aldehydes are coupled with N-sulfinyl imines to give N-sulfinyl amino alcohols in the presence of SmI2.1674 Hemiaminals are coupled to give 1,2-diamines with TiI4/Zn.1675 Amides are converted to 1,2-diamines with Cp2TiF2 and PhMeSiH2.1676 Samarium(II) iodide was used to couple iminium salts, giving the 1,2-diamine.1677 Ketones can be treated with Yb, and then an imine to give amino alcohols.1678
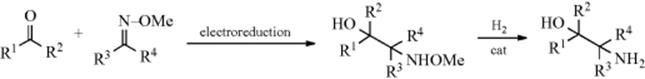
The N-methoxyamino alcohol could then be reduced to the amino alcohol.1671 A photochemical coupling has also been reported.1679 A variation of this reaction treats an imine with Yb in THF/HMPA, and then an aldehyde to give a 1,2-bis(imine).1680
OS I, 459; II, 71; X, 312; 81, 26.
19-77 Bimolecular Reduction of Aldehydes or Ketones to Alkenes
De-oxygen-coupling
![]()
Aldehydes and ketones, both aromatic and aliphatic (including cyclic ketones), can be converted in high yields to dimeric alkenes by treatment with low valent Ti,1681 initially generated with TiCl3 and a Zn–Cu couple.1682 This is called the McMurry reaction.1683 The reagent produced in this way is called a low-valent titanium reagent, and the reaction has also been accomplished1684 with low-valent Ti reagents prepared in other ways, for example, from Mg and a TiCl3–THF complex;1685 from TiCl4 and Zn or Mg;1686 from TiCl3 and LiAlH4;1687 and from TiCl3 and K or Li;1688 and with certain compounds prepared from WCl6 and either lithium, lithium iodide, LiAlH4, or an organolithium1689 (see Reaction 17-18). Microwave irradiation has been used to facilitate the coupling.1690 The reaction has been used to convert dialdehydes and diketones to cycloalkenes.1691 Rings of 3–16 and 22 members have been closed in this way, for example,1692
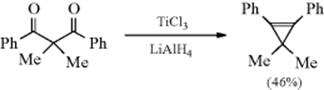
The same reaction on a keto ester gives a cycloalkanone.1693
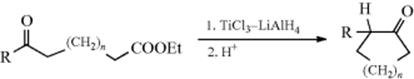
Indoles have been prepared form o-acyl amides with Ti(powder) and Me3SiCl1694 or with TiCl3–C8K.1695 Benzofurans have been prepared by a closely related reaction.1696
Unsymmetrical alkenes can be prepared from a mixture of two ketones in a cross-coupling reaction, if one is in excess.1697 An aldehyde and a ketone were cross-coupled using Yb(OTf)3, for example.1698 The mechanism consists of initial coupling of two radical species to give a 1,2-dioxygen compound (a titanium pinacolate), which is then deoxygenated.1699
OS VII, 1.
19-78 Acyloin Ester Condensation
When carboxylic esters are heated with sodium in refluxing ether or benzene, a bimolecular reduction takes place, and the product is an α-hydroxy ketone (called an acyloin).1700 The reaction, called the acyloin ester condensation(or just acyloin condensation),1701 is quite successful when R is alkyl. Acyloins with long chains have been prepared in this way (e.g., R = C17H35), but for high-molecular-weight esters, toluene or xylene is used as the solvent. Modifications to this procedure have been reported, including an ultrasound-promoted acyloin condensation in ether,1702 which improved the yields of four-, five-, and six-membered rings, and Olah's procedure, which was also done in ether.1703
The acyloin condensation has been used with great success, in boiling xylene, to prepare cyclic acyloins from diesters.1704 The yields are 50–60% for the preparation of 6- and 7-membered rings, 30–40% for 8- and 19-membered, and 60–95% for rings of 10–20 members. Even larger rings have been closed in this manner. Indeed, this is one of the best ways of closing rings of 10 members or more. The reaction has been used to close 4-membered rings,1705although this is generally unsuccessful. For larger rings, the presence of double or triple bonds does not interfere.1706 Even a benzene ring can be present, and many paracyclophane derivatives (44) with n = 9 or more have been synthesized in this manner.1707
Yields in the acyloin condensation can be improved by running the reaction in the presence of chlorotrimethylsilane (Me3SiCl), in which case the dianion (43) is converted to the bis (silyl) enol ether (45), which can be isolated and subsequently hydrolyzed to the acyloin with aq acid.1708 This is now the standard way to conduct the acyloin condensation. Among other things, this method inhibits the Dieckmann condensation1709 (Reaction 16-85), which otherwise competes with the acyloin condensation when a five-, six-, or seven-membered ring can be closed (note that the ring formed by a Dieckmann condensation is always one carbon atom smaller than that formed by an acyloin condensation of the same substrate). The Me3SiCl method is especially good for the closing of four-membered rings.1710
The mechanism is usually presumed to have a diketone (RCOCOR) as an intermediate,1711 since small amounts of it are usually isolated as side products, and when it is resistant to reduction (e.g., t-Bu–COCO–t-Bu), it is the major product. A possible sequence (analogous to that of Reaction 19-76) is
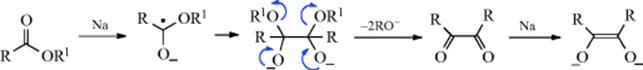
A large surface area for the Na is usually required for good results in this coupling, consistent with a surface reaction. In order to account for the ready formation of large rings, which means that the two ends of the chain must approach each other even though this is conformationally unfavorable for long chains, it may be postulated that the two ends become attached to nearby sites on the surface1712 of the Na. Although high dilution techniques are not always necessary, effective stirring (high-speed stirrer at 2000–2500 rpm) is usually required to generate “sodium sand”. Highly pure Na gives poorer results because the presence of a small percentage of K is important. Up to 50% potassium (1 : 1 Na/K)1713 has been used in acyloin condensations.
In a related reaction, aromatic carboxylic acids were condensed to α-diketones (2ArCOOH → ArCOCOAr) on treatment with excess Li in dry THF in the presence of ultrasound.1714
The acyloin condensation was used in an ingenious manner to prepare the first reported catenane (see Sec. 3.D).1715 This synthesis of a catenane produced only a small yield and relied on chance for threading the molecules before ring closure.
OS II, 114; IV, 840; VI, 167.
19-79 Reduction of Nitro to Azoxy Compounds
Nitro-azoxy reductive transformation
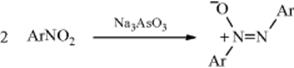
Azoxy compounds can be obtained from nitro compounds with certain reducing agents, notably sodium arsenite, sodium ethoxide, NaTeH,1716 and glucose. The most probable mechanism with most reagents is that one molecule of nitro compound is reduced to a nitroso compound and another to a hydroxylamine (Reaction 19-46), and these combine (Reaction 12-51). The combination step is rapid compared to the reduction process.1717 Nitroso compounds can be reduced to azoxy compounds with triethylphosphite or triphenylphosphine1718 or with an alkaline aqueous solution of an alcohol.1719
OS II, 57.
19-80 Reduction of Nitro to Azo Compounds
N-De-bisoxygen-coupling
![]()
Nitro compounds can be reduced to azo compounds with various reducing agents, of which LiAlH4 and Zn and alkali are the most common. A combination of triethylammonium formate and lead in methanol is also effective.1720With many of these reagents, slight differences in conditions can lead either to the azo or azoxy (Reaction 19-79) compound. By analogy to Reaction 19-79, this reaction may be looked on as a combination of ArN=O and ArNH2(13-24). However, when the reducing agent was NaBH4,1721 it was shown that azoxy compounds were intermediates. Nitroso compounds can be reduced to azo compounds with LiAlH4. Dicarborane, with a catalytic amount of acetic acid, reduces aromatic nitro compounds to the amine.1722
Nitro compounds can be further reduced to hydrazo compounds with Zn and sodium hydroxide, with hydrazine hydrate and Raney nickel,1723 or with LiAlH4 mixed with a metal chloride (e.g., TiCl4 or VCl3).1724 The reduction has also been accomplished electrochemically.
OS III, 103.
G. Reactions in which an Organic Substrate is Both Oxidized and Reduced
Some reactions that belong in this category have been considered in earlier chapters. Among these are the Tollens' condensation (Reaction 16-43), the benzil–benzilic acid rearrangement (Reaction 18-6), and the Wallach rearrangement (Reaction 18-43).
19-81 The Cannizzaro Reaction
Cannizzaro Aldehyde Disproportionation
![]()
Aromatic aldehydes, and aliphatic ones with no α hydrogen, give the Cannizzaro reaction when treated with NaOH or other strong bases.1725 Reaction with triethylamine and MgBr2 gave a room temperature Cannizzaro reaction.1726 The reaction is mediated by organobases.1727 In this reaction, one molecule of aldehyde oxidizes another to the acid and is itself reduced to the primary alcohol. Aldehydes with an α hydrogen do not give the reaction, because when these compounds are treated with base, the aldol reaction (16-34) is much faster.1728 Normally, the best yield of acid or alcohol is 50% each, but this can be altered in certain cases. Solvent-free reactions are known.1729On the other hand, high yields of alcohol can be obtained from almost any aldehyde by running the reaction in the presence of formaldehyde.1730 In this case, the formaldehyde reduces the aldehyde to alcohol and is itself oxidized to formic acid. In such a case, where the oxidant aldehyde differs from the reductant aldehyde, the reaction is called the crossed-Cannizzaro reaction.1731 The Tollens' condensation (Reaction 16-43) includes a crossed-Cannizzaroreaction as its last step. A Cannizzaro reaction with 1,4-dialdehydes (note that α-hydrogen atoms are present here) and a Rh catalyst gives ring closure, for example,1732
![]()
The product is the lactone derived from the hydroxy acid that would result from a normal Cannizzaro reaction. Chiral additives have been used, but with bis(oxazolidine) derivatives, the reaction proceeded with poor enantioselectivity.1733
α-Keto aldehydes give internal Cannizzaro reactions1734:
This product is also obtained on alkaline hydrolysis of compounds of the formula RCOCHX2. Similar reactions have been performed on α-keto acetals1735 and γ-keto aldehydes.
The mechanism1736 of the Cannizzaro reaction1737 involves a hydride shift (an example of mechanism type 2, Sec. 19.A). First −OH adds to the C=O to give 46, which may lose a proton in the basic solution to give the diion (47).
![]()
The strong electron-donating character of O− greatly facilitates the ability of the aldehyde hydrogen to leave with its electron pair. Of course, this effect is even stronger in 49. Hydride is transferred to another molecule of aldehyde. The hydride can come from 48 or 49:
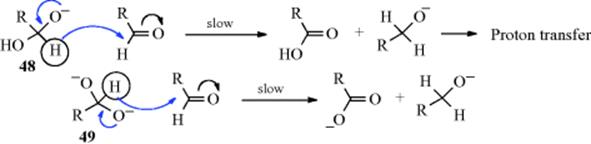
If the hydride ion comes from 48, the final step is a rapid proton transfer. In the other case, the acid salt is formed directly, and the alkoxide ion acquires a proton from the solvent. Evidence for this mechanism is (1) The reaction can be first order in base and second order in substrate (thus going through 48) or, at higher base concentrations, second order in each (going through 49); and (2) when the reaction was run in D2O, the recovered alcohol contained no α deuterium,1738 indicating that the hydrogen comes from another equivalent of aldehyde and not from the medium.1739
OS I, 276; II, 590; III, 538; IV, 110.
19-82 The Tishchenko Reaction
Tishchenko aldehyde-ester disproportionation
![]()
When aldehydes, with or without an α hydrogen, are treated with aluminum ethoxide, one molecule is oxidized and another is reduced, as in Reaction 19-81, but here they are found as the ester. The process is called the Tishchenko reaction.1740 Crossed-Tishchenko reactions are also possible. With more strongly basic alkoxides (e.g., Mg or sodium alkoxides), aldehydes with an a hydrogen give the aldol reaction. Treatment of a dialdehyde [e.g., phthalic dicarboxaldehyde (phthalaldehyde) with CaO] leads to a lactone.1741 Like Reaction 19-81, this reaction has a mechanism that involves hydride transfer.1742 The Tishchenko reaction can also be catalyzed1743 by Ru complexes,1744 organoactinides,1745 alkaline earth amides,1746 by Cp2ZrH21747 and, for aromatic aldehydes, by disodium tetracarbonylferrate [Na2Fe(CO)4].1748 Both CaO (noted above) and SrO have been used as catalysts.1749 A bis(phenylenedioxy) bis(aluminum) catalyst has been used to convert aliphatic aldehydes to the corresponding ester.1750 The bis Al(O-iPr)2 derivative of catechol has also been used as a catalyst.1751
A Tishchenko–aldol transfer reaction was reported using β-hydroxy ketones and an aldehydes with an AlMe3 catalyst, giving a monoacyl diol.1752
OS I, 104.
19-83 The Pummerer Rearrangement1753
Pummerer methyl sulfoxide rearrangement
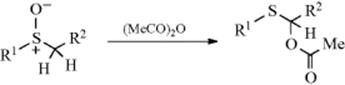
When sulfoxides bearing an α hydrogen are treated with acetic anhydride, the product is an α-acetoxy sulfide. This is one example of the Pummerer rearrangement,1754 in which the sulfur is reduced while an adjacent carbon is oxidized.1755 The product is readily hydrolyzed (Reaction 10-6) to the aldehyde (R2CHO).1756 Besides acetic anhydride, other anhydrides and acyl halides give similar products. Inorganic acids (e.g., HCl), also give the reaction, and RSOCH2R′ can be converted to RSCHClR′ in this way. Sulfoxides can also be converted to α-halo sulfides1757 by other reagents, including sulfuryl chloride, NBS, and NCS. Enantioselective Pummerer rearrangements are known.1758 Uncatalyzed thermal rearrangements are also known.1759
The following 4-step mechanism has been proposed for the reaction between acetic anhydride and DMSO1760:
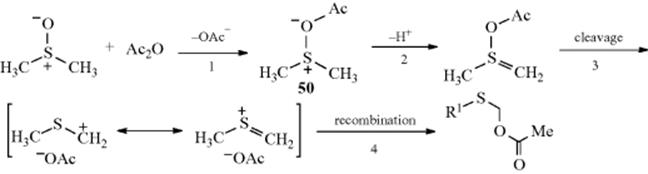
For DMSO and acetic anhydride, step 4 is intermolecular, as shown by 18O isotopic labeling studies.1761 With other substrates, however, step 4 can be inter- or intramolecular, depending on the structure of the sulfoxide.1762Depending on the substrate and reagent, any of the first three steps can be rate determining. In the case of Me2SO treated with (F3CCO)2O, the intermediate corresponding to 501763 could be isolated at low temperature, and on warming gave the expected product.1764 There is also an abundance of other evidence for this mechanism.1765
A sila-Pummerer rearrangement has been reported.1766
19-84 The Willgerodt Reaction
Willgerodt carbonyl transformation
![]()
In the Willgerodt reaction, a straight- or branched-chain aryl alkyl ketone is converted to the amide and/or the ammonium salt of the acid by heating with ammonium polysulfide.1767 The carbonyl group of the product is always at the end of the chain. Thus ArCOCH2CH3 gives the amide and the salt of ArCH2CH2CO2H and ArCOCH2CH2CH3 gives derivatives of ArCH2CH2CH2CO2H. However, yields sharply decrease with increasing length of chain. The reaction has also been carried out on vinylic and ethynyl aromatic compounds and on aliphatic ketones, but yields are usually lower in these cases. Unlike the Pummerer rearrangement (Reaction 19-83), which involves transposition of an oxygen from S to C, the Willgerodt reaction involves oxygen migration and oxidation of the organic species. The use of sulfur and a dry primary or secondary amine (or ammonia), as the reagent is called the Kindler modification of the Willgerodt reaction.1768 The product in this case is Ar(CH2)nCSNR2,1769 which can be hydrolyzed to the acid. Particularly good results are obtained with morpholine as the amine. For volatile amines, the HCl salts can be used instead, with NaOAc in DMF at 100 °C.1770 Dimethylamine has also been used in the form of dimethylammonium dimethylcarbamate (Me2NCOO− Me2NH2+).1771 The Kindler modification has also been applied to aliphatic ketones.1772 Thioamides have been prepared from ketones in a base-catalyzed reaction.1773
Alkyl aryl ketones can be converted to arylacetic acid derivatives in an entirely different manner. The reaction consists of treatment of the substrate with silver nitrate and I2 or Br2.1774
The mechanism of the Willgerodt reaction is not completely known, but some conceivable mechanisms can be excluded. Thus, one might suppose that the alkyl group becomes completely detached from the ring, and then attacks it with its other end. However, this possibility is ruled out by experiments, such as the following: When isobutyl phenyl ketone (51) is subjected to the Willgerodt reaction, the product is 52, not 53, which would arise if the end carbon of the ketone became bonded to the ring in the product1775:
This also excludes a cyclic-intermediate mechanism similar to that of the Claisen rearrangement (Reaction 18-33). Another important fact is that the reaction is successful for singly branched side chains (e.g., 52), but not for doubly branched side chains, as in PhCOCMe3.1775 Still another piece of evidence is that compounds oxygenated along the chain give the same products; thus PhCOCH2CH3, PhCH2COMe, and PhCH2CH2CHO all give PhCH2CH2CONH2.1776 All these facts point to a mechanism consisting of consecutive oxidations and reductions along the chain, although just what form these take is not certain. Initial reduction to the hydrocarbon can be ruled out, since alkylbenzenes do not give the reaction. In certain cases, imines1777 or enamines1778 have been isolated from primary and secondary amines, respectively, and these have been shown to give the normal products, leading to the suggestion that they may be reaction intermediates.
Notes
1. For more extensive tables, see Soloveichik, S.; Krakauer, H. J. Chem. Educ. 1966, 43, 532.
2. See Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 16, Elsevier, NY, 1980; Oxidation in Organic Chemistry, Academic Press, NY, pt. A [Wiberg, K.B.], 1965, pts. B, C, and D [Trahanovsky, W.S.], 1973, 1978, 1982; Waters, W.A. Mechanisms of Oxidation of Organic Compounds, Wiley, NY, 1964; Stewart, R. Oxidation Mechanisms; W. A. Benjamin, NY, 1964. For a review, see Stewart, R. Isot. Org. Chem. 1976, 2, 271.
3. Wiberg, K.B. Surv. Prog. Chem. 1963, 1, 211.
4. See Eberson, L. Electron Transfer Reactions in Organic Chemistry; Springer, NY, 1987; Eberson, L. Adv. Phys. Org. Chem. 1982, 18, 79; Deuchert, K.; Hünig, S. Angew. Chem. Int. Ed. 1978, 17, 875.
5. Littler, J.S.; Sayce, I.G. J. Chem. Soc. 1964, 2545.
6. See Mihailovic, M.Lj.; Cekovic, Z. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pp. 505–592.
7. For a review, see Watt, C.I.F. Adv. Phys. Org. Chem. 1988, 24, 57.
8. See Nenitzescu, C.D. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, pp. 463–520.
9. See Hudlicky, M. J. Chem. Educ. 1977, 54, 100.
10. See Mijs, W.J.; de Jonge, C.R.J.I. Organic Synthesis by Oxidation with Metal Compounds, Plenum, NY, 1986; Cainelli, G.; Cardillo, G. Chromium Oxidations in Organic Chemistry, Springer, NY, 1984; Arndt, D. Manganese Compounds as Oxidizing Agents in Organic Chemistry, Open Court Publishing Company, La Salle, IL, 1981; Lee, D.G. The Oxidation of Organic Compounds by Permanganate Ion and Hexavalent Chromium; Open Court Publishing Company: La Salle, IL, 1980. For some reviews, see Curci, R. Adv. Oxygenated Processes 1990, 2, 1 (dioxiranes); Adam, W.; Curci, R.; Edwards, J.O. Acc. Chem. Res. 1989, 22, 205 (dioxiranes); Murray, R.W. Chem. Rev. 1989, 89, 1187(dioxiranes); Ley, S.V. in Liotta, D.C. Organoselenium Chemistry, Wiley, NY, 1987, pp. 163–206 (seleninic anhydrides and acids); Fatiadi, A.J. Synthesis 1987, 85 (KMnO4); Rubottom, G.M. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. D, Academic Press, NY, 1982, pp. 1–145 (lead tetraacetate); Fatiadi, A.J. in Pizey, J.S. Synthetic Reagents, Vol. 4, Wiley, NY, 1981, pp. 147–335; Synthesis 1974, 229 (HIO4); Fatiadi, A.J. Synthesis 1976, 65, 133 (MnO2); Pizey, J.S. Synthetic Reagents, Vol. 2, Wiley, NY, 1974, pp. 143–174 (MnO2); George, M.V.; Balachandran, K.S. Chem. Rev. 1975, 75, 491 (nickel peroxide); Courtney, J.L.; Swansborough, K.F. Rev. Pure Appl. Chem. 1972, 22, 47 (ruthenium tetroxide); Ho, T.L. Synthesis 1973, 347 (ceric ion); Aylward, J.B. Q. Rev. Chem. Soc. 1971, 25, 407 (lead tetraacetate); Sklarz, B. Q. Rev. Chem. Soc. 1967, 21, 3 (HIO4); Korshunov, S.P.; Vereshchagin, L.I. Russ. Chem. Rev. 1966, 35, 942 (MnO2);. For reviews of the behavior of certain reducing agents, see Keefer, L.K.; Lunn, G. Chem. Rev. 1989, 89, 459 (Ni-Al alloy); Málek, J. Org. React. 1988, 36, 249; 1985, 34, 1–317 (metal alkoxyaluminum hydrides); Caubère, P. Angew. Chem. Int. Ed. 1983, 22, 599 (modified sodium hydride); Nagai, Y. Org. Prep. Proced. Int. 1980, 12, 13 (hydrosilanes); Pizey, J.S. Synthetic Reagents, Vol. 1, Wiley, NY, 1974, pp. 101–294 (LiAlH4); Winterfeldt, E. Synthesis 1975, 617 (diisobutylaluminum hydride and triisobutylaluminum); Hückel, W. Fortschr. Chem. Forsch. 1966, 6, 197 (metals in ammonia or amines). See also Ref. 9.
11. For books on oxidation reactions, see Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, 1990; Haines, A.H. Methods for the Oxidation of Organic Compounds, 2 vols., Academic Press, NY, 1985, 1988 [The first volume pertains to hydrocarbon substrates; the second mostly to oxygen- and nitrogen-containing substrates]; Chinn, L.J. Selection of Oxidants in Synthesis, Marcel Dekker, NY, 1971; Augustine, R.L.; Trecker, D.J. Oxidation, 2 Vols., Marcel Dekker, NY, 1969, 1971.
12. See Haines, A.H. Methods for the Oxidation of Organic Compounds, Academic Press, NY, 1985, pp. 16–22, 217–222; Fu, P.P.; Harvey, R.G. Chem. Rev. 1978, 78, 317; Valenta, Z. in Bentley, K.W.; Kirby, G.W. Elucidation of Chemical Structures by Physical and Chemical Methods (Vol. 4 of Weissberger, A. Techniques of Chemistry), 2nd ed., pt. 2, Wiley, NY, 1973, pp. 1–76; House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 34–44.
13. See Rylander, P.N. Organic Synthesis with Noble Metal Catalysts, Academic Press, NY, 1973, pp. 1–59.
14. See Cossy, J.; Belotti, D. Org. Lett. 2002, 4, 2557. See Cho, C.S.; Patel, D.B.; Shim, S.C. Tetrahedron 2005, 61, 9490.
15. Bercaw, J.E.; Hazari, N.; Labinger, J.A. J. Org. Chem. 2008, 73, 8654.
16. See Tsai, M.; Friend, C.M.; Muetterties, E.L. J. Am. Chem. Soc. 1982, 104, 2539. See also, Augustine, R.L.; Thompson, M.M. J. Org. Chem. 1987, 52, 1911.
17. Land, D.P.; Pettiette-Hall, C.L.; McIver Jr., R.T.; Hemminger, J.C. J. Am. Chem. Soc. 1989, 111, 5970.
18. Srinivas, G.; Periasamy, M. Tetrahedron Lett. 2002, 43, 2785.
19. Marshall, J.L.; Miiller, D.E.; Ihrig, A.M. Tetrahedron Lett. 1973, 3491.
20. Nakamichi, N.; Kawabata, H.; Hiyashi, M. J. Org. Chem. 2003, 68, 8272.
21. Silverwood, H.A.; Orchin, M. J. Org. Chem. 1962, 27, 3401.
22. Becker, H.; Turner, A.B. in Patai, S.; Rappoport, Z. The Chemistry of the Quinonoid Compounds, Vol. 2, pt. 2, Wiley, NY, 1988, pp. 1351–1384; Becker, H. in Patai, S. The Chemistry of the Quinonoid Compounds, Vol. 1, pt. 1, Wiley, NY, 1974, pp. 335–423.
23. See Turner, A.B. in Pizey, J.S. Synthetic Reagents, Vol. 3, Wiley, NY, 1977, pp. 193–225; Walker, D.; Hiebert, J.D. Chem. Rev. 1967, 67, 153.
24. Trost, B.M. J. Am. Chem. Soc. 1967, 89, 1847. See also, Radtke, R.; Hintze, H.; Rösler, K.; Heesing, A. Chem. Ber. 1990, 123, 627; Höfler, C.; Rüchardt, C. Liebigs Ann. Chem. 1996, 183. See also Ref. 22.
25. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 187–191.
26. See Leffingwell, J.C.; Bluhm, H.J. Chem. Commun. 1969, 1151.
27. Tanaka, H.; Ikeno, T.; Yamada, T. Synlett 2003, 576.
28. Varma, R.S.; Kumar, D. Tetrahedron Lett. 1998, 39, 9113.
29. Damavandi, J.A.; Zolfigol, M.A.; Karami, B. Synth. Commun. 2001, 31, 3183.
30. Zolfigol, M.A.; Kiany-Borazjani, M.; Sadeghi, M.M.; Mohammadpoor-Baltork, I.; Memarian, H.R. Synth. Comm. 2000, 30, 551.
31. Memarian, H.R.; Sadeghi, M.M.; Momeni, A.R. Synth. Commun. 2001, 31, 2241.
32. Lu, J.; Bai, Y.; Wang, Z.; Yang, B.Q.; Li, W. Synth. Commun. 2001, 31, 2625.
33. Cai, X.-h.; Yang, H.-j.; Zhang, G.-l. Can. J. Chem. 2005, 83, 273.
34. Heravi, M.M.; Behbahani, F.K.; Oskooie, H.A.; Shoar, R.H. Tetrahedron Lett. 2005, 46, 2775.
35. DeKimpe, N.; Keppens, M.; Fonck, G. Chem. Commun. 1996, 635.
36. Bigdeli, M.A.; Rahmati, A.; Abbasi-Ghadim, H.; Mahdavinia, G.H. Tetrahedron Lett. 2007, 48, 4575.
37. Locatelli, F.; Candy, J.-P.; Didillon, B.; Niccolai, G.P.; Uzio, D.; Basset, J.-M. J. Am. Chem. Soc. 2001, 123, 1658.
38. Göttker-Schnetmann, I.; White, P.; Brookhart, M. J. Am. Chem. Soc. 2004, 126, 1804.
39. Takai, K.; Kokumai, R.; Toshikawa, S. Synlett 2002, 1164.
40. See Haines, A.J. Methods for the Oxidation of Organic Compounds, Vol. 1, Academic Press, NY, 1985, pp. 6–16, 206–216. For lists of examples, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 251–256.
41. See Leonard, N.J.; Musker, W.K. J. Am. Chem. Soc. 1959, 81, 5631; 1960, 82, 5148.
42. See Haynes, L.W.; Cook, A.G. in Cook, A.G. Enamines, 2nd ed. Marcel Dekker, NY, 1988, pp. 103–163; Lee, D.G. in Augustine, R.L.; Trecker, D.J. Oxidation, Vol. 1, Marcel Dekker, NY, 1969, pp. 102–107.
43. Bolig, A.D.; Brookhart, M. J. Am. Chem. Soc. 2007, 129, 14544.
44. Zhang, X.; Fried, A.; Knapp, S.; Goldman, A.S. Chem. Commun. 2003, 2060.
45. See Back, T.G. in Patai, S. The Chemistry of Organic Selenium and Tellurium Compounds, pt. 2, Wiley, NY, 1987, pp. 91–213, 110–114; Jerussi, R.A. Sel. Org. Transform. 1970, 1, 301, see pp. 315–321.
46. Bernstein, S.; Littell, R. J. Am. Chem. Soc. 1960, 82, 1235.
47. See Barnes, C.S.; Barton, D.H.R. J. Chem. Soc. 1953, 1419.
48. Tokunaga, M.; Harada, S.; Iwasawa, T.; Obora, Y.; Tsuji, Y. Tetrahedron Lett. 2007, 48, 6860. For a review, see Muzart, J. Eur. J. Org. Chem. 2010, 3779.
49. See Mukaiyama, T.; Ohshima, M.; Nakatsuka, T. Chem. Lett. 1983, 1207. See also, Heck, R.F. Palladium Reagents in Organic Synthesis, Academic Press, NY, 1985, pp. 103–110.
50. Cardinale, G.; Laan, J.A.M.; Russell, S.W.; Ward, J.P. Recl. Trav. Chim. Pays-Bas 1982, 101, 199.
51. Barton, D.H.R.; Hui, R.A.H.F.; Ley, S.V.; Williams, D.J. J. Chem. Soc. Perkin Trans. 1 1982, 1919; Barton, D.H.R.; Godfrey, C.R.A.; Morzycki, J.W.; Motherwell, W.B.; Ley, S.V. J. Chem. Soc. Perkin Trans. 1 1982, 1947.
52. Jung, M.E.; Pan, Y.; Rathke, M.W.; Sullivan, D.F.; Woodbury, R.P. J. Org. Chem. 1977, 42, 3961.
53. Ryu, I.; Murai, S.; Hatayama, Y.; Sonoda, N. Tetrahedron Lett. 1978, 3455. Also see Tsuji, J.; Minami, I.; Shimizu, I. Tetrahedron Lett. 1983, 24, 5635, 5639.
54. Evans, P.A.; Longmire, J.M.; Modi, D.P. Tetrahedron Lett. 1995, 36, 3985.
55. Larock, R.C.; Hightower, T.R.; Kraus, G.A.; Hahn, P.; Zheng, O. Tetrahedron Lett. 1995, 36, 2423.
56. See Maguire, J.A.; Boese, W.T.; Goldman, A.S. J. Am. Chem. Soc. 1989, 111, 7088; Sakakura, T.; Ishida, K.; Tanaka, M. Chem. Lett. 1990, 585, and references cited therein.
57. See Breslow, R. Chemtracts: Org. Chem. 1988, 1, 333; Acc. Chem. Res. 1980, 13, 170; Isr. J. Chem. 1979, 18, 187; Chem. Soc. Rev. 1972, 1, 553.
58. Baldwin, J.E.; Bhatnagar, A.K.; Harper, R.W. Chem. Commun. 1970, 659.
59. See Czekay, G.; Drewello, T.; Schwarz, H. J. Am. Chem. Soc. 1989, 111, 4561. See also, Bégué, J. J. Org. Chem. 1982, 47, 4268; Nagata, R.; Saito, I. Synlett 1990, 291; Breslow, R.; Brandl, M.; Hunger, J.; Adams, A.D. J. Am. Chem. Soc. 1987, 109, 3799; Batr, R.; Breslow, R. Tetrahedron Lett. 1989, 30, 535; Orito, K.; Ohto, M.; Suginome, H. J. Chem. Soc. Chem. Commun. 1990, 1076.
60. Akiyama, S.; Nakatsuji, S.; Nomura, K.; Matsuda, K.; Nakashima, K. J. Chem. Soc. Chem. Commun. 1991, 948.
61. Tilstam, U.; Harre, M.; Heckrodt, T.; Weinmann, H. Tetrahedron Lett. 2001, 42, 5385.
62. Paquette, L.A.; Doherty, A.M. Polyquinane Chemistry, Springer, NY, 1987. See in Olah, G.A. Cage Hydrocarbons, Wiley, NY, 1990, the reviews by Paquette, L.A. pp. 313–352, and by Fessner, W.; Prinzbach, H. pp. 353–405; Paquette, L.A. Chem. Rev. 1989, 89, 1051; in Lindberg, T. Strategies and Tactics in Organic Synthesis, Academic Press, NY, 1984, pp. 175–200.
63. Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, pp. 114–126, 132–149; Haines, A.M. Methods for the Oxidation of Organic Compounds, Vol. 2, Academic Press, NY, 1988, pp. 5–148, 326–390; Müller, P. in Patai, S. The Chemistry of Functional Groups, Supplement E, Wiley, NY, 1980, pp. 469–538. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1234–1250.
64. See Lee, D.G. in Augustine, R.L.; Trecker, D.J. Oxidation, Vol. 2, Marcel Dekker, NY, 1971, pp. 56–81; House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 257–273.
65. See Cainelli, G.; Cardillo, G. Chromium Oxidations in Organic Chemistry, Open Court Pub. Co., La Salle, IL, 1981, pp. 118–216; Fieser, L.F.; Fieser, M. Reagents for Organic Synthesis Vol. 1, Wiley, NY, 1967, pp. 142–147, 1059–1064, and subsequent volumes in this series.
66. Bowers, A.; Halsall, T.G.; Jones, E.R.H.; Lemin, A.J. J. Chem. Soc. 1953, 2548. Also see, Ali, M.H.; Wiggin, C.J. Synth. Commun. 2001, 31, 1389; Ali, M.H.; Wiggin, C.J. Synth. Commun. 2001, 31, 3383.
67. See Djerassi, C.; Hart, P.A.; Warawa, E.J. J. Am. Chem. Soc. 1964, 86, 78.
68. Lou, J.-D.; Gao, C.-L.; Ma, Y.-C.; Huang, L.-H.; Li, L. Tetrahedron Lett. 2006, 47, 311.
69. Lou, J.-D.; Xu, Z.-N. Tetrahedron Lett. 2002, 43, 6095.
70. González-Núñez, M.E.; Mello, R.; Olmos, A.; Acerete, R.; Asensio, G. J. Org. Chem. 2006, 71, 1039.
71. Moiseenkov, A.M.; Cheskis, B.A.; Veselovskii, A.B.; Veselovskii, V.V.; Romanovich, A.Ya.; Chizhov, B.A. J. Org. Chem. USSR 1987, 23, 1646.
72. Singh, J.; Sharma, M.; Chhibber, M.; Kaur, J.; Kad, G.L. Synth. Commun. 2000, 30, 3941. Also see Heravi, M.M.; Ajami, D.; Tabar-Hydar, K. Synth. Commun. 1999, 29, 163; Mirza-Ayhayan, M.; Heravi, M.M. Synth. Commun. 1999, 29, 785
73. For a review, see Patel, S.; Mishra, B.K. Tetrahedron 2007, 63, 4367.
74. Xu, L.; Trudell, M.L. Tetrahedron Lett. 2003, 44, 2553.
75. Harding, K.E.; May, L.M.; Dick, K.F. J. Org. Chem. 1975, 40, 1664.
76. Though ketones are much less susceptible to further oxidation than aldehydes, such oxidation is possible (19-8), and care must be taken to avoid it, usually by controlling the temperature and/or the oxidizing agent.
77. Fernandes, R.A.; Kumar, P. Tetrahedron Lett. 2003, 44, 1275.
78. Also see Nishiguchi, T.; Asano, F. J. Org. Chem. 1989, 54, 1531; Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1234–1250.
79. See Warrener, R.N.; Lee, T.S.; Russell, R.A.; Paddon-Row, M.N. Aust. J. Chem. 1978, 31, 1113.
80. Collins, J.C.; Hess, W.W. Org. Synth. VI, 644; Sharpless, K.B.; Akashi, K. J. Am. Chem. Soc. 1975, 97, 5927.
81. Corey, E.J.; Suggs, J.W. Tetrahedron Lett. 1975, 2647. See Luzzio, F.A.; Guziec, Jr., F.S. Org. Prep. Proced. Int. 1988, 20, 533; Piancatelli, G.; Scettri, A.; D'Auria, M. Synthesis 1982, 245; Agarwal, S.; Tiwari, H.P.; Sharma, J.P. Tetrahedron 1990, 46, 4417; Salehi, P.; Firouzabadi, H.; Farrokhi, A.; Gholizadeh, M. Synthesis 2001, 2273.
82. See Czernecki, S.; Georgoulis, C.; Stevens, C.L.; Vijayakumaran, K. Tetrahedron Lett. 1985, 26, 1699.
83. See Ren, S.-K.; Wang, F.; Dou, H.-N.; Fan, C.-A.; He, L.; Song, Z.-L.; Xia, W.-J.; Li, D-R.; Jia, Y.-X.; Li, X.; Tu, Y.-Q. Synthesis 2001, 2384.
84. Hajipour, A.R.; Mallakpour, S.E.; Khoee, S. Synlett 2000, 740.
85. Khodaei, M.M.; Salehi, P.; Goodarzi, M. Synth. Commun. 2001, 31, 1253.
86. Rajkumar, G.A.; Arabindoo, B.; Murugesan, V. Synth. Commun. 1999, 29, 2105.
87. Hajipour, A.R.; Mallakpour, S.E.; Backnejad, H. Synth. Commun. 2000, 30, 3855.
88. For a review, see McKillop, A.; Young, D.W. Synthesis 1979, 401. See also, Shirini, F.; Dabiri, M.; Dezyani, S.; Jalili, F. Russ. J. Org. Chem. 2005, 41, 390.
89. Cainelli, G.; Cardillo, G.; Orena, M.; Sandri, S. J. Am. Chem. Soc. 1976, 98, 6737; Santaniello, E.; Ponti, F.; Manzocchi, A. Synthesis 1978, 534. See also, San Filippo, Jr., J.; Chern, C. J. Org. Chem. 1977, 42, 2182.
90. Srinivasan, R.; Balasubramanian, K. Synth. Commun. 2000, 30, 4397.
91. Hajipour, A.R.; Safaei, S.; Ruoho, A.E. Synth. Commun. 2009, 39, 3687.
92. See Müller, P. Chimia 1977, 31, 209; Wiberg, K.B. in Wiberg, K.B. Oxidation in Organic Chemistry, pt. A, Academic Press, NY, 1965, pp. 142–170; Waters, W.A. Mechanisms of Oxidation of Organic Compounds, Wiley, NY, 1964, pp. 49–71; Stewart, R. Oxidation Mechanisms, W.A. Benjamin, NY, 1964, pp. 37–48; Sengupta, K.K.; Samanta, T.; Basu, S.N. Tetrahedron 1985, 41, 205.
93. Westheimer, F.H. Chem. Rev. 1949, 45, 419, see p. 434; Holloway, F.; Cohen, M.; Westheimer, F.H. J. Am. Chem. Soc. 1951, 73, 65.
94. Kwart, H.; Nickle, J.H. J. Am. Chem. Soc. 1979, 98, 2881 and cited rererences; Sengupta, K.K.; Samanta, T.; Basu, S.N. Tetrahedron 1986, 42, 681. See also, Agarwal, S.; Tiwari, H.P.; Sharma, J.P. Tetrahedron 1990, 46, 1963.
95. Westheimer, F.H.; Nicolaides, N. J. Am. Chem. Soc. 1949, 71, 25. Also see Wiberg, K.B.; Schäfer, H. J. Am. Chem. Soc. 1969, 91, 927, 933; Lee, D.G.; Raptis, M. Tetrahedron 1973, 29, 1481.
96. Doyle, M.P.; Swedo, R.J.; Rocek, J. J. Am. Chem. Soc. 1973, 95, 8352; Wiberg, K.B.; Mukherjee, S.K. J. Am. Chem. Soc. 1974, 96, 1884, 6647.
97. See Cockerill, A.F.; Harrison, R.G. in Patai, S. The Chemistry of Functional Groups, Supplement A pt. 1, Wiley, NY, 1977, pp. 264–277.
98. Barter, R.M.; Littler, J.S. J. Chem. Soc. B 1967, 205. See Moodie, R.B.; Richards, S.N. J. Chem. Soc. Perkin Trans. 2 1986, 1833; Ross, D.S.; Gu, C.; Hum, G.P.; Malhotra, R. Int. J. Chem. Kinet. 1986, 18, 1277.
99. Bonthrone, W.; Reid, D.H. J. Chem. Soc. 1959, 2773.
100. Walling, C.; Camaioni, D.M. J. Org. Chem. 1978, 43, 3266; Clerici, A.; Minisci, F.; Ogawa, K.; Surzur, J. Tetrahedron Lett. 1978, 1149; Beylerian, N.M.; Khachatrian, A.G. J. Chem. Soc. Perkin Trans. 2 1984, 1937.
101. Littler, J.S.; Waters, W.A. J. Chem. Soc. 1959, 4046.
102. Littler, J.S. J. Chem. Soc. 1962, 2190.
103. See Takemoto, T.; Yasuda, K.; Ley, S.V. Synlett 2001, 1555. For oxidation in an ionic liquid, see Kumar, A.; Jain, N.; Chauhan, S.M.S. Synth. Commun. 2004, 34, 2835.
104. Kim, K.S.; Chung, S.; Cho, I.H.; Hahn, C.S. Tetrahedron Lett. 1989, 30, 2559. See Lee, D.G. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. D Academic Press, NY, 1982, pp. 147–206.
105. See Landini, D.; Montanari, F.; Rolla, F. Synthesis 1979, 134; Pletcher, D.; Tait, S.J.D. J. Chem. Soc. Perkin Trans. 2 1979, 788.
106. Morris Jr., P.E.; Kiely, D.E. J. Org. Chem. 1987, 52, 1149.
107. Yamawaki, J.; Sumi, S.; Ando, T.; Hanfusa, T. Chem. Lett. 1983, 379.
108. See Noureldin, N.A.; Lee, D.G. Tetrahedron Lett. 1981, 22, 4889. See also, Menger, F.M.; Lee, C. J. Org. Chem. 1979, 44, 3446.
109. Lou, J.D.; Xu, Z.-N. Tetrahedron Lett. 2002, 43, 6149.
110. Taylor, R.J.K.; Reid, M.; Foot, J.; Raw, S.A. Acc. Chem. Res. 2005, 38 851. See Varma, R.S.; Saini, R.K.; Dahiya, R. Tetrahedron Lett. 1997, 38, 7823.
111. Firouzabadi, H.; Etemadi, S.; Karimi, B.; Jarrahpour, A.A. Synth. Commun. 1999, 29, 4333.
112. Pearson, A.J.; Kwak, Y. Tetrahedron Lett. 2005, 46, 5417.
113. Shaabani, A.; Karimi, A.-R. Synth. Commun. 2001, 31, 759.
114. Tajbakhsh, M.; Heravi, M.M.; Habibzadeh, S.; Ghassemzadeh, M. J. Chem. Res. (S) 2001, 39.
115. Guan, B.; Xing, D.; Cai, G.; Wan, X.; Yu, N.; Fang, Z.; Yang, L.; Shi, Z. J. Am. Chem. Soc. 2005, 127, 18004.
116. Adair, G.R.A.; Williams, J.M.J. Tetrahedron Lett. 2005, 46, 8233.
117. Griffith, W.P.; Ley, S.V. Aldrichimica Acta 1990, 23, 13; Chandler, W.D.; Wang, Z.; Lee, D.G. Can. J. Chem. 2005, 83, 1212.
118. With organotrifluoroborates: Molander, G.A.; Petrillo, D.E. J. Am. Chem. Soc. 2006, 128, 9634.
119. Lenz, R.; Ley, S.V. J. Chem. Soc. Perkin Trans. 1 1997, 3291.
120. Hinzen, B.; Lenz, R.; Ley, S.V. Synthesis 1998, 977. Also see Brown, D.S.; Kerr, W.J.; Lindsay, D.M.; Pike, K.G.; Ratcliffe, P.D. Synlett 2001, 1257.
121. Ley, S.V.; Ramarao, C.; Smith, M.D. Chem. Commun. 2001, 2278.
122. See Lee, D.G.; van den Engh, M. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. B Academic Press, NY, 1973, pp. 197–222.
123. Divalentin, C.; Gandolfi, R.; Gisdakis, P.; Rösch, N. J. Am. Chem. Soc. 2001, 123, 2365; Jain, S.L.; Sharma, V.B.; Sain, B. Tetrahedron Lett. 2004, 45, 1233.
124. Barrett, A.G.M.; Braddock, D.C.; McKinnell, R.M.; Waller, F.J. Synlett 1999, 1489.
125. Martín, S.E.; Garrone, A. Tetrahedron Lett. 2003, 44, 549.
126. Mehdi, H.; Bodor, A.; Lantos, D.; Horváth, I.T.; De Vos, D.E.; Binnemans, K. J. Org. Chem. 2007, 72, 517.
127. Matano, Y.; Nomura, H. J. Am. Chem. Soc. 2001, 123, 6443. With tert-butylhydroperoxide, see Malik, P.; Chakraborty, D. Synthesis 2010, 3736.
128. See Schultz, M.J.; Sigman, M.S. Tetrahedron 2006, 62, 8227; Lenoir, D. Angew. Chem. Int. Ed. 2006, 45, 3206; Zhan, B.-Z.; Thompson, A. Tetrahedron 2004, 60, 2917; Mallat, T.; Baiker, A. Chem. Rev. 2004, 104, 3037; Uma, R.; Crévisy, C.; Grée, R. Chem. Rev. 2003, 103, 27. Catalysts of Au: Abad, A.; Almela, C.; Corma, A.; García, H. Tetrahedron 2006, 62, 6666; Li, H.; Guan, B.; Wang, W.; Xing, D.; Fang, Z.; Wan, X.; Yang, L.; Shi, Z. Tetrahedron 2007, 63, 8430; Miyamura, H.; Matsubara, R.; Miyazaki, Y.; Kobayashi, S. Angew. Chem. Int. Ed. 2007, 46, 4151; Abad, A.; Almela, C.; Corma, A.; García, H. Chem. Commun. 2006, 3178; Kim, S.; Bae, S.W.; Lee, J.S.; Park, J. Tetrahedron 2009, 65, 1461. Cu: Lipshutz, B.H.; Shimizu, H. Angew. Chem. Int. Ed. 2004, 43, 2228; Jiang, N.; Ragauskas, A.J. Org. Lett. 2005, 7, 3689. Co: Jiang, N.; Ragauskas, A.J. J. Org. Chem. 2006, 71, 7087. Mo: Velusamy, S.; Ahamed, M.; Punniyamurthy, T. Org. Lett. 2004, 6, 4821. Pd: Nielsen, R.J.; Goddard III, W.A. J. Am. Chem. Soc. 2006, 128, 9651; Steinhoff, B.A.; King, A.E.; Stahl, S.S. J. Org. Chem. 2006, 71, 1861; Batt, F.; Bourcet, E.; Kassab, Y.; Fache, F. Synlett 2007, 1869. Ru: Yamaguchi, K.; Mizuno, N. Angew. Chem. Int. Ed. 2002, 41, 4538. V: Jiang, N.; Ragauskas, A.J. Tetrahedron Lett. 2007, 48, 273.
129. Zhan, B.-Z.; White, M.A.; Sham, T.-K.; Pincock, J.A.; Doucet, R.J.; Rao, K.V.R.; Robertson, K.N.; Cameron, T.S. J. Am. Chem. Soc. 2003, 125, 2195.
130. Heravi, M.M.; Ajami, D.; Aghapoor, K.; Ghassemzadeh, M. Chem. Commun. 1999, 833.
131. For a review, see Arterburn, J.B. Tetrahedron 2001, 57, 9765.
132. See Sakata, Y.; Ishii, Y. J. Org. Chem. 1991, 56, 6233.
133. Park, H.J.; Lee, J.C. Synlett 2009, 79.
134. Tomioka, H.; Takai, K.; Oshima, K.; Nozaki, H. Tetrahedron Lett. 1981, 22, 1605.
135. Maione, A.M.; Romeo, A. Synthesis 1984, 955.
136. Doyle, M.P.; Dow, R.L.; Bagheri, V.; Patrie, W.J. J. Org. Chem. 1983, 48, 476.
137. For a list of references, see Kulkarni, M.G.; Mathew T.S. Tetrahedron Lett. 1990, 31, 4497.
138. Nakano, T.; Terada, T.; Ishii, Y.; Ogawa, M. Synthesis 1986, 774.
139. Tymonko, S.A; Nattier, B.A.; Mohan, R.S. Tetrahedron Lett. 1999, 40, 7657.
140. Zhou, Y.-M.; Ye, X.-R.; Xin, X.-Q. Synth. Commun. 1999, 29, 2229.
141. Kirahara, M.; Ochiai, Yy.; Takizawa, S.; Takahata, H.; Nemoto, H. Chem. Commun. 1999, 1387. See also, Sigman, M.S.; Jensen, D.R. Acc. Chem. Res. 2006, 39, 221.
142. Onomura, O.; Arimoto, H.; Matsumura, Y.; Demizu, Y. Tetrahedron Lett. 2007, 48, 8668.
143. See Djerassi, C. Org. React. 1951, 6, 207. See Ooi, T.; Miura, T.; Itagaki, Y.; Ichikawa, H.; Maruoka, K. Synthesis 2002, 279; Graves, C.R.; Zeng, B.-S.; Nguyen. S.T. J. Am. Chem. Soc. 2006, 128, 12596.
144. Suzuki, T.; Morita, K.; Tsuchida, M.; Hiroi, K. J. Org. Chem. 2003, 68, 1601.
145. Ajjou, A.N. Tetrahedron Lett. 2001, 42, 13.
146. Ajjou, A.N.; Pinet, J.-L. Can. J. Chem. 2005, 83, 702.
147. Sominsky, L.; Rozental, E.; Gottlieb, H.; Gedanken, A.; Hoz, S. J. Org. Chem. 2004, 69, 1492.
148. Omura, K.; Swern, D. Tetrahedron 1978, 34, 1651. See Ohsugi, S.-i.; Nishide, K.; Oono, K.; Okuyama, K.; Fudesaka, M.; Kodama, S.; Node, M. Tetrahedron 2003, 59, 8393.
149. For a mechanism study, see Giagou T.; Meyer, M.P. J. Org. Chem. 2010, 75, 8088.
150. Fang, X.; Bandarage, U.K.; Wang, T.; Schroeder, J.D.; Garvey, D.S. J. Org. Chem. 2001, 66, 4019.
151. Nishida, K.; Ohsugi, S.-i.; Fudesaka, M.; Kodama, S.; Node, M. Tetrahedron Lett. 2002, 43, 5177.
152. Crich, D.; Neelamkavil, S. Tetrahedron 2002, 58, 3865.
153. Choi, M.K.W.C.; Toy, P.H. Tetrahedron 2003, 59, 7171.
154. For a review, see Mancuso, A.J.; Swern, D. Synthesis 1981, 165.
155. Albright, J.D.; Goldman, L. J. Am. Chem. Soc. 1967, 89, 2416.
156. Parikh, J.R.; Doering, W. von E. J. Am. Chem. Soc. 1967, 89, 5507.
157. Huang, S.L.; Omura, K.; Swern, D. Synthesis 1978, 297.
158. Dubey. A.; Kandula, S.R.V.; Kumar, P. Synth. Commun. 2008, 38, 746.
159. Albright, J.D. J. Org. Chem. 1974, 39, 1977.
160. Bisai, A.; Chandrasekhar, M.; Singh, V.K. Tetrahedron Lett. 2002, 43, 8355.
161. Godfrey, A.G.; Ganem, B. Tetrahedron Lett. 1990, 31, 4825.
162. Khenkin, A.M.; Neumann, R. J. Org. Chem. 2002, 67, 7075.
163. Li, C.; Xu, Y.; Lu, M.; Zhao, Z.; Liu, L.; Zhao, Z.; Cui, Y.; Zheng, P.; Ji, X.; Gao, G. Synlett 2002, 2041.
164. The DCC is converted to dicyclohexylurea, which in some cases is difficult to separate from the product. One way to avoid this problem is to use a carbodiimide linked to an insoluble polymer: Weinshenker, N.M.; Shen, C. Tetrahedron Lett. 1972, 3285.
165. Fenselau, A.H.; Moffatt, J.G. J. Am. Chem. Soc. 1966, 88, 1762; Albright, J.D.; Goldman, L. J. Org. Chem. 1965, 30, 1107.
166. Rychnovsky, S.D.; Vaidyanathan, R. J. Org. Chem. 1999, 64, 310.
167. Mn/Co: Cecchetto, A.; Fontana, F.; Minisci, F.; Recupero, F. Tetrahedron Lett. 2001, 42, 6651. Mo: Ben-Daniel, R.; Alssteers, P.; Neumann, R. J. Org. Chem. 2001, 66, 8650. Ru: Dijksman, A.; Marino-González, A.; Payeras, A.M.; Arends, I.W.C.E.; Sheldon, R.A. J. Am. Chem. Soc. 2001, 123, 6826.
168. Xie, Y.; Mo, W.; Xu, D.; Shen, Z.; Sun, N.; Hu, B.; Hu, X. J. Org. Chem. 2007, 72, 4288.
169. Gamez, P.; Arends, I.W.C.E.; Reedijk, J.; Sheldon, R.A. Chem. Commun. 2003, 2414.
170. Betzemeier, B.; Cavazzini, M.; Quici, S.; Knochel, P. Tetrahedron Lett. 2000, 41, 4343.
171. Liu, R.; Dong, C.; Liang, X.; Wang, X.; Hu, X. J. Org. Chem. 2005, 70, 729.
172. Fabbrini, M.; Galli, C.; Gentili, P.; Macchitella, D. Tetrahedron Lett. 2001, 42, 7551.
173. Guin, J.; Sarkar, S.D.; Grimme, S.; Studer, A. Angew. Chem. Int. Ed. 2008, 47, 8727.
174. Wang, X.; Liu, R.; Jin, Y.; Liang, X. Chemistry: European J. 2008, 14, 2679. For an Fe–TEMPO system, see Wang, N.; Liu, R.; Chen, J.; Liang, X. Chem. Commun. 2005, 5322.
175. Lei, M.; Hu, R.-J.; Wang, Y.-G. Tetrahedron 2006, 62, 8928.
176. Kim, S.S.; Nehru, K. Synlett 2002, 616.
177. Bolm, C.; Fey, T. Chem. Commun. 1999, 1795.
178. Fey, T.; Fischer, H.; Bachmann, S.; Albert, K.; Bolm, C. J. Org. Chem. 2002, 66, 8154.
179. Benaglia, M.; Puglisi, A.; Holczknecht, O.; Quici, S.; Pozzi, G. Tetrahedron 2005, 61, 12058. See Miao, C.-X.; He, L.-N.; Wang, J.-Q.; Gao, J. Synlett 2009, 3291.
180. Wu, X.-E.; Ma, L.; Ding, M.-X.; Gao, L.-X. Synlett 2005, 607.
181. Fall, A.; Sene, M.; Gaye, M.; Gómez, G.; Fall, Y. Tetrahedron Lett. 2010, 51, 4501.
182. Sakuratani, K.; Togo, H. Synthesis 2003, 21.
183. Ansar, I.A.; Gree, R. Org. Lett. 2002, 4, 1507.
184. Qian, W.; Jin, E.; Bao, W.; Zhang, Y. Tetrahedron 2006, 62, 556.
185. de Nooy, A.E.J.; Besemer, A.C.; van Bekkum, H. Synthesis 1996, 1153; Gilhespy, M.; Lok, M.; Baucherel, X. Chem. Commun. 2005, 1085.
186. See Merbouh, N.; Bobbitt, J.M.; Brückner, C. Org. Prep. Proceed. Int. 2004, 36, 1.
187. Bailey, W.F.; Bobbitt, J.M.; Wiberg, K.B. J. Org. Chem. 2007, 72, 4504.
188. See Su, J.T.; Goddard, III, W.A. J. Am. Chem. Soc. 2005, 127, 14146; Wirth, T. Angew. Chem. Int. Ed. 2005, 44, 3656. See Duschek, A.; Kirsch, S.F. Chemistry: Eur. J. 2009, 15, 10713.
189. See Lin, C.-K.; Lu, T.-J. Tetrahedron 2010, 66, 9688.
190. Dess, D.B.; Martin, J.C. J. Am. Chem. Soc. 1991, 113, 7277. See Frigerio, M.; Santagostino, M.; Sputore, S. J. Org. Chem. 1999, 64, 4537.
191. See Frigerio, M.; Santagostino, M. Tetrahedron Lett. 1994, 35, 8019.
192. Meyer, S.D.; Schreiber, S.L. J. Org. Chem. 1994, 59, 7549. In aqueous β–cyclodextrin–acetone solution, see Surendra, K.; Krishnaveni, N.S.; Reddy, M.A.; Nageswar, Y.V.D.; Rao, K.R. J. Org. Chem. 2003, 68, 2058.
193. Gallen, M.J.; Goumont, R.; Clark, T.; Terrier, F.; Williams, C.M. Angew. Chem. Int. Ed. 2006, 45, 2929.
194. Thottumkara, A.P.; Vinod, T.K. Tetrahedron Lett. 2001, 43, 569.
195. Van Arman, S.A. Tetrahedron Lett. 2009, 50, 4693.
196. Plumb, J.B.; Harper, D.J. Chem. Eng. News, 1990, July 16, p. 3. For an improved procedure, see Ireland, R.E.; Liu, L. J. Org. Chem. 1993, 58, 2899.
197. Karade, N.N.; Tiwari, G.B.; Huple, D.B. Synlett 2005, 2039.
198. Moriarty, R.M.; Prakash, O. Accts. Chem. Res. 1986, 19, 244; Dohi, T.; Takenaga, N.; Fukushima, K.-i.; Uchiyama, T.; Kato, D.; Motoo, S.; Fujioka, H.; Kita, Y. Chem. Commun. 2010, 7697.
199. DeMico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. J. Org. Chem. 1997, 62, 6974.
200. Adam, W.; Hajra, S.; Herderich, M.; Saha-Möller, C.R. Org. Lett. 2000, 2, 2773.
201. Varma, R.S.; Saini, R.K.; Dahiya, R. J. Chem. Res. (S) 1998, 120.
202. Qian, W.; Jin, E.; Bao, W.; Zhang, Y. Angew. Chem. Int. Ed. 2005, 44, 952.
203. Lee, J.C.; Lee, J.Y.; Lee, S.J. Tetrahedron Lett. 2004, 45, 4939.
204. Liu, Z.; Chen, Z.-C.; Zheng, Q.-C. Org. Lett. 2003, 5, 3321; Karthikeyan, G.; Perumal, P.T. Synlett 2003, 2249.
205. Moorthy, J.N.; Singhal, N.; Venkatakrishnan, P. Tetrahedron Lett. 2004, 45, 5419.
206. Uyanik, M.; Akakura, M.; Ishihara, K. J. Am. Chem. Soc. 2009, 131, 251.
207. Muldoon, J.; Brown, S.N. Org. Lett. 2002, 4, 1043.
208. Takahashi, M.; Oshima, K.; Matsubara, S. Tetrahedron Lett. 2003, 44, 9201.
209. Meijer, R.H.; Ligthart, G.B.W.L.; Meuldijk, J.; Vekemans, J.A.J.M.; Hulshof, L.A.; Mills, A.M.; Kooijman, H.; Spek, A.L. Tetrahedron 2004, 60, 1065.
210. Krafft, M.E.; Zorc, B. J. Org. Chem. 1986, 51, 5482.
211. See Mandal, S.K.; Jensen, D.R.; Pugsley, J.S.; Sigman, M.S. J. Org. Chem. 2003, 68, 4600. See also, Mandal, S.K.; Sigman, M.S. J. Org. Chem. 2003, 68, 7535; Guram, A.S.; Bei, X.; Turner, H.W. Org. Lett. 2003, 5, 2485. For a review, see Muzart, J. Tetrahedron 2003, 59, 5789.
212. Choudary, B.M.; Reddy, N.P.; Kantam, M.L.; Jamil, Z. Tetrahedron Lett. 1985, 26, 6257.
213. Tanaka, K.; Fu, G.C. J. Org. Chem. 2001, 66, 8177.
214. Miyata, A.; Murakami, M.; Irie, R.; Katsuki, T. Tetrahedron Lett. 2001, 42, 7067; Csjernyik, G.; Ell, A.H.; Fadini, L.; Pugin, B.; Bäckvall, J.-E. J. Org. Chem. 2002, 67, 1657.
215. Maeda, Y.; Kakiuchi, N.; Matsumura, S.; Nishimura, T.; Uemura, S. Tetrahedron Lett. 2001, 42, 8877.
216. Samajdar, S.; Becker, F.F.; Banik, B.K. Synth. Commun. 2001, 31, 2691.
217. See Orbegozo, T.; de Vries, J.G.; Kroutil, W. Eur. J. Org. Chem. 2010, 3445; Orbegozo, T.; Lavandera, I.; Fabian, W.M.F.; Mautner, B.; de Vries, J.G.; Kroutil, W. Tetrahedron 2009, 65, 6805.
218. See Sheldon, R.A.; Arends, I.W.C.E.; ten Brink, G.-J.; Dijksman, A. Acc. Chem. Res. 2002, 35, 774.
219. Strazzolini, P.; Runcio, A. Eur. J. Org. Chem. 2003, 526.
220. Itoh, A.; Kodama, T.; Masaki, Y. Chem. Lett. 2001, 686.
221. Khurana, J.M.; Kandpal, B.M. Tetrahedron Lett. 2003, 44, 4909.
222. Gogoi, P.; Sarmah, G.K.; Konwar, D. J. Org. Chem. 2004, 69, 5153.
223. Bacilus stearothermophilus: Fantin, G.; Fogagnolo, M.; Giovannini, P.P.; Medici, A.; Pedrini, P.; Poli, S. Tetrahedron Lett. 1995, 36, 441. Chloroperoxidase: Hu, S.; Dordick, J.S. J. Org. Chem. 2002, 67, 314. Gluconobaccter oxydans DSM 2343: Villa, R.; Romano, A.; Gandolfi, R.; Gargo, J.V.S.; Molinari, F. Tetrahedron Lett. 2002, 43, 6059.
224. Liu, Z.-Q.; Zhao, Y.; Luo, H.; Chai, L.; Sheng, Q. Tetrahedron Lett. 2007, 48, 3017.
225. See Deubel, D.V. J. Org. Chem. 2001, 66, 3790.
226. Baumstark, A.L.; Kovac, F.; Vasquez, P.C. Can. J. Chem. 1999, 77, 308. See Angelis, Y.S.; Hatzakis, N.S.; Smonou, I.; Orfanopoulos, M. Tetrahedron Lett. 2001, 42, 3753.
227. D'Accolti, L.; Fusco, C.; Annese, C.; Rella, M.R.; Turteltaub, J.S.; Williard, P.G.; Curci, R. J. Org. Chem. 2004, 69, 8510.
228. Balicki, R. Synth. Commun. 2001, 31, 2195.
229. Liu, C.; Han, J.; Wang, J. Synlett 2007, 643.
230. Adam, W.; Saha-Möller, C.R.; Zhao, C.-G. J. Org. Chem. 1999, 64, 7492.
231. Döbler, C.; Mehltretter, G.M.; Sundermeier, U.; Eckert, M.; Militzer, H.-C.; Beller, M. Tetrahedron Lett. 2001, 42, 8447.
232. Son, Y.-C.; Makwana, V.D.; Howell, A.R.; Suib, S.L. Angew. Chem. Int. Ed. 2001, 40, 4280.
233. Hunsen, M. Tetrahedron Lett. 2005, 46, 1651. See also, Zhang, S.; Xu, L.; Trudell, M.L. Synthesis 2005, 1757.
234. Stevens, R.V.; Chapman, K.T.; Weller, H.N. J. Org. Chem. 1980, 45, 2030. See also, Mohrig, J.R.; Nienhuis, D.M.; Linck, C.F.; van Zoeren, C.; Fox, B.G.; Mahaffy, P.G. J. Chem. Educ. 1985, 62, 519; Xie, H.; Zhang, S.; Duan, H. Tetrahedron Lett. 2004, 45, 2013.
235. Ji, H.-B.; Shi, D.-P.; Shao, M.; Li, Z.; Wang, L.-F. Tetrahedron Lett. 2005, 46, 2517.
236. Mojtahedi, M.M.; Saidi, M.R.; Bolourtchian, M.; Shirzi, J.S. Monat. Chem. 2001, 132, 655.
237. Sharma, V.B.; Jain, S.L.; Sain, B. Synlett 2005, 173.
238. Peng, K.; Chen, F.; She, X.; Yang, C.; Cui, Y.; Pan, X. Tetrahedron Lett. 2005, 46, 1217.
239. Kuwabara, K.; Itoh, A. Synthesis 2006, 1949.
240. Gogoi, P.; Konwar, D. Org. Biomol. Chem. 2005, 3, 3473.
241. Shirini, F.; Zolfigol, M.A.; Mollarazi, E. Synth. Commun. 2005, 35, 1541.
242. Wu, S.; Ma, H.; Le, Z. Tetrahedron 2010, 66, 8641.
243. Friedrich, H.B.; Khan, F.; Singh, N.; van Staden, M. Synlett 2001, 869.
244. Hajipour, A.R.; Mallakpour, S.E.; Samimi, H.A. Synlett 2001, 1735.
245. Morey, J.; Dzielenziak, A.; Saá, J.M. Chem. Lett. 1985, 263.
246. Karimi, B.; Rajabi, J. Org. Lett. 2004, 6, 2841.
247. Hajipour, A.R.; Mallakpour, S.E.; Baltork, I.M.; Adibi, H. Synth. Commun. 2001, 31, 1625.
248. Heravi, M.M.; Ajami, D.; Ghassemzadeh, M.; Tabar-Hydar, K. Synth. Commun. 2001, 31, 2097.
249. Mohajerani, B.; Heravi, M.M.; Ajami, D. Monat. Chem. 2001, 132, 871.
250. Trost, B.M.; Richardson, J.; Yong, K. J. Am. Chem. Soc. 2006, 128, 2540.
251. Narender, M.; Reddy, M.S.; Rao, K.R. Synthesis 2004, 1741. See Reddy, M.S.; Narender, M.; Nageswar, Y.V.D.; Rao, K.R. Synthesis 2005, 714.
252. See Haines, A.H. Methods for the Oxidation of Organic Compounds, Vol. 2, Academic Press, NY, 1988, pp. 305–323, 438–447; Naruta, Y.; Maruyama, K. in Patai, S.; Rappoport, Z. The Chemistry of the Quinoid Compounds, Vol. 2, pt. 1, Wiley, NY, 1988, pp. 247–276; Thomson, R.H. in Patai, S. The Chemistry of the Quinoiid Compounds, Vol. 1, pt. 1, Wiley, NY, 1974, pp. 112–132.
253. See Cainelli, G.; Cardillo, G. Chromium Oxiations in Organic Chemistry, Open Court Pub. Co., La Salle, IL, 1981, pp. 92–117.
254. Kim, D.W.; Choi, H.Y.; Lee, K.Y.; Chi, D.Y. Org. Lett. 2001, 3, 445.
255. Adam, W.; Schönberger, A. Tetrahedron Lett. 1992, 33, 53.
256. See Hashemi, M.M.; Beni, Y.A. J. Chem. Res. (S) 1998, 138.
257. Cossy, J.; Belotti, S. Tetrahedron Lett. 2001, 42, 4329.
258. See Zimmer, H.; Lankin, D.C.; Horgan, S.W. Chem. Rev. 1971, 71, 229.
259. Yakura, T.; Konishi, T. Synlett 2007, 765.
260. Zalomaeva, O.V.; Sorokin, A.B. New J. Chem. 2006, 30, 1768,
261. Adler, E.; Falkehag, I.; Smith, B. Acta Chem. Scand. 1962, 16, 529.
262. This mechanism is an example of category 4 (Sec. 19.A).
263. Bock, H.; Jaculi, D. Angew. Chem. Int. Ed. 1984, 23, 305.
264. For an example, see Rathore, R.; Bosch, E.; Kochi, J.K. Tetrahedron Lett. 1994, 35, 1335.
265. Sheldon, R.A.; Kochi, J.K. Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, NY, 1981, pp. 368–381; Walling, C. Free Radicals in Solution, Wiley, NY, 1957, pp. 457–461.
266. Iida, S.; Togo, H. Synlett 2006, 2633.
267. Chiampanichayakul, S.; Pohmakotr, M.; Reutrakul, V.; Jaipetch, T.; Kuhakarn, C. Synthesis 2008, 2045.
268. Yamazaki, S. Synth. Commun. 1997, 27, 3559; Jursic, B. J. Chem. Res. (S) 1988, 168.
269. Gao, S.; Herzig, D.; Wang, B. Synthesis 2001, 544.
270. Yamaguchi, K.; Mizuno, N. Angew. Chem. Int. Ed. 2003, 42, 1480.
271. Capdevielle, P.; Lavigne, A.; Maumy, M. Tetrahedron 1990, 2835; Capdevielle, P.; Lavigne, A.; Sparfel, D.; Baranne-Lafont, J.; Cuong, N.K.; Maumy, M. Tetrahedron Lett. 1990, 31, 3305.
272. Iida, S.; Togo, H. Tetrahedron 2007, 63, 8274.
273. Biondini, D.; Brinchi, L.; Germani, R.; Goracci, L.; Savelli, G. Eur. J. Org. Chem. 2005, 3060.
274. See Dayagi, S.; Degani, Y. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 117–124.
275. See Cornejo, J.J.; Larson, K.D.; Mendenhall, G.D. J. Org. Chem. 1985, 50, 5382; Nishinaga, A.; Yamazaki, S.; Matsuura, T. Tetrahedron Lett. 1988, 29, 4115.
276. Müller, P.; Gilabert, D.M. Tetrahedron 1988, 44, 7171.
277. Keirs, D.; Overton, K. J. Chem. Soc. Chem. Commun. 1987, 1660.
278. Murahashi, S.; Naot, T.; Taki, H. J. Chem. Soc. Chem. Commun. 1985, 613.
279. Wolfe, J.P.; Ney, J.E. Org. Lett. 2003, 5, 4607.
280. Sezen, B.; Sames, D. J. Am. Chem. Soc. 2004, 126, 13244.
281. See Larsen, J.; J![]() rgensen, K.A. J. Chem. Soc. Perkin Trans. 2 1992, 1213. Also see, Yamaguchi, J.; Takeda, T. Chem. Lett. 1992, 1933; Yamazaki, S. Chem. Lett. 1992, 823.
rgensen, K.A. J. Chem. Soc. Perkin Trans. 2 1992, 1213. Also see, Yamaguchi, J.; Takeda, T. Chem. Lett. 1992, 1933; Yamazaki, S. Chem. Lett. 1992, 823.
282. Murahashi, S.; Yoshimura, N.; Tsumiyama, T.; Kojima, T. J. Am. Chem. Soc. 1983, 105, 5002. See also, Wilson, Jr., R.B.; Laine, R.M. J. Am. Chem. Soc. 1985, 107, 361.
283. Sasson, R.; Rozen, S. Org. Lett. 2005, 7, 2177.
284. Dimroth, K.; Tüncher, W. Synthesis 1977, 339.
285. Wang, C.-L.; Wang, X.-X.; Wang, X.-Y.; Xiao, J.-P.; Wang, Y.-L. Synth. Commun. 1999, 29, 3435.
286. Hyatt, J.A. Tetrahedron Lett. 1977, 141.
287. See Newbold, B.T. in Patai, S. The Chemistry of the Hydrazo, Azo, and Azoxy Groups, pt. 1, Wiley, NY, 1975, pp. 543–557, 564–573.
288. See Mannen, S.; Itano, H.A. Tetrahedron 1973, 29, 3497.
289. Patel, S.; Mishra, B.K. Tetrahedron Lett. 2004, 45, 1371.
290. For a review, see Regitz, M.; Maas, G. Diazo Compounds, Academic Press, NY, 1986, pp. 233–256.
291. Mobbs, D.B.; Suschitzky, H. Tetrahedron Lett. 1971, 361.
292. Fernández, R.; Gasch, C.; Lassaletta, J.-M.; Llera, J.-M.; Vázquez, J. Tetrahedron Lett. 1993, 34, 141.
293. Altamura, A.; D'Accolti, L.; Detomaso, A.; Dinoi, A.; Fiorentino, M.; Fusco, C.; Curci, R. Tetrahedron Lett. 1998, 39, 2009.
294. Ramalingam, T.; Reddy, B.V.S.; Srinivas, R.; Yadav, J.S. Synth. Commun. 2000, 30, 4507.
295. Barman, D.C.; Thakur, A.J.; Prajapati, D.; Sandhu, J.S. Chem. Lett. 2000, 1196.
296. Firouzabadi, H.; Mostafavipoor, Z. Bull. Chem. Soc. Jpn. 1983, 56, 914.
297. Kosswig, K. Liebigs Ann. Chem. 1971, 749, 206.
298. Cicchi, S.; Corsi, M.; Goti, A. J. Org. Chem. 1999, 64, 7243.
299. Forcato, M.; Nugent, W.A.; Licini, G. Tetrahedron Lett. 2003, 44, 49.
300. See Bentley, K.W. in Bentley, K.W.; Kirby, G.W. Elucidation of Chemical Structures by Physical and Chemical Methods (Vol. 4 of Weissberger, A. Techniques of Chemistry), 2nd ed., pt. 2, Wiley, NY, 1973, pp. 137–254.
301. See Haines, A.H. Methods for the Oxidation of Organic Compounds, Vol. 2, Academic Press, NY, 1988, pp. 277–301, 432–437; House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 3353–363; Perlin, A.S. in Augustine, R.L. Oxidation, Vol. 1, Marcel Dekker, NY, 1969, pp. 189–212; Bunton, C.A. in Wiberg, K.B. in Wiberg, K.B. Oxidation in Organic Chemistry, pt. A, Academic Press, NY, 1965, pp. 367–407. Also see Rubottom, G.M. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. D Academic Press, NY, 1982, p. 1; Aylward, J.B. Q. Rev. Chem. Soc. 1971, 25, 407; Fatiadi, A.J. Synthesis 1976, 65,133.
302. Yu, W.; Mei, Y.; Kang, Y.; Hua, Z.; Jin, Z. Org. Lett. 2004, 6, 3217.
303. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1250–1255.
304. Khurana, J.M.; Sharma, P.; Gogia, A.; Kandpal, B.M. Org. Prep. Proceed. Int. 2007, 39, 185.
305. See Ohloff, G.; Giersch, W. Angew. Chem. Int. Ed. 1973, 12, 401.
306. Takezawa, E.; Sakaguchi, S.; Ishii, Y. Org. Lett. 1999, 1, 713.
307. Cisneros, A.; Fernández, S.; Hernández, J.E. Synth. Commun. 1982, 12, 833.
308. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1650–1652.
309. Lena, J.I.C.; Sesenoglu, Ö.; Birlirakis, N.; Arseniyadis, S. Tetrahedron Lett. 2001, 42, 21.
310. Gagnon, J.L.; Zajac Jr., W.W. Tetrahedron Lett. 1995, 36, 1803.
311. See Ogata, Y.; Sawaki, Y.; Shiroyama, M. J. Org. Chem. 1977, 42, 4061.
312. Nagarkatti, J.P.; Ashley, K.R. Tetrahedron Lett. 1973, 4599.
313. Kore, A.R.; Sagar, A.D.; Salunkhe, M.M. Org. Prep. Proceed. Int. 1995, 27, 373.
314. Criegee, R.; Kraft, L.; Rank, B. Liebigs Ann. Chem. 1933, 507, 159. For reviews, see Waters, W.A. Mechanisms of Oxidation of Organic Compounds, Wiley, NY, 1964, pp. 72–81; Stewart, R. Oxidation Mechanisms, W.A. Benjamin, NY, 1964, pp. 97–106.
315. See Criegee, R.; Höger, E.; Huber, G.; Kruck, P.; Marktscheffel, F.; Schellenberger, H. Liebigs Ann. Chem. 1956, 599, 81.
316. Buist, G.J.; Bunton, C.A.; Hipperson, W.C.P. J. Chem. Soc. B 1971, 2128.
317. Angyal, S.J.; Young, R.J. J. Am. Chem. Soc. 1959, 81, 5251.
318. See Trahanovsky, W.S. Methods Free-Radical Chem. 1973, 4, 133–169; Verter, H.S. in Zabicky, J. The Chemistry of the Carbonyl Group, pt. 2, Wiley, NY, 1970, pp. 71–156.
319. Gurunath, S.; Sudalai, A. Synlett 1999, 559.
320. Minisci, F.; Recupero, F.; Fontana, F.; Bj![]() rsvik, H.-R.; Liguori, L. Synlett 2002, 610.
rsvik, H.-R.; Liguori, L. Synlett 2002, 610.
321. Lee, J.C.; Choi, J.-H.; Lee, Y.C. Synlett 2001, 1563.
322. Hajipour, A.R.; Mohammadpoor-Baltork, I.; Niknam, K. Org. Prep. Proceed. Int. 1999, 31, 335.
323. Ashford, S.W.; Grega, K.C. J. Org. Chem. 2001, 66, 1523.
324. He, L.; Horiuchi, C.A. Bull. Chem. Soc. Jpn. 1999, 72, 2515.
325. Bj![]() rsvik, H.-R.; Liguori, L.; González, R.R.; Merinero, J.A.V. Tetrahedron Lett. 2002, 43, 4985. See also, Osowska-Pacewicka, K.; Alper, H. J. Org. Chem. 1988, 53, 808.
rsvik, H.-R.; Liguori, L.; González, R.R.; Merinero, J.A.V. Tetrahedron Lett. 2002, 43, 4985. See also, Osowska-Pacewicka, K.; Alper, H. J. Org. Chem. 1988, 53, 808.
326. Sotiriou, C.; Lee, W.; Giese, R.W. J. Org. Chem. 1990, 55, 2159.
327. Yoshida, J.; Itoh, M.; Matsunaga, S.; Isoe, S. J. Org. Chem. 1992, 57, 4877.
328. Wolfrom, M.L.; Bobbitt, J.M. J. Am. Chem. Soc. 1956, 78, 2489.
329. Yan, J.; Travis, B.R.; Borhan, B. J. Org. Chem. 2004, 69, 9299.
330. Pappas, J.J.; Keaveney, W.P.; Gancher, E.; Berger, M. Tetrahedron Lett. 1966, 4273.
331. See Razumovskii, S.D.; Zaikov, G.E. Ozone and its Reactions with Organic Compounds; Elsevier, NY, 1984; Bailey, P.S. Ozonation in Organic Chemistry, 2 Vols., Academic Press, NY, 1978, 1982. For reviews, see Odinokov, V.N.; Tolstikov, G.A. Russ. Chem. Rev. 1981, 50, 636; Belew, J.S. in Augustine, R.L.; Trecker, D.J. Oxidation, Vol. 1, Marcel Dekker, NY, 1969, pp. 259–335; Menyailo, A.T.; Pospelov, M.V. Russ. Chem. Rev. 1967, 36, 284.
332. Hon, Y.-S.; Lin, S.-W.; Chen, Y.-J. Synth. Commun. 1993, 23, 1543.
333. Knowles, W.S.; Thompson, Q.E. J. Org. Chem. 1960, 25, 1031.
334. Gupta, D.; Soman, R.; Dev, S. Tetrahedron 1982, 38, 3013.
335. See Flippin, L.A.; Gallagher, D.W.; Jalali-Araghi, K. J. Org. Chem. 1989, 54, 1430.
336. See White, R.W.; King, S.W.; O'Brien, J.L. Tetrahedron Lett. 1971, 3591.
337. Neumeister, J.; Keul, H.; Saxena, M.P.; Griesbaum, K. Angew. Chem. Int. Ed. 1978, 17, 939. See also, Cardinale, G.; Grimmelikhuysen, J.C.; Laan, J.A.M.; Ward, J.P. Tetrahedron 1984, 40, 1881.
338. Drug synthesis: see Van Ornum, S.G.; Champeau, R.M.; Pariza, R. Chem. Rev. 2006, 106, 2990.
339. Schiaffo, C.E.; Dussault, P.H. J. Org. Chem. 2008, 73, 4688.
340. Pryor, W.A.; Giamalva, D.; Church, D.F. J. Am. Chem. Soc. 1985, 107, 2793. See Kuczkowski, R.L. Adv. Oxygenated Processes 1991, 3, 1; Gillies, C.W.; Kuczkowski, R.L. Isr. J. Chem. 1983, 23, 446.
341. See Bailey, P.S.; Hwang, H.H.; Chiang, C. J. Org. Chem. 1985, 50, 231.
342. Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R.; Cuerva, J.M.; Segovia, A. Synlett. 2000, 1269.
343. See Pryor, W.A.; Govindan, C.K.; Church, D.F. J. Am. Chem. Soc. 1982, 104, 7563.
344. See Williamson, D.G.; Cvetanovic, R.J. J. Am. Chem. Soc. 1968, 90, 4248; Razumovskii, S.D.; Zaikov, G.E. J. Org. Chem. USSR 1972, 8, 468, 473; Klutsch, G.; Fliszár, S. Can. J. Chem. 1972, 50, 2841.
345. See O'Murchu, C. Synthesis 1989, 880.
346. Jung, M.E.; Davidov, P. Org. Lett. 2001, 3, 627.
347. See Kuczkowski, R.L. Acc. Chem. Res. 1983, 16, 42; Criegee, R. Angew. Chem. Int. Ed. 1975, 14, 745; Murray, R.W. Acc. Chem. Res. 1968, 1, 313.
348. Also see Ponec, R.; Yuzhakov, G.; Haas, Y.; Samuni, U. J. Org. Chem. 1997, 62, 2757.
349. Gillies, J.Z.; Gillies, C.W.; Suenram, R.D.; Lovas, F.J. J. Am. Chem. Soc. 1988, 110, 7991. See also, Kohlmiller, C.K.; Andrews, L. J. Am. Chem. Soc. 1981, 103, 2578; McGarrity, J.F.; Prodolliet, J. J. Org. Chem. 1984, 49,4465.
350. See Fajgar, R.; Vítek, J.; Haas, Y.; Pola, J. Tetrahedron Lett. 1996, 37, 3391.
351. See Sander, W. Angew. Chem. Int. Ed. 1990, 29, 344; Brunelle, W.H. Chem. Rev. 1991, 91, 335.
352. Fliszár, S.; Chylinska, J.B. Can. J. Chem. 1967, 45, 29; 1968, 46, 783.
353. See Griesbaum, K.; Volpp, W.; Greinert, R.; Greunig, H.; Schmid, J.; Henke, H. J. Org. Chem. 1989, 54, 383.
354. See Criegee, R.; Korber, H. Chem. Ber. 1971, 104, 1812.
355. Kamata, M.; Komatsu, K.i.; Akaba, R. Tetrahedron Lett. 2001, 42, 9203. For a report of an isolable ozonide, see dos Santos, C.; de Rosso, C.R.S.; Imamura, P.M. Synth. Commun. 1999, 29, 1903.
356. Riezebos, G.; Grimmelikhuysen, J.C.; van Dorp, D.A. Recl. Trav. Chim. Pays-Bas 1963, 82, 1234; Privett, O.S.; Nickell, E.C. J. Am. Oil Chem. Soc. 1964, 41, 72.
357. Loan, L.D.; Murray, R.W.; Story, P.R. J. Am. Chem. Soc. 1965, 87, 737; Lorenz, O.; Parks, C.R. J. Org. Chem. 1965, 30, 1976.
358. Murray, R.W.; Williams, G.J. J. Org. Chem. 1969, 34, 1891.
359. See Wojciechowski, B.J.; Chiang, C.; Kuczkowski, R.L. J. Org. Chem. 1990, 55, 1120; Paryzek, Z.; Martynow, J.; Swoboda, W. J. Chem. Soc. Perkin Trans. 1 1990, 1220; Murray, R.W.; Morgan, M.M. J. Org. Chem. 1991, 56, 684, 6123.
360. Higley, D.P.; Murray, R.W. J. Am. Chem. Soc. 1974, 96, 3330.
361. See Murray, R.W.; Williams, G.J. J. Org. Chem. 1969, 34, 1896.
362. See Kolsaker, P. Acta Chem. Scand. Ser. B 1978, 32, 557.
363. Murray, R.W.; Youssefyeh, R.D.; Story, P.R. J. Am. Chem. Soc. 1966, 88, 3143, 3655; Story, P.R.; Murray, R.W.; Youssefyeh, R.D. J. Am. Chem. Soc. 1966, 88, 3144. Also see, Choe, J.; Srinivasan, M.; Kuczkowski, R.L. J. Am. Chem. Soc. 1983, 105, 4703.
364. Keul, H.; Kuczkowski, R.L. J. Am. Chem. Soc. 1985, 50, 3371.
365. See Choe, J.; Painter, M.K.; Kuczkowski, R.L. J. Am. Chem. Soc. 1984, 106, 2891.
366. See Mazur, U.; Kuczkowski, R.L. J. Org. Chem. 1979, 44, 3185.
367. Mile, B.; Morris, G.M. J. Chem. Soc. Chem. Commun. 1978, 263.
368. See Samuni, U.; Fraenkel, R.; Haas, Y.; Fajgar, R.; Pola, J. J. Am. Chem. Soc. 1996, 118, 3687.
369. Fong, G.D.; Kuczkowski, R.L. J. Am. Chem. Soc. 1980, 102, 4763.
370. Suenram, R.D.; Lovas, F.J. J. Am. Chem. Soc. 1978, 100, 5117. See, however, Ishiguro, K.; Hirano, Y.; Sawaki, Y. J. Org. Chem. 1988, 53, 5397.
371. Ferrer, M.; Sánchez-Baeza, F.; Casas, J.; Messeguer, A. Tetrahedron Lett. 1994, 35, 2981.
372. See Atkinson, R.; Carter, W.P.L. Chem. Rev. 1984, 84, 437.
373. See Atkinson, R.; Carter, W.P.L. Chem. Rev. 1984, 84, 437, pp. 452–454; Martinez, R.I.; Herron J.T. J. Phys. Chem. 1988, 92, 4644.
374. See Henry, P.M.; Lange, G.L. in Patai, S. The Chemistry of Functional Groups, Supplement A pt. 1, Wiley, NY, 1977, pp. 965–1098; Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, D.C., 1990, pp. 77–84, 96–98; Badanyan, Sh.O.; Minasyan, T.T.; Vardapetyan, S.K. Russ. Chem. Rev. 1987, 56, 740; Cainelli, G.; Cardillo, G. Chromium Oxiations in Organic Chemistry, Open Court Pub. Co., La Salle, IL, 1981, pp. 59–92. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 1634.
375. Sam, D.J.; Simmons, H.E. J. Am. Chem. Soc. 1972, 94, 4024. See also, Lee, D.G.; Chang, V.S. J. Org. Chem. 1978, 43, 1532.
376. von Rudloff, E. Can. J. Chem. 1955, 33, 1714; 1956, 34, 1413; 1965, 43, 1784.
377. Lee, D.G.; van den Engh, M. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. B Academic Press, NY, 1973, pp. 186–192. See Cainelli, G.; Contento, M.; Manescalchi, F.; Plessi, L. Synthesis 1989, 47.
378. Shoair, A.G.F.; Mohamed, R.H. Synth. Commun. 2006, 36, 59.
379. Göksu, S.; Altunda![]() , R.; Sütbeyaz, Y. Synth. Commun. 2000, 30, 1615.
, R.; Sütbeyaz, Y. Synth. Commun. 2000, 30, 1615.
380. Griffith, W.P.; Shoair, A.G.; Suriaatmaja, M. Synth. Commun. 2000, 30, 3091.
381. Henry, J.R.; Weinreb, S.M. J. Org. Chem. 1993, 58, 4745.
382. Travis, B.R.; Narayan, R.S.; Borhan, B. J. Am. Chem. Soc. 2002, 124, 3824. See also, Whitehead, D.C.; Travis, B.R.; Borhan, B. Tetrahedron Lett. 2006, 47, 3797.
383. Yang, D.; Chen, F.; Dong, Z.-M.; Zhang, D.-W. J. Org. Chem. 2004, 69, 2221.
384. Ranganathan, S.; Ranganathan, D.; Singh, S.K. Tetrahedron Lett. 1985, 26, 4955.
385. Viski, P.; Szeverényi, Z.; Simándi, L.I. J. Org. Chem. 1986, 51, 3213.
386. Pappo, R.; Allen, Jr., D.S.; Lemieux, R.U.; Johnson, W.S. J. Org. Chem. 1956, 21, 478.
387. Taylor, R. J. Chem. Res. (S) 1987, 178. See Torii, S.; Inokuchi, T.; Kondo, K. J. Org. Chem. 1985, 50, 4980.
388. Liu, S.-T.; Reddy, K.V.; Lai, R.-Y. Tetrahedron 2007, 63, 1821.
389. See Lee, D.G.; Spitzer, U.A. J. Org. Chem. 1976, 41, 3644; Lee, D.G.; Chang, V.S.; Helliwell, S. J. Org. Chem. 1976, 41, 3644, 3646.
390. There is evidence for an epoxide intermediate: Rocek, J.; Drozd, J.C. J. Am. Chem. Soc. 1970, 92, 6668.
391. McKillop, A.; Oldenziel, O.H.; Swann, B.P.; Taylor, E.C.; Robey, R.L. J. Am. Chem. Soc. 1973, 95, 1296.
392. Moriarty, R.M.; Penmasta, R.; Awasthi, A.K.; Prakash, I. J. Org. Chem. 1988, 53, 6124.
393. See Nuñez, M.T.; Martín, V.S. J. Org. Chem. 1990, 55, 1928.
394. Spitzer, U.A.; Lee, D.G. J. Org. Chem. 1974, 39, 2468.
395. Caputo, J.A.; Fuchs, R. Tetrahedron Lett. 1967, 4729.
396. Klein, H.; Steinmetz, A. Tetrahedron Lett. 1975, 4249. See Liotta, R.; Hoff, W.S. J. Org. Chem. 1980, 45, 2887; Chakraborti, A.K.; Ghatak, U.R. J. Chem. Soc. Perkin Trans. 1 1985, 2605.
397. See Pyatnitskii, Yu.I. Russ. Chem. Rev. 1976, 45, 762.
398. Nakagawa, K.; Onoue, H. Tetrahedron Lett. 1965, 1433; Chem. Commun. 1966, 396.
399. Kajimoto, T.; Takahashi, H.; Tsuji, J. J. Org. Chem. 1976, 41, 1389.
400. Tsuji, J.; Takayanag, H.i Tetrahedron 1978, 34, 641; Bankston, D. Org. Synth. 66, 180.
401. Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, pp. 105–109; Lee, D.G. The Oxidation of Organic Compounds by Permanganate Ion and Hexavalent Chromium, Open-Court Publishing Co., La Salle, IL, 1980, pp. 43–64. See Cainelli, G.; Cardillo, G. Chromium Oxidations in Organic Chemistry, Open Court Pub. Co., La Salle, IL, 1981, pp. 23–33.
402. Brandenberger, S.G.; Maas, L.W.; Dvoretzky, I. J. Am. Chem. Soc. 1961, 83, 2146.
403. Friedman, L.; Fishel, D.L.; Shechter, H. J. Org. Chem. 1965, 30, 1453.
404. Yamazaki, S. Synth. Commun. 1999, 29, 2211.
405. Itoh, A.; Kodama, T.; Hashimoto, S.; Masaki, Y. Synthesis 2003, 2289.
406. Onopchenko, A.; Schulz, J.G.D.; Seekircher, R. J. Org. Chem. 1972, 37, 1414.
407. See Foster, G.; Hickinbottom, W.J. J. Chem. Soc. 1960, 680; Ferguson, L.N.; Wims, A.I. J. Org. Chem. 1960, 25, 668.
408. Hermans, I.; Peeters, J.; Jacobs, P.A. J. Org. Chem. 2007, 72, 3057.
409. Pines, H.; Stalick, W.M. Base-Catalyzed Reactions of Hydrocarbons and Related Compounds, Academic Press, NY, 1977, pp. 508–543.
410. Wiberg, K.B.; Evans, R.J. Tetrahedron 1960, 8, 313.
411. Kaeding, W.W. J. Org. Chem. 1961, 26, 3144. See Lee, D.G.; van den Engh, M. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. B Academic Press, NY, 1973, pp. 91–94.
412. For a convenient procedure, see Hocking, M.B. Can. J. Chem. 1973, 51, 2384.
413. See Schubert, W.M.; Kintner, R.R. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 749–752.
414. See Hocking, M.B.; Bhandari, K.; Shell, B.; Smyth, T.A. J. Org. Chem. 1982, 47, 4208.
415. Hocking, M.B.; Ko, M.; Smyth, T.A. Can. J. Chem. 1978, 56, 2646.
416. Matsumoto, M.; Kobayashi, H.; Hotta, Y. J. Org. Chem. 1984, 49, 4740.
417. Zambrano, J.L.; Dorta, R. Synlett 2003, 1545.
418. See Serguchev, Yu.A.; Beletskaya, I.P. Russ. Chem. Rev. 1980, 49, 1119; Sheldon, R.A.; Kochi, J.K. Org. React. 1972, 19, 279.
419. See Davies, D.I.; Waring, C. J. Chem. Soc. C 1968, 1865, 2337.
420. Ogibin, Yu.N.; Katzin, M.I.; Nikishin, G.I. Synthesis 1974, 889.
421. See Trahanovsky, W.S.; Cramer, J.; Brixius, D.W. J. Am. Chem. Soc. 1974, 96, 1077; Kochi, J.K. Organometallic Mechanisms and Catalysis, Academic Press, NY, 1978, pp. 99–106. See also,; Fristad, W.E.; Fry, M.A.; Klang, J.A. J. Org. Chem. 1983, 48, 3575; Barton, D.H.R.; Crich, D.; Motherwell, W.B. J. Chem. Soc. Chem. Commun. 1984, 242.
422. For another method, see Barton, D.H.R.; Bridon, D.; Zard, S.Z. Tetrahedron 1989, 45, 2615.
423. See Cantello, B.C.C.; Mellor, J.M.; Scholes, G. J. Chem. Soc. Perkin Trans. 2 1974, 348; Beckwith, A.L.J.; Cross, R.T.; Gream, G.E. Aust. J. Chem. 1974, 27, 1673, 1693.
424. Kochi, J.K.; Bacha, J.D. J. Org. Chem. 1968, 33, 2746; Torssell, K. Ark. Kemi, 1970, 31, 401.
425. Santaniello, E.; Ponti, F.; Manzocchi, A. Tetrahedron Lett. 1980, 21, 2655. Also see Doleschall, G.; Tóth, G. Tetrahedron 1980, 36, 1649.
426. Smushkevich, Yu.I.; Usorov, M.I.; Suvorov, N.N. J. Org. Chem. USSR 1975, 11, 653.
427. Corey, E.J.; Casanova, J. J. Am. Chem. Soc. 1963, 85, 165.
428. For a review, see De Lucchi, O.; Modena, G. Tetrahedron 1984, 40, 2585, pp. 2591–2608.
429. Radlick, P.; Klem, R.; Spurlock, S.; Sims, J.J.; van Tamelen, E.E.; Whitesides, T. Tetrahedron Lett. 1968, 5117; Westberg, H.H.; Dauben Jr., H.J. Tetrahedron Lett. 1968, 5123. For additional references, see Fry, A.J. Synthetic Organic Electrochemistry, 2nd ed., Wiley, NY, 1989, pp. 253–254.
430. See Salomon, R.G.; Roy, S.; Salomon, R.G. Tetrahedron Lett. 1988, 29, 769.
431. Tufariello, J.J.; Kissel, W.J. Tetrahedron Lett. 1966, 6145.
432. For other methods of achieving this conversion, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 1260.
433. See Kulp, S.S.; McGee, M.J. J. Org. Chem. 1983, 48, 4097.
434. Chinn, L.J. Selection of Oxidants in Synthesis, Marcel Dekker, NY, 1971, pp. 7–11; Lee, D.G. in Augustine, R.L. Oxidation, Vol. 1, Marcel Dekker, NY, 1969, pp. 2–6; Hill, C.L. Activation and Functionalization of Alkanes, Wiley, NY, 1989.
435. Cohen, Z.; Keinan, E.; Mazur, Y.; Varkony, T.H. J. Org. Chem. 1975, 40, 2141; Org. Synth. VI, 43; Keinan, E.; Mazur, Y. Synthesis 1976, 523; McKillop, A.; Young, D.W. Synthesis 1979, 401, see pp. 418–419.
436. Cainelli, G.; Cardillo, G. Chromium Oxidations in Organic Chemistry, Springer, NY, 1984, pp. 8–23.
437. Bakke, J.M.; Braenden, J.E. Acta Chem. Scand. 1991, 45, 418.
438. Lee, J.C.; Park, C.; Choi, Y. Synth. Commun. 1997, 27, 4079.
439. Collman, J.P.; Tanaka, H.; Hembre, R.T.; Brauman, J.I. J. Am. Chem. Soc. 1990, 112, 3689.
440. Bales, B.C.; Brown, P.; Dehestani, A.; Mayer, J.M. J. Am. Chem. Soc. 2005, 127, 2832.
441. Schneider, H.; Müller, W. J. Org. Chem. 1985, 50, 4609; Tori, M.; Sono, M.; Asakawa, Y. Bull. Chem. Soc. Jpn. 1985, 58, 2669. See also, Querci, C.; Ricci, M. Tetrahedron Lett. 1990, 31, 1779.
442. Deno, N.C.; Jedziniak, E.J.; Messer, L.A.; Meyer, M.D.; Stroud, S.G.; Tomezsko, E.S. Tetrahedron 1977, 33, 2503.
443. For other procedures, see Sharma, S.N.; Sonawane, H.R.; Dev, S. Tetrahedron 1985, 41, 2483; Nam, W.; Valentine, J.S. New J. Chem. 1989, 13, 677.
444. See Groves, J.T.; Nemo, T.E. J. Am. Chem. Soc. 1983, 105, 6243.
445. Groves, J.T.; Viski, P. J. Org. Chem. 1990, 55, 3628.
446. Wiberg, K.B.; Eisenthal, R. Tetrahedron 1964, 20, 1151.
447. Stewart, R.; Spitzer, U.A. Can. J. Chem. 1978, 56, 1273.
448. See Rabjohn, N. Org. React. 1976, 24, 261; Jerussi, R.A. Sel. Org. Transform. 1970, 1, 301; Trachtenberg, E.N. in Augustine, R.L. Oxidation, Vol. 1, Marcel Dekker, NY, 1969, pp. 123–153.
449. Singh, J.; Sharma, M.; Kad, G.L.; Chhabra, B.R. J. Chem. Res. (S) 1997, 264.
450. Arigoni, D.; Vasella, A.; Sharpless, K.B.; Jensen, H.P. J. Am. Chem. Soc. 1973, 95, 7917; Woggon, W.; Ruther, F.; Egli, H. J. Chem. Soc. Chem. Commun. 1980, 706. Also see Stephenson, L.M.; Speth, D.R. J. Org. Chem.1979, 44, 4683.
451. Umbreit, M.A.; Sharpless, K.B. J. Am. Chem. Soc. 1977, 99, 5526. See also Singh, J.; Sabharwal, A.; Sayal, P.K.; Chhabra, B.R. Chem. Ind. (London) 1989, 533.
452. See Sabol, M.R.; Wiglesworth, C.; Watt, D.S. Synth. Commun. 1988, 18, 1.
453. Warpehoski, M.A.; Chabaud, B.; Sharpless, K.B. J. Org. Chem. 1982, 47, 2897.
454. Chabaud, B.; Sharpless, K.B. J. Org. Chem. 1979, 44, 4202.
455. For a review, see Andrus, M.B.; Lashley, J.C. Tetrahedron 2002, 58, 845.
456. Koltun, E.S.; Kass, S.R. Synthesis 2000, 1366.
457. Brodsky, B.H.; Du Bois, J. J. Am. Chem. Soc. 2005, 127, 15391.
458. Drees, M.; Strassner, T. J. Org. Chem. 2006, 71, 1755.
459. Davies, T.E.; García, T.; Solsona, B.; Taylor, S.H. Chem. Commun. 2006, 3417.
460. Shul'pina, L.S.; Kirillova, M.V.; Pombeiro, A.J.L.; Shul'pin, G.B. Tetrahedron 2009, 65, 2424.
461. Hamada, T.; Irie, R.; Mihara, J.; Hamachi, K.; Katsuki, T. Tetrahedron 1998, 54, 10017.
462. Kawasaki, K.; Tsumura, S.; Katsuki, T. Synlett 1995, 1245.
463. Dick, A.R.; Hull, K.L.; Sanford, M.S. J. Am. Chem. Soc. 2004, 126, 2300.
464. Xie, Y.-Y.; Chen, Z.-C. Synth. Commun. 2002, 32, 1875.
465. Laali, K.K.; Herbert, M.; Cushnyr, B.; Bhatt, A.; Terrano, D. J. Chem. Soc., Perkin Trans. 1 2001, 578.
466. Carloni, S.; Frullanti, B.; Maggi, R.; Mazzacani, A.; Bigi, F.; Sartori, G. Tetrahedron Lett. 2000, 41, 8947.
467. LeBras, J.; Muzart, J. Tetrahedron Asymmetry 2003, 14, 1911; Fache, F.; Piva, O. Synlett 2002, 2035.
468. Lee, W.-S.; Kwong, H.-L.; Chan, H.-L.; Choi, W.-W.; Ng, L.-Y. Tetrahedron Asymmetry 2001, 12, 1007.
469. Sekar, G.; Datta Gupta, A.; Singh, V.K. J. Org. Chem. 1998, 63, 2961; Kohmura, Y.; Katsuki, T. Tetrahedron Lett. 2000, 41, 3941.
470. Södergren, M.J.; Andersson, P.G. Tetrahedron Lett. 1996, 37, 7577; Rispens, M.T.; Zondervan, C.; Feringa, B.L. Tetrahedron Asymmetry, 1995, 6, 661.
471. Levina, A.; Muzart, J. Tetrahedron Asymmetry, 1995, 6, 147.
472. Covell, D.J.; White, M.C. Angew. Chem. Int. Ed. 2008, 47, 6448.
473. Chen, M.S.; White, M.C. J. Am. Chem. Soc. 2004, 126, 1346.
474. Adam, W.; Lukacs, Z.; Saha-Möller, C.R.; Weckerle, B.; Schreier, P. Eur. J. Org. Chem. 2000, 2923.
475. Hamada, H.; Tanaka, T.; Furuya, T.; Takahata, H.; Nemoto, H. Tetrahedron Lett. 2001, 42, 909.
476. Adam, W.; Lukacs, Z.; Harmsen, D.; Saha-Möller, C.R.; Schreier, P. J. Org. Chem. 2000, 65, 878.
477. Adam, W.; Boland, W.; Hartmann-Schreier, J.; Humpf, H.-U.; Lazarus, M.; Saffert, A.; Saha-Möller, C.R.; Schreier, P. J. Am. Chem. Soc. 1998, 120, 11044.
478. Curci, R.; D'Accolti, L.; Fusco, C. Tetrahedron Lett. 2001, 42, 7087.
479. Asensio, G.; Mello, R.; González-Nuñez, M.E.; Castellano, G.; Corral, J. Angew. Chem. Int. Ed. 1996, 35, 217.
480. Komiya, N.; Noji, S.; Murahashi, S.-I. Chem. Commun. 2001, 65.
481. Murray, R.W.; Gu, D. J. Chem. Soc. Perkin Trans. 2 1994, 451.
482. Tanemura, K.; Suzuki, T.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. J. Chem. Soc., Perkin Trans. 1 2001, 3230.
483. Freccero, M.; Gandolfi, R.; Sarzi-Amadé, M.; Rastelli, A. Tetrahedron 2001, 57, 9843.
484. Camaioni, D.M.; Bays, J.T.; Shaw, W.J.; Linehan, J.C.; Birnbaum, J.C. J. Org. Chem. 2001, 66, 789.
485. Sheu, C.; Richert, S.A.; Cofré, P.; Ross Jr., B.; Sobkowiak, A.; Sawyer, D.T.; Kanofsky, J.R. J. Am. Chem. Soc. 1990, 112, 1936. See also, Sheu, C.; Sobkowiak, A.; Jeon, S.; Sawyer, D.T. J. Am. Chem. Soc. 1990, 112, 879; Tung, H.; Sawyer, D.T. J. Am. Chem. Soc. 1990, 112, 8214.
486. Named for Gif-sur-Yvette, France, where it was discovered. See Schuchardt, U.; Jannini, M.J.D.M.; Richens, D.T.; Guerreiro, M.C.; Spinacé, E.V. Tetrahedron 2001, 57, 2685.
487. About-Jaudet, E.; Barton, D.H.R.; Csuhai, E.; Ozbalik, N. Tetrahedron Lett. 1990, 31, 1657. For a review of the mechanism, see Barton, D.H.R. Chem. Soc. Rev. 1996, 25, 237.
488. See Barton, D.H.R.; Csuhai, E.; Ozbalik, N. Tetrahedron 1990, 46, 3743 and references cited therein.
489. Barton, D.H.R.; Csuhai, E.; Doller, D.; Ozbalik, N.; Senglet, N. Tetrahedron Lett. 1990, 31, 3097. For mechanistic studies, see Barton, D.H.R.; Doller, D.; Geletii, Y.V. Tetrahedron Lett. 1991, 32, 3911 and references cited therein; Knight, C.; Perkins, M.J. J. Chem. Soc. Chem. Commun. 1991, 925. Also see, Minisci, F.; Fontana, F. Tetrahedron Lett. 1994, 35, 1427; Barton, D.H.R.; Hill, D.R. Tetrahedron Lett. 1994, 35, 1431.
490. D'Accolti, L.; Dinoi, A.; Fusco, C.; Russo, A.; Curci, R. J. Org. Chem. 2003, 68, 7806.
491. Li, Z.; Xiu, C.-G.; Xu, C.-Z. Tetrahedron Lett. 2003, 44, 9229.
492. Che, C.-M.; Cheng, K.-W.; Chan, M.C.W.; Lau, T.-C.; Mak, C.-K. J. Org. Chem. 2000, 65, 7996.
493. See Moriarty, R.M.; Prakash, O. Acc. Chem. Res. 1986, 19, 244. Also see, Reddy, D.R.; Thornton, E.R. J. Chem. Soc. Chem. Commun. 1992, 172.
494. Moriarty, R.M.; Hou, K.; Prakash, O.; Arora, S.K. Org. Synth. VII, 263.
495. Masui, M.; Ando, A.; Shioiri, T. Tetrahedron Lett. 1988, 29, 2835.
496. Curci, R.; D'Accolti, L.; Fusco, C. Acc. Chem. Res. 2006, 39, 1.
497. Christoffers, J. J. Org. Chem. 1999, 64, 7668.
498. Christoffers, J.; Werner, T. Synlett 2002, 119.
499. Monguchi, Y.; Takahashi, T.; Iida, Y.; Fujiwara, Y.; Inagaki, Y.; Maegawa, T.; Sajiki, H. Synlett 2008, 2291.
500. Landwehr, M.; Hochrein, L.; Otey, C.R.; Kasrayan, A.; Bäckvall, J.-E.; Arnold, F.H. J. Am. Chem. Soc. 2006, 128, 6058.
501. Paju, A.; Kanger, T.; Pehk, T.; Lopp, M. Tetrahedron 2002, 58, 7321.
502. Zhang, Y.; Shen, Z.; Tang, J.; Zhang, Y.; Kong, L.; Zhang, Y. Org. Biomol. Chem. 2006, 4, 1478.
503. Huang, H.-Y.; Hou, R.-S.; Wang, H.-M.; Chen, L.-C. Org. Prep. Proceed. Int. 2006, 38, 473.
504. Vedejs, E.; Larsen, S. Org. Synth. VII, 277; Gamboni, R.; Tamm, C. Tetrahedron Lett. 1986, 27, 3999; Helv. Chim. Acta 1986, 69, 615. See also, Hara, O.; Takizawa, J.-i.; Yamatake, T.; Makino, K.; Hamada, Y. Tetrahedron Lett. 1999, 40, 7787.
505. Wasserman, H.H.; Lipshutz, B.H. Tetrahedron Lett. 1975, 1731. For another method, see Pohmakotr, M.; Winotai, C. Synth. Commun. 1988, 18, 2141.
506. Cuvigny, T.; Valette, G.; Larcheveque, M.; Normant, H. J. Organomet. Chem. 1978, 155, 147.
507. Rubottom, G.M.; Gruber, J.M.; Juve, Jr., H.D.; Charleson, D.A. Org. Synth. VII, 282. See also, Horiguchi, Y.; Nakamura, E.; Kuwajima, I. Tetrahedron Lett. 1989, 30, 3323.
508. Davis, F.A.; Vishwakarma, L.C.; Billmers, J.M.; Finn, J. J. Org. Chem. 1984, 49, 3241.
509. For formation of α−benzyloxy lactones, see Brodsky, B.H.; DuBois, J. Org. Lett. 2004, 6, 2619.
510. Davis, F.A.; Vishwakarma, L.C. Tetrahedron Lett. 1985, 26, 3539.
511. Davis, F.A.; Sheppard, A.C.; Chen, B.; Haque, M.S. J. Am. Chem. Soc. 1990, 112, 6679; Davis, F.A.; Weismiller, M.C. J. Org. Chem. 1990, 55, 3715.
512. Guertin, K.R.; Chan, T.H. Tetrahedron Lett. 1991, 32, 715.
513. Schulz, M.; Kluge, R.; Schüßler, M.; Hoffmann, F. Tetrahedron 1995, 51, 3175.
514. Adam, W.; Müller, M.; Prechtl, F. J. Org. Chem. 1994, 59, 2358.
515. Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Hibino, K.; Shoji, M. J. Org. Chem. 2004, 69, 5966; Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Shoji, M. Angew. Chem. Int. Ed. 2004, 43, 1112.
516. B![]() gevig, A.; Sundén, H.; Córdova, A. Angew. Chem. Int. Ed. 2004, 43, 1109.
gevig, A.; Sundén, H.; Córdova, A. Angew. Chem. Int. Ed. 2004, 43, 1109.
517. Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Shoji, M. Tetrahedron Lett. 2003, 44, 8293.
518. Chemla, F.; Julia, M.; Uguen, D. Bull. Soc. Chim. Fr. 1993, 130, 547; 1994, 131, 639.
519. See Davis, F.A.; Sheppard, A.C. J. Org. Chem. 1987, 52, 954; Takai, T.; Yamada, T.; Rhode, O.; Mukaiyama, T. Chem. Lett. 1991, 281.
520. Stankovic, S.; Espenson, J.H. J. Org. Chem. 1998, 63, 4129.
521. Moriarty, R.M.; Epa, W.R.; Penmasta, R.; Awasthi, A.K. Tetrahedron Lett. 1989, 30, 667.
522. Stankovic, S.; Espenson, J.H. J. Org. Chem. 2000, 65, 5528.
523. Demir, A.S.; Jeganathan, A. Synthesis 1992, 235.
524. Punniyamurthy, T.; Katsuki, T. Tetrahedron 1999, 55, 9439.
525. Desai, L.; Hull, K.L.; Sanford, M.S. J. Am. Chem. Soc. 2004, 126, 9542.
526. Tanyeli, C.; Özdemïrhan, D.; Sezen, B. Tetrahedron 2002, 58, 9983.
527. Demir, A.S.; Reis, Ö.; Igdir, A.C. Tetrahedron 2004, 60, 3427.
528. Tanyeli, C.; Iyigün, C. Tetrahedron 2003, 59, 7135.
529. Sheng, J.; Li, X.; Tang, M.; Gao, B.; Huang, G. Synthesis 2007, 1165.
530. Lee, J.C.; Jin, Y.S.; Choi, J.-H. Chem. Commun. 2001, 956.
531. Lee, J.C.; Choi, Y. Tetrahedron Lett. 1998, 39, 3171.
532. Nabana, T.; Togo, H. J. Org. Chem. 2002, 67, 4362. See Yamamoto, Y.; Togo, H. Synlett 2006, 708; Richardson, R.D.; Page, T.K.; Altermann, S.; Paradine, S.M.; French, A.N.; Wirth, T. Synlett 2007, 538; Akiike, J.; Yamamoto, Y.; Togo, H. Synlett 2007, 2168.
533. John, O.R.S.; Killeen, N.M.; Knowles, D.A.; Yau, S.C.; Bagley, M.C.; Tomkinson, N.C.O. Org. Lett. 2007, 9, 4009.
534. Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K.J. Am. Chem. Soc. 2005, 127, 12244.
535. Díaz-Requejo, M.M.; Belderraín, T.R.; Nicasio, M.C.; Trofimenko, S.; Pérez, P.J. J. Am. Chem. Soc. 2003, 125, 12078.
536. Yu, X.-Q.; Huang, J.-S.; Zhou, X.-G.; Che, C.-M. Org. Lett. 2000, 2, 2233.
537. Au, S.-M.; Huang, J.-S.; Che, C.-M.; Yu, W.-Y. J. Org. Chem. 2000, 65, 7858.
538. Juhl, K.; J![]() rgensen, K.A. J. Am. Chem. Soc. 2002, 124, 2420.
rgensen, K.A. J. Am. Chem. Soc. 2002, 124, 2420.
539. Li, Z.; Feiten, H.-J.; van Beilen, J.B.; Duetz, W.; Witholt, B. Tetrahedron Asymmetry 1999, 10, 1323.
540. Li, Z.; Feiten, H.-J.; Chang, D.; Duetz, W.A.; Beilen, J.B.; Witholt, B. J. Org. Chem. 2001, 66, 8424.
541. Chang, D.; Witholt, B.; Li, Z. Org. Lett. 2000, 2, 3949.
542. Chang, D.; Feiten, H.-J.; Engesser, K.-H.; van Beilen, J.; Witholt, B.; Li, Z. Org. Lett. 2002, 4, 1859.
543. Cho, S.-D.; Kim, H.-J.; Ahn, C.; Falck, J.R.; Shin, D.-S. Tetrahedron Lett. 1999, 40, 8215.
544. Wermeckes, B.; Beck, F.; Schulz, H. Tetrahedron 1987, 43, 577.
545. Miyafuji, A.; Katsuki, T. Synlett 1997, 836.
546. Ishii, Y.; Matsunaka, K.; Sakaguchi, S. J. Am. Chem. Soc. 200,122, 7390.
547. See Krief, A.; Hevesi, L. Organoselenium Chemistry I, Springer, NY, 1988, pp. 115–180; Krongauz, E.S. Russ. Chem. Rev. 1977, 46, 59; Rabjohn, N. Org. React. 1976, 24, 261; Trachtenberg, E.N. in Augustine, R.L.; Trecker, D.J. Oxidation, Marcel Dekker, NY, pp. 119–187.
548. See Wasserman, H.H.; Ives, J.L. J. Org. Chem. 1985, 50, 3573.
549. Lee, J.C.; Park, H.-J.; Park, J.Y. Tetrahedron Lett. 2002, 43, 5661.
550. Rüedi, G.; Oberli, M.A.; Nagel, M.; Weymuth, C.; Hansen, H.-J. Synlett 2004, 2315.
551. Rangarajan, R.; Eisenbraun, E.J. J. Org. Chem. 1985, 50, 2435.
552. Borkar, S.D.; Khadilkar, B.M. Synth. Commun. 1999, 29, 4295.
553. Rathore, R.; Saxena, N.; Chandrasekaran, S. Synth. Commun. 1986, 16, 1493.
554. Lee, H.; Harvey, R.G. J. Org. Chem. 1988, 53, 4587.
555. Wei, H.-X.; Jasoni, R.L.; Shao, H.; Hu, J.; Paré, P.W. Tetrahedron 2004, 60, 11829.
556. Shaabani, A.; Lee, D.G. Tetrahedron Lett. 2001, 42, 5833.
557. Meciarova, M.; Toma, S.; Heribanová, A. Tetrahedron 2000, 56, 8561.
558. Komiya, N.; Noji, S.; Murahashi, S.-I. Tetrahedron Lett. 1998, 39, 7921; Lee, N.H.; Lee, C.-S.; Jung, D.-S. Tetrahedron Lett. 1998, 39, 1385.
559. Murahashi, S.-I.; Komiya, N.; Oda, Y.; Kuwabara, T.; Naota, T. J. Org. Chem. 2000, 65, 9186.
560. Velusamy, S.; Punniyamurthy, T. Tetrahedron Lett. 2003, 44, 8955.
561. Ma, D.; Xia, C.; Tian, H. Tetrahedron Lett. 1999, 40, 8915.
562. Khan, A.T.; Parvin, T.; Choudhury, L.H.; Ghosh, S. Tetrahedron Lett. 2007, 48, 2271.
563. Crich, D.; Zou, Y. J. Org. Chem. 2005, 70, 3309.
564. See Muzart, J. Bull. Soc. Chim. Fr. 1986, 65. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1207–1210.
565. Muzart, J. Tetrahedron Lett. 1987, 28, 2131; Chidambaram, N.; Chandrasekaran, S. J. Org. Chem. 1987, 52, 5048.
566. Yu, J.-Q.; Corey, E.J. J. Am. Chem. Soc. 2003, 125, 3232.
567. Catino, A.J.; Forslund, R.E.; Doyle, M.P. J. Am. Chem. Soc. 2004, 126, 13622.
568. Ferraz, H.M.C.; Longo, Jr., L.S.; Zukerman-Schpector, J. J. Org. Chem. 2002, 67, 3518.
569. Pérollier, C.; Sorokin, A.B. Chem. Commun. 2002, 1548.
570. McLaughlin, E.C.; Doyle, M.P. J. Org. Chem. 2008, 73, 4317.
571. Ajjou, A.N.; Ferguson, G. Tetrahedron Lett. 2006, 47, 3719.
572. Li, S.-J.; Wan, Y.-G. Tetrahedron Lett. 2005, 46, 8013.
573. Sharma, N.K.; Ganesh, K.N. Tetrahedron Lett. 2004, 45, 1403.
574. Markgraf, J.H.; Stickney, C.A. J. Heterocyclic Chem. 2000, 37, 109.
575. Markgraf, J.H.; Sangani, P.K.; Finkelstein, M. Synth. Commun. 1995, 27, 1285.
576. Wenkert, E.; Angell, E.C. Synth. Commun. 1988, 18, 1331.
577. Doumaux Jr., A.R.; Trecker, D.J. J. Org. Chem. 1970, 35, 2121.
578. Minisci, F.; Punta, C.; Recupero, F.; Fontana, F.; Pedulli, G.F. J. Org. Chem. 2002, 67, 2671.
579. Bakke, J.M.; Fr![]() haug, Az. Acta Chem. Scand. B 1995, 49, 615; Lee, D.G.; van den Engh, M. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. B, Academic Press, NY, 1973, pp. 222–225; Carlsen, P.H.J.; Katsuki, T.; Martin, V.S.; Sharpless, K.B. J. Org. Chem. 1981, 46, 3936.
haug, Az. Acta Chem. Scand. B 1995, 49, 615; Lee, D.G.; van den Engh, M. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. B, Academic Press, NY, 1973, pp. 222–225; Carlsen, P.H.J.; Katsuki, T.; Martin, V.S.; Sharpless, K.B. J. Org. Chem. 1981, 46, 3936.
580. Minakata, S.; Imai, E.; Ohshima, Y.; Inaki, K.; Ryu, I.; Komatsu, M.; Ohshiro, Y. Chem. Lett. 1996, 19.
581. Miyamoto, M.; Minami, Y.; Ukaji, Y.; Kinoshita, H.; Inomata, K. Chem. Lett. 1994, 1149.
582. See Ferraz, H.M.C.; Longo, Jr., L.S. Org. Lett. 2003, 5, 1337.
583. Shahi, S.P.; Gupta, A.; Pitre, S.V.; Reddy, M.V.R.; Kumareswaran, R.; Vankar, Y.D. J. Org. Chem. 1999, 64, 4509.
584. Metsger, L.; Bittner, S. Tetrahedron 2000, 56, 1905.
585. Harrison, I.T.; Harrison, S. Chem. Commun. 1966, 752.
586. Schmidt, H.; Schäfer, H.J. Angew. Chem. Int. Ed. 1979, 18, 69.
587. Corey, E.J.; Schaefer, J.P. J. Am. Chem. Soc. 1960, 82, 918.
588. Sharpless, K.B.; Gordon, K.M. J. Am. Chem. Soc. 1976, 98, 300.
589. Breslow, R.; Scholl, P.C. J. Am. Chem. Soc. 1971, 93, 2331. See also, Breslow, R.; Heyer, D. Tetrahedron Lett. 1983, 24, 5039.
590. See also, Beckwith, A.L.J.; Duong, T. J. Chem. Soc. Chem. Commun. 1978, 413.
591. Mitani, M.; Tamada, M.; Uehara, S.; Koyama, K. Tetrahedron Lett. 1984, 25, 2805. For an alternative photochemical procedure, see Negele, S.; Wieser, K.; Severin, T. J. Org. Chem. 1998, 63, 1138.
592. Nikishin, G.I.; Troyansky, E.I.; Lazareva, M.I. Tetrahedron Lett. 1984, 25, 4987.
593. Tsunoi, S.; Ryu, I.; Okuda, T.; Tanaka, M.; Komatsu, M.; Sonoda, N. J. Am. Chem. Soc. 1998, 120, 8692. Also see, Tsunoi, S.; Ryu, I.; Sonoda, N. J. Am. Chem. Soc. 1994, 116, 5473.
594. The name Étard reaction is often applied to any oxidation with chromyl chloride, for example, oxidation of glycols (19-7), alkenes (19-10), and so on.
595. See Hartford, W.H.; Darrin, M. Chem. Rev. 1958, 58, 1, see pp. 25–53.
596. See Steckhan, E. Top. Curr. Chem. 1987, 142, 1; pp. 12–17.
597. Trahanovsky, W.S.; Young, L.B. J. Org. Chem. 1966, 31, 2033; Syper, L. Tetrahedron Lett. 1967, 4193. See Ganin, E.; Amer, I. Synth. Commun. 1995, 25, 3149.
598. Hosseinzadeh, R.; Tajbakhsh, M.; Vahedi, H. Synlett 2005, 2769.
599. Nicolaou, K.C.; Baran, P.S.; Zhong, Y.-L. J. Am. Chem. Soc. 2001, 123, 3183.
600. Bonvin, Y.; Callens, E.; Larrosa, I.; Henderson, D.A.; Oldham, J.; Burton, A.J.; Barrett, A.G.M. Org. Lett. 2005, 7, 4549.
601. Paul, S.; Nanda, P.; Gupta, R. Synlett 2004, 531.
602. Rajabi, F.; Clark, J.H.; Karimi, B.; Macquarrie, D.J. Org. Biomol. Chem. 2005, 3, 725.
603. For a review, see Nenitzescu, C.D. Bull. Soc. Chim. Fr. 1968, 1349.
604. Wheeler, O.H. Can. J. Chem. 1960, 38, 2137. See also, Makhija, R.C.; Stairs, R.A. Can. J. Chem. 1968, 46, 1255.
605. Stairs, R.A. Can. J. Chem. 1964, 42, 550.
606. Wiberg, K.B.; Eisenthal, R. Tetrahedron 1964, 20, 1151. See also, Gragerov, I.P.; Ponomarchuk, M.P. J. Org. Chem. USSR 1969, 6, 1125.
607. Necsoiu, I.; Przemetchi, V.; Ghenciulescu, A.; Rentea, C.N.; Nenitzescu, C.D. Tetrahedron 1966, 22, 3037.
608. Duffin, H.C.; Tucker, R.B. Chem. Ind. (London) 1966, 1262; Tetrahedron 1968, 24, 6999.
609. Naruta, Y.; Maruyama, K. in Patai, S.; Rappoport, Z. The Chemistry of the Quinoid Compounds, Vol. 2, pt. 1, Wiley, NY, 1988, pp. 242–247; Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, pp. 94–96; Haines, A.H. Methods for the Oxidation of Organic Compounds, Vol. 1, Academic Press, NY, 1985, pp. 182–185, 358–360; Thomson, R.H. in Patai, S. The Chemistry of the Quinoid Compounds, Vol. 1, pt. 1, Wiley, NY, 1974, pp. 132–134.
610. Periasamy, M.; Bhatt, M.V. Synthesis 1977, 330; Balanikas, G.; Hussain, N.; Amin, S.; Hecht, S.S. J. Org. Chem. 1988, 53, 1007.
611. See Ito, S.; Katayama, R.; Kunai, A.; Sasaki, K. Tetrahedron Lett. 1989, 30, 205.
612. Yamazaki, S. Tetrahedron Lett. 2001, 42, 3355.
613. Tomatsu, A.; Takemura, S.; Hashimoto, K.; Nakata, M. Synlett 1999, 1474.
614. For reviews, see Tidwell, T.T. Org. React. 1990, 39, 297; Synthesis 1990, 857; Haines, A.H. Methods for the Oxidation of Organic Compounds, Vol. 2, Academic Press, NY, 1988, pp. 171–181, 402–406; Durst, T. Adv. Org. Chem. 1969, 6, 285, pp. 343–356; Epstein, W.W.; Sweat, F.W. Chem. Rev. 1967, 67, 247; Moffatt, J.G. in Augustine, R.L.; Trecker, D.J. Oxidation, Vol. 2, Marcel Dekker, NY, 1971, pp. 1–64. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, NY, 1999, pp. 1222–1225.
615. Nace, H.R.; Monagle, J.J. J. Org. Chem. 1959, 24, 1792; Kornblum, N.; Jones, W.J.; Anderson, G.J. J. Am. Chem. Soc. 1959, 81, 4113. Also see Villemin, D.; Hammadi, M. Synth. Commun. 1995, 25, 3141.
616. Kornblum, N.; Powers, J.W.; Anderson, G.J.; Jones, W.J.; Larson, H.O.; Levand, O.; Weaver, W.M. J. Am. Chem. Soc. 1957, 79, 6562. Mg–Al hydrotalcites have been used as heterogeneous basic catalysts: see Kshirsagar, S.W.; Patil, N.R.; Samant, S.D. Tetrahedron Lett. 2008, 49, 1160.
617. Baizer, M.M. J. Org. Chem., 1960, 25, 670.
618. Kornblum, N.; Jones, W.J.; Anderson, G.J. J. Am. Chem. Soc. 1959, 81, 4113.
619. Guo, Z.; Sawyer, R.; Prakash, I. Synth. Commun. 2001, 31, 667; Guo, Z.; Sawyer, R.; Prakash, I. Synth. Commun. 2001, 31, 3395.
620. Goswami, S.; Jana, S.; Dey, S.; Adak, A.K. Chem. Lett. 2005, 34, 194.
621. Ali Shaikh, T.M.; Emmanuvel, L.; Sudalai, A. Synth. Commun. 2007, 37, 2641.
622. Tang, J.; Zhu, J.; Shen, Z.; Zhang, Y. Tetrahedron Lett. 2007, 48, 1919.
623. Chen, D.X.; Ho, C.M.; Wu, Q.Y.R.; Wu, P.R.; Wong, F.M.; Wu, W. Tetrahedron Lett. 2008, 49, 4147.
624. See Johnson, C.R.; Phillips, W.G. J. Org. Chem. 1967, 32, 1926; Torssell, K. Acta Chem. Scand. 1967, 21, 1.
625. Khuddus, M.A.; Swern, D. J. Am. Chem. Soc. 1973, 95, 8393.
626. For an alternateive, see Moffatt, J.G. J. Org. Chem. 1971, 36, 1909 and references cited therein.
627. See Katayama, S.; Fukuda, K.; Watanabe, T.; Yamauchi, M. Synthesis 1988, 178.
628. See Angyal, S.J. Org. React. 1954, 8, 197.
629. Franzen, V.; Otto, S. Chem. Ber. 1961, 94, 1360. For the use of other amine oxides, see Suzuki, S.; Onishi, T.; Fujita, Y.; Misawa, H.; Otera, J. Bull. Chem. Soc. Jpn. 1986, 59, 3287.
630. Barbry, D.; Champagne, P. Tetrahedron Lett. 1996, 37, 7725.
631. See Olah, G.A.; Vankar, Y.D.; Arvanaghi, M. Tetrahedron Lett. 1979, 3653.
632. Santosusso, T.M.; Swern, D. J. Org. Chem. 1975, 40, 2764.
633. See Haines, A.H. Methods for the Oxidation of Organic Compounds, Vol. 2, Academic Press, NY, 1988, pp. 200–220, 411–415.
634. For lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1225–1227; Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, p. 240.
635. Banerji, K.K. Bull. Chem. Soc. Jpn. 1988, 61, 3717.
636. Lee, G.A.; Freedman, H.H. Tetrahedron Lett. 1976, 1641.
637. See Babler, J.H.; Invergo, B.J. J. Org. Chem. 1981, 46, 1937.
638. Deno, N.C.; Fruit Jr., R.E. J. Am. Chem. Soc. 1968, 90, 3502.
639. Rawalay, S.S.; Shechter, H. J. Org. Chem. 1967, 32, 3129. For another procedure, see Monkovic, I.; Wong, H.; Bachand, C. Synthesis 1985, 770.
640. Knowles, D.A.; Mathews, C.J.; Tomkinson, N.C.O. Synlett 2008, 2769.
641. Doyle, M.P.; Siegfried, B. J. Chem. Soc. Chem. Commun. 1976, 433.
642. Ballini, R.; Petrini, M. Tetrahedron Lett. 1989, 30, 5329.
643. Tokunaga, Y.; Ihara, M.; Fukumoto, K. J. Chem. Soc. Perkin Trans. 1 1997, 207.
644. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1227–1228.
645. See Rankin, K.N.; Liu, Q.; Hendry, J.; Yee, H.; Noureldin, N.A.; Lee, D.G. Tetrahedron Lett. 1998, 39, 1095.
646. See Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, pp. 127–132; Haines, A.H. Methods for the Oxidation of Organic Compounds, Vol. 2, Academic Press, NY, 1988, 148–165, 391–401. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1646–1650.
647. Zhao, M.; Li, J.; Song, Z.; Desmond, R.; Tschaen, D.M.; Grabowski, E.J.J.; Reider, P.J. Tetrahedron Lett. 1998, 39, 5323
648. Bogdal, D.; Lukasiewicz, M. Synlett 2000, 143.
649. See DeLuca, L.; Giacomelli, G.; Masala, S.; Porcheddu, A. J. Org. Chem. 2003, 68, 4999.
650. Zhao, M.; Li, J.; Mano, E.; Song, Z.; Tschaen, D.M.; Grabowski, E.J.J.; Reider, P.J. J. Org. Chem. 1999, 64, 2564.
651. Grill, J.M.; Ogle, J.W.; Miller, S.A. J. Org. Chem. 2006, 71, 9291.
652. Prashad, M.; Lu, Y.; Kim, H.-Y.; Hu, B.; Repic, O.; Blacklock, T.J. Synth. Commun. 1999, 29, 2937.
653. Das, S.; Punniyamurthy, T. Tetahedron Lett. 2003, 44, 6033.
654. Shibuya, M.; Sato, T.; Tomizawa, M.; Iwabuchi, Y. Chem. Commun. 2009, 1739.
655. Craig, J.C.; Horning, E.C. J. Org. Chem. 1960, 25, 2098. See also, Nwaukwa, S.O.; Keehn, P.M. Tetrahedron Lett. 1982, 23, 35.
656. Schulze, A.; Pagona, G.; Giannis, A. Synth. Commun. 2006, 36, 1147.
657. Bhar, S.; Chaudjuri, S.K. Tetrahedron 2003, 59, 3493.
658. Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. J. Am. Chem. Soc. 2005, 127, 10840.
659. Mori, N.; Togo, H. Tetrahedron 2005, 61, 5915.
660. Owston, N.A.; Parker, A.J.; Williams, J.M.J. Chem. Commun. 2008, 624.
661. Koo, B.-S.; Kim, E.-H.; Lee, K.-J. Synth. Commun. 2002, 32, 2275.
662. Foot, J.S.; Kanno, H.; Giblin, G.M.P.; Taylor, R.J.K. Synlett 2002, 1293.
663. Hiegel, G.A.; Gilley, C.B. Synth. Commun. 2003, 33, 2003.
664. See Ito, M.; Osaku, A.; Shiibashi, A.; Ikariya, T. Org. Lett. 2007, 9, 1821. For a list of reagents used to effect this conversion, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1650–1652.
665. Yanagisawa, Y.; Kashiwagi, Y.; Kurashima, F.; Anzai, J.; Osa, T.; Bobbitt, J.M. Chem. Lett. 1996, 1043.
666. Stavber, S.; Planinsek, Z.; Zupan, M. Tetrahedron Lett. 1989, 30, 6095.
667. Fujita, K.-i.; Takahashi, Y.; Owaki, M.; Yamamoto, K.; Yamaguchi, R. Org. Lett. 2004, 6, 2785.
668. See Haines, A.H. Methods for the Oxidation of Organic Compounds, Academic Press, NY, 1988, pp. 241–263, 423–428; Chinn, L.J. Selection of Oxidants in Synthesis, Marcel Dekker, NY, 1971, pp. 63–70; Lee, D.G. in Augustine, R.L. Oxidation, Vol. 1, Marcel Dekker, NY, 1969, pp. 81–86.
669. See Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, pp. 174–180; Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1653–1661; Srivastava, R.G.; Venkataramani, P.S. Synth. Commun. 1988, 18, 2193. See also, Haines, A.H. Methods for the Oxidation of Organic Compounds, Academic Press, NY, 1988.
670. See Cainelli, G.; Cardillo, G. Chromium Oxidations in Organic Chemistry, Springer, NY, 1984, pp. 217–225.
671. See Travis, B.R.; Sivakumar, M.; Hollist, G.O.; Borhan, B. Org. Lett. 2003, 5, 1031.
672. See Nigh, W.G. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. B, Academic Press, NY, 1973, pp. 31–34.
673. Dalcanale, E.; Montanari, F. J. Org. Chem. 1986, 51, 567. See also, Bayle, J.P.; Perez, F.; Courtieu, J. Bull. Soc. Chim. Fr. 1990, 565.
674. See Swern, D. in Swern, D. Organic Peroxides, Vol. 1, Wiley, NY, 1970, pp. 313–516.
675. For reviews of the autoxidation of aldehydes, see Vardanyan, I.A.; Nalbandyan, A.B. Russ. Chem. Rev. 1985, 54, 532 (gas phase); Sajus, L.; Sérée de Roch, I. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 16, Elsevier, NY, 1980, pp. 89–124 (liquid phase); Maslov, S.A.; Blyumberg, E.A. Russ. Chem. Rev. 1976, 45, 155 (liquid phase). See Niclause, M.; Lemaire, J.; Letort, M. Adv. Photochem. 1966, 4, 25. See Larkin, D.R. J. Org. Chem. 1990, 55, 1563.
676. Lim, M.; Yoon, C.M.; An, G.; Rhee, H. Tetrahedron Lett. 2007, 48, 3835.
677. Sato, K.; Hyodo, M.; Takagi, J.; Aoki, M.; Noyori, R. Tetrahedron Lett. 2000, 41, 1439.
678. Wójtowicz, H.; Brzaszcz, M.; Kloc, K.; Mlochowski, J. Tetrahedron 2001, 57, 9743.
679. Tashino, Y.; Togo, H. Synlett 2004, 2010.
680. Chakraborty, D.; Gowda, R.R.; Malik, P. Tetrahedron Lett. 2009, 50, 6553.
681. Kon, Y.; Imao, D.; Nakashima, T.; Sato, K. Chem .Lett. 2009, 38, 430.
682. Gopinath, R.; Patel, B.K. Org. Lett. 2000, 2, 577.
683. Chavan, S.P.; Dantale, S.W.; Govande, C.A.; Venkatraman, M.S.; Praveen, C. Synlett 2002, 267.
684. Kiyooka, S.-i.; Wada, Y.; Ueno, M.; Yokoyama, T.; Yokoyama, R. Tetrahedron 2007, 63, 12695.
685. Kiran, Y.B.; Ikeda, R.; Sakai, N.; Konakahara, T. Synthesis 2010, 276.
686. Qin, C.; Wu, H.; Chen, J.; Liu, M.; Cheng, J.; Su, W.; Ding, J. Org. Lett. 2008, 10, 1537.
687. Maki, B.E.; Scheidt, K.A. Org. Lett. 2008, 10, 4331. Using boronic acdis: see Rosa, J.N.; Reddy, R.S.; Candeias, N.R.; Cal, P.M.S.D.; Gois, P.M.P. Org. Lett. 2010, 12, 2686.
688. Al Neirabeyeh, M.; Pujol, M.D. Tetrahedron Lett. 1990, 31, 2273; Kennedy, K.; Kirkpatrick, E.; Leathers, T.; Vanemon, P. J. Org. Chem. 1989, 54, 1212. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1661–1669.
689. Seo, S.Y.; Marks, T.J. Org. Lett. 2008, 10, 317.
690. See Rocek, J., in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 461–505.
691. Wiberg, K.B.; Szeimies, G. J. Am. Chem. Soc. 1974, 96, 1889. See also, Sen Gupta, S.; Dey, S.; Sen Gupta, K.K. Tetrahedron 1990, 46, 2431.
692. See Rocek, J.; Ng, C. J. Org. Chem. 1973, 38, 3348.
693. See Freeman, F.; Lin, D.K.; Moore, G.R. J. Org. Chem. 1982, 47, 56; Jain, A.L.; Banerji, K.K. J. Chem. Res. (S) 1983, 60.
694. Freeman, F.; Brant, J.B.; Hester, N.B.; Kamego, A.A.; Kasner, M.L.; McLaughlin, T.G.; Paul, E.W. J. Org. Chem. 1970, 35, 982.
695. Huang, R.; Espenson, J.H. J. Org. Chem. 1999, 64, 6935.
696. See Swern, D. in Swern, D. Organic Peroxides, Vol. 1, Wiley, NY, 1970, pp. 313–516.
697. Dussault, P.; Sahli, A. J. Org. Chem. 1992, 57, 1009.
698. See Henry, P.M. Palladium Catalyzed Oxidation of Hydrocarbons, D. Reidel Publishing Co., Dordrecht, 1980; Tsuji, J. Organic Synthesis with Palladium Compounds, Springer, NY, 1980, pp. 6–12; Synthesis 1990, 739; Heck, R.F. Palladium Reagents in Organic Syntheses, Academic Press, NY, 1985, pp. 59–80; Sheldon, R.A.; Kochi, J.K. Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, NY, 1981, pp. 189–193, 299–303; Jira, R.; Freiesleben, W. Organomet. React. 1972, 3, 1, pp. 1–44; Khan, M.M.T.; Martell, A.E. Homogeneous Catalysis by Metal Complexes, Vol. 2, Academic Press, NY, 1974, pp. 77–91; Hüttel, R. Synthesis 1970, 225, see pp. 225–236; Bird, C.W. Transition Metal Intermediates in Organic Synthesis, Academic Press, NY, 1967, pp. 88–111.
699. Smidt, J.; Hafner, W.; Jira, R.; Sieber, R.; Sedlmeier, J.; Sabel, A. Angew. Chem. Int. Ed. 1962, 1, 80; Jira, R.; Freiesleben, W. Organomet. React. 1972, 3, 1; The Merck Index, 14th ed., Merck & Co., Inc., Whitehouse Station, New Jersey, 2006, p ONR-98; Mundy, B.P.; Ellerd, M.G.; Favaloro, Jr., F.G. Name Reactions and Reagents in Organic Synthesis, 2nd ed., Wiley–Interscience, New Jersey, 2005, pp. 676–677. See Muzart, J. Tetrahedron 2007, 63,7505.
700. See Cornell, C.N.; Sigman, M.S. Org. Lett. 2006, 8, 4117.
701. See Cornell, C.N.; Sigman, M.S. J. Am. Chem. Soc. 2005, 127, 2796; Keith, J.A.; Nielsen, R.J.; Oxgaard, J.; Goddard III, W.A. J. Am. Chem. Soc. 2007, 129, 12342.
702. Hart, H.; Lerner, L.R. J. Org. Chem. 1967, 32, 2669.
703. Kikuchi, H.; Kogure, K.; Toyoda, M. Chem. Lett. 1984, 341.
704. Lethbridge, A.; Norman, R.O.C.; Thomas, C.B. J. Chem. Soc. Perkin Trans. 1 1973, 35.
705. Roussel, M.; Mimoun, H. J. Org. Chem. 1980, 45, 5387.
706. Alper, H.; Januszkiewicz, K.; Smith, D.J.H. Tetrahedron Lett. 1985, 26, 2263.
707. Nair, V.; Nair, L.G.; Panicker, S.B.; Sheeba, V.; Augustine, A. Chem. Lett. 2000, 584.
708. Srinivasan, N.S.; Lee, D.G. Synthesis 1979, 520. See also, Baskaran, S.; Das, J.; Chandrasekaran, S. J. Org. Chem. 1989, 54, 5182.
709. Sharpless, K.B.; Teranishi, A.Y. J. Org. Chem. 1973, 38, 185. See also, Kageyama, T.; Tobito, Y.; Katoh, A.; Ueno, Y.; Okawara, M. Chem. Lett. 1983, 1481; Lee, J.G.; Ha, D.S. Tetrahedron Lett. 1989, 30, 193.
710. Evans, R.D.; Schauble, J.H. Synthesis 1986, 727.
711. Jensen, H.P.; Sharpless, K.B. J. Org. Chem. 1974, 39, 2314.
712. Piancatelli, G.; Scettri, A.; D'Auria, M. Tetrahedron Lett. 1977, 3483. See Baskaran, S.; Islam, I.; Raghavan, M.; Chandrasekaran, S. Chem. Lett. 1987, 1175.
713. Davis, F.A.; Sheppard, A.C. Tetrahedron Lett. 1988, 29, 4365.
714. Abrams. S. R. Can. J. Chem. 1983, 61, 2423.
715. Liu, G.; Stahl, S.S. J. Am. Chem. Soc. 2007, 129, 6328; Zhang, Z.; Tan, J.; Wang, Z. Org. Lett. 2008, 10, 173.
716. See Haines, A.H. Methods for the Oxidation of Organic Compounds,Vol. 1, Academic Press, NY, 1985, pp. 153–162, 332–338; Simándi, L.I. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt. 1, Wiley, NY, 1983, pp. 513–570.
717. See Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, p. 92.
718. See Tatlock, J.H. J. Org. Chem. 1995, 60, 6221.
719. Vasil'eva, V.P.; Khalfina, I.L.; Karpitskaya, L.G.; Merkushev, E.B. J. Org. Chem. USSR 1987, 23, 1967.
720. See Al-Rashid, Z.F.; Johnson, W.L.; Hsung, R.P.; Wei, Y.; Yao, P.-Y.; Liu, R.; Zhao, K. J. Org. Chem. 2008, 73, 8780.
721. Zhu, Z.; Espenson, J.H. J. Org. Chem. 1995, 60, 7728.
722. Ren, W.; Xia, Y.; Ji, S.-J.; Zhang, Y.; Wan, X.; Zhao, J. Org. Lett. 2009, 11, 1841.
723. Che, C.-M.; Yu, W.-Y.; Chan, P.-M.; Cheng, W.-C.; Peng, S.-M.; Lau, K.-C.; Li, W.-K. J. Am. Chem. Soc. 2000, 122, 11380.
724. Chu, J.H.; Chen, Y.-J.; Wu, M.-J. Synthesis 2009, 2115.
725. Sonoda, N.; Yamamoto, Y.; Murai, S.; Tsutsumi, S. Chem. Lett. 1972, 229.
726. Wan, Z.; Jones, C.D.; Mitchell, D.; Pu, J.Y.; Zhang, T.Y. J. Org. Chem. 2006, 71, 826.
727. Rosenblatt, D.H.; Burrows, E.P. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 2, Wiley, NY, 1982, pp. 1085–1149; Challis, B.C.; Butler, A.R. in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, pp. 320–338; Hedayatullah, M. Bull. Soc. Chim. Fr. 1972, 2957.
728. Holmes, R.R.; Bayer, R.P. J. Am. Chem. Soc. 1960, 82, 3454.
729. Zajac, Jr., W.W.; Darcy, M.G.; Subong, A.P.; Buzby, J.H. Tetrahedron Lett. 1989, 30, 6495.
730. Dewkar, G.K.; Nikalje, M.D.; Ali, I.S.; Paraskar, A.S.; Jagtap, H.S.; Sadalai, A. Angew. Chem. Int. Ed. 2001, 40, 405.
731. Dirk, S.M.; Mickelson, E.T.; Henderson, J.C.; Tour, J.M. Org. Lett. 2002, 2, 3405.
732. Corey, E.J.; Gross, A.W. Org. Synth. 65, 166.
733. See Kahr, K.; Berther, C. Chem. Ber. 1960, 93, 132.
734. Gragerov, I.P.; Levit, A.F. J. Gen Chem. USSR 1960, 30, 3690.
735. Murray, R.W.; Singh, M. Synth. Commun. 1989, 19, 3509. This reagent also oxidizes primary amines to hydroxylamines: Wittman, M.D.; Halcomb, R.L.; Danishefsky, S.J. J. Org. Chem. 1990, 55, 1981.
736. Biloski, A.J.; Ganem, B. Synthesis 1983, 537.
737. Fields, J.D.; Kropp, P.J. J. Org. Chem. 2000, 65, 5937.
738. Colonna, S.; Pironti, V.; Carrea, G.; Pasta, P.; Zambianchi, F. Tetahedron 2004, 60, 569.
739. Detomaso, A.; Curci, R. Tetrahedron Lett. 2001, 42, 755.
740. Iranpoor, N.; Firouzabadi, H.; Pourali, A.R. Synthesis 2003, 1591.
741. Larson, H.O. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, Vol. 1, Wiley, NY, 1969, pp. 306–310. See also, Barnes, M.W.; Patterson, J.M. J. Org. Chem. 1976, 41, 733. For reviews of oxidations of nitrogen compounds, see Butler, R.N. Chem. Rev. 1984, 84, 249; Boyer, J.H. Chem. Rev. 1980, 80, 495.
742. Murray, R.W.; Rajadhyaksha, S.N.; Mohan, L. J. Org. Chem. 1989, 54, 5783. See also, Zabrowski, D.L.; Moorman, A.E.; Beck, Jr., K.R. Tetrahedron Lett. 1988, 29, 4501.
743. See Keinan, E.; Mazur, Y. J. Org. Chem. 1977, 42 844
744. See Gilbert, K.E.; Borden, W.T. J. Org. Chem. 1979, 44, 659.
745. See Cardona, F.; Soldaini, G.; Goti, A. Synlett 2004, 1553.
746. Webb, K.S.; Seneviratne, V. Tetrahedron Lett. 1995, 36, 2377.
747. Howe, G.R.; Hiatt, R.R. J. Org. Chem. 1970, 35, 4007. See also, Nielsen, A.T.; Atkins, R.L.; Norris, W.P.; Coon, C.L.; Sitzmann, M.E. J. Org. Chem. 1980, 45, 2341.
748. McKillop, A.; Tarbin, J.A. Tetrahedron 1987, 43, 1753.
749. Harel, T.; Rozen, S. J. Org. Chem. 2007, 72, 6500.
750. Eaton, P.E.; Wicks, G.E. J. Org. Chem. 1988, 53, 5353.
751. Olah, G.A.; Ramaiah, P.; Lee, G.K.; Prakash, G.K.S. Synlett 1992, 337.
752. Cicchi, S.; Marradi, M.; Goti, A.; Brandi, A. Tetrahedron Lett. 2001, 42, 6503.
753. Corey, E.J.; Samuelsson, B.; Luzzio, F.A. J. Am. Chem. Soc. 1984, 106, 3682.
754. Carmeli, M.; Rozen, S. J. Org. Chem. 2006, 71, 4585.
755. See Boyer, J.H. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, Vol. 1, Wiley, NY, 1969, pp. 264–265.
756. Albini, A.; Pietra, S. Heterocyclic N-Oxides, CRC Press, Boca Raton, FL, 1991, pp. 31–41; Katritzky, A.R.; Lagowski, J.M. Chemistry of the Heterocyclic N-Oxides, Academic Press, NY, 1971, pp. 21–72, 539–542.
757. Balicki, R.; Golinski, J. Synth. Commun. 2000, 30, 1529.
758. Ogata, Y.; Tabushi, I. Bull. Chem. Soc. Jpn. 1958, 31, 969.
759. Choudary, B.M.; Bharathi, B.; Reddy, Ch.V.; Kantam, M.L.; Raghavan, K.V. Chem. Commun. 2001, 1736.
760. Colacino, E.; Nun, P.; Maria Colacino, F.; Martinez, J.; Lamaty, F. Tetrahedron 2008, 64, 5569.
761. Jain, S.L.; Sain, B. Chem. Commun. 2002, 1040.
762. Sharma, V.B.; Jain, S.L.; Sain, B. Tetrahedron Lett. 2004, 45, 4281.
763. Wang, F.; Zhang, H.; Song, G.; Lu, X. Synth. Commun. 1999, 29, 11.
764. Jain, S.L.; Sain, B. Angew. Chem. Int. Ed. 2003, 42, 1265.
765. Mielniczak, G.; Lopusinski, A. Synlett 2001, 505.
766. Uziel, J.; Darcel, C.; Moulin, D.; Bauduin, C.; Juge, S. Tetrahedron Asymmetry 2001, 12, 1441.
767. Bergin, E.; O'Connor, C.T.; Robinson, S.B.; McGarrigle, E.M.; O'Mahony, C.P.; Gilheany, D.G. J. Am. Chem. Soc. 2007, 129, 9566.
768. See Savige, W.E.; Maclaren, J.A. in Kharasch, N.; Meyers, C.Y. Organic Sulfur Compounds, Vol. 2; pp. 367–402, Pergamon, NY, 1966.
769. See Capozzi, G.; Modena, G. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, pp. 785–839; Gilbert, E.E. Sulfonation and Related Reactions, Wiley, NY, 1965, pp. 217–239.
770. Gu, D.; Harpp, D.N. Tetrahedron Lett. 1993, 34, 67. See Ballistreri, F.P.; Tomaselli, G.A.; Toscano, R.M. Tetrahedron Lett. 2008, 49, 3291.
771. Wallace, T.J.; Schriesheim, A. Tetrahedron 1965, 21, 2271.
772. See Gilbert, E.E. Sulfonation and Related Reactions, Wiley, NY, 1965, pp. 202–214.
773. See Hudlicky, M. Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990, pp. 252–263; Drabowicz, J.; Kielbasinski, P.; Mikolajczyk, M. in Patai, S.; Rappoport, Z.; Stirling, C. The Chemistry of Sulphones and Sulphoxides, Wiley, NY, 1988, pp. 233–378, pp. 235–255; Madesclaire, M. Tetrahedron 1986, 42, 5459; Drabowicz, J.; Mikolajczyk, M. Org. Prep. Proced. Int. 1982, 14, 45; Oae, S. in Oae, S. The Organic Chemistry of Sulfur, Plenum, NY, 1977, pp. 385–390; Holland, H.L. Chem. Rev. 1988, 88, 473.
774. Hiskey, R.G.; Harpold, M.A. J. Org. Chem. 1967, 32, 3191; Varma, R.S.; Saini, R.K.; Meshram, H.M. Tetrahedron Lett. 1997, 38, 6525.
775. Matteucci, M.; Bhalay, G.; Bradley, M. Org. Lett. 2003, 5, 235.
776. Lakouraj, M.M.; Tajbakhsh, M.; Shirini, F.; Asady Tamami, M.V. Synth. Commun. 2005, 35, 775.
777. Colonna, S.; Gaggero, N. Tetrahedron Lett. 1989, 30, 6233. For a discussion of the mechanism, see González-Núñez, M.E.; Mello, R.; Royo, J.; Ríos, J.V.; Asensio, G. J. Am. Chem. Soc. 2002, 124, 9154.
778. See Choi, S.; Yang, J.-D.; Ji, M.; Choi, H.; Kee, M.; Ahn, K.-H.; Byeon, S.-H.; Baik, W.; Koo, S. J. Org. Chem. 2001, 66, 8192.
779. See Ali, M.H.; Kriedelbaugh, D.; Wencewicz, T. Synthesis 2007, 3507.
780. Chen, Y.-J.; Huang, Y.-P. Tetrahedron Lett. 2000, 41, 5233.
781. Shaabani, A.; Teimouri, M.B.; Safaei, H.R. Synth. Commun. 2000, 30, 265. See Kowalski, P.; Mitka, K.; Ossowska, K.; Kolarska, Z. Tetrahedron 2005, 61, 1933.
782. Kim, S.S.; Nehru, K.; Kim, S.S.; Kim, D.W.; Jung, H.C. Synthesis 2002, 2484.
783. See Koposov, A.Y.; Zhdankin, V.V. Synthesis 2005, 22.
784. See Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, p. 16.
785. See Schank, K. in Patai, S.; Rappoport, Z.; Stirling, C. The Chemistry of Sulphones and Sulphoxides, Wiley, NY, 1988, pp. 165–231, pp. 205–213.
786. Guertin, K.R.; Kende, A.S. Tetrahedron Lett. 1993, 34, 5369.
787. Kaczorowska, K.; Kolarska, Z.; Mitka, K.; Kowalski, P. Tetrahedron 2005, 61, 8315. See Velusamy, S.; Kumar, A.V.; Saini, R.; Punniyamurthy, T. Tetrahedron Lett. 2005, 46, 3819.
788. Margues, A.; Marin, M.; Ruasse, M.-F. J. Org. Chem. 2001, 66, 7588.
789. Bahrami, K. Tetrahedron Lett. 2006, 47, 2009.
790. Kirihara, M.; Yamamoto, J.; Noguchi, T.; Hirai, Y. Tetrahedron Lett. 2009, 50, 1180.
791. Trivedi, R.; Lalitha, P. Synth. Commun. 2006, 36, 3777.
792. Yuan, Y.; Bian, Y. Tetrahedron Lett. 2007, 48, 8518.
793. Jeyakumar, K.; Chand, D.K. Tetrahedron Lett. 2006, 47, 4573; Gamelas, C.A; Lourenço, T.; da Costa, A.P.; Simplício, A.L.; Royo, B.; Romão, C.C. Tetrahedron Lett. 2008, 49, 4708.
794. Lindén, A.A.; Johansson, M.; Hermanns, N.; Bäckvall, J-E. J. Org. Chem. 2006, 71, 3849.
795. Balicki, R. Synth. Commun. 1999, 29, 2235.
796. Iranpoor, N.; Mohajer, D.; Rezaeifard, A.-R. Tetrahedron Lett. 2004, 45, 3811.
797. Hajipour, A.R.; Kooshki, B.; Ruoho, A.E. Tetrahedron Lett. 2005, 46, 2643.
798. SeeVenier, C.G.; Barager, III, H.J. Org. Prep. Proced. Int. 1974, 6, 77, pp. 85–86.
799. For reviews, see Kagan, H.B.; Rebiere, F. Synlett 1990, 643; Drabowicz, J.; Kielbasinski, P.; Mikolajczyk, M. Org. Prep. Proceed. Int. 1982, 14, 45, see p. 288.
800. See Shibata, N.; Matsunaga, M.; Nakagawa, M.; Fukuzumi, T.; Nakamura, S.; Toru, T. J. Am. Chem. Soc. 2005, 127, 1374; Egami, H.; Katsuki, T. J. Am. Chem. Soc. 2007, 129, 8940; del Río, R.E.; Wang, B.; Achab, S.; Bohé, L. Org. Lett. 2007, 9, 2265; Gao, J.; Guo, H.; Liu, S.; Wang, M. Tetrahedron Lett. 2007, 48, 8453; Yamaguchi, T.; Matsumoto, K.; Saito, B.; Katsuki, T. Angew. Chem. Int. Ed. 2007, 46, 4729; Matsumoto, K.; Yamaguchi, T.; Katsuki, T. Chem. Commun. 2008, 1704; Kelly, P.; Lawrence, S.E.; Maguire, A.R. Synlett 2007, 1501; Jurok, R.; Cibulka, R.; Dvo![]() áková, H.; Hampl, F.; Hoda
áková, H.; Hampl, F.; Hoda![]() ová, J. Eur. J. Org. Chem. 2010, 5217; Dieva, S.A.; Eliseenkova, R.M.; Efremov, Yu.Ya.; Sharafutdinova, D.R.; Bredikhin, A.A. Russ. J. Org. Chem. 2006, 42, 12.
ová, J. Eur. J. Org. Chem. 2010, 5217; Dieva, S.A.; Eliseenkova, R.M.; Efremov, Yu.Ya.; Sharafutdinova, D.R.; Bredikhin, A.A. Russ. J. Org. Chem. 2006, 42, 12.
801. Colonna, S.; Gaggero, N.; Pasta, P.; Ottolina, G. Chem. Commun. 1996, 2303; Pasta, P.; Carrea, G.; Holland, H.L.; Dallavalle, S. Tetrahedron Asymmetry, 1995, 6, 933.
802. Ozaki, S.-i.; Watanabe, S.; Hayasaka, S.; Konuma, M. Chem. Commun. 2001, 1654.
803. Mikolajczyk, M.; Drabowicz, J.; Kielbasinski, P. Chiral Sulfur Reagents, CRC Press, Boca Raton, FL, 1997.
804. Gabbi, C.; Ghelfi, F.; Grandi, R. Synth. Commun. 1997, 27, 2857.
805. See Jennings, W.B.; O'Shea, J.H.; Schweppe, A. Tetrahedron Lett. 2001, 42, 101.
806. See Reich, H.J. in Trahanovsky, W.S. Oxidations in Organic Chemistry, pt. C, Academic Press, NY, 1978, pp. 7–13; Kobayashi, M.; Ohkubo, H.; Shimizu, T. Bull. Chem. Soc. Jpn. 1986, 59, 503.
807. Blum, S.A.; Bergman, R.G.; Ellman, J.A. J. Org. Chem. 2003, 68, 150.
808. See Agarwal, A.; Bhatt, P.; Banerji, K.K. J. Phys. Org. Chem. 1990, 3, 174; Lee, D.G.; Chen, T. J. Org. Chem. 1991, 56, 5346.
809. Modena, G.; Todesco, P.E. J. Chem. Soc. 1962, 4920, and references cited therein.
810. Curci, R.; Di Furia, F.; Modena, G. J. Chem. Soc. Perkin Trans. 2 1978, 603 and references cited therein. See also, Akasaka, T.; Ando, W. J. Chem. Soc. Chem. Commun. 1983, 1203.
811. See Henbest, H.B.; Khan, S.A. Chem. Commun. 1968, 1036.
812. See Saunders Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 548–554.
813. See Reisdorf, D.; Normant, H. Organomet. Chem. Synth. 1972, 1, 375; Hanna, S.B.; Wideman, L.G. Chem. Ind. (London) 1968, 486. Also see Bethell, D.; Bird, R. J. Chem. Soc. Perkin Trans. 2 1977, 1856.
814. Vernigor, E.M.; Shalaev, V.K.; Luk'yanets, E.A. J. Org. Chem. USSR 1981, 17, 317.
815. Buckles, R.E.; Matlack, G.M. Org. Synth. IV, 914.
816. Khurana, J.M.; Maikap, G.C.; Mehta, S. Synthesis 1990, 731.
817. See Aurell, M.J.; Gil, S.; Tortajada, A.; Mestres, R. Synthesis 1990, 317.
818. Rathke, M.W.; Lindert, A. J. Am. Chem. Soc. 1971, 93, 4605; Baudin, J.; Julia, M.; Rolando, C.; Verpeaux, J. Bull. Soc. Chim. Fr. 1987, 493.
819. Nakayama, J.; Tanuma, M.; Honda, Y.; Hoshino, M. Tetrahedron Lett. 1984, 25, 4553.
820. Ito, Y.; Konoike, T.; Saegusa, T. J. Am. Chem. Soc. 1975, 97, 649.
821. Moriarty, R.; Prakash, O.; Duncan, M.P. J. Chem. Soc. Perkin Trans. 1 1987, 559.
822. Baciocchi, E.; Casu, A.; Ruzziconi, R. Tetrahedron Lett. 1989, 30, 3707.
823. Moriarty, R.M.; Penmasta, R.; Prakash, I. Tetrahedron Lett. 1987, 28, 873.
824. Inaba, S.; Ojima, I. Tetrahedron Lett. 1977, 2009. See also, Totten, G.E.; Wenke, G.; Rhodes, Y.E. Synth. Commun. 1985, 15, 291, 301.
825. Frazier, Jr., R.H.; Harlow, R.L. J. Org. Chem. 1980, 45, 5408.
826. See Capozzi, G.; Modena, G. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, pp. 785–839; Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978.
827. See, however, Evans, B.J.; Doi, J.T.; Musker, W.K. J. Org. Chem. 1990, 55, 2337.
828. Ali, M.H.; McDermott, M. Tetrahedron Lett. 2002, 43, 6271.
829. McKillop, A.; Koyunçu, D. Tetrahedron Lett. 1990, 31, 5007.
830. Zhan, Z.-P.; Lang, K.; Liu, F.; Hu, L.-m. Synth. Commun. 2004, 34, 3203.
831. Tanaka, K.; Ajiki, K. Tetahedron Lett. 2004, 45, 25.
832. Patel, S.; Mishra, B.K. Tetrahedron Lett. 2004, 45, 1371. See also, Tajbakhsh, M.; Hosseinzadeh, R.; Shakoori, A. Tetrahedron Lett. 2004, 45, 1889.
833. Kesavan, V.; Bonnet-Delpon, D.; Bégué, J.-P. Synthesis 2000, 223.
834. Firouzabadi, H.; Abbassi, M.; Karimi, B. Synth. Commun. 1999, 129, 2527.
835. Salehi, P.; Farrokhi, A.; Gholizadeh, M. Synth. Commun. 2001, 31, 2777.
836. Leino, R.; Lönnqvist, J.-E. Tetrahedron Lett. 2004, 45, 8489.
837. Joshi, A.V.; Bhusare, S.; Baidossi, M.; Qafisheh, N.; Sasson, Y. Tetrahedron Lett. 2005, 46, 3583.
838. Tarbell, D.S. in Kharasch, N. Organic Sulfur Compounds, Pergamon, Elmsford, NY, 1961, pp. 97–102.
839. Wallace, T.J.; Schriesheim, A.; Bartok, W. J. Org. Chem. 1963, 28, 1311.
840. Mukaiyama, T.; Takahashi, K. Tetrahedron Lett. 1968, 5907.
841. Also see Boustany, K.S.; Sullivan, A.B. Tetrahedron Lett. 1970, 3547; Oae, S.; Fukushima, D.; Kim, Y.H. J. Chem. Soc. Chem. Commun. 1977, 407.
842. Bruni, R.; Fantin, G.; Medici, A.; Pedrini, P.; Sacchetti, G. Tetrahedron Lett. 2002, 43, 3377.
843. See Hudlicky, M. Reductions in Organic Chemistry, Wiley, NY, 1984; Augustine, R.L. Reduction, Marcel Dekker, NY, 1968; Candlin, J.P.; Rennie, R.A.C. in Bentley, K.W.; Kirby, G.W. Elucidation of Chemical Structures by Physical and Chemical Methods (Vol. 4 of Weissberger, A. Techniques of Chemistry), 2nd ed., pt. 2, Wiley, NY, 1973, pp. 77–135.
844. See Brown, H.C.; Krishnamurthy, S. Tetrahedron 1979, 35, 567; Walker, E.R.H. Chem. Soc. Rev. 1976, 5, 23; Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 209–251; Rerick, M.N. in Augustine, R.L. Reduction, Marcel Dekker, NY, 1968.
845. See Rylander, P.N. Aldrichimica Acta 1979, 12, 53. See also, Rylander, P.N. Hydrogenation Methods, Academic Press, NY, 1985.
846. Table 19.2 is from House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, p. 9. Tables 19.3 and 19.4 are from Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 213 and 232, respectively.
847. The first 10 columns are from Brown, H.C.; Krishnamurthy, S. Tetrahedron 1979, 35, 567, p. 604. The column on (i-Bu)2AlH is from Yoon, N.M.; Gyoung, Y.S. J. Org. Chem. 1985, 50, 2443; the one on NaAlEt2H2 from Stinson, S.R. Chem. Eng. News, Nov. 3, 1980, 58, No. 44, 19; and the one on LiBEt3H from Brown, H.C.; Kim, S.C.; Krishnamurthy, S. J. Org. Chem. 1980, 45, 1. Also see Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, p. 129; Hajós, A. Complex Hydrides, Elsevier, NY, 1979, pp. 16–17; Hudlicky, M. Reductions in Organic Chemistry, Wiley, NY, 1984, pp. 177–200.
848. See also, the table in Hudlicky, M. J. Chem. Educ. 1977, 54, 100.
849. See Pizey, J.S. Synthetic Reagents, Vol. 1, Wiley, NY, 1974, pp. 101–194.
850. See Málek, J. J. Org. Chem. 1988, 36, 249; 1985, 34, 1; Málek, J.; Cerny, M. Synthesis 1972, 217.
851. Brown, H.C.; Bigley, D.B.; Arora, S.K.; Yoon, N.M. J. Am. Chem. Soc. 1970, 92, 7161. For reductions with thexylborane, see Brown, H.C.; Heim, P.; Yoon, N.M. J. Org. Chem. 1972, 37, 2942.
852. Brown, H.C.; Krishnamurthy, S.; Yoon, N.M. J. Org. Chem. 1976, 41, 1778.
853. See Yoon, N.M.; Brown, H.C. J. Am. Chem. Soc.1968, 90, 2927.
854. Brown, H.C.; Kim, S.C.; Krishnamurthy, S. J. Org. Chem. 1980, 45, 1. See Brown, H.C.; Singaram, B.; Singaram, S. J. Organomet. Chem. 1982, 239, 43.
855. See Brown, H.C.; Heim, P.; Yoon, N.M. J. Am. Chem. Soc. 1970, 92, 1637; Cragg, G.M.L. Organoboranes in Organic Synthesis, Marcel Dekker, NY, 1973, pp. 319–371. Also see Wade, R.C. J. Mol. Catal., 1983, 18, 273; Lane, C.F. Chem. Rev. 1976, 76, 773; Aldrichimica Acta 1977, 10, 41; Brown, H.C.; Krishnamurthy, S. Aldrichimica Acta 1979, 12, 3; Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, pp. 125–164; Pelter, A. Chem. Ind. (London) 1976, 888.
856. Reduced to a hydroxylamine (Reaction 19-46).
857. See Brown, H.C.; Heim, P.; Yoon, N.M. J. Am. Chem. Soc. 1970, 92, 1637; Cragg, G.M.L. Organoboranes in Organic Synthesis, Marcel Dekker, NY, 1973, pp. 319–371. For reviews of reductions with BH3, see Wade, R.C. J. Mol. Catal. 1983, 18, 273 (BH3 and a catalyst); Lane, C.F. Chem. Rev. 1976, 76, 773; Aldrichimica Acta 1977, 10, 41; Brown, H.C.; Krishnamurthy, S. Aldrichimica Acta 1979, 12, 3. For reviews of reduction with borane derivatives, see Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, pp. 125–164; Pelter, A. Chem. Ind. (London) 1976, 888.
858. Reacts with solvent, reduced in aprotic solvents.
859. Reduced to an aldehyde (Reaction 19–44).
860. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1019–1027.
861. See Healy, E.F.; Lewis, J.D.; Minniear, A.B. Tetrahedron Lett. 1994, 35, 6647.
862. Brown, H.C.; Ikegami, S.; Kawakami, J.H. J. Org. Chem. 1970, 35, 3243.
863. See Rylander, P.N. Catalytic Hydrogenation over Platinum Metals, Academic Press, NY, 1967, pp. 478–485; Oshima, M.; Yamazaki, H.; Shimizu, I.; Nizar, M.; Tsuji, J. J. Am. Chem. Soc. 1989, 111, 6280.
864. Molander, G.A.; La Belle, B.E.; Hahn, G. J. Org. Chem. 1986, 51, 5259; Otsubo, K.; Inanaga, J.; Yamaguchi, M. Tetrahedron Lett. 1987, 28, 4437. See also, Miyashita, M.; Hoshino, M.; Suzuki, T.; Yoshikoshi, A. Chem. Lett.1988, 507.
865. Noguchi, Y.; Yamada, T.; Uchiro, H.; Kobayashi, S. Tetrahedron Lett. 2000, 41, 7493, 7499.
866. Gao, Y.; Sharpless, K.B. J. Org. Chem. 1988, 53, 4081.
867. Krishnamurthy, S.; Schubert, R.M.; Brown, H.C. J. Am. Chem. Soc. 1973, 95, 8486.
868. Laxmi, Y.R.S.; Iyengar, D.S. Synth. Commun. 1997, 27, 1731.
869. Ley, S.V.; Mitchell, C.; Pears, D.; Ramarao, C.; Yu, J.Q.; Zhou, W. Org. Lett. 2003, 5, 4665.
870. See Cragg, G.M.L. Organoboranes in Organic Synthesis, Marcel Dekker, NY, 1973, pp. 345–348. See also, Yamamoto, Y.; Toi, H.; Sonoda, A.; Murahashi, S. J. Chem. Soc. Chem. Commun. 1976, 672.
871. Jankowska, R.; Liu, H.-J.; Mhehe, G.L Chem. Commun. 1999, 1581.
872. Hardouin, C.; Chevallier, F.; Rousseau, B.; Doris, E. J. Org. Chem. 2001, 66, 1046.
873. Concellón, J.M.; Bardales, E. Org. Lett. 2003, 5, 4783.
874. Uenishi, J.; Kubo, Y. Tetrahedron Lett. 1994, 35, 6697.
875. van Tamelen, E.E.; Gladys, J.A. J. Am. Chem. Soc. 1974, 96, 5290.
876. Fry, J.L.; Mraz, T.J. Tetrahedron Lett. 1979, 849.
877. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 347–422.
878. See Hudlicky, M. Reductions in Organic Chemistry, Ellis Horwood, Chichester, 1984, pp. 96–129. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1075–1113.
879. See Abdel-Magid, A.F. (Ed.) Reductions in Organic Synthesis American Chemical Society Washington, 1996; Seyden-Penne, J. Reductions by the Alumino- and Borohydrides, VCH, NY, 1991; Hajos, A. Complex Hydrides, Elsevier, NY, 1979; House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 49–71; Wheeler, O.H. in Patai, S. The Chemistry of the Carbonyl Group, pt. 1, Wiley, NY, 1966, pp. 507–566.
880. See Meta, C.T.; Koide, K. Org. Lett. 2004, 6, 1785.
881. Hoye, T.R.; Aspaas, A.W.; Eklov, B.M.; Ryba, T.D. Org. Lett. 2005, 7, 2205.
882. See Toda, F.; Kiyoshige, K.; Yagi, M. Angew. Chem. Int. Ed. 1989, 28, 320.
883. Hajipour, A.R.; Mallakpour, S.E. Synth. Commun. 2001, 31, 1177.
884. Cho, B.T.; Kang, S.K.; Kim, M.S.; Ryu, S.R.; An, D.K. Tetrahedron 2006, 62, 8164.
885. Tamami, B.; Mahdavi, H. Tetrahedron 2003, 59, 821.
886. Varma, R.S.; Saini, R.K. Tetrahedron Lett. 1997, 38, 4337.
887. Liu, W.-y.; Xu, Q.-h.; Ma, Y.-x. Org. Prep. Proceed. Int. 2000, 32, 596.
888. Fieser, L.F.; Fieser, M. Reagents for Organic Synthesis Vol. 2, Wiley, New York, 1969, p. 382; Fieser, L.F.; Fieser, M. Reagents for Organic Synthesis Vol. 3, Wiley, New York, 1972, p. 260; Vit, J.; Cásensky, B.; Machácek, J. Fr. Pa., 1,515,582 1968 [Chem. Abstr. 70: 115009x, 1967].
889. Capka, M.; Chvalovsky, V.; Kochloefl, K.; Kraus, M. Collect. Czech. Chem. Commun. 1969, 34, 118; Stotter, P.L.; Friedman, M.D.; Minter, D.E. J. Org. Chem. 1985, 50, 29.
890. Zurflüh, R.; Dunham. L.L.; Spain, V.L.; Siddall, J.B. J. Am. Chem. Soc. 1970, 92, 425.
891. Markezich, R.L.; Willy, W.E.; McCarry, B.E.; Johnson, W.S. J. Am. Chem. Soc. 1973, 95, 4414; McCarry, B.E.; Markezich, R.L.; Johnson, W.S J. Am. Chem. Soc. 1973, 95, 4416.
892. See Kesenheimer, C.; Groth, U. Org. Lett. 2006, 8, 2507; White, J.D.; Choi, Y. Org. Lett. 2000, 2, 2373; Gao, Y.; Sharpless, K.B. J. Org. Chem. 1988, 53, 4081. Maloney, D.J.; Hecht, S.M. Org. Lett. 2005, 7, 4297
893. See Keinan, E.; Greenspoon, N. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 2, Wiley, NY, 1989, pp. 923–1022.
894. Dilling, W.L.; Plepys, R.A. J. Org. Chem. 1970, 35, 2971.
895. See Fukuzawa, S.; Fujinami, T.; Yamauchi, S.; Sakai, S. J. Chem. Soc. Perkin Trans. 1 1986, 1929. See also, Chênevert, R.; Ampleman, G. Chem. Lett. 1985, 1489; Varma, R.S.; Kabalka, G.W. Synth. Commun. 1985, 15, 985.
896. Ohtsuka, Y.; Koyasu, K.; Ikeno, T.; Yamada, T. Org. Lett. 2001, 3, 2543.
897. Halimjani, A.Z.; Saidi, M.R. Synth. Commun. 2005, 35, 2271.
898. Khurana, J.M.; Chauhan, S. Synth. Commun. 2001, 31, 3485.
899. Nutaitis, C.F.; Bernardo, J.E. J. Org. Chem. 1989, 54, 5629.
900. For a review of the reactivity of this reagent, see Ranu, B. Synlett 1993, 885.
901. Sreekumar, R.; Padmakumar, R.; Rugmini, P. Tetrahedron Lett. 1998, 39, 5151.
902. Ojima, I.; Kogure, T. Organometallics 1982, 1, 1390.
903. Johnson, M.R.; Rickborn, B. J. Org. Chem. 1970, 35, 1041.
904. Chaikin, S.W.; Brown, W.G. J. Am. Chem. Soc. 1949, 71, 122.
905. Ranu, B.C.; Samanta, S. Tetrahedron 2003, 59, 7901.
906. See Luibrand, R.T.; Taigounov, I.R.; Taigounov, A.A. J. Org. Chem. 2001, 66, 7254.
907. See Borbaruah, M.; Barua, N.C.; Sharma, R.P. Tetrahedron Lett. 1987, 28, 5741.
908. Gribble, G.W.; Ferguson, D.C. J. Chem. Soc. Chem. Commun. 1975, 535. See also, Nutaitis, C.F.; Gribble, G.W. Tetrahedron Lett. 1983, 24, 4287.
909. Blanton, J.R. Synth. Commun. 1997, 27, 2093.
910. Ranu, B.C.; Chakraborty, R. Tetrahedron Lett. 1990, 31, 7663; See Ranu, B. Synlett 1993, 885.
911. Fuller, J.C.; Stangeland, E.L.; Jackson, T.C.; Singaram, B. Tetrahedron Lett. 1994, 35, 1515. See also, Mogali, S.; Darville, K.; Pratt, L.M. J. Org. Chem. 2001, 66, 2368.
912. Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R.; Meneses, R. Synlett 2000, 197.
913. See Li, K.; Hamann, L.G.; Koreeda, M. Tetrahedron Lett. 1992, 33, 6569.
914. Lansbury, P.T.; Peterson, J.O. J. Am. Chem. Soc. 1962, 84, 1756.
915. Ward, D.E.; Rhee, C.K.; Zoghaib, W.M. Tetrahedron Lett. 1988, 29, 517.
916. Sarkar, D.C.; Das, A.R.; Ranu, B.C. J. Org. Chem. 1990, 55, 5799.
917. Ward, D.E.; Rhee, C.K. Can. J. Chem. 1989, 67, 1206.
918. Maruoka, K.; Araki, Y.; Yamamoto, H. J. Am. Chem. Soc. 1988, 110, 2650.
919. For lists, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1089–1092, and references given in Ward, D.E.; Rhee, C.K. Can. J. Chem. 1989, 67, 1206.
920. Ravikumar, K.S.; Sinha, S.; Chandrasekaran, S. J. Org. Chem. 1999, 64, 5841.
921. Krishnamurthy, S.; Brown, H.C. J. Am. Chem. Soc. 1976, 98, 3383.
922. K-Selectride: Lawson, E.C.; Zhang, H.-C.; Maryanoff, B.E. Tetrahedron Lett. 1999, 40, 593.
923. Caro, B.; Boyer, B.; Lamaty, G.; Jaouen, G. Bull. Soc. Chim. Fr. 1983, II-281; Boone, J.R.; Ashby, E.C. Top. Stereochem. 1979, 11, 53; Wigfield, D.C. Tetrahedron 1979, 35, 449; Tramontini, M. Synthesis 1982, 605.
924. See Mukherjee, D.; Wu, Y.; Fronczek, F.R.; Houk, K.N. J. Am. Chem. Soc. 1988, 110, 3328.
925. Boireau, G.; Deberly, A.; Toneva, R. Synlett 1993, 585
926. See Feoktistov, L.G.; Lund, H. in Baizer, M.M.; Lund, H. Organic Electochemistry, Marcel Dekker, NY, 1983, pp. 315–358, 315–326. See also, Coche, L.; Moutet, J. J. Am. Chem. Soc. 1987, 109, 6887.
927. Abdel-Magid, A.F., Ed., Reductions in Organic Synthesis American Chemical Society, Washington, DC, 1996, pp. 31–50; Parker, D. in Hartley, F.R. The Chemistry of the Metal-Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 979–1047; Tanaka, K. in Cerveny, L. Catalytic Hydrogenation, Elsevier, NY, 1986, pp. 79–104; Rylander, P.N. Hydrogenation Methods, Academic Press, NY, 1985, pp. 66–77; Rylander, P.N. Catalytic Hydrogenation over Platinum Metals, Academic Press, NY., 1967, pp. 238–290.
928. Hedberg, C.; Källström, K.; Arvidsson, P.I.; Brandt, P.; Andersson, P.G. J. Am. Chem. Soc. 2005, 127, 15083.
929. See Heck, R.F. Organotransition Metal Chemistry, Academic Press, NY, 1974, pp. 65–70; Enthaler, S.; Hagemann, B.; Erre, G.; Junge, K.; Beller, M. Chemistry: Asian J. 2006, 1, 598.
930. See Narasimhan, C.S.; Deshpande, V.M.; Ramnarayan, K. J. Chem. Soc. Chem. Commun. 1988, 99.
931. See, however, Pavlenko, N.V. Russ. Chem. Rev. 1989, 58, 453.
932. See House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 152–160.
933. Pradhan, S.K. Tetrahedron 1986, 42, 6351; Huffman, J.W. Acc. Chem. Res. 1983, 16, 399. See Rautenstrauch, V. Tetrahedron 1988, 44, 1613; Song, W.M.; Dewald, R.R. J. Chem. Soc. Perkin Trans. 2, 1989, 269; Rassat, A. Pure Appl. Chem. 1977, 49, 1049.
934. House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, p. 151. See, however, Giordano, C.; Perdoncin, G.; Castaldi, G. Angew. Chem. Int. Ed. 1985, 24, 499.
935. See Rautenstrauch, V.; Geoffroy, M. J. Am. Chem. Soc. 1976, 98, 5035; 1977, 99, 6280.
936. Rees, N.V.; Baron, R.; Kershaw, N.M.; Donohoe, T.J.; Compton, R.G. J. Am. Chem. Soc. 2008, 130, 12256.
937. Kreiser, W. Liebigs Ann. Chem. 1971, 745, 164.
938. Kardile, G.B.; Desai, D.G.; Swami, S.S. Synth. Commun. 1999, 29, 2129.
939. Bhar, S.; Guha, S. Tetrahedron Lett. 2004, 45, 3775.
940. Keck, G.E.; Wager, C.A.; Sell, T.; Wager, T.T. J. Org. Chem. 1999, 64, 2172.
941. See Prasad, E.; Flowers II, R.A. J. Am. Chem. Soc. 2002, 124, 6895.
942. Dahlén, A.; Hilmersson, G. Tetrahedron Lett. 2002, 43, 7197.
943. Fukuzawa, S.-i.; Nakano, N.; Saitoh, T. Eur. J. Org. Chem. 2004, 2863.
944. Sadavarte, V.S.; Swami, S.S.; Desai, D.G. Synth. Commun. 1998, 28, 1139.
945. Kim, J.Y.; Kim, H.D.; Seo, M.J.; Kim, H.R.; No, Z.; Ha, D.-C.; Lee, G.H. Tetrahedron Lett. 2006, 47, 9; Chopade, P.R.; Davis, T.A.; Prasad, E.; Flowers, II, R.A. Org. Lett. 2004, 6, 2685.
946. Hayakawa, R.; Sahara, T.; Shimizu, M. Tetrahedron Lett. 2000, 41, 7939.
947. Clerici, A.; Pastori, N.; Porta, O. Eur. J. Org. Chem. 2001, 2235.
948. See Maruoka, K.; Saito, S.; Concepcion, A.B.; Yamamoto, H. J. Am. Chem. Soc. 1993, 115, 1183. For a microwave-induced version of this reaction, see Barbry, D.; Torchy, S. Tetrahedron Lett. 1997, 38, 2959.
949. Bagnell, L.; Strauss, C.R. Chem. Commun. 1999, 287.
950. See Hutton, J. Synth. Commun. 1979, 9, 483; Namy, J.L.; Souppe, J.; Collin, J.; Kagan, H.B. J. Org. Chem. 1984, 49, 2045; Okano, T.; Matsuoka, M.; Konishi, H.; Kiji, J. Chem. Lett. 1987, 181.
951. Corma, A.; Domine, M.E.; Nemeth, L.; Valencia, S. J. Am. Chem. Soc. 2002, 124, 3194.
952. Campbell, E.J.; Zhou, H.; Nguyen, S.T. Org. Lett. 2001, 3, 2391. See Albrecht, M.; Crabtree, R.H.; Mata, J.; Peris, E. Chem. Commun. 2002, 32.
953. Zhu, Y.; Chuah, G.; Jaenicke, S. Chem. Commun. 2003, 2734.
954. Kazemi, F.; Kiasat, A.R. Synth. Commun. 2002, 32, 2255.
955. Yin, J.; Huffman, M.A.; Conrad, K.M.; Armstrong, III, J.D. J. Org. Chem. 2006, 71, 840.
956. Dong, Z.-R.; Li, Y.-Y.; Chen, J.-S.; Li, B.-Z.; Xing, Y.; Gao, J.-X. Org. Lett. 2005, 7, 1043; Onodera, G.; Nishibayashi, Y.; Uemura, S. Angew. Chem. Int. Ed. 2006, 45, 3819.
957. Campbell, E.J.; Zhou, H.; Nguyen, S.T. Angew. Chem. Int. Ed. 2002, 41, 1020.
958. See, however, Ashby, E.C.; Argyropoulos, J.N. J. Org. Chem. 1986, 51, 3593; Yamataka, H.; Hanafusa, T. Chem. Lett. 1987, 643.
959. See Warnhoff, E.W.; Reynolds-Warnhoff, P.; Wong, M.Y.H. J. Am. Chem. Soc. 1980, 102, 5956.
960. Moulton, W.N.; Van Atta, R.E.; Ruch, R.R. J. Org. Chem. 1961, 26, 290.
961. Galian, R.E.; Litwinienko, G.; Pérez-Prieto, J.; Ingold, K.U. J. Am. Chem. Soc. 2007, 129, 9280.
962. Shiner, Jr., V.J.; Whittaker, D. J. Am. Chem. Soc. 1969, 91, 394.
963. Kamitanaka, T.; Matsuda, T.; Harada, T. Tetrahedron 2007, 63, 1429.
964. See Cragg, G.M.L. Organoboranes in Organic Synthesis, Marcel Dekker, NY, 1973, pp. 324–335. See Cha, J.S.; Moon, S.J.; Park, J.H. J. Org. Chem. 2001, 66, 7514.
965. Brown, H.C.; Subba Rao, B.C. J. Am. Chem. Soc. 1960, 82, 681; Brown, H.C.; Korytnyk, W. J. Am. Chem. Soc. 1960, 82, 3866.
966. See Bae, J.W.; Lee, S.H.; Jung, Y.J.; Yoon, C.-O.M.; Yoon, C.M. Tetrahedron Lett. 2001, 42, 2137.
967. Krishnamurthy, S.; Brown, H.C. J. Org. Chem. 1975, 40, 1864; Lane, C.F. Aldrichimica Acta 1976, 9, 31.
968. Mincione, E. J. Org. Chem. 1978, 43, 1829.
969. Bartoli, G.; Bosco, M.; Bellucci, M.C.; Daplozzo, R.; Marcantoni, E.; Sambri, L. Org. Lett. 2000, 2, 45.
970. Kabalka, G.W.; Malladi, R.R. Chem. Commun. 2000, 2191.
971. Du, D.-M.; Fang, T.; Xu, J.; Zhang, S.-W. Org. Lett. 2006, 8, 1327; Krzemi![]() ski, M.P.; Wojtczak, A. Tetrahedron Lett. 2005, 46, 8299.
ski, M.P.; Wojtczak, A. Tetrahedron Lett. 2005, 46, 8299.
972. Stepanenko, V.; De Jesús, M.; Correa, W.; Guzmán, I.; Vázquez, C.; de la Cruz, W.; Ortiz-Marciales, M.; Barnes, C.L. Tetrahedron Lett. 2007, 48, 5799.
973. Eagon, S.; Kim, J.; Yan, K.; Haddenham, D.; Singaram, B. Tetrahedron Lett. 2007, 48, 9025.
974. Nakamura, S.; Kuroyanagi, M.; Watanabe, Y.; Toru, T. J. Chem. Soc. Perkin Trans. 1 2000, 3143.
975. Kamiura, K.; Wada, M. Tetrahedron Lett. 1999, 40, 9059; Adams, C.M.; Schemenaur, J.E. Synth. Commun. 1990, 20, 2359. For a review, see Kuivila, H.G. Synthesis 1970, 499.
976. Yu, H.; Wang, B. Synth. Commun. 2001, 31, 2719.
977. Kamiya, I.; Ogawa, A. Tetrahedron Lett. 2002, 43, 1701.
978. Sasaki, K.; Komatsu, N.; Shivakawa, S.; Maruoka, K. Synlett 2002, 575.
979. Perchyonok, V.T. Tetrahedron Lett. 2006, 47, 5163.
980. Ison, E.A.; Trivedi, E.R.; Corbin, R.A.; Abu-Omar, M.M. J. Am. Chem. Soc. 2005, 127, 15374.
981. For a review, see Díez-González, S.; Nolan, S.P. Org. Prep. Proceed. Int. 2007, 39, 523. See Chen, T.; Liu, X.-G.; Shi, M. Tetrahedron 2007, 63, 4874; Fernandes, A.C.; Fernandes, R.; Romão, C.R.; Royo, B. Chem. Commun.2005, 213; Comte, V.; Balan, C.; Le Gendre, P.; Moïse, C. Chem. Commun. 2007, 713; Nishiyama, H.; Furuta, A. Chem. Commun. 2007, 760.
982. See Yamamoto, Y.; Matsuoka, K.; Nemoto, H. J. Am. Chem. Soc. 1988, 110, 4475.
983. Smonou, I. Tetrahedron Lett. 1994, 35, 2071.
984. Iwasaki, F.; Onomura, O.; Mishima, K.; Maki, T.; Matsumura, Y. Tetrahedron Lett. 1999, 40, 7507.
985. Enholm, E.J.; Schulte II, J.P. J. Org. Chem. 1999, 64, 2610.
986. Nadkarni, D.; Hallissey, J.; Mojica, C. J. Org. Chem. 2003, 68, 594.
987. Babler, J.H.; Sarussi, S.J. J. Org. Chem. 1981, 46, 3367.
988. Babler, J.H.; Invergo, B.J. Tetrahedron Lett. 1981, 22, 621.
989. Morris, D.J.; Hayes, A.M.; Wills, M. J. Org. Chem. 2006, 71, 7035.
990. Wu, X.; Li, X.; King, F.; Xiao, J. Angew. Chem. Int. Ed. 2005, 44, 3407.
991. For a review, see Sih, C.J.; Chen, C. Angew. Chem. Int. Ed. 1984, 23, 570.
992. See Wolfson, A.; Dlugy, C.; Tavor, D.; Blumenfeld, J.; Shotland, Y. Tetrahedron Asymmetry 2006, 17, 2043; Yadav, J.S.; Reddy, G.S.K.K.; Sabitha, G.; Krishna, A.D.; Prasad, A.R.; Rahaman, H.U.R.; Rao, K.V.; Rao, A.B. Tetrahedron Asymmetry 2007, 18, 717.
993. See Moore, J.C.; Pollard, D.J.; Kosjek, B.; Devine, P.N. Acc. Chem. Res. 2007, 40, 1412; Ema, T.; Yagasaki, H.; Okita, N.; Takeda, M.; Sakai, T. Tetrahedron 2006, 62, 6143; Hoyos, P.; Sansottera, G.; Fernández, M.; Molinari, F.; Sinisterra, J.V.; Alcántara, A.R. Tetrahedron 2008, 64, 7929; Utsukihara, T.; Misumi, O.; Kato, N.; Kuroiwa, T.; Horiuchi, C.A. Tetrahedron Asymmetry 2006, 17, 1179; Zhu, D.; Malik, H.T.; Hua, L. Tetrahedron Asymmetry 2006, 17, 3010; Lavandera, I.; Höller, B.; Kern, A.; Ellmer, U.; Glieder, A.; de Wildeman, S.; Kroutil, W. Tetrahedron Asymmetry 2008, 19, 1954. For enzymatic reduction of thio ketones, see Nielsen, J.K.; Madsen, J.Ø. Tetrahedron Asymmetry, 1994, 5, 403.
994. See Nakamura, K.; Yamanaka, R.; Matsuda, T.; Harada, T. Tetrahedron Asymmetry 2003, 14, 2659.
995. Matsuda, T.; Yamagishi, Y.; Koguchi, S.; Iwai, N.; Kitazume, T. Tetrahedron Lett. 2006, 47, 4619; Bräutigam, S.; Bringer-Meyer, S.; Weuster-Botz, D. Tetrahedron Asymmetry 2007, 18, 1883.
996. Matsuda, T.; Marukado, R.; Mukouyama, M.; Harada, T.; Nakamura, K. Tetrahedron Asymmetry 2008, 19, 2272.
997. See Brown, H.C.; Wang, K.K.; Chandrasekharan, J. J. Am. Chem. Soc. 1983, 105, 2340.
998. See Caro, B.; Boyer, B.; Lamaty, G.; Jaouen, G. Bull. Soc. Chim. Fr. 1983, II-281; Boone, J.R.; Ashby, E.C. Top. Stereochem. 1979, 11, 53; Wigfield, D.C. Tetrahedron 1979, 35, 449.
999. Ashby, E.C.; Boone, J.R. J. Am. Chem. Soc. 1976, 98, 5524.
1000. Pierre, J.; Handel, H. Tetrahedron Lett. 1974, 2317. See also, Loupy, A.; Seyden-Penne, J.; Tchoubar, B. Tetrahedron Lett. 1976, 1677; Ashby, E.C.; Boone, J.R. J. Am. Chem. Soc. 1976, 98, 5524.
1001. Wigfield, D.C.; Gowland, F.W. J. Org. Chem. 1977, 42, 1108. See, however, Adams, C.; Gold, V.; Reuben, D.M.E. J. Chem. Soc. Perkin Trans. 2 1977, 1466, 1472; Kayser, M.M.; Eliev, S.; Eisenstein, O. Tetrahedron Lett.1983, 24, 1015.
1002. Haubenstock, H.; Eliel, E.L. J. Am. Chem. Soc. 1962, 84, 2363; Malmvik, A.; Obenius, U.; Henriksson, U. J. Chem. Soc. Perkin Trans. 2 1986, 1899, 1905.
1003. Malmvik, A.; Obenius, U.; Henriksson, U. J. Org. Chem. 1988, 53, 221.
1004. For reviews of reductions with alkoxyaluminum hydrides, see Málek, J. Org. React. 1988, 36, 249; 1985, 34, 1; Málek, J.; Cerny, M. Synthesis 1972, 217.
1005. Levine, S.G.; Eudy, N.H. J. Org. Chem. 1970, 35, 549; Heusler, K.; Wieland, P.; Meystre, C. Org. Synth. V, 692.
1006. Brown, C.A.; Krishnamurthy, S.; Kim, S.C. J. Chem. Soc. Chem. Commun. 1973, 391.
1007. Brown, H.C.; Park, W.S.; Cho, B.T.; Ramachandran, P.V. J. Org. Chem. 1987, 52, 5406.
1008. Wang, Z.; La, B.; Fortunak, J.M.; Meng, X.-J.; Kabalka, G.W. Tetrahedron Lett. 1998, 39, 5501.
1009. See Singh, V.K. Synthesis 1992, 605; Midland, M.M. Chem. Rev. 1989, 89, 1553; Nógrádi, M. Stereoselective Synthesis, VCH, NY, 1986, pp. 105–130; in Morrison, J.D. Asymmetric Synthesis, Academic Press, NY, 1983, the articles by Midland, M.M. Vol. 2, pp. 45–69, and Grandbois, E.R.; Howard, S.I.; Morrison, J.D. Vol. 2, pp. 71–90; Haubenstock, H. Top. Stereochem. 1983, 14, 231.
1010. For a list of many of these reducing agents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1097–1111.
1011. See Midland, M.M.; Kazubski, A.; Woodling, R.E. J. Org. Chem. 1991, 56, 1068.
1012. Kaselj, M.; Gonikberg, E.M.; le Noble, W.J. J. Org. Chem. 1998, 63, 3218.
1013. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 391–411.
1014. See Liang, Y.; Jing, Q.; Li, X.; Shi, L.; Ding, K. J. Am. Chem. Soc. 2005, 127, 7694; Ohkuma, T.; Sandoval, C.A.; Srinivasan, R.; Lin, Q.; Wei, Y.; Muñiz, K.; Noyori, R. J. Am. Chem. Soc. 2005, 127, 8288; Huang, H.; Okuno, T.; Tsuda, K.; Yoshimura, M.; Kitamura, M. J. Am. Chem. Soc. 2006, 128, 8716; Xie, J.-H.; Zhou, Z.-T.; Kong, W.-L.; Zhou, Q.-L. J. Am. Chem. Soc. 2007, 129, 1868; Xie, J.-H.; Liu, S.; Huo, X.-H.; Cheng, X.; Duan, H.-F.; Fan, B.-M.; Wang, L.-X.; Zhou, Q.-L. J. Org. Chem. 2005, 70, 2867; Truppo, M.D.; Pollard, D.; Devine, P. Org. Lett. 2007, 9, 335; Ngo, H.L.; Hu, A.; Lin, W. Tetrahedron Lett. 2005, 46, 595; Lu, S.-M.; Bolm, C. Angew. Chem. Int. Ed. 2008, 47, 8920; Jiang, H.-y.; Yang, C.-f.; Li, C.; Fu, H.-y.; Chen, H.; Li, R.-x.; Li, X.-j. Angew. Chem. Int. Ed. 2008, 47, 9240; Burk, S.; Franciò, G.; Leitner, W. Chem. Commun. 2005, 3460; Li, W.; Sun, X.; Zhou, L.; Hou, G.; Shichao Yu, S.; Zhang, X. J. Org. Chem. 2009, 74, 1397; Martins, J.,E,D,; Wills, M. Tetrahedron 2009, 65, 5782; Li, W.; Hou, G.; Wang, C.; Jiang, Y.; Zhang, X. Chem. Commun. 2010, 3979.
1015. See Noyori, R. Science 1990, 248, 1194; Noyori, R.; Takaya, H. Acc. Chem. Res. 1990, 23, 345; Takaya, H.; Akutagawa, S.; Noyori, R. Org. Synth. 67, 20.
1016. Taber, D.F.; Silverberg, L.J. Tetrahedron Lett. 1991, 32, 4227. See also, Kitamura, M.; Ohkuma, T.; Inoue, S.; Sayo, N.; Kumobayashi, H.; Akutagawa, S.; Ohta, T.; Takaya, H.; Noyori, R. J. Am. Chem. Soc. 1988, 110, 629.
1017. See Sun, L.; Tang, M.; Wang, H.; Wei, D.; Liu, L. Tetrahedron Asymmetry 2008, 19, 779; Ohkuma, T.; Hattori, T.; Ooka, H.; Inoue, T.; Noyori, R. Org. Lett. 2004, 6, 2681; Lei, A.; Wu, S.; He, M.; Zhang, X. J. Am. Chem. Soc. 2004, 126, 1626; Sun, Y.; Wan, X.; Guo, M.; Wang, D.; Dong, X.; Pan, Y.; Zhang, Z. Tetrahedron Asymmetry 2004, 15, 2185. Also see Sandoval, C.A.; Ohkuma, T.; Muñiz, K.; Noyori, R. J. Am. Chem. Soc. 2003, 125, 13490.
1018. Ngo, H.L.; Hu, A.; Lin, W. Chem.. Commun. 2003, 1912.
1019. See Ford, A.; Woodward, S. Angew. Chem. Int. Ed. 1999, 38, 335.
1020. See Santhi, V.; Rao, J.M. Tetrahedron Asymmetry 2000, 11, 3553; Jones, S.; Atherton, J.C.C. Tetrahedron Asymmetry 2000, 11, 4543; Cho, B.T.; Kim, D.J. Tetrahedron Asymmetry 2001, 12, 2043; Jiang, B.; Feng, Y.; Hang, J.-F. Tetrahedron Asymmetry 2001, 12, 2323; Gilmore, N.J.; Jones, S.; Muldowney, M.P. Org. Lett. 2004, 6, 2805; Huertas, R.E.; Corella, J.A.; Soderquist, J.A. Tetrahedron Lett. 2003, 44, 4435.
1021. See Brunel, J.M.; Legrand, O.; Buono, G. Eur. J. Org. Chem. 2000, 3313; Kawanami, Y.; Murao, S.; Ohga, T.; Kobayashi, N. Tetrahedron 2003, 59, 8411; Basavaiah, D.; Reddy, G.J.; Chandrashekar, V. Tetrahedron Asymmetry 2004, 15, 47; Zhang, Y.-X.; Du, D.-M.; Chen, X.; Lü, S.-F.; Hua, W.-T. Tetrahedron Asymmetry 2004, 15, 177; Lindsay. D.M.; McArthur, D. Chem. Commun. 2010, 2474.
1022. Price, M.D.; Sui, J.K.; Kurth, M.J.; Schore, N.E. J. Org. Chem. 2002, 67, 8086.
1023. Bolm, C.; Derrien, N.; Seger, A. Chem. Commun. 1999, 2087.
1024. Hu, J.-b.; Zhao, G.; Yang, G.-s.; Ding, Z.-d. J. Org. Chem. 2001, 66, 303; Zhou, H.; Lü, S.; Xie, R.; Chan, A.S.C.; Yang, T.-K. Tetrahedron Lett. 2001, 42, 1107; Basavaiah, D.; Reddy, G.J.; Chandrashekar, V. Tetrahedron Asymmetry 2001, 12, 685.
1025. Li, G.-Q.; Yan, Z.-Y.; Niu, Y.-N.; Wu, L.-Y.; Wei, H.-L.; Liang, Y.-M. Tetrahedron Asymmetry 2008, 19, 816.
1026. Ren, Y.; Tian, X.; Sun, K.; Xu, J.; Xu, X.; Lu, S. Tetrahedron Lett. 2006, 47, 463.
1027. Lange, D.A.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2006, 62, 3704. In an ionic liquid: see Xiao, Y.; Malhotra, S.V. Tetrahedron Asymmetry 2006, 17, 1062.
1028. For a review, see Daverio, P.; Zanda, M. Tetrahedron Asymmetry 2001, 12, 2225.
1029. Kim, J.; Singaram, B. Tetrahedron Lett. 2006, 47, 3901.
1030. Molvinger, K.; Lopez, M.; Court, J. Tetrahedron Lett. 1999, 40, 8375.
1031. Nozaki, K.; Kobori, K.; Uemura, T.; Tsutsumi, T.; Takaya, H.; Hiyama, T. Bull. Chem. Soc. Jpn. 1999, 72, 1109.
1032. Jiang, B.; Feng, Y.; Zheng, J. Tetrahedron Lett. 2000, 41, 10281.
1033. Zhao, G.; Hu, J.-b.; Qian, Z.-s.; Yin, X.-x. Tetrahedron Asymmetry 2002, 13, 2095.
1034. Wu, X.; Li, X.; Zanotti-Gerosa, A.; Pettman, A.; Liu, J.; Mills, A.J.; Xiao, J. Chemistry: European J. 2008, 14, 2209; Kawasaki, I.; Tsunoda, K.; Tsuji, T.; Yamaguchi, T.; Shibuta, H.; Uchida, N.; Yamashita, M.; Ohta, S. Chem. Commun. 2005, 2134; Li, X.; Blacker, J.; Houson, I.; Wu, X.; Xiao, J. Synlett 2006, 1155; Ito, M.; Shibata, Y.; Watanabe, A.; Ikariya, T. Synlett 2009, 1621; Zani, L.; Eriksson, L.; Adolfsson, H. Eur. J. Org. Chem. 2008, 4655.
1035. Hayashi, T.; Hayashi, C.; Uozumi, Y. Tetrahedron Asymmetry, 1995, 6, 2503.
1036. Liu, P.N.; Gu, P.M.; Wang, F.; Tu, Y.Q. Org. Lett. 2004, 6, 169; Wu, X.; Li, X.; Hems, W.; King, F.; Xiao, J. Org. Biomol. Chem. 2004, 2, 1818; Schlatter, A.; Kundu, M.K.; Woggon, W.-D. Angew. Chem. Int. Ed. 2004, 43, 6731.
1037. Shaikh, N.S.; Enthaler, S.; Junge, K.; Beller, M. Angew. Chem. Int. Ed. 2008, 47, 2497; Inagaki, T.; Yamada, Y.; Phong, L.T.; Furuta, A.; Ito, J.-i.; Nishiyama, H. Synlett 2009, 253; Ghoshal, A.; Sarkar, A.R.; Manickam, G.; Kumaran, R.S.; Jayashankaran, J. Synlett 2010, 1459.
1038. Lipshutz, B.H.; Noson, K.; Chrisman, W.; Lower, A. J. Am. Chem. Soc. 2003, 125, 8779.
1039. Gade, L.H.; César, V.; Bellemin-Laponnaz, S. Angew. Chem. Int. Ed. 2004, 43, 1014.
1040. Cecchetto, A.; Fontana, F.; Minisci, F.; Recupero, F. Tetrahedron Lett. 2001, 42, 6651.
1041. Zhou, L.; Wang, Z.; Wei, S.; Sun, J. Chem. Commun. 2007, 2977.
1042. Ohno, K.; Kataoka, Y.; Mashima, K. Org. Lett. 2004, 6, 4695.
1043. Yang, T.-K.; Lee, D.-S. Tetrahedron Asymmetry 1999, 10, 405.
1044. See, however, Li, X.; List, B. Chem. Commun. 2007, 1739. See also, Giacomini, D.; Galletti, P.; Quintavalla, A.; Gucciardo, G.; Paradisi, F. Chem. Commun. 2007, 4038.
1045. Midland, M.M.; Greer, S.; Tramontano, A.; Zderic, S.A. J. Am. Chem. Soc. 1979, 101, 2352. See also, Midland, M.M.; Zderic, S.A. J. Am. Chem. Soc. 1982, 104, 525.
1046. Ramachandran, P.V.; Pitre, S.; Brown, H.C. J. Org. Chem. 2002, 67, 5315. For a discussion of the sources of stereoselectivity, see Xu, J.; Wei, T.; Zhang, Q. J. Org. Chem. 2004, 69, 6860.
1047. Dalton, D.M.; Gladysz, J.A. J. Organomet. Chem. 1989, 370, C17.
1048. See Bloch, R.; Gilbert, L.; Girard, C. Tetrahedron Lett. 1988, 53, 1021; Evans, D.A.; Chapman, K.T.; Carreira, E.M. J. Am. Chem. Soc. 1988, 110, 3560.
1049. See Nógrádi, M. Stereoselective Synthesis VCH, NY, 1986, pp. 131–148; Oishi, T.; Nakata, T. Acc. Chem. Res. 1984, 17, 338.
1050. See Yamamoto, Y.; Matsuoka, K.; Nemoto, H. J. Am. Chem. Soc. 1988, 110, 4475.
1051. See Gaylord, N.G. Reduction with Complex Metal Hydrides, Wiley, NY, 1956, pp. 322–373.
1052. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1114–1116. Zinc borohydride has also been used; see Narashimhan, S.; Madhavan, S.; Prasad, K.G. J. Org. Chem. 1995, 60, 5314.
1053. See, however, Fujisawa, T.; Mori, T.; Sato, T. Chem. Lett. 1983, 835.
1054. Tale, R.H.; Patil, K.M.; Dapurkar, S.E. Tetrahedron Lett. 2003, 44, 3427.
1055. Zhou, Y.; Gao, G.; Li, H.; Qu, J. Tetrahedron Lett. 2008, 49, 3260.
1056. Narashimhan, S.; Swarnalakshmi, S.; Balakumar, R. Synth. Commun. 2000, 30, 941.
1057. See Rylander, P.N. Hydrogenation Methods, Academic Press, NY, 1985, pp. 78–79.
1058. Brown, H.C.; Stocky, T.P. J. Am. Chem. Soc. 1977, 99, 8218; Chen, M.H.; Kiesten, E.I.S.; Magano, J.; Rodriguez, D.; Sexton, K.E.; Zhang, J.; Lee, H.T. Org. Prep. Proceed. Int. 2002, 34, 665.
1059. Yoon, N.M.; Cho, B.T. Tetrahedron Lett. 1982, 23, 2475.
1060. Kamochi, Y.; Kudo, T. Bull. Chem. Soc. Jpn. 1992, 65, 3049.
1061. Kamochi, Y.; Kudo, T. Tetrahedron 1992, 48, 4301.
1062. Kamochi, Y.; Kudo, T. Chem. Lett. 1993, 1495.
1063. McKennon, M.J.; Meyers, A.I.; Drauz, K.; Schwarm, M. J. Org. Chem. 1993, 58, 3568.
1064. For a review, see Gaylord, N.G. Reduction with Complex Metal Hydrides, Wiley, NY, 1956, pp. 391–531.
1065. For a ring size-selective reduction using SmI2–H2O, see Duffy, L.A.; Matsubara, H.; Procter, D.J. J. Am. Chem. Soc. 2008, 130, 1136.
1066. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1116–1120.
1067. Ayers, T.A. Tetrahedron Lett. 1999, 40, 5467.
1068. Brown, H.C.; Choi, Y.M. Synthesis 1981, 439; Brown, H.C.; Choi, Y.M.; Narasimhan, S. J. Org. Chem. 1982, 47, 3153.
1069. Takahashi, S.; Cohen, L.A. J. Org. Chem. 1970, 35, 1505.
1070. For example, see Brown, M.S.; Rapoport, H. J. Org. Chem. 1963, 28, 3261; Boechat, N.; da Costa, J.C.S.; Mendonca, J.de S.; de Oliveira, P.S.M.; DeSouza, M.V.N. Tetrahedron Lett. 2004, 45, 6021.
1071. See also, Soai, K.; Oyamada, H.; Takase, M.; Ookawa, A. Bull. Chem. Soc. Jpn. 1984, 57, 1948; Guida, W.C.; Entreken, E.E.; Guida, W.C. J. Org. Chem. 1984, 49, 3024.
1072. Zanka, A.; Ohmori, H.; Okamoto, T. Synlett 1999, 1636.
1073. Feng, J.-C.; Liu, B.; Dai, L.; Yang, X.-L.; Tu, S.-J. Synth. Commun. 2001, 31, 1875.
1074. For a review, see Adkins, H. Org. React. 1954, 8, 1.
1075. Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. Angew. Chem. Int. Ed. 2006, 45, 1113.
1076. Chablay, E. Compt. Rend 1913, 156, 1020; Bouveault, L.; Blanc, G. Bull. Soc. Chim. Fr. 1904, 31, 666; Bouveault, L.; Blanc, G. Compt. Rend. 1903, 136, 1676. See Bodnar, B.S.; Vogt, P.F. J. Org. Chem. 2009, 74, 2598.
1077. Ohta, T.; Kamiya, M.; Kusui, K.; Michibata, T.; Nobutomo, M.; Furukawa, I. Tetrahedron Lett. 1999, 40, 6963.
1078. Matsubara, K.; Iura, T.; Maki, T.; Nagashima, H. J. Org. Chem. 2002, 67, 4985.
1079. See Fuson, R.C. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 211–232; Wheeler, O.H. in Patai, S. The Chemistry of Acyl Halides, Wiley, NY, 1972, pp. 231–251.
1080. Cha, J.S.; Brown, H.C. J. Org. Chem. 1993, 58, 4732 and references cited therein.
1081. See Rylander, P.N. Catalytic Hydrogenation Over Platinum Metals, Academic Press, NY, 1967, pp. 398–404; Maier, W.F.; Chettle, S.J.; Rai, R.S.; Thomas, G. J. Am. Chem. Soc. 1986, 108, 2608.
1082. Peters, J.A.; van Bekkum, H. Recl. Trav. Chim. Pays-Bas 1981, 100, 21. See also, Burgstahler, A.W.; Weigel, L.O.; Shaefer, C.G. Synthesis 1976, 767.
1083. See Leblanc, J.C.; Moise, C.; Tirouflet, J. J. Organomet. Chem. 1985, 292, 225; Corriu, R.J.P.; Lanneau, G.F.; Perrot, M. Tetrahedron Lett. 1988, 29, 1271. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1265–1266.
1084. See Lusztyk, J.; Lusztyk, E.; Maillard, B.; Ingold, K.U. J. Am. Chem. Soc. 1984, 106, 2923.
1085. Inoue, K.; Yasuda, M.; Shibata, I.; Baba, A. Tetrahedron Lett. 2000, 41, 113.
1086. Babler, J.H. Synth. Commun. 1982, 12, 839. See Entwistle, I.D.; Boehm, P.; Johnstone, R.A.W.; Telford, R.P. J. Chem. Soc. Perkin Trans. 1 1980, 27.
1087. Jia, X.; Liu, X.; Li, J.; Zhao, P.; Zhang, Y. Tetrahedron Lett. 2007, 48, 971.
1088. Shamsuddin, K.M.; Zubairi, Md.O.; Musharraf, M.A. Tetrahedron Lett. 1998, 39, 8153.
1089. Lee, K.; Maleczka Jr., R.E. Org. Lett. 2006, 8, 1887.
1090. For a review, see Cha, J.S. Org. Prep. Proced. Int. 1989, 21, 451.
1091. See Lanneau, G.F.; Perrot, M. Tetrahedron Lett. 1987, 28, 3941; Cha, J.S.; Kim, J.E.; Yoon, M.S.; Kim, Y.S. Tetrahedron Lett. 1987, 28, 6231. See also, the lists, in Larock, R.C. Comprehensive Organic Transformations,2nd ed., Wiley–VCH, NY, 1999, pp. 1265–1268.
1092. Bedenbaugh, A.O.; Bedenbaugh, J.H.; Bergin, W.A.; Adkins, J.D. J. Am. Chem. Soc. 1970, 92, 5774.
1093. Chloro - see Brown, H.C.; Cha, J.S.; Yoon, N.M.; Nazer, B. J. Org. Chem. 1987, 52, 5400; Bromo, see Cha, J.S.; Kim, J.E.; Lee, K.W. J. Org. Chem. 1987, 52, 5030.
1094. Fujisawa, T.; Mori, T.; Tsuge, S.; Sato, T. Tetrahedron Lett. 1983, 24, 1543.
1095. Cha, J.S.; Kim, J.M.; Jeoung, M.K.; Kwon, O.O.; Kim, E.J. Org. Prep. Proceed. Int. 1995, 27, 95.
1096. Gooßen, L.J.; Ghosh, K. Chem. Commun. 2002, 836.
1097. Zakharkin, L.I.; Sorokina, L.P. J. Gen. Chem. USSR 1967, 37, 525. See Song, J.I.; An, D.K. Chem. Lett. 2007, 36, 886.
1098. Cha, J.S.; Kim, J.M.; Jeoung, M.K.; Kwon, O.O.; Kim, E.J. Org. Prep. Proceed. Int. 1995, 27, 95.
1099. Zakharkin, L.I.; Gavrilenko, V.V.; Maslin, D.N. Bull. Acad. Sci. USSR Div. Chem. Sci. 1964, 867; Weissman, P.M.; Brown, H.C. J. Org. Chem. 1966, 31, 283.
1100. Chandrasekhar, S.; Kumar, M.S.; Muralidhar, B. Tetrahedron Lett. 1998, 39, 909.
1101. Fukuyama, T.; Lin, S.; Li, L. J. Am. Chem. Soc. 1990, 112, 7050.
1102. Penn, J.H.; Owens, W.H. Tetrahedron Lett. 1992, 33, 3737.
1103. Watanabe, Y.; Yamashita, M.; Mitsudo, T.; Igami, M.; Takegami, Y. Bull. Chem. Soc. Jpn. 1975, 48, 2490; Watanabe, Y.; Yamashita, M.; Mitsudo, T.; Igami, M.; Tomi, K.; Takegami, Y. Tetrahedron Lett. 1975, 1063.
1104. Shi, Z.; Gu, H. Synth. Commun. 1997, 27, 2701.
1105. Malanga, C.; Mannucci, S.; Lardicci, L. Tetrahedron Lett. 1997, 38, 8093.
1106. Fuson, R.C. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 220–225.
1107. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp.1269–1271.
1108. Zakharkin, L.I.; Khorlina, I.M. Bull. Acad. Sci. USSR Div. Chem. Sci. 1959, 2046.
1109. Muraki, M.; Mukaiyama, T. Chem. Lett. 1975, 875.
1110. Godjoian, G.; Singaram, B. Tetrahedron Lett. 1997, 38, 1717.
1111. White, J.M.; Tunoori, A.R.; Georg, G.I. J. Am. Chem. Soc. 2000, 122, 11995.
1112. See Craig, J.C.; Ekwurieb, N.N.; Fu, C.C.; Walker, K.A.M. Synthesis 1981, 303.
1113. See Popp, F.D.; Uff, B.C. Heterocycles 1985, 23, 731; Popp, F.D. Bull. Soc. Chim. Belg. 1981, 90, 609; Adv. Heterocycl. Chem. 1979, 24, 187; 1968, 9, 1. See Bridge, A.W.; Hursthouse, M.B.; Lehmann, C.W.; Lythgoe, D.J.; Newton, C.G. J. Chem. Soc. Perkin Trans. 1 1993, 1839 for isoquinoline Reissert salts.
1114. Dudman, C.C.; Grice, P.; Reese, C.B. Tetrahedron Lett. 1980, 21, 4645.
1115. See Cacchi, S.; Paolucci, G. Gazz. Chem. Ital. 1974, 104, 221; Matin, S.B.; Craig, J.C.; Chan, R.P.K. J. Org. Chem. 1974, 39, 2285.
1116. For a review, see Staab, H.A.; Rohr, W. Newer Methods Prep. Org. Chem. 1968, 5, 61.
1117. Nahm, S.; Weinreb, S.M. Tetrahedron Lett. 1981, 22, 3815; Mundy, B.P.; Ellerd, M.G.; Favaloro Jr., F.G. Name Reactions and Reagents in Organic Synthesis, 2nd Ed. Wiley–Interscience, New Jersey, 2005, p. 866. See Sibi, M. P. Org. Prep. Proceed. Int. 1993, 25, 15; Mentzel, M.; Hoffmann, H.M.R. J. Prakt. Chem. 1997, 339, 517.
1118. Spletstoser, J.T.; White, J.M.; Tunoori, A.R.; Georg, G.I. J. Am. Chem. Soc. 2007, 129, 3408; White, J.M.; Tunoori, A.R.; Georg, G.I. J. Am. Chem. Soc. 2000, 122, 11995; Wang, J.; Xu, H.; Gao, H.; Su, C.-Y.; Phillips, D.L. Organometallics 2010, 29, 42; Gondi, V.B.; Hagihara, K.; Rawal, V.H. Chem. Commun. 2010, 46, 904.
1119. See Verdaguer, X.; Lange, U.E.W.; Buchwald, S.L. Angew. Chem. Int. Ed. 1998, 37, 1103.
1120. See Burk, M.J.; Feaster, J.E. J. Am. Chem. Soc. 1992, 114, 6266.
1121. Bhattacharyya, S.; Neidigh, K.A.; Avery, M.A.; Williamson, J.S. Synlett 1999, 1781.
1122. See Harada, K. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 276–293; Rylander, P.N. Catalytic Hydrogenation over Platinum Metals, Academic Press, NY, 1967, pp. 123–138.
1123. Chase, P.A.; Welch, G.C.; Jurca, T.; Stephan, D.W. Angew. Chem. Int. Ed. 2007, 46, 8050.
1124. Zhang, Z.; Schreiner, P.R. Synlett 2007, 1455.
1125. Banik, B.K.; Zegrocka, O.; Banik, I.; Hackfeld, L.; Becker, F.F. Tetrahedron Lett. 1999, 40, 6731.
1126. Banik, B.K.; Hackfeld, L.; Becker, F.F. Synth. Commun. 22001, 31, 1581.
1127. Shibata, I.; Moriuchi-Kawakami, T.; Tanizawa, D.; Suwa, T.; Sugiyama, E.; Matsuda, H.; Baba, A. J. Org. Chem. 1998, 63, 383.
1128. Paukstelis, J.V.; Cook, A.G. in Cook, A.G. Enamines, 2nd ed., Marcel Dekker, NY, 1988, pp. 275–356.
1129. See Malkov, A.V.; Mariani, A.; MacDougall, K.N.; Kocovsky, P. Org. Lett. 2004, 6, 2253.
1130. Blackwell, J.M.; Sonmor, E.R.; Scoccitti, T.; Piers, W.E. Org. Lett. 2000, 2, 3921.
1131. Shimizu, M.; Sahara, T.; Hayakawa, R. Chem. Lett. 2001, 792.
1132. Knettle, B.W.; Flowers II, R.A. Org. Lett. 2001, 3, 2321.
1133. Samec, J.S.M.; Bäckvall, J.-E. Chem. Eur. J. 2002, 8, 2955.
1134. Moghaddam, F.M.; Khakshoor, O.; Ghaffarzadeh, M. J. Chem. Res. (S) 2001, 525.
1135. Davies, I.W.; Taylor, M.; Marcoux, J.-F.; Matty, L.; Wu, J.; Hughes, D.; Reider, P.J. Tetrahedron Lett. 2000, 41, 8021.
1136. See Bolm, C.; Felder, M. Synlett 1994, 655; Williams, D.R.; Osterhout, M.H.; Reddy, J.P. Tetrahedron Lett. 1993, 34, 3271.
1137. Kawase, M.; Kikugawa, Y. J. Chem. Soc. Perkin Trans. 1 1979, 643.
1138. See Hutchins, R.O.; Natale, N.R. Org. Prep. Proced. Int. 1979, 11, 201; Lane, C.F. Synthesis 1975, 135.
1139. Ueda, M.; Miyabe, H.; Namba, M.; Nakabayashi, T.; Naito, T. Tetrahedron Lett. 2002, 43, 4369.
1140. Gowda, S.; Abiraj, K.; Gowda, D.C. Tetrahedron Lett. 2002, 43, 1329.
1141. See Denmark, S.E.; Nakajima, N.; Nicaise, O. J.-C. J. Am. Chem. Soc. 1994, 116, 8797; Fuller, J.C.; Belisle, C.M.; Goralski, C.T.; Singaram, B. Tetrahedron Lett. 1994, 35, 5389. For a review of asymmetric reductions involving the C=N unit, see Zhu, Q.-C.; Hutchins, R.O. Org. Prep. Proceed. Int. 1994, 26, 193.
1142. Hou, G.; Gosselin, F.; Li, W.; McWilliams, J.C.; Sun, Y.; Weisel, M.; O'Shea, P.D.; Chen, C.-y.; Davies, I.W.; Zhang, X. J. Am. Chem. Soc. 2009, 131, 9882.
1143. See Li, C.; Wang, C.; Villa-Marcos, B.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 14450; Vargas, S.; Rubio, M.; Suárez, A.; Pizzano, A. Tetrahedron Lett. 2005, 46, 2049.
1144. Nolin, K.A.; Ahn, R.W.; Toste, F.D. J. Am. Chem. Soc. 2005, 127, 12462.
1145. Li, C.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 13208.
1146. Abe, H.; Amii, H.; Uneyama, K. Org. Lett. 2001, 3, 313.
1147. Magee, M.P.; Norton, J.R. J. Am. Chem. Soc. 2001, 123, 1778.
1148. Dunsmore, C.J.; Carr, R.; Fleming, T.; Turner, N.J. J. Am. Chem. Soc. 2006, 128, 2224.
1149. Li, G.; Liang, Y.; Antilla, J.C. J. Am. Chem. Soc. 2007, 129, 5830.
1150. Tararov, V.I.; Kadyrov, R.; Riermeier, T.H.; Holz, J.; Börner, A. Tetrahedron Lett. 2000, 41, 2351.
1151. Choi, Y.K.; Kim, M.J.; Ahn, Y.; Kim, M.-J. Org. Lett. 2001, 3, 4099.
1152. Rueping, M.; Sugiono, E.; Azap, C.; Theissmann, T.; Bolte, M. Org. Lett. 2005, 7, 3781.
1153. Hayashi, T.; Ishigedani, M. Tetrahedron 2001, 57, 2589.
1154. Lipshutz, B.H.; Shimizu, H. Angew. Chem. Int. Ed. 2004, 43, 2228.
1155. Hansen, M.C.; Buchwald, S.L. Org. Lett. 2000, 2, 713.
1156. For a review, see Guizzetti, S.; Benaglia, M. Eur. J. Org. Chem. 2010, 5529. See Onomura, O.; Kouchi, Y.; Iwasaki, F.; Matsumura, Y. Tetrahedron Lett. 2006, 47, 3751; Malkov, A.V.; Ston![]() ius, S.; MacDougall, K.N.; Mariani, A.; McGeoch, G.D.; Ko
ius, S.; MacDougall, K.N.; Mariani, A.; McGeoch, G.D.; Ko![]() ovský, P. Tetrahedron 2006, 62, 264; Wang, C.; Wu, X.; Zhou, L.; Sun, J. Chemistry: European J. 2008, 14, 8789; Malkov, A.V.; Figlus, M.; Cooke, G.; Caldwell, S.T.; Rabani, G.; Prestly, M.R.; Ko
ovský, P. Tetrahedron 2006, 62, 264; Wang, C.; Wu, X.; Zhou, L.; Sun, J. Chemistry: European J. 2008, 14, 8789; Malkov, A.V.; Figlus, M.; Cooke, G.; Caldwell, S.T.; Rabani, G.; Prestly, M.R.; Ko![]() ovský, P. Org. Biomol. Chem. 2009, 7, 1878; Malkov, A.V.; Vranková, K.; Sigerson, R.C.; Ston
ovský, P. Org. Biomol. Chem. 2009, 7, 1878; Malkov, A.V.; Vranková, K.; Sigerson, R.C.; Ston![]() ius, S.; Ko
ius, S.; Ko![]() ovský, P. Tetrahedron 2009, 65, 9481.
ovský, P. Tetrahedron 2009, 65, 9481.
1157. Vaijayanthi, T.; Chadha, A. Tetrahedron Asymmetry 2008, 19, 93.
1158. Chu, Y.; Shan, Z.; Liu, D.; Sun, N. J. Org. Chem. 2006, 71, 3998; Huang, K.; Merced, F.G.; Ortiz-Marciales, M.; Meléndez, H.J.; Correa, W.; De Jesús, M. J. Org. Chem. 2008, 73, 4017.
1159. Howell, H.G. Synth. Commun. 1983, 13, 635.
1160. Park, H.S.; Lee, I.S.; Kim, Y.H. Chem. Commun. 1996, 1805.
1161. See Rabinovitz, M. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 307–340; Enthaler, S.; Addis, D.; Junge, K.; Erre, G.; Beller, M. Chemistry: European J. 2008, 14, 9491. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 875–878.
1162. See Brown, H.C.; Choi, Y.M.; Narasimhan, S. Synthesis 1981, 605.
1163. Satoh, T.; Suzuki, S. Tetrahedron Lett. 1969, 4555. For a discussion of the mechanism, see Heinzman, S.W.; Ganem, B. J. Am. Chem. Soc. 1982, 104, 6801.
1164. Khurana, J.M.; Kukreja, G. Synth. Commun. 2002, 32, 1265.
1165. Egli, R.A. Helv. Chim. Acta 1970, 53, 47.
1166. Thomas, S.; Collins, C.J.; Cuzens, J.R.; Spieciarich, D.; Goralski, C.T.; Singaram, B. J. Org. Chem. 2001, 66, 1999.
1167. See Reguillo, R.; Grellier, M.; Vautravers, N.; Vendier, L.; Sabo-Etienne, S. J. Am. Chem. Soc. 2010, 132, 7854.
1168. See Galán, A.; de Mendoza, J.; Prados, P.; Rojo, J.; Echavarren, A.M. J. Org. Chem. 1991, 56, 452.
1169. See Gould, F.E.; Johnson, G.S.; Ferris, A.F. J. Org. Chem. 1960, 25, 1658.
1170. For example, see Freifelder, M. J. Am. Chem. Soc. 1960, 82, 2386.
1171. Tanaka, K.; Nagasawa, M.; Kasuga, Y.; Sakamura, H.; Takuma, Y.; Iwatani, K. Tetrahedron Lett. 1999, 40, 5885.
1172. Takamizawa, S.; Wakasa, N.; Fuchikami, T. Synlett 2001, 1623.
1173. Borch, R.F. Chem. Commun. 1968, 442.
1174. Rabinovitz, M. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, p. 307. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1271–1272.
1175. Zil'berman, E.N.; Pyryalova, P.S. J. Gen. Chem. USSR 1963, 33, 3348.
1176. Xi, F.; Kamal, F.; Schenerman, M.A. Tetrahedron Lett. 2002, 43, 1395.
1177. Brown, H.C.; Shoaf, C.J. J. Am. Chem. Soc. 1964, 86, 1079. For a review of reductions with this and related reagents, see Málek, J. Org. React. 1988, 36, 249, see pp. 287–289, 438–448.
1178. Cha, J.S.; Lee, S.E.; Lee, H.S. Org. Prep. Proceed. Int. 1992, 24, 331. Also see, Cha, J.S.; Jeoung, M.K.; Kim, J.M.; Kwon, O.O.; Lee, J.C. Org. Prep. Proceed. Int. 1994, 26, 583.
1179. Marshall, J.A.; Andersen, N.H.; Schlicher, J.W. J. Org. Chem. 1970, 35, 858.
1180. See Ioffe, S.L.; Tartakovskii, V.A.; Novikov, S.S. Russ. Chem. Rev. 1966, 35, 19.
1181. For reviews, see Rylander, P.N. Hydrogenation Methods, Academic Press, NY, 1985, pp. 104–116, Catalytic Hydrogenation over Platinum Metals, Academic Press, NY, 1967, pp. 168–202. See Deshpande, R.M.; Mahajan, A.N.; Diwakar, M.M.; Ozarde, P.S.; Chaudhari, R.V. J. Org. Chem. 2004, 69, 4835.
1182. Takasaki, M.; Motoyama, Y.; Higashi, K.; Yoon, S.-H.; Mochida, I.; Nagashima, H. Org. Lett. 2008, 10, 1601. See also Gelder, E.A.; Jackson, S.D.; Lok, C.M. Chem. Commun. 2005, 522; Chen, Y.; Wang, C.; Liu, H.; Qiu, J.; Bao, X. Chem. Commun. 2005, 5298.
1183. Soltani, O.; Ariger, M.A.; Carreira, E.M. Org. Lett. 2009, 11, 4196.
1184. Banik, B.K.; Suhendra, M.; Banik, I.; Becker, F.F. Synth. Commun. 2000, 30, 3745.
1185. Lee, J.G.; Choi, K.I.; Koh, H.Y.; Kim, Y.; Kang, Y.; Cho, Y.S. Synthesis 2001, 81.
1186. Kim, B.H.; Han, R.; Piao, F.; Jun, Y.M.; Baik, W.; Lee, B.M. Tetrahedron Lett. 2003, 44, 77.
1187. Basu, M.K.; Becker, F.F.; Banik, B.K. Tetrahedron Lett. 2000, 41, 5603.
1188. Ankner, T.; Hilmersson, G. Tetrahedron Lett. 2007, 48, 5707.
1189. Fitch, R.W.; Luzzio, F.A. Tetrahedron Lett. 1994, 35, 6013.
1190. Rai, G.; Jeong, J.M.; Lee, Y.-S.; Kim, H.W.; Lee, D.S.; Chung, J.-K.; Lee, M.C. Tetrahedron Lett. 2005, 46, 3987.
1191. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 821–828.
1192. Brinkman, H.R. Synth. Commun. 1996, 26, 973.
1193. Entwistle, I.D.; Jackson, A.E.; Johnstone, R.A.W.; Telford, R.P. J. Chem. Soc. Perkin Trans. 1 1977, 443. See also, Terpko, M.O.; Heck, R.F. J. Org. Chem. 1980, 45, 4992; Babler, J.H.; Sarussi, S.J. Synth. Commun. 1981, 11, 925.
1194. Gowda, D.C.; Gowda, A.S.P.; Baba, A.R.; Gowda, S. Synth. Commun. 2000, 30, 2889.
1195. For a review of the Zinin reduction, see Porter, H.K. Org. React. 1973, 20, 455.
1196. Ram, S.; Ehrenkaufer, R.E. Tetrahedron Lett. 1984, 25, 3415; Abiraj, K.; Srinivasa, G.R.; Gowda, D.C. Synth. Commun. 2005, 35, 223.
1197. Barrett, A.G.M.; Spilling, C.D. Tetrahedron Lett. 1988, 29, 5733.
1198. See He, Y.; Zhao, H.; Pan, X.; Wang, S. Synth. Commun. 1989, 19, 3047 and references cited therein.
1199. Chary, K.P.; Ram, S.R.; Iyengar, D.S. Synlett 2000, 683.
1200. Yoon, N.M.; Choi, J. Synlett 1993, 135.
1201. Severin, T.; Schmitz, R. Chem. Ber. 1962, 95, 1417; Severin, T.; Adam, M. Chem. Ber. 1963, 96, 448.
1202. Kaplan, L.A. J. Am. Chem. Soc. 1964, 86, 740. See also, Swanwick, M.G.; Waters, W.A. Chem. Commun. 1970, 63.
1203. See Ono, A.; Terasaki, S.; Tsuruoka, Y. Chem. Ind. (London) 1983, 477; Ayyangar, N.R.; Kalkote, U.R.; Lugad, A.G.; Nikrad, P.V.; Sharma, V.K. Bull. Chem. Soc. Jpn. 1983, 56, 3159.
1204. Baik, W.; Han, J.L.; Lee, K.C.; Lee, N.H.; Kim, B.H.; Hahn, J.-T. Tetrahedron Lett. 1994, 35, 3965.
1205. Meshram, H.M.; Ganesh, Y.S.S.; Sekhar, K.C.; Yadav, J.S. Synlett 2000, 993.
1206. Vass, A.; Dudás, J.; Tóth, J.; Varma, R.S. Tetrahedron Lett. 2001, 42, 5347.
1207. Gowda, S.; Gowda, D.C. Tetrahedron 2002, 58, 2211.
1208. Kumar, J.S.D.; Ho, M.M.; Toyokuni, T. Tetrahedron Lett. 2001, 42, 5601.
1209. House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, p. 211.
1210. Petersen, W.C.; Letsinger, R.L. Tetrahedron Lett. 1971, 2197; Vink, J.A.J.; Cornelisse, J.; Havinga, E. Recl. Trav. Chim. Pays-Bas 1971, 90, 1333.
1211. Lamoureux, C.; Moinet, C. Bull. Soc. Chim. Fr. 1988, 59.
1212. Bieber, L.W.; da Costa, R.C.; da Silva, M.F. Tetahedron Lett. 2000, 41, 4827.
1213. Kang, K.H.; Choi, K.I.; Koh, H.Y.; Kim, Y.; Chung, B.Y.; Cho, Y.S. Synth. Commun. 2001, 31, 2277.
1214. See Entwistle, I.D.; Gilkerson, T.; Johnstone, R.A.W.; Telford, R.P. Tetrahedron 1978, 34, 213.
1215. Kende, A.S.; Mendoza, J.S. Tetrahedron Lett. 1991, 32, 1699.
1216. Oxley, P.W.; Adger, B.M.; Sasse, M.J.; Forth, M.A. Org. Synth. 67, 187.
1217. Ren, P.D-D.; Pan, X.-W.; Jin, Q.-H.; Yao, Z.-P. Synth. Commun. 1997, 27, 3497.
1218. Feuer, H.; Bartlett, R.S.; Vincent, Jr., B.F.; Anderson, R.S. J. Org. Chem. 1965, 31, 2880.
1219. See Fry, A.J. Synthetic Organic Electrochemistry, 2nd ed., Wiley, NY, 1989, pp. 188–198; Lund, H. in Baizer, M.M.; Lund, H. Organic Electrochemistry, Marcel Dekker, NY, 1983, pp. 285–313.
1220. Yu, Y.; Srogl, J.; Liebeskind, L.S. Org. Lett. 2004, 6, 2631.
1221. Schwartz, M.A.; Gu, J.; Hu, X. Tetrahedron Lett. 1992, 33, 1687.
1222. Cicchi, S.; Bonanni, M.; Cardona, F.; Revuelta, J.; Goti, A. Org. Lett. 2003, 5, 1773.
1223. See Lunn, G.; Sansone, E.B.; Keefer, L.K. J. Org. Chem. 1984, 49, 3470.
1224. Felkin, H. C.R. Acad. Sci. 1950, 230, 304.
1225. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 845–846.
1226. Feuer, H.; Braunstein, D.M. J. Org. Chem. 1969, 34, 1817.
1227. Leeds, J.P.; Kirst, H.A. Synth. Commun. 1988, 18, 777.
1228. Chandrasekhar, S.; Reddy, M.V.; Chandraiah, L. Synlett 2000, 1351.
1229. See Sugden, J.K.; Patel, J.J.B. Chem. Ind. (London) 1972, 683.
1230. See Rylander, P.N. Catalytic Hydrogenation over Platinum Metals, Academic Press, NY, 1967, pp. 139–159.
1231. Harrison, J.R.; Moody, C.J.; Pitts, M.R. Synlett 2000, 1601.
1232. Gibbs, D.E.; Barnes, D. Tetrahedron Lett. 1990, 31, 5555.
1233. Brunner, H.; Becker, R.; Gauder, S. Organometallics 1986, 5, 739; Takei, I.; Nishibayashi, Y.; Ishii, Y.; Mizobe, Y.; Uemura, S.; Hidai, M. Chem. Commun. 2001, 2360.
1234. Fontaine, E.; Namane, C.; Meneyrol, J.; Geslin, M.; Serva, L.; Russey, E.; Tissandié, S.; Maftouh, M.; Roger, P. Tetrahedron Asymmetry 2001, 12, 2185; Huang, X.; Ortiz-Marciales, M.; Huang, K.; Stepanenko, V.; Merced, F.G.; Ayala, A.M.; Correa, W.; De Jesús, M. Org. Lett. 2007, 9, 1793.
1235. Sasatani, S.; Miyazaki, T.; Maruoka, K.; Yamamoto, H. Tetrahedron Lett. 1983, 24, 4711.
1236. For a review, see Kotera, K.; Kitahonoki, K. Org. Prep. Proced. 1969, 1, 305. See Tatchell, A.R. J. Chem. Soc. Perkin Trans. 1 1974, 1294; Ferrero, L.; Rouillard, M.; Decouzon, M.; Azzaro, M. Tetrahedron Lett. 1974, 131; Diab, Y.; Laurent, A.; Mison, P. Tetrahedron Lett. 1974, 1605.
1237. Barton, D.H.R.; Motherwell, W.B.; Simon, E.S.; Zard, S.Z. J. Chem. Soc. Chem. Commun. 1984, 337.
1238. Akazome, M.; Tsuji, Y.; Watanabe, Y. Chem. Lett. 1990, 635.
1239. Boruah, M.; Konwar, D. Synlett 2001, 795.
1240. Johnson, K.; Degering, E.F. J. Am. Chem. Soc. 1939, 61, 3194.
1241. Albanese, D.; Landini, D.; Penso, M. Synthesis 1990, 333.
1242. Hanson, J.R. Synthesis 1974, 1, pp. 7–8.
1243. Zeilstra, J.J.; Engberts, J.B.F.N. Synthesis 1974, 49.
1244. Yadav, J.S.; Subba Reddy, B.V.; Srinivas, R.; Ramalingam, T. Synlett 2000, 1447.
1245. See Kabalka, G.W.; Pace, E.D.; Wadgaonkar, P.P. Synth. Commun. 1990, 20, 2453; Sera, A.; Yamauchi, H.; Yamada, H.; Itoh, K. Synlett 1990, 477.
1246. El Kaim, L.; Gacon, A. Tetrahedron Lett. 1997, 38, 3391.
1247. For a review, see Scriven, E.F.V.; Turnbull, K. Chem. Rev. 1988, 88, 297, see pp. 321–327. For lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 815–820; Rolla, F. J. Org. Chem. 1982, 47, 4327.
1248. Ram, S.R.; Chary, K.P.; Iyengar, D.S. Synth. Commun. 2000, 30, 4495.
1249. Fringuelli, F.; Pizzo, F.; Vaccaro, L. Synthesis 2000, 646.
1250. Chary, K.P.; Ram, S.R.; Salahuddin, S.; Iyengar, D.S. Synth. Commun. 2000, 30, 3559.
1251. Maiti, S.N.; Spevak, P.; Narender Reddy, A.V. Synth. Commun. 1988, 18, 1201.
1252. Wu, H.; Chen, R.; Zhang, Y. Synth. Commun. 2002, 32, 189.
1253. Huang, Y.; Zhang, Y.; Wang, Y. Tetrahedron Lett. 1997, 38, 1065.
1254. Bartoli, G.; Di Antonio, G.; Giovannini, R.; Giuli, S.; Lanari, S.; Paoletti, M.; Marcantoni, E. J. Org. Chem. 2008, 73, 1919.
1255. Lin, W.; Zhang, X.; He, Z.; Jin, Y.; Gong, L.; Mi, A. Synth. Commun. 2002, 32, 3279.
1256. Kamal, A.; Damayanthi, Y.; Reddy, B.S.N.; Lakminarayana, B.; Reddy, B.S.P. Chem. Commun. 1997, 1015; Baruah, M.; Boruah, A.; Prajapati, D.; Sandhu, J.S. Synlett 1996, 1193.
1257. Reddy, G.V.; Rao, G.V.; Iyengar, D.S. Tetrahedron Lett. 1999, 40, 3937.
1258. Benati, L.; Bencivenni, G.; Leardini, R.; Minozzi, M.; Nanni, D.; Scialpi, R.; Spagnolo, P.; Zanardi, G. J. Org. Chem. 2006, 71, 5822.
1259. Staudinger, H.; Meyer, J. Helv. Chim. Acta 1919, 2, 635. See Golobov, Y.G.; Zhmurova, I.N.; Kasukhin, L.F. Tetrahedron 1981, 37, 437; Tian, W.Q.; Wang, Y.A. J. Org. Chem. 2004, 69, 4299; Lin, F.L.; Hoyt, H.M.; van Halbeek, H.; Bergman, R.G.; Bertozzi, C.R. J. Am. Chem. Soc. 2005, 127, 2686. For a modification that leads to β-lactams, see Jiao, L.; Liang, Y.; Xu, J. J. Am. Chem. Soc. 2006, 128, 6060.
1260. Kato, H.; Ohmori, K.; Suzuki, K. Synlett 2001, 1003.
1261. Hu, L.; Wang, Y.; Li, B.; Du, D.-M.; Xu, J. Tetrahedron 2007, 63, 9387.
1262. Zhang, Y.-R.; He, L.; Wu, X.; Shao, P.-L.; Ye, S. Org. Lett. 2008, 10, 277.
1263. See Koziara, A.; Osowska-Pacewicka, K.; Zawadzki, S.; Zwierzak, A. 1987, 487. The Reactions 10-48, 10-43, and 19-50 have also been accomplished in one laboratory step: Koziara, A. J. Chem. Res. (S) 1989, 296.
1264. Reagen, M.T.; Nickon, A. J. Am. Chem. Soc. 1968, 90, 4096.
1265. Lee, Y.; Closson, W.D. Tetrahedron Lett. 1974, 381.
1266. See Newbold, B.T. in Patai, S. The Chemistry of the Hydrazo, Azo, and Azoxy Groups, pt. 2, Wiley, NY, 1975, pp. 601, 604–614.
1267. See Ioffe, B.V.; Sergeeva, Z.I.; Dumpis,Yu.Ya. J. Org. Chem. USSR 1969, 5, 1683.
1268. Huisgen, R.; Lux, R. Chem. Ber. 1960, 93, 540.
1269. Enders, D.; Hassel, T.; Pieter, R.; Renger, B.; Seebach, D. Synthesis 1976, 548.
1270. Jeyaraman, R.; Ravindran, T. Tetrahedron Lett. 1990, 31, 2787.
1271. Kano, S.; Tanaka, Y.; Sugino, E.; Shibuya, S.; Hibino, S. Synthesis 1980, 741.
1272. Fridman, A.L.; Mukhametshin, F.M.; Novikov, S.S. Russ. Chem. Rev. 1971, 40, 34, pp. 41–42.
1273. Kindler, K.; Lührs, K. Chem. Ber. 1966, 99, 227; Liebigs Ann. Chem. 1967, 707, 26.
1274. See also, Brown, G.R.; Foubister, A.J. Synthesis 1982, 1036.
1275. Khurana, J.M.; Singh, S. J. Chem. Soc., Perkin Trans. 1 1999, 1893.
1276. Poliskie, G.M.; Mader, M.M.; van Well, R. Tetrahedron Lett. 1999, 40, 589.
1277. See Hudlicky, M. Reductions in Organic Chemistry, Ellis Horwood, Chichester, 1984, pp. 62–67, 181; Pinder, A.R. Synthesis 1980, 425. For a list of reagents, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 29–39.
1278. See Pizey, J.S. Synthetic reagents, Vol. 1, Wiley, NY, 1974, pp. 101–294; Seyden-Penne, J. Reductions by the Alumino- and Borohydrides, VCH, NY, 1991; Hajós, A. Complex Hydrides, Elsevier, NY, 1979.
1279. Krishnamurthy, S.; Brown, H.C. J. Org. Chem. 1982, 47, 276.
1280. Krishnamurthy, S.; Brown, H.C. J. Org. Chem. 1980, 45, 849; 1983, 48, 3085.
1281. Masamune, S.; Bates, G.S.; Georghiou, P.E. J. Am. Chem. Soc. 1974, 96, 3686.
1282. Hutchins, R.O.; Bertsch, R.J.; Hoke, D. J. Org. Chem. 1971, 36, 1568.
1283. Hutchins, R.O.; Kandasamy, D.; Dux III, F.; Maryanoff, C.A.; Rotstein, D.; Goldsmith, B.; Burgoyne, W.; Cistone, F.; Dalessandro, J.; Puglis, J. J. Org. Chem. 1978, 43, 2259.
1284. See Bergbreiter, D.E.; Blanton, J.R. J. Org. Chem. 1987, 52, 472.
1285. Inoue, K.; Sawada, A.; Shibata, I.; Baba, A. J. Am. Chem. Soc. 2002, 124, 906.
1286. Yoon, N.M.; Lee, H.J.; Ahn, J.H.; Choi, J. J. Org. Chem. 1994, 59, 4687.
1287. See Kirwan, J.N.; Roberts, B.P.; Willis, C.R. J. Chem. Soc. Perkin Trans. 1 1991, 103; Hudlicky, M. Reductions in Organic Chemistry, Ellis Horwood, Chichester, 1984, pp. 62–67, 181; Pinder, A.R. Synthesis 1980, 425. For a list of reagents, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 29–39.
1288. Doyle, M.P.; McOsker, C.C.; West, C.T. J. Org. Chem. 1976, 41, 1393; Parnes, Z.N.; Romanova, V.S.; Vol'pin, M.E. J. Org. Chem. USSR 1988, 24, 254.
1289. Yang, J.; Brookhart, M. J. Am. Chem. Soc. 2007, 129, 12656.
1290. Miura, K.; Tomita, M.; Yamada, Y.; Hosomi, A. J. Org. Chem. 2007, 72, 787.
1291. Hirao, T.; Kohno, S.; Ohshiro, Y.; Agawa, T. Bull. Chem. Soc. Jpn. 1983, 56, 1881.
1292. Downie, I.M.; Lee, J.B. Tetrahedron Lett. 1968, 4951.
1293. Seyferth, D.; Yamazaki, H.; Alleston, D.L. J. Org. Chem. 1963, 28, 703. For a novel trialkyltin hydride, see Gastaldi, S.; Stein, D. Tetrahedron Lett. 2002, 43, 4309.
1294. See Neumann, W.P. Synthesis 1987, 665; Kuivila, H.G. Synthesis 1970, 499, Acc. Chem. Res. 1968, 1, 299; Uenishi, J.; Kawahama, R.; Shiga, Y.; Yonemitsu, O.; Tsuji, J. Tetrahedron Lett. 1996, 37, 6759.
1295. Hayashi, N.; Shibata, I.; Baba, A. Org. Lett. 2004, 6, 4981.
1296. Light, J.; Breslow, R. Tetrahedron Lett. 1990, 31, 2957.
1297. Mebane, R.C.; Grimes, K.D.; Jenkins, S.R.; Deardorff, J.D.; Gross, B.H. Synth. Commun. 2002, 32, 2049.
1298. Wang, Y.-C.; Yan, T.-H. Chem. Commun. 2000, 545.
1299. Rylander, P.N. Hydrogenation Methods, Academic Press, NY, 1985; Kantam, M.L.; Rahman, A.; Bandyopadhyay, T.; Haritha, Y. Synth. Commun. 1999, 29, 691. See Ye, P.; Gellman, A.J. J. Am. Chem. Soc. 2008, 130, 8518.
1300. See Marquié, J.; Laporterie, A.; Dubac, J.; Roques, N. Synlett 2001, 493.
1301. Ohkuma, T.; Tsutsumi, K.; Utsumi, N.; Arai, N.; Noyori, R.; Murata, K. Org. Lett. 2007, 9, 255.
1302. See Fieser, L.F.; Sachs, D.H. J. Org. Chem. 1964, 29, 1113; Berkowitz, D.B. Synthesis 1990, 649.
1303. See Gassman, P.G.; Aue, D.H.; Patton, D.S. J. Am. Chem. Soc. 1968, 90, 7271; Gassman, P.G.; Marshall, J.L. Org. Synth. V, 424.
1304. Li, J.; Ye, D.; Liu, H.; Luo, X.; Jiang, H. Synth. Commun. 2008, 38, 567.
1305. Khurana, J.M.; Kandpal, B.M.; Kukreja, G.; Sharma, P. Can. J. Chem. 2006, 84, 1019.
1306. See Claesson, A.; Olsson, L. J. Am. Chem. Soc. 1979, 101, 7302.
1307. See Noyori, R.; Hayakawa, Y. Org. React. 1983, 29, 163.
1308. Lee, S.H.; Jung, Y.J.; Cho, Y.J.; Yoon, C.-O.M.; Hwang, H.-J.; Yoon, C.M. Synth. Commun. 2001, 31, 2251.
1309. Ren, P.-D.; Hin, Q.-H.; Yao, Z.-P. Synth. Commun. 1997, 27, 2577.
1310. Park, L.; Keum, G.; Kang, S.B.; Kim, K.S.; Kim, Y. J. Chem. Soc. Perkin Trans. 1 2000, 4462.
1311. Oriyama, T.; Mukaiyama, T. Chem. Lett. 1984, 2069.
1312. Ranu, B.C.; Chattopadhyay, K.; Jana, R. Tetrahedron 2007, 63, 155.
1313. Lee, S.H.; Cho, M.Y.; Nam, M.H.; Park, Y.S.; Yoo, B.W.; Lee, C.-W.; Yoon, C.M. Synth. Commun. 2005, 35, 1335.
1314. Aelterman, W.; Eeckhaut, A.; De Kimpe, N. Synlett 2000, 1283.
1315. Kim, S.; Ko, J.S. Synth. Commun. 1985, 15, 603.
1316. Hutchins, R.O.; Kandasamy, D.; Maryanoff, C.A.; Masilamani, D.; Maryanoff, B.E. J. Org. Chem. 1977, 42, 82.
1317. See Ohsawa, T.; Takagaki, T.; Haneda, A.; Oishi, T. Tetrahedron Lett. 1981, 22, 2583. See also, Brandänge, S.; Dahlman, O.; Ölund, J. Acta Chem. Scand. Ser. B 1983, 37, 141.
1318. Bryce-Smith, D.; Wakefield, B.J.; Blues, E.T. Proc. Chem. Soc. 1963, 219.
1319. See Curran, D.P. Synthesis 1988, 417, 489.
1320. Abbas S.; Hayes, C.J.; Worden, S. Tetrahedron Lett. 2000, 41, 3215.
1321. Ranu, B.C.; Samanta, S.; Guchhait, S.K. J. Org. Chem. 2001, 66, 4102.
1322. Kdota, I.; Ueno, H.; Ohno, A.; Yamamoto, Y. Tetrhaedron Lett. 2003, 44, 8645.
1323. See Kraus, W.; Chassin, C. Tetrahedron Lett. 1970, 1443. See Omoto, M.; Kato, N.; Sogon, T.; Mori, A. Tetrahedron Lett. 2001, 42, 939.
1324. Ashby, E.C.; Deshpande, A.K. J. Org. Chem. 1994, 59, 3798. See however Park, S.; Chung, S.; Newcomb, M. J. Org. Chem. 1987, 52, 3275.
1325. Chung, S. J. Org. Chem. 1980, 45, 3513.
1326. Hatem, J.; Waegell, B. Tetrahedron 1990, 46, 2789.
1327. Bell, H.M.; Brown, H.C. J. Am. Chem. Soc. 1966, 88, 1473.
1328. Matsumura, S.; Tokura, N. Tetrahedron Lett. 1969, 363.
1329. Hutchins, R.O.; Bertsch, R.J.; Hoke, D. J. Org. Chem. 1971, 36, 1568.
1330. For an exception, see Carey, F.A.; Tramper, H.S. Tetrahedron Lett. 1969, 1645.
1331. Tanner, D.D.; Singh, H.K. J. Org. Chem. 1986, 51, 5182.
1332. For a review, see Müller, P. in Patai, S. The Chemistry of Functional Groups, Supplement E, pt. 1, Wiley, NY, 1980, pp. 515–522.
1333. See Rylander, P.N. Hydrogenation Methods, Academic Prss, NY, 1985, pp. 157–163, Catalytic Hydrogenation over Platinum Metals, Academic Press, NY, 1967, pp. 449–468. For a review of the stereochemistry of hydrogenolysis, see Klabunovskii, E.I. Russ. Chem. Rev. 1966, 35, 546.
1334. Avendaño, C.; de Diego, C.; Elguero, J. Monatsh. Chem. 1990, 121, 649.
1335. See Gribble, G.W.; Nutaitis, C.F. Org. Prep. Proced. Int. 1985, 17, 317. Also see, Nutaitis, C.F.; Bernardo, J.E. Synth. Commun. 1990, 20, 487.
1336. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 44–46.
1337. Cain, G.A.; Holler, E.R. Chem. Commun. 2001, 1168.
1338. Orfanopoulos, M.; Smonou, I. Synth. Commun. 1988, 18, 833; Smonou, I.; Orfanopoulos, M. Tetrahedron Lett. 1988, 29, 5793; Wustrow, D.J.; Smith, III, W.J.; Wise, L.D. Tetrahedron Lett. 1994, 35, 61.
1339. Kusuda, K.; Inanaga, J.; Yamaguchi, M. Tetrahedron Lett. 1989, 30, 2945.
1340. Yasuda, M.; Onishi, Y.; Ueba, M.; Miyai, T.; Baba, A. J. Org. Chem. 2001, 66, 7741.
1341. Krafft, M.E.; Crooks, III, W.J. J. Org. Chem. 1988, 53, 432. For another catalyst, see Parnes, Z.N.; Shaapuni, D.Kh.; Kalinkin, M.I.; Kursanov, D.N. Bull. Acad. Sci. USSR Div. Chem. Sci. 1974, 23, 1592.
1342. See Elphimoff-Felkin, I.; Sarda, P. Org. Synth. VI, 769; Tetrahedron 1977, 33, 511. For another reagent, see Lee, J.; Alper, H. Tetrahedron Lett. 1990, 31, 4101.
1343. Ho, T.L.; Wong, C.M. Synthesis 1975, 161.
1344. Ho, T.L. Synth. Commun. 1979, 9, 665.
1345. Corey, E.J.; Achiwa, K. J. Org. Chem. 1969, 34, 3667.
1346. Morita, T.; Okamoto, Y.; Sakurai, H. Synthesis 1981, 32.
1347. See Garbisch, Jr., E.W.; Schreader, L.; Frankel, J.J. J. Am. Chem. Soc. 1967, 89, 4233; Mitsui, S.; Imaizumi, S.; Esashi, Y. Bull. Chem. Soc. Jpn. 1970, 43, 2143.
1348. Mitsui, S.; Imaizumi, S.; Esashi, Y. Bull. Chem. Soc. Jpn. 1970, 43, 2143.
1349. Shukun, H.; Yougun, S.; Jindong, Z.; Jian, S. J. Org. Chem. 2001, 66, 4487.
1350. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 44–52ff.
1351. Shuikin, N.I.; Erivanskaya, L.A. Russ. Chem. Rev. 1960, 29, 309, see pp. 313–315. See also, Bagnell, L.J.; Jeffery, E.A. Aust. J. Chem. 1981, 34, 697.
1352. Cacchi, S.; Ciattini. P.G.; Morera, E.; Ortar, G. Tetrahedron Lett. 1986, 27, 5541. See also, Cabri, W.; De Bernardinis, S.; Francalanci, F.; Penco, S. J. Org. Chem. 1990, 55, 350.
1353. Wang, F.; Chiba, K.; Tada, M. J. Chem. Soc. Perkin Trans. 1 1992, 1897.
1354. Welch, S.C.; Walters, M.E. J. Org. Chem. 1978, 43, 4797. See also, Rossi, R.A.; Bunnett, J.F. J. Org. Chem. 1973, 38, 2314.
1355. Sajiki, H.; Mori, A.; Mizusaki, T.; Ikawa, T.; Maegawa, T.; Hirota, K. Org. Lett. 2006, 8, 987.
1356. Mori, A.; Mizusaki, T.; Ikawa, T.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Tetrahedron 2007, 63, 1270.
1357. Ranu, B.C.; Bhar, S. Org. Prep. Proceed. Int. 1996, 28, 371.
1358. Bailey, W.J.; Marktscheffel, F. J. Org. Chem. 1960, 25, 1797.
1359. Krishnamurthy, S.; Brown, H.C. J. Org. Chem. 1979, 44, 3678.
1360. For a review of ether reduction, see Müller, P. in Patai, S. The Chemisry of Functional Groups, Supplement E, pt. 1, Wiley, NY, 1980, pp. 522–528.
1361. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1013–1019.
1362. Rao, G.V.; Reddy, D.S.; Mohan, G.H.; Iyengar, D.S. Synth. Commun. 2000, 30, 3565.
1363. Tweedie, V.L.; Barron B.G. J. Org. Chem. 1960, 25, 2023. See also, Hutchins, R.O.; Learn, K. J. Org. Chem. 1982, 47, 4380.
1364. Shi, L.; Xia, W.J.; Zhang, F.M.; Tu, Y.Q. Synlett 2002, 1505. See also, Olivero, S.; Duñach, E. Tetrahedron Lett. 1997, 38, 6193.
1365. Majetich, G.; Zhang, Y.; Wheless, K. Tetrahedron Lett. 1994, 35, 8727.
1366. Lautens, M.; Chiu, P.; Ma, S.; Rovis, T. J. Am. Chem. Soc. 1995, 117, 532.
1367. Mikami, K.; Yamaoka, M.; Yoshida, A. Synlett 1998, 607.
1368. See Rerick, M.N. in Augustine, R.L. Reduction, Marcel Dekker, NY, 1968, pp. 1–94.
1369. Eliel, E.L.; Badding, V.G.; Rerick, M.N. J. Am. Chem. Soc. 1962, 84, 2371.
1370. Ashby, E.C.; Prather, J. J. Am. Chem. Soc. 1966, 88, 729; Diner, U.E.; Davis, H.A.; Brown, R.K. Can. J. Chem. 1967, 45, 207.
1371. See Takano, S.; Akiyama, M.; Sato, S.; Ogasawara, K. Chem. Lett. 1983, 1593.
1372. See Hojo, M.; Ushioda, N.; Hosomi, A. Tetrahedron Lett. 2004, 45, 4499; Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 931–942.
1373. Srikrishna, A.; Viswajanani, R. Synlett 1995, 95.
1374. For a list of substrate types and reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 46–52.
1375. See Goodenough, K.M.; Moran, W.J.; Raubo, P.; Harrity, J.P.A. J. Org. Chem. 2005, 70, 207.
1376. Hutchins, R.O.; Hoke, D.; Keogh, J.; Koharski, D. Tetrahedron Lett. 1969, 3495.
1377. Janssen, C.G.M.; Hendriks, A.H.M.; Godefroi, E.F. Recl. Trav. Chim. Pays-Bas 1984, 103, 220.
1378. Ueno, Y.; Tanaka, C.; Okawara, M. Chem. Lett. 1983, 795.
1379. Kogan, V. Tetrahedron Lett. 2006, 47, 7515.
1380. Krishnamurthy, S. J. Org. Chem. 1980, 45, 2550.
1381. Barton, D.H.R.; Jaszberenyi, J.Cs.; Tang, D. Tetrahedron Lett. 1993, 34, 3381.
1382. See Robins, M.J.; Wilson, J.S.; Hansske, F. J. Am. Chem. Soc. 1983, 105, 4059; Lopez, R.M.; Hays, D.S.; Fu, G.C. J. Am. Chem. Soc. 1997, 119, 6949; The Merck Index, 14th Ed. Merck & Co., Inc., Whitehouse Station, New Jersey, 2006, p ONR-6; Mundy, B.P.; Ellerd, M.G.; Favaloro, Jr., F.G. Name Reactions and Reagents in Organic Synthesis, 2nd Ed. Wiley-Interscience, New Jersey, 2005, pp. 68–69.
1383. Barton, D.H.R.; Jang, D.O.; Jaszberenyi, J.Cs. Tetrahedron 1993, 49, 2793.
1384. Barton, D.H.R.; Jang, D.O.; Jaszberenyi, J.Cs. Tetrahedron 1993, 49, 7193.
1385. Lopez, R.M.; Hays, D.S.; Fu, G.C. J. Am. Chem. Soc. 1997, 119, 6949.
1386. Oba, M.; Nishiyama, K. Tetrahedron 1994, 50, 10193.
1387. Spiegel, D.A.; Wiberg, K.B.; Schacherer, L.N.; Medeiros, M.R.; Wood, J.L. J. Am. Chem. Soc. 2005, 127, 12513.
1388. Park, H.S.; Lee, H.Y.; Kim, Y.H. Org. Lett. 2005, 7, 3187.
1389. See Hartwig, W. Tetrahedron 1983, 39, 2609.
1390. Barrett, A.G.M.; Godfrey, C.R.A.; Hollinshead, D.M.; Prokopiou, P.A.; Barton, D.H.R.; Boar, R.B.; Joukhadar, L.; McGhie, J.F.; Misra, S.C. J. Chem. Soc. Perkin Trans. 1 1981, 1501. See Garst, M.E.; Dolby, L.J.; Esfandiari, S.; Fedoruk, N.A.; Chamberlain, N.C.; Avey, A.A. J. Org. Chem. 2000, 65, 7098.
1391. Barrett, A.G.M.; Prokopiou, P.A.; Barton, D.H.R.; Boar, R.B.; McGhie, J.F. J. Chem. Soc. Chem. Commun. 1979, 1173.
1392. Deshayes, H.; Pete, J. Can. J. Chem. 1984, 62, 2063.
1393. Also see, Barton, D.H.R.; Crich, D. J. Chem. Soc. Perkin Trans. 1 1986, 1603.
1394. Jackson, R.A.; Malek, F. J. Chem. Soc. Perkin Trans. 1 1980, 1207.
1395. See Barton, D.H.R.; Jang, D.O.; Jaszberenyi, J.C. Tetrahedron Lett. 1990, 31, 4681, and references cited therein. For similar methods, see Nozaki, K.; Oshima, K.; Utimoto, K. Bull. Chem. Soc. Jpn. 1990, 63, 2578; Kirwan, J.N.; Roberts, B.P.; Willis, C.R. Tetrahedron Lett. 1990, 31, 5093.
1396. Oba, M.; Nishiyama, K. Synthesis 1994, 624.
1397. Hutchins, R.O.; Learn, K.; Fulton, R.P. Tetrahedron Lett. 1980, 21, 27. See also, Ipaktschi, J. Chem. Ber. 1984, 117, 3320.
1398. Tabuchi, T.; Inanaga, J.; Yamaguchi, M. Tetrahedron Lett. 1986, 27, 601, 5237. See also, Kusuda, K.; Inanaga, J.; Yamaguchi, M. Tetrahedron Lett. 1989, 30, 2945.
1399. Ballestri, M.; Chatgilialoglu, C.; Cardi, N.; Sommazzi, A. Tetrahedron Lett. 1992, 33, 1787.
1400. See Rowley, A.G. in Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis, Academic Press, NY, 1979, pp. 318–320.
1401. Rebeller, M.; Clément, G. Bull. Soc. Chim. Fr. 1964, 1302.
1402. Freerksen, R.W.; Selikson, S.J.; Wroble, R.R.; Kyler, K.S.; Watt, D.S. J. Org. Chem. 1983, 48, 4087.
1403. Dai, P.; Dussault, P.H.; Trullinger, T.K. J. Org. Chem. 2004, 69, 2851.
1404. Horner, L.; Jurgeleit, W. Liebigs Ann. Chem. 1955, 591, 138. See also, Rowley, A.G. in Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis, Academic Press, NY, 1979, pp. 320–322.
1405. Harpp, D.N.; Gleason, J.G. J. Am. Chem. Soc. 1971, 93, 2437. For another method, see Comasseto, J.V.; Lang, E.S.; Ferreira, J.T.B.; Simonelli, F.; Correi, V.R. J. Organomet. Chem. 1987, 334, 329.
1406. See Reusch, W. in Augustine, R.L. Reduction, Marcel Dekker, NY, 1968, pp. 171–211.
1407. See, however, Bailey, K.E.; Davis, B.R. Aust. J. Chem. 1995, 48, 1827. Also see, Rosnati, V. Tetrahedron Lett. 1992, 33, 4791.
1408. Seee Vedejs, E. Org. React. 1975, 22, 401. For a discussion of experimental conditions, see Fieser, L.F.; Fieser, M. Reagents for Organic Synthesis, Vol. 1, Wiley, NY, 1967, pp. 1287–1289.
1409. See Todd, D. Org. React. 1948, 4, 378.
1410. Huang-Minlon J. Am. Chem. Soc. 1946, 68, 2487; 1949, 71, 3301.
1411. Jaisankar, P.; Pal, B.; Giri, V.S. Synth. Commun. 2002, 32, 2569.
1412. Cram, D.J.; Sahyun, M.R.V.; Knox, G.R. J. Am. Chem. Soc. 1962, 84, 1734.
1413. Gadhwal, S.; Baruah, M.; Sandhu, J.S. Synlett 1999, 1573.
1414. Toda, M.; Hayashi, M.; Hirata, Y.; Yamamura, S. Bull. Chem. Soc. Jpn. 1972, 45, 264.
1415. See Buchanan, J.G.S.; Woodgate, P.D. Q. Rev. Chem. Soc. 1969, 23, 522.
1416. Galton, S.A.; Kalafer, M.; Beringer, F.M. J. Org. Chem. 1970, 35, 1.
1417. Pyrazolines can be converted to cyclopropanes; see Reaction 17-34.
1418. See, however, Banerjee, A.K.; Alvárez, J.; Santana, M.; Carrasco, M.C. Tetrahedron 1986, 42, 6615.
1419. Barton, D.H.R.; Ives, D.A.J.; Thomas, B.R. J. Chem. Soc. 1955, 2056.
1420. For a list, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 61–66.
1421. Yato, M.; Homma, K.; Ishida, A. Heterocycles 1995, 41, 17.
1422. Bandgar, B.P.; Nikat, S.M.; Wadgaonkar, P.P. Synth. Commun. 1995, 25, 863.
1423. Olah, G.A.; Wang, Q.; Prakash, G.K.S. Synlett 1992, 647.
1424. Pashkovsky, F.S.; Lokot, I.P.; Lakhvich, F.A. Synlett 2001, 1391.
1425. Ishimoto, K.; Mitoma, Y.; Negashima, S.; Tashiro, H.; Prakash, G.K.S.; Olah, G.A.; Tahshiro, M. Chem. Commun. 2003, 514.
1426. Ram, S.; Spicer, L.D. Tetrahedron Lett. 1988, 29, 3741.
1427. West, C.T.; Donnelly, S.J.; Kooistra, D.A.; Doyle, M.P. J. Org. Chem. 1973, 38, 2675. See also, Olah, G.A.; Arvanaghi, M.; Ohannesian, L. Synthesis 1986, 770.
1428. Zaccheria, F.; Ravasio, N.; Ercoli, M.; Allegrini, P. Tetrahedron Lett. 2005, 46, 7743.
1429. Gevorgyan, V.; Rubin, M.; Liu, J.-X.; Yamamoto, Y. J. Org. Chem. 2001, 66, 1672.
1430. Li, Z.; Deng, G.; Li, Y.-C. Synlett 2008, 3053.
1431. van Tamelen, E.E.; Gladys, J.A. J. Am. Chem. Soc. 1974, 96, 5290.
1432. See Zahalka, H.A.; Alper, H. Organometallics 1986, 5, 1909.
1433. Mayer, R.; Hiller, G.; Nitzschke, M.; Jentzsch, J. Angew. Chem. Int. Ed. 1963, 2, 370.
1434. Reusch, W.; LeMahieu, R. J. Am. Chem. Soc. 1964, 86, 3068.
1435. Meyer, K.H. Org. Synth. I, 60; Macleod, L.C.; Allen, C.F.H. Org. Synth. II, 62.
1436. Cormier, R.A.; McCauley, M.D. Synth. Commun. 1988, 18, 675.
1437. Hatano, B.; Tagaya, H. Tetraehedron Lett. 2003, 44, 6331.
1438. Hutchins, R.O.; Natale, N.R. J. Org. Chem. 1978, 43, 2299; Greene, A.E. Tetrahedron Lett. 1979, 63.
1439. Szmant, H.H. Angew. Chem. Int. Ed. 1968, 7, 120. Also see Taber, D.F.; Stachel, S.J. Tetrahedron Lett. 1992, 33, 903.
1440. See Di Vona, M.L.; Rosnati, V. J. Org. Chem. 1991, 56, 4269.
1441. Burdon, J.; Price, R.C. J. Chem. Soc. Chem. Commun. 1986, 893.
1442. Talukdar, S.; Banerji, A. Synth. Commun, 1996, 26, 1051.
1443. Ager, D.J.; Sutherland, I.O. J. Chem. Soc. Chem. Commun. 1982, 248. See also, Dias, J.R.; Pettit, G.R. J. Org. Chem. 1971, 36, 3485.
1444. Baldwin, S.W.; Haut, S.A. J. Org. Chem. 1975, 40, 3885. See also, Kraus, G.A.; Frazier, K.A.; Roth, B.D.; Taschner, M.J.; Neuenschwander, K. J. Org. Chem. 1981, 46, 2417.
1445. Kim, J.-G.; Cho, D.H.; Jang, D.O. Tetrahedron Lett. 2004, 45, 3031.
1446. Yato, M.; Homma, K.; Ishida, A. Tetrahedron 2001, 57, 5353.
1447. Morra, N.A.; Pagenkopf, B.L. Synthesis 2008, 511.
1448. Sakai, N.; Moriya, T.; Fujii, K.; Konakahara, T. Synthesis 2008, 3533.
1449. Iwanami, K.; Seo, H.; Tobita, Y.; Oriyama, T. Synthesis 2005, 183.
1450. See Pettit, G.R.; Kasturi, T.R.; Green, B.; Knight, J.C. J. Org. Chem. 1961, 26, 4773; Edward, J.T.; Ferland, J.M. Chem. Ind. (London) 1964, 975.
1451. Hansen, M.C.; Verdaguer, X.; Buchwald, S.L. J. Org. Chem. 1998, 63, 2360.
1452. Baxter, S.L.; Bradshaw, J.S. J. Org. Chem. 1981, 46, 831.
1453. Jang, D.O.; Song, S.H. Synlett 2000, 811; Jang, D.O.; Song, S.H.; Cho, D.H. Tetrahedron 1999, 55, 3479.
1454. Eliel, E.L.; Daignault, R.A. J. Org. Chem. 1964, 29, 1630; Bublitz, D.E. J. Org. Chem. 1967, 32, 1630.
1455. Morand, P.; Kayser, M.M. J. Chem. Soc. Chem. Commun. 1976, 314. See also, Hara, Y.; Wada, K. Chem. Lett. 1991, 553.
1456. Bailey, D.M.; Johnson, R.E. J. Org. Chem. 1970, 35, 3574.
1457. Bloomfield, J.J.; Lee, S.L. J. Org. Chem. 1967, 32, 3919.
1458. Matsuki, K.; Inoue, H.; Takeda, M. Tetrahedron Lett. 1993, 34, 1167.
1459. See Soucy, C.; Favreau, D.; Kayser, M.M. J. Org. Chem. 1987, 52, 129.
1460. Soai, K.; Yokoyama, S.; Mochida, K. Synthesis 1987, 647.
1461. See Wheeler, O.H. in Patai, S. The Chemistry of Acyl Halides, Wiley, NY, 1972, pp. 231–251. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1263–1264.
1462. Myers, A.G.; Yang, B.H.; Kopecky, D.J. Tetrahedron Lett. 1996, 37, 3623.
1463. Moody, H.M.; Kaptein, B.; Broxterman, Q.B.; Boesten, W.H.J.; Kamphuis, J. Tetrahedron Lett. 1994, 35, 1777.
1464. Sharma, R.; Voynov, G.H.; Ovaska, T.V.; Marquez, V.E. Synlett 1995, 839.
1465. See Wann, S.R.; Thorsen, P.T.; Kreevoy, M.M. J. Org. Chem. 1981, 46, 2579; Mandal, S.B.; Giri, V.S.; Pakrashi, S.C. Synthesis 1987, 1128; Akabori, S.; Takanohashi, Y. Chem. Lett. 1990, 251.
1466. Prasad, A.S.B.; Kanth, J.V.B.; Periasamy, M. Tetrahedron 1992, 48, 4623.
1467. Tanaka, H.; Ogasawara, K. Tetrahedron Lett. 2002, 43, 4417.
1468. See Bonnat, M.; Hercourt, A.; Le Corre, M. Synth. Commun. 1991, 21, 1579.
1469. Bhandari, K.; Sharma, V.L.; Chatterjee, S.K. Chem. Ind. (London) 1990, 547.
1470. Brown, H.C.; Kim, S.C. Synthesis 1977, 635.
1471. Hutchins, R.O.; Learn, K.; El-Telbany, F.; Stercho, Y.P. J. Org. Chem. 1984, 49, 2438.
1472. See Challis, B.C.; Challis, J.A. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 795–801; Gaylord, N.G. Reduction with Complex Metal Hydrides, Wiley, NY, 1956, p. 544. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 869–872.
1473. Aoun, R.; Renaud, J.-L.; Dixneuf, P.H.; Bruneau, C. Angew. Chem. Int. Ed. 2005, 44, 2021; Magro, A.A.N.; Eastham, G.R.; Cole-Hamilton, D.J. Chem. Commun. 2007, 3154.
1474. Nagata, Y.; Dohmaru, T.; Tsurugi, J. Chem. Lett. 1972, 989. See also, Benkeser, R.A.; Li, G.S.; Mozdzen, E.C. J. Organomet. Chem. 1979, 178, 21.
1475. Igarashi, M.; Fuchikami, T. Tetrahedron Lett. 2001, 42, 1945.
1476. Hanada, S.; Motoyama, Y.; Nagashima, H. Tetrahedron Lett. 2006, 47, 6173.
1477. Sakai, N.; Fujii, K.; Konakahara, T. Tetrahedron Lett. 2008, 49, 6873.
1478. Das, S.; Addis, D.; Zhou, S.; Junge, K.; Beller, M. J. Am. Chem. Soc. 2010, 132, 1770.
1479. Hanada, S.; Ishida, T.; Motoyama, Y.; Nagashima, H. J. Org. Chem. 2007, 72, 7551.
1480. Franco, D.; Duñach, E. Tetrahedron Lett. 2000, 41, 7333.
1481. Barbe, G.; Charette, A.B. J. Am. Chem. Soc. 2008, 130, 18.
1482. Pedregal, C.; Ezquerra, J.; Escribano, A.; Carreño, M.C.; García Ruano, J.L.G. Tetrahedron Lett. 1994, 35, 2053.
1483. Colllins, C.J.; Lanz, M.; Singaram, B. Tetrahedron Lett. 1999, 40, 3673.
1484. Flaniken, J.M.; Collins, C.J.; Lanz, M.; Singaram, B. Org. Lett. 1999, 1, 799.
1485. See Akula, M.R.; Kabalka, G.W. Org. Prep. Proceed. Int. 1999, 31, 214.
1486. Witkop, B.; Patrick, J.B. J. Am. Chem. Soc. 1952, 74, 3861.
1487. See Issa, F.; Fischer, J.; Turner, P.; Coster, M.J. J. Org. Chem. 2006, 71, 4703.
1488. Benkeser, R.A.; Foley, K.M.; Gaul, J.M.; Li, G.S. J. Am. Chem. Soc. 1970, 92, 3232.
1489. Benkeser, R.A.; Ehler, D.F. J. Org. Chem. 1973, 38, 3660.
1490. Benkeser, R.A.; Mozdzen, E.C.; Muth, C.L. J. Org. Chem. 1979, 44, 2185.
1491. Cerny, M.; Málek, J. Collect. Czech. Chem. Commun. 1970, 35, 2030.
1492. Chatani, N.; Tatamidani, H.; Ie, Y.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 2001, 123, 4849.
1493. See Le Deit, H.; Cron S.; Le Corre, M. Tetrahedron Lett. 1991, 32, 2759.
1494. Attanasi, O.; Caglioti, L.; Gasparrini, F.; Misiti, D. Tetrahedron 1975, 31, 341, and references cited therein.
1495. Sassaman, M.B. Tetrahedron 1996, 52, 10835.
1496. Kang, H.-Y.; Hong, W.S.; Cho, Y.S.; Koh, H.Y. Tetrahedron Lett. 1995, 36, 7661.
1497. Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. J. Am. Chem. Soc. 2009, 131, 3174.
1498. Hartung, W.H.; Simonoff, R. Org. React. 1953, 7, 263.
1499. du Vigneaud, V.; Behrens, O.K. J. Biol. Chem. 1937, 117, 27.
1500. Bull, S.D.; Davies, S.G.; Fenton, G.; Mulvaney, A.W.; Prasad, R.S.; Smith, A.D. J. Chem. Soc. Perkin Trans. 1 2000, 3765.
1501. Doldouras, G.A.; Kollonitsch, J. J. Am. Chem. Soc. 1978, 100, 341.
1502. Nickon, A.; Hill, R.H. J. Am. Chem. Soc. 1964, 86, 1152.
1503. Guziec, Jr., F.S.; Wei, D. J. Org. Chem. 1992, 57, 3772.
1504. Chandrasekhar, S.; Ahmed, M. Tetahedron Lett. 1999, 40, 9325.
1505. Hutchins, R.O.; Cistone, F.; Goldsmith, B.; Heuman, P. J. Org. Chem. 1975, 40, 2018.
1506. See Katritzky, A.R.; Bravo-Borja, S.; El-Mowafy, A.M.; Lopez-Rodriguez, G. J. Chem. Soc. Perkin Trans. 1 1984, 1671.
1507. Molander, G.A.; Stengel, P.J. Tetrahedron 1997, 53, 8887.
1508. Schwan, A.L.; Refvik, M.D. Tetrahedron Lett. 1993, 34, 4901.
1509. Coulter, J.M.; Lewis, J.W.; Lynch, P.P. Tetrahedron 1968, 24, 4489.
1510. Singaram, B.; Goralski, C.T.; Rangaishenvi, M.V.; Brown, H.C. J. Am. Chem. Soc. 1989, 111, 384.
1511. For example, see Pojer, P.M.; Ritchie, E.; Taylor, W.C. Aust. J. Chem. 1968, 21, 1375.
1512. Cooke, Jr., M.P.; Parlman, R.M. J. Org. Chem. 1975, 40, 531.
1513. See Fessard, T.C.; Motoyoshi, H.; Carreira, E.M. Angew. Chem. Int. Ed. 2007, 46, 2078.
1514. Kornblum, N.; Carlson, S.C.; Smith, R.G. J. Am. Chem. Soc. 1979, 101, 647; Kornblum, N.; Widmer, J.; Carlson, S.C. J. Am. Chem. Soc. 1979, 101, 658.
1515. See Ono, N. in Feuer, H.; Nielsen, A.T. Nitro Compounds: Recent Advances in Synthesis and Chemistry, VCH, NY, 1990, pp. 1–135, 1–45; Rosini, G.; Ballini, R. Synthesis 1988, 833, see pp. 835–837; Ono, N.; Kaji, A. Synthesis 1986, 693. See Kamimura, A.; Ono, N. Bull. Chem. Soc. Jpn. 1988, 61, 3629.
1516. Tanner, D.D.; Harrison, D.J.; Chen, J.; Kharrat, A.; Wayner, D.D.M.; Griller, D.; McPhee, D.J. J. Org. Chem. 1990, 55, 3321; Bowman, W.R.; Crosby, D.; Westlake, P.J. J. Chem. Soc. Perkin Trans. 2 1991, 73.
1517. Suzuki, H.; Takaoka, K.; Osuka, A. Bull. Chem. Soc. Jpn. 1985, 58, 1067.
1518. See Kniel, P. Helv. Chim. Acta 1968, 51, 371. For another method, see Ono, N.; Tamura, R.; Kaji, A. J. Am. Chem. Soc. 1983, 105, 4017.
1519. Azzena, U.; Dessanti, F.; Melloni, G.; Pisano, L. Tetrahedron Lett. 1999, 40, 8291.
1520. Geoffroy, O.J.; Morinelli, T.A.; Meier, G.B. Tetrahedron Lett. 2001, 42, 5367.
1521. Wang, Y.; Guziec Jr., F.S. J. Org. Chem. 2001, 66, 8293.
1522. Barton, D.H.R.; Bringmann, G.; Motherwell, W.B. Synthesis 1980, 68.
1523. See Yadav, J.S.; Reddy, P.S.; Joshi, B.V. Tetrahedron Lett. 1988, 44, 7243.
1524. Ohsawa, T.; Mitsuda, N.; Nezu, J.; Oishi, T. Tetrahedron Lett. 1989, 30, 845.
1525. Kamimura, A.; Kurata, K.; Ono, N. Tetrahedron Lett. 1989, 30, 4819.
1526. See Albini, A.; Pietra, S. Heterocyclic N-Oxides, CRC Press, Boca Raton, FL, 1991, pp. 120–134; Katritzky, A.R.; Lagowski, J.M. Chemistry of the Heterocyclic N-Oxides, Academic Press, NY, 1971, pp. 166–231.
1527. See Newbold, B.T. in Patai, S. The Chemistry of the Hydrazo, Azo, and Azoxy Groups, pt. 2, Wiley, NY, 1975, pp. 602–603, 614–624.
1528. See Rowley, A.G. in Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis, Academic Press, NY, 1979, pp. 295–350.
1529. Ram, S.R.; Chary, K.P.; Iyengar, D.S. Synth. Commun. 2000, 30, 3511.
1530. Han, J.H.; Choi, K.I.; Kim, J.H.; Yoo, B.W. Synth. Commun. 2004, 34, 3197.
1531. Yoo, B.W.; Choi, K.H.; Choi, K.I.; Kim, J.H. Synth. Commun. 2003, 33, 4185.
1532. Yadav, J.S.; Reddy, B.V.S.; Reddy, M.M. Tetrahedron Lett. 2000, 41, 2663.
1533. Balicki, R.; Maciejewski, G. Synth. Commun. 2002, 32, 1681.
1534. Balicki, R.; Cybulski, M.; Maciejewski, G. Synth. Commun. 2003, 33, 4137.
1535. Bj![]() rsvik, H.-R.; Gambarotti, C.; Jensen, V.R.; González, R.R. J. Org. Chem. 2005, 70, 3218.
rsvik, H.-R.; Gambarotti, C.; Jensen, V.R.; González, R.R. J. Org. Chem. 2005, 70, 3218.
1536. Yoo, B.W.; Choi, J.W.; Yoon, C.M. Tetrahedron Lett. 2006, 47, 125.
1537. Ilias, Md.; Barman, D.C.; Prajapati, D.; Sandhu, J.S. Tetrahedron Lett. 2002, 43, 1877.
1538. See Torssell, K.B.G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis, VCH, NY, 1988, pp. 55–74; Grundmann, C. Fortschr. Chem. Forsch. 1966, 7, 62.
1539. Grundmann, C.; Frommeld, H.D. J. Org. Chem. 1965, 30, 2077.
1540. Baldwin, J.E.; Derome, A.E.; Riordan, P.D. Tetrahedron 1983, 39, 2989.
1541. Imamoto, T.; Kikuchi, S.-i.; Miura, T.; Wada, Y. Org. Lett. 2001, 3, 87.
1542. Lee, H.; Sabila, P.; Saha, A.; Sarvestani, M.; Shen, S.; Varsolona, R.; Wei, X.; Senanayake, C.H. Org. Lett. 2005, 7, 4277.
1543. Wu, H.-C.; Yu, J.-Q.; Spencer, J.B. Org. Lett. 2004, 6, 4675.
1544. See Zollinger, H. in Patai, S.; Rappoport, Z. The Chemistry of Functinal Groups, Supplement C pt. 1, Wiley, NY, 1983, pp. 603–669.
1545. For lists of some of these, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 39–41; Tröndlin, F.; Rüchardt, C. Chem. Ber. 1977, 110, 2494.
1546. Shono, T.; Matsumura, Y.; Tsubata, K. Chem. Lett. 1979, 1051.
1547. See Korzeniowski, S.H.; Blum, L.; Gokel, G.W. J. Org. Chem. 1977, 42, 1469.
1548. Nakayama, J.; Yoshida, M.; Simamura, O. Tetrahedron 1970, 26, 4609.
1549. Hendrickson, J.B. J. Am. Chem. Soc. 1961, 83, 1251. See also, Threadgill, M.D.; Gledhill, A.P. J. Chem. Soc. Perkin Trans. 1 1986, 873.
1550. Doyle, M.P.; Dellaria, Jr., J.F.; Siegfried, B.; Bishop, S.W. J. Org. Chem. 1977, 42, 3494.
1551. Cadogan, J.I.G.; Molina, G.A. J. Chem. Soc. Perkin Trans. 1 1973, 541.
1552. See Broxton, T.J.; Bunnett, J.F.; Paik, C.H. J. Org. Chem. 1977, 42, 643.
1553. See Levit, A.F.; Kiprianova, L.A.; Gragerov, I.P. J. Org. Chem. USSR 1975, 11, 2395.
1554. König, E.; Musso, H.; Záhorszky, U.I. Angew. Chem. Int. Ed. 1972, 11, 45.
1555. Smith III, M.R.; Hillhouse, G.L. J. Am. Chem. Soc. 1988, 110, 4066.
1556. See König, E.; Musso, H.; Záhorszky, U.I.; König, E.; Musso, H.; Záhorszky, U.I. Angew. Chem. Int. Ed. 1972, 11, 45; Broxton, T.J.; McLeish, M.J. Aust. J. Chem. 1983, 36, 1031.
1557. Shinkai, S.; Mori, S.; Araki, K.; Manabe, O. Bull. Chem. Soc. Jpn. 1987, 60, 3679.
1558. For a review of the reduction of thioethers, see Block, E. in Patai, S. The Chemistry of Functional Groups, Supplement E, pt. 1, Wiley, NY, 1980, pp. 585–600.
1559. For reviews, see Belen'kii, L.I. in Belen'kii, L.I. Chemistry of Organosulfur Compounds, Ellis Horwood, Chichester, 1990, pp. 193–228; Pettit, G.R.; van Tamelen, E.E. Org. React. 1962, 12, 356; Hauptmann, H.; Walter, W.F. Chem. Rev. 1962, 62, 347.
1560. See Baxter, S.L.; Bradshaw, J.S. J. Org. Chem. 1981, 46, 831.
1561. See Nakata, D.; Kusaka, C.; Tani, S.; Kunishima, M. Tetrahedron Lett. 2001, 42, 415.
1562. Fishman, J.; Torigoe, M.; Guzik, H. J. Org. Chem. 1963, 28, 1443.
1563. For lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 53–60. See Luh, T.; Ni, Z. Synthesis 1990, 89; Becker, S.; Fort, Y.; Vanderesse, R.; Caubère, P. J. Org. Chem. 1989, 54, 4848.
1564. See Ikeshita, K.-i.; Kihara, N.; Ogawa, A. Tetrahedron Lett. 2005, 46, 8773.
1565. Schoenebeck, F.; Murphy, J.A.; Zhou, S.-z.; Uenoyama, Y.; Miclo, Y.; Tuttle, T. J. Am. Chem. Soc. 2007, 129, 13368.
1566. Smith, M.B.; Wolinsky, J. J. Chem. Soc. Perkin Trans. 2 1998, 1431.
1567. Back, T.G.; Baron D.L.; Yang, K. J. Org. Chem. 1993, 58, 2407.
1568. Guo, H.; Ye, S.; Wang, J.; Zhang, Y. J. Chem. Res. (S) 1997, 114.
1569. Wang, J.; Zhang, Y. Synth. Commun. 1996, 26, 1931.
1570. Schut, J.; Engberts, J.B.F.N.; Wynberg, H. Synth. Commun. 1972, 2, 415.
1571. Kikugawa, Y. J. Chem. Soc. Perkin Trans. 1 1984, 609.
1572. Clive, D.L.J.; Chittattu, G.; Wong, C.K. J. Chem. Soc. Chem. Commun. 1978, 41.
1573. Back, T.G. J. Chem. Soc. Chem. Commun. 1984, 1417.
1574. Ogawa, A.; Ohya, S.; Doi, M.; Sumino, Y.; Sonoda, N.; Hirao, T. Tetrahedron Lett. 1998, 39, 6341.
1575. For a review, see Bonner, W.A.; Grimm, R.A. in Kharasch, N.; Meyers, C.Y. The Chemistry of Organic Sulfur Compounds, Vol. 2, Pergamon, NY, 1966, pp. 35–71, 410–413. Also see Friend, C.M.; Roberts, J.T. Acc. Chem. Res. 1988, 21, 394.
1576. Owens, P.J.; Ahmberg, C.H. Can. J. Chem. 1962, 40, 941.
1577. See Wardell, J.L. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, pp. 216–220.
1578. Oae, S.; Togo, H. Bull. Chem. Soc. Jpn. 1983, 56, 3802; 1984, 57, 232.
1579. See Narayana, C.; Padmanabhan, S.; Kabalka, G.W. Synlett 1991, 125.
1580. Meshram, H.M.; Bandyopadhyay, A.; Reddy, G.S.; Yadav, J.S. Synth. Commun. 1999, 29, 2705.
1581. Yoo, B.W.; Baek, H.S.; Keum, S.R.; Yoon, C.M.; Nam. G.S.; Kim, S.H.; Kim, J.H. Synth. Commun. 2000, 30, 4317.
1582. Wang, L.; Li, P.; Zhou, L. Tetrahedron Lett. 2002, 43, 8141.
1583. See Kukushkin, V.Yu. Russ. Chem. Rev. 1990, 59, 844; Madesclaire, M. Tetrahedron 1988, 44, 6537; Drabowicz, J.; Togo, H.; Mikolajczyk, M.; Oae, S. Org. Prep. Proced. Int. 1984, 16, 171; Drabowicz, J.; Numata, T.; Oae, S. Org. Prep. Proced. Int. 1977, 9, 63. See Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978.
1584. Kozuka, S.; Furumai, S.; Akasaka, T.; Oae, S. Chem. Ind. (London) 1974, 496.
1585. Ogura, K.; Yamashita, M.; Tsuchihashi, G. Synthesis 1975, 385.
1586. Khurana, J.M.; Ray, A.; Singh, S. Tetrahedron Lett. 1998, 39, 3829.
1587. Karimi, B.; Zareyee, D. Synthesis 2003, 335.
1588. Harrison, D.J.; Tam, N.C.; Vogels, C.M.; Langler, R.F.; Baker, R.T.; Decken, A.; Westcott, S.A. Tetrahedron Lett. 2004, 45, 8493.
1589. Fernandes, A.C.; Romão, C.C. Tetrahedron Lett. 2007, 48, 9176.
1590. Yoo, B.W.; Song, M.S.; Park, M.C. Synth. Commun. 2007, 37, 3089.
1591. See Yoo, B.W.; Choi, K.H.; Kim, D.Y.; Choi, K.I.; Kim, J.H. Synth. Commun.2003, 33, 53.
1592. Yadav, J.S.; Subba Reddy, B.V.; Srinivas, C.; Srihari, P. Synlett 2001, 854.
1593. Hua, G.; Woollins, J.D. Tetrahedron Lett. 2007, 48, 3677.
1594. Gardner, J.N.; Kaiser, S.; Krubiner, A.; Lucas, H. Can. J. Chem. 1973, 51, 1419.
1595. See Weber, W.P.; Stromquist, P.; Ito, T.I. Tetrahedron Lett. 1974, 2595.
1596. Kiso, S.; Oae, S. Bull. Chem. Soc. Jpn. 1967, 40, 1722. See also, Oae, S.; Nakai, M.; Tsuchida, Y.; Furukawa, N. Bull. Chem. Soc. Jpn. 1971, 44, 445.
1597. Still, I.W.J.; Ablenas, F.J. J. Org. Chem. 1983, 48, 1617.
1598. See Denis, J.N.; Krief, A. J. Chem. Soc. Chem. Commun. 1980, 544.
1599. Taniguchi, T.; Ogasawara, K. Tetrahedron Lett. 1998, 39, 4679.
1600. Lemaire-Audoire, S.; Savignac, M.; Dupuis, C.; Genêt, J.-P. Bull. Soc. Chim. Fr. 1995, 132, 1157; Lemaire-Audoire, S.; Savignac, M.; Genêt, J.-P.; Bernard, J.-M. Tetrahedron Lett. 1995, 36, 1267.
1601. Talukdar, S.; Banerji, A. Synth. Commun. 1995, 25, 813.
1602. Murahashi, S.-I.; Naota, T.; Miyaguchi, N.; Nakato, T. Tetrahedron Lett. 1992, 33, 6991.
1603. Bull, S.D.; Davies, S.G.; Mulvaney, A.W.; Prasad, R.S.; Smith, A.D.; Fenton, G. Chem. Commun. 2000, 337.
1604. Haddach, A.A.; Kelleman, A.; Deaton-Rewoliwski, M.V. Tetrahedron Lett. 2002, 43, 399.
1605. Routier, S.; Saugé, L.; Ayerbe, N,; Couderet, G.; Mérour, J.-Y. Tetrahedron Lett. 2002, 43, 589. For a related debenzylation see Meng, G.; He, Y.-P.; Chen, F.-E. Synth. Commun. 2003, 33, 2593.
1606. Kok, G.B.; Pye, C.C.; Singer, R.D.; Scammells, P.J. J. Org. Chem. 2010, 75, 4806.
1607. Nandi, P.; Dye, J.L.; Jackson, J.E. Tetrahedron Lett. 2009, 50, 3864.
1608. Baker, S.R.; Parsons, A.F.; Wilson, M. Tetrahedron Lett. 1998, 39, 331.
1609. Mahalingam, A.K.; Wu, X.; Alterman, M. Tetrahedron Lett. 2006, 47, 3051.
1610. Katohgi, M.; Togo, H. Tetrahedron 2001, 57, 7481.
1611. Xu, L.; Zhang, S.; Trudell, M.L. Synlett 2004, 1901.
1612. Wan, Y.; Wu, X.; Kannan, M.A.; Alterman, M. Tetrahedron Lett. 2003, 44, 4523.
1613. See Newbold, B.T. in Patai, S. The Chemistry of Hydrazo, Azo, and Azoxy Groups, pt. 2, Wiley, NY, 1975, pp. 629–637.
1614. Tan, Z.; Qu, Z.; Chen, B.; Wang, J. Tetrahedron 2000, 56, 7457.
1615. Brown, H.C.; Subba Rao, B.C. J. Am. Chem. Soc. 1960, 82, 681.
1616. Zhang, C.-R.; Wang, Y.-L. Synth. Commun. 2003, 33, 4205.
1617. Mobinikhaledi, A.; Foroughifar, N.; Jirandehi, H.F. Monat. Chemie 2007, 138, 755.
1618. See Wardell, J.L. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, pp. 220–229.
1619. Reddy, G.V.S.; Rao, G.V.; Iyengar, D.S. Synth. Commun. 2000, 30, 859.
1620. Overman, L.E.; Smoot, J.; Overman, J.D. Synthesis 1974, 59.
1621. See Danehy, J.P.; Hunter, W.E. J. Org. Chem. 1967, 32, 2047.
1622. Chary, K.P.; Rajaram, S.; Iyengar, D.S. Synth. Commun. 2000, 30, 3905.
1623. Sridhar, M.; Vadivel, S.K.; Bhalerao, U.T. Synth. Commun. 1997, 27, 1347.
1624. Maiti, S.N.; Spevak, P.; Singh, M.P.; Micetich, R.G.; Narender Reddy, A.V. Synth. Commun. 1988, 18, 575.
1625. Huang, X.; Wu, L.-L.; Xu, X.-H. Synth. Commun. 2001, 31, 1871.
1626. See Fürstner, A.; Csuk, R.; Rohrer, C.; Weidmann, H. J. Chem. Soc. Perkin Trans. 1 1988, 1729. Also see Bian, Y.-J.; Liu, S.-M.; Li, J.-T.; Li, T.-S. Synth. Commun. 2002, 32, 1169.
1627. Gomberg, M.; Bachmann, W.E. J. Am. Chem. Soc. 1927, 49, 236; The Merck Index, 14th Ed. Merck & Co., Inc., Whitehouse Station, New Jersey, 2006, p ONR-74; Mundy, B.P.; Ellerd, M.G.; Favaloro, Jr., F.G. Name Reactions and Reagents in Organic Synthesis, 2nd Ed. Wiley–Interscience, New Jersey, 2005, pp. 512–513. See Wang, J.-S.; Li, J.-T.; Lin, Z.-P.; Li, T.-S. Synth. Commun. 2005, 35, 1419; Wang, S.-X.; Wang, K.; Li, J.-T. Synth. Commun. 2005, 35, 2387; Li, J.-T.; Chen, Y.-X.; Li, T.-S. Synth. Commun. 2005, 35, 2831.
1628. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp.1111–1114.
1629. Hélion, F.; Lannou, M.-I.; Namy, J.-L. Tetrahedron Lett. 2003, 44, 5507. See Banik, B.K.; Banik, I.; Aounallah, N.; Castillo, M. Tetrahedron Lett. 2005, 46, 7065.
1630. Aspinall, H.C.; Greeves, N.; Valla, C. Org. Lett. 2005, 7, 1919.
1631. Drapo, J.R.; Priefer, R. Synth. Commun. 2009, 39, 85.
1632. Hou, Z.; Takamine, K.; Fujiwara, Y.; Taniguchi, K. Chem. Lett. 1987, 2061.
1633. Lim, H.J.; Keum, G.; Kang, S.B.; Chung, B.Y.; Kim, Y. Tetrahedron Lett. 1998, 39, 4367.
1634. Mori, K.; Ohtaka, S.; Uemura, S. Bull. Chem. Soc. Jpn. 2001, 74, 1497.
1635. Wang, C.; Pan, Y.; Wu, A. Tetrahedron 2007, 63, 429.
1636. Li, J.-T.; Lin, Z.-P.; Qi, N.; Li, T.-S. Synth. Commun. 2004, 34, 4339.
1637. Xu, X.; Hirao, T. J. Org. Chem. 2005, 70, 8594.
1638. Rieke, R.D.; Kim, S.-H. J. Org. Chem. 1998, 63, 5235. See Groth, U.; Jeske, M. Synlett 2001, 129.
1639. Hekmatshoar, R.; Yavari, I.; Beheshtiha, Y.S.; Heravi, M.M. Monat. Chem. 2001, 132, 689.
1640. For a discussion of the mechanism, see Hashimoto, Y.; Mizuno, U.; Matsuoka, H.; Miyahara, T.; Takakura, M.; Yoshimoto, M.; Oshima, K.; Utimoto, K.; Matsubara, S. J. Am. Chem Soc. 2001, 123, 1503. See Duan, X.-F.; Feng, J.-X.; Zi, G.-F.; Zhang, Z.-B. Synthesis 2009, 277. Also see Li, T.; Cui, W.; Liu, J.; Zhao, J.; Wang, Z. Chem. Commun. 2000, 139; Kagayama, A.; Igarashi, K.; Mukaiyama, T. Can. J. Chem. 2000, 78, 657.
1641. Clerici, A.; Porta, O. J. Org. Chem. 1982, 47, 2852; Tetrahedron 1983, 39, 1239. See Delair, P.; Luche, J. J. Chem. Soc. Chem. Commun. 1989, 398; Takahara, P.M.; Freudenberger, J.H.; Konradi, A.W.; Pedersen, S.F. Tetrahedron Lett. 1989, 30, 7177.
1642. Freudenberger, J.H.; Konradi, A.W.; Pedersen, S.F. J. Am. Chem. Soc. 1989, 111, 8014.
1643. Zhang, W.-C.; Li, C.-J. J. Chem. Soc. Perkin Trans. 1 1998, 3131.
1644. Gansäuer, A.; Bauer, D. Eur. J. Org. Chem. 1998, 2673. Also see, Barden, M.C.; Schwartz, J. J. Am. Chem. Soc. 1996, 118, 5484; Gansäuer, A. Chem. Commun. 1997, 457; Gansäuer, A. Synlett 1997, 363.
1645. Tsuritani, T.; Ito, S.; Shinokubo, H.; Oshima, K. J. Org. Chem.2000, 65, 5066.
1646. Hayakawa, R.; Shimizu, M. Chem. Lett. 2000, 724. For a syn-selective coupling with conjugated aldehydes see Shimizu, M.; Goto, H.; Hayakawa, R. Org. Lett. 2002, 4, 4097.
1647. Szymoniak, J.; Besançon, J.; Moïse, C. Tetrahedron 1994, 50, 2841.
1648. Chatterjee, A.; Bennur, T.H.; Joshi, N.N. J. Org. Chem. 2003, 68, 5668.
1649. Shi, L.; Fan, C.-A.; Tu, Y.-Q.; Wang, M.; Zhang, F.-M. Tetrahedron 2004, 60, 2851. See also Luanphaisarnnont, T.; Ndubaku, C.O.; Jamison, T.F. Org. Lett. 2005, 7, 2937.
1650. Matsukawa, S.; Hinakubo, Y. Org. Lett. 2003, 5, 1221.
1651. Dunlap, M.S.; Nicholas, K.M. Synth. Commun. 1999, 29, 1097.
1652. For a review, see Chatterjee, A.; Joshi, N.N. Tetrahedron 2006, 62, 12137.
1653. Enders, D.; Ullrich, E.C. Tetrahedron Asymmetry 2000, 11, 3861.
1654. Kumagai, N.; Matsunaga, S.; Kinoshita, T.; Harada, S.; Okada, S.; Sakamoto, S.; Yamaguchi, K.; Shibasaki, M. J. Am. Chem. Soc. 2003, 125, 2169.
1655. Yang, H.; Wang, H.; Zhu, C. J. Org. Chem. 2007, 72, 10029.
1656. Maekawa, H.; Yamamoto, Y.; Shimada, H.; Yonemura, K.; Nishiguchi, I. Tetraheron Lett. 2004, 45, 3869.
1657. See Takenaka, N.; Xia, G.; Yamamoto, H. J. Am. Chem. Soc. 2004, 126, 13198.
1658. See Handa, S.; Kachala, M.S.; Lowe, S.R. Tetrahedron Lett. 2004, 45, 253.
1659. McMurry, J.E.; Siemers, N.O. Tetrahedron Lett. 1993, 34, 7891. See Raw, A.S.; Pedersen, S.F. J. Org. Chem. 1991, 56, 830; Chiara, J.L.; Cabri, W.; Hanessian, S. Tetrahedron Lett. 1991, 32, 1125.
1660. Baek, H.S.; Lee, S.J.; Yoo, B.W.; Ko, J.J.; Kim, S.H.; Kim, J.H. Tetrahedron Lett. 2000, 41, 8097.
1661. Studer, A.; Curran, D.P. Synlett 1996, 255.
1662. See Schönberg, A. Preparative Organic Photochemistry; Springer, NY, 1968, pp. 203–217; Neckers, D.C. Mechanistic Organic Photochemistry, Reinhold, NY, 1967, pp. 163–177; Calvert, J.G.; Pitts, Jr., J.N. Photochemistry, Wiley, NY, 1966, pp. 532–536; Turro, N.J. Modern Molecular Photochemistry, W.A. Benjamin, NY, 1978, pp. 363–385; Kan, R.O. Organic Photochemisty, McGraw-Hill, NY, 1966, pp. 222–229.
1663. See Cohen, S.G.; Parola, A.; Parsons, Jr., G.H. Chem. Rev. 1973, 73, 141.
1664. See Huyser, E.S.; Neckers, D.C. J. Am. Chem. Soc. 1963, 85, 3641.
1665. Porter, G.; Suppan, P. Proc. Chem. Soc. 1964, 191.
1666. Elinson, M.N.; Feducovich, S.K.; Dorofeev, A.S.; Vereshchagin, A.N.; Nikishin, G.I. Tetrahedron 2000, 56, 9999. See Fry, A.J. Synthetic Organic Electrochemistry, 2nd ed., Wiley, NY, 1989, pp. 174–180; Shono, T. Electroorganic Chemistry as a New Tool in Organic Synthesis, Springer, NY, 1984, pp. 137–140; Baizer, M.M.; Petrovich, J.P. Prog. Phys. Org. Chem. 1970, 7, 189; Baizer, M.M. in Baizer, M.M.; Lund, H. Organic Electrochemistry, Marcel Dekker, NY, 1983, pp. 639–689.
1667. See Alexakis, A.; Aujard, I.; Mangeney, P. Synlett 1998, 873, 875.
1668. Kalyanam, N.; Rao, G.V. Tetrahedron Lett. 1993, 34, 1647.
1669. Dutta, M.P.; Baruah, B.; Boruah, A.; Prajapati, D.; Sandu, J.S. Synlett 1998, 857.
1670. Zhong, Y.-W.; Izumi, K.; Xu, M.-H.; Lin, G.-Q. Org. Lett. 2004, 6, 4747.
1671. Shono, T.; Kise, N.; Fujimoto, T. Tetrahedron Lett. 1991, 32, 525.
1672. Kise, N.; Ueda, N. Tetrahedron Lett. 2001, 42, 2365.
1673. Mojtahedi, M.M.; Saidi, M.R.; Shirzi, J.S.; Bolourtchian, M. Synth. Commun. 2001, 31, 3587.
1674. Zhong, Y.-W.; Dong, Y.-Z.; Fang, K.; Izumi, K.; Xu, M.-H.; Lin, G.-Q. J. Am. Chem. Soc. 2005, 127, 11956.
1675. Yoshimura, N.; Mukaiyama, T. Chem. Lett. 2001, 1334.
1676. Selvakumar, K.; Harrod, J.F. Angew. Chem. Int. Ed. 2001, 40, 2129.
1677. Kim, M.; Knettle, B.W.; Dahlén, A.; Hilmersson, G.; Flowers III, R.A. Tetrahedron 2003, 59, 10397.
1678. Su, W.; Yang, B. Synth. Commun. 2003, 33, 2613.
1679. Ortega, M.; Rodríguez, M.A.; Campos, P.J. Tetrahedron 2004, 60, 6475.
1680. Jin, W.; Makioka, Y.; Kitamura, T.; Fujiwara, Y. J. Org. Chem. 2001, 66, 514.
1681. See Rele, S.; Chattopadhyay, S.; Nayak, S.K. Tetrahedron Lett. 2001, 42, 9093.
1682. McMurry, J.E.; Fleming, M.P.; Kees, K.L.; Krepski, L.R. J. Org. Chem. 1978, 43, 3255. For an optimized procedure, see McMurry, J.E.; Lectka, T.; Rico, J.G. J. Org. Chem. 1989, 54, 3748.
1683. See McMurry, J.E. Chem. Rev. 1989, 89, 1513; Acc. Chem. Res. 1983, 16, 405; Lenoir, D. Synthesis 1989, 883; Betschart, C.; Seebach, D. Chimia 1989, 43, 39; Lai, Y. Org. Prep. Proceed. Int. 1980, 12, 363. For related reviews, see Kahn, B.E.; Rieke, R.D. Chem. Rev. 1988, 88, 733; Pons, J.; Santelli, M. Tetrahedron 1988, 44, 4295. See Duan, X.-F.; Zeng, J.; Lü, J.-W.; Zhang, Z.-B. J. Org. Chem. 2006, 71, 9873.
1684. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 305–308.
1685. Tyrlik, S.; Wolochowicz, I. Bull. Soc. Chim. Fr. 1973, 2147.
1686. Carroll, A.R.; Taylor, W.C. Aust. J. Chem. 1990, 43, 1439.
1687. Dams, R.; Malinowski, M.; Geise, H.J. Bull. Soc. Chim. Belg. 1982, 91, 149, 311; Bottino, F.A.; Finocchiaro, P.; Libertini, E.; Reale, A.; Recca, A. J. Chem. Soc. Perkin Trans. 2 1982, 77. This reagent has been reported to give capricious results; see McMurry, J.E.; Fleming, M.P. J. Org. Chem. 1976, 41, 896.
1688. Rele, S.; Talukdar, S.; Banerji, A.; Chattopadhyay, S. J. Org. Chem. 2001, 66, 2990.
1689. Dams, R.; Malinowski, M.; Geise, H.J. Bull. Soc. Chim. Belg. 1982, 19, 149, 311. See also, Chisholm, M.H.; Klang, J.A. J. Am. Chem. Soc. 1989, 111, 2324.
1690. Stuhr-Hansen, N. Tetrahedron Lett. 2005, 46, 5491.
1691. McMurry, J.E.; Fleming, M.P.; Kees, K.L.; Krepski, L.R. J. Org. Chem. 1978, 43, 3255.
1692. Baumstark, A.L.; McCloskey, C.J.; Witt, K.E. J. Org. Chem. 1978, 43, 3609.
1693. McMurry, J.E.; Miller, D.D. J. Am. Chem. Soc. 1983, 105, 1660.
1694. Fürstner, A.; Hupperts, A. J. Am. Chem. Soc. 1995, 117, 4468.
1695. Fürstner, A.; Hupperts, A.; Ptock, A.; Janssen, E. J. Org. Chem. 1994, 59, 5215.
1696. Fürstner, A.; Jumbam, D.N. Tetrahedron 1992, 48, 5991.
1697. See Chisholm, M.H.; Klang, J.A. J. Am. Chem. Soc. 1989, 111, 2324.
1698. Curini, M.; Epifano, F.; Maltese, F.; Marcotullio, M.C. Eur. J. Org. Chem. 2003, 1631.
1699. Dams, R.; Malinowski, M.; Westdorp, I.; Geise, H.Y. J. Org. Chem. 1982, 47, 248. See Villiers, C.; Ephritikhine, M. Angew. Chem,. Int. Ed. 1997, 36, 2380; Stahl, M.; Pindur, U.; Frenking, G. Angew. Chem. Int. Ed. 1997, 36, 2234.
1700. See Bloomfield, J.J.; Owsley, D.C.; Nelke, J.M. Org. React. 1976, 23, 259. For a list of reactions, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1313–1315.
1701. Also see Daynard, T.S.; Eby, P.S.; Hutchinson, J.H. Can. J. Chem. 1993, 71, 1022.
1702. Fadel, A.; Canet, J,-L.; Salaün, J. Synlett 1990, 89.
1703. Olah, G.A.; Wu, A. Synthesis 1991, 1177.
1704. See Finley, K.T. Chem. Rev. 1964, 64, 573.
1705. Bloomfield, J.J.; Irelan, J.R.S. J. Org. Chem. 1966, 31, 2017.
1706. Cram, D.J.; Gaston, L.K. J. Am. Chem. Soc. 1960, 82, 6386.
1707. For a review, see Cram, D.J. Rec. Chem. Prog. 1959, 20, 71.
1708. Schräpler, U.; Rühlmann, K. Chem. Ber. 1964, 97, 1383. See Rühlmann, K. Synthesis 1971, 236.
1709. Bloomfield, J.J. Tetrahedron Lett. 1968, 591.
1710. Bloomfield, J.J.; Martin, R.A.; Nelke, J.M. J. Chem. Soc. Chem. Commun. 1972, 96.
1711. Another mechanism has been proposed: Bloomfield, J.J.; Owsley, D.C.; Ainsworth, C.; Robertson, R.E. J. Org. Chem. 1975, 40, 393.
1712. For the preparation of high-surface sodium, see Makosza, M.; Grela, K. Synlett 1997, 267.
1713. Vogel, I.A. A Textbook of Practical Organic Chemistry, 3rd ed, Wiley, NY, 1966, p. 856.
1714. Karaman, R.; Fry, J.L. Tetrahedron Lett. 1989, 30, 6267.
1715. For reviews of the synthesis of catenanes, see Sauvage, J. Acc. Chem. Res. 1990, 23, 319; Nouv. J. Chim. 1985, 9, 299; Dietrich-Buchecker, C.O.; Sauvage, J. Chem. Rev. 1987, 87, 795.
1716. Osuka, A.; Shimizu, H.; Suzuki, H. Chem. Lett. 1983, 1373. See Ohe, K.; Uemura, S.; Sugita, N.; Masuda, H.; Taga, T. J. Org. Chem. 1989, 54, 4169.
1717. Ogata, Y.; Mibae, J. J. Org. Chem. 1962, 27, 2048.
1718. Bunyan, P.J.; Cadogan, J.I.G. J. Chem. Soc. 1963, 42.
1719. See Hutton, J.; Waters, W.A. J. Chem. Soc. B 1968, 191. See also, Porta, F.; Pizzotti, M.; Cenini, S. J. Organomet. Chem. 1981, 222, 279.
1720. Srinavasa, G.R.; Abiraj, K.; Gowda, D.C. Tetrahedron Lett. 2003, 44, 5835.
1721. Hutchins, R.O.; Lamson, D.W.; Rufa, L.; Milewski, C.; Maryanoff, B. J. Org. Chem. 1971, 36, 803.
1722. Bae, J.W.; Cho, Y.J.; Lee, S.H.; Yoon, C.M. Tetrahedron Lett. 2000, 41, 175.
1723. Furst, A.; Moore, R.E. J. Am. Chem. Soc. 1957, 79, 5492.
1724. Olah, G.A. J. Am. Chem. Soc. 1959, 81, 3165.
1725. See Geissman, T.A. Org. React. 1944, 2, 94.
1726. Abaee, M.S.; Sharifi, R.; Mojtahedi, M.M. Org. Lett. 2005, 7, 5893.
1727. Basavaiah, D.; Sharada, D.S.; Veerendhar, A. Tetrahedron Lett. 2006, 47, 5771.
1728. An exception is cyclopropanecarboxaldehyde: van der Maeden, F.P.B.; Steinberg, H.; de Boer, T.J. Recl. Trav. Chim. Pays-Bas 1972, 91, 221.
1729. Yoshizawa, K.; Toyota, S.; Toda, F. Tetrahedron Lett. 2001, 42, 7983.
1730. See Thakuria, J.A.; Baruah, M.; Sandhu, J.S. Chem. Lett. 1999, 995.
1731. See Reddy, B.V.S.; Srinivas, R.; Yadav, J.S.; Ramalingam, T. Synth. Commun. 2002, 32, 219.
1732. Bergens, S.H.; Fairlie, D.P.; Bosnich, B. Organometallics 1990, 9, 566.
1733. Russell, A.E.; Miller, S.P.; Morken, J.P. J. Org. Chem. 2000, 65, 8381.
1734. Russell, G.A.; Mikol, G.J. J. Am. Chem. Soc. 1966, 88, 6498; Prey, V.; Berbdk, H.; Steinbauer, E. Monatch. Chem. 1960, 91, 1196; 1962, 93, 237.
1735. Thompson, J.E. J. Org. Chem. 1967, 32, 3947.
1736. See Ashby, E.C.; Coleman, III, D.T.; Gamasa, M.P. J. Org. Chem. 1987, 52, 4079; Fuentes, A.; Marinas, J.M.; Sinisterra, J.V. Tetrahedron Lett. 1987, 28, 2947.
1737. See Swain, C.G.; Powell, A.L.; Sheppard, W.A.; Morgan, C.R. J. Am. Chem. Soc. 1979, 101, 3576; Watt, C.I.F. Adv. Phys. Org. Chem. 1988, 24, 57, pp. 81–86.
1738. Fredenhagen, H.; Bonhoeffer, K.F. Z. Phys. Chem. Abt. A 1938, 181, 379; Hauser, C.R.; Hamrick, Jr., P.J.; Stewart, A.T. J. Org. Chem. 1956, 21, 260.
1739. See Swain, C.G.; Powell, A.L.; Lynch, T.J.; Alpha, S.R.; Dunlap, R.P. J. Am. Chem. Soc. 1979, 101, 3584. See, however, Chung, S. J. Chem. Soc. Chem. Commun. 1982, 480.
1740. For a review, see Seki, T.; Nakajo, T.; Onaka, M. Chem. Lett. 2006, 35, 824.
1741. Seki, T.; Hattori, H. Chem. Commun. 2001, 2510.
1742. See Saegusa, T.; Ueshima, T.; Kitagawa, S. Bull. Chem. Soc. Jpn. 1969, 42, 248; Ogata, Y.; Kishi, I. Tetrahedron 1969, 25, 929.
1743. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 1653–1655.
1744. Ito, T.; Horino, H.; Koshiro,Y.; Yamamoto, A. Bull. Chem. Soc. Jpn. 1982, 55, 504.
1745. Andrea, T.; Barnea, E.; Eisen, M.S. J. Am. Chem. Soc. 2008, 130, 2454.
1746. Crimmin, M.R.; Barrett, A.G.M.; Hill, M.S.; Procopiou, P.A. Org. Lett. 2007, 9, 331.
1747. DeMico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. J. Org. Chem. 1997, 62, 6974.
1748. Yamashita, A.; Watanabe, Y.; Mitsudo, T.; Takegami, Y. Bull. Chem. Soc. Jpn. 1976, 49, 3597.
1749. Seki, T.; Akutsu, K.; Hattori, H. Chem. Commun. 2001, 1000.
1750. Ooi, T.; Miura, T.; Takaya, K.; Maruoka, K. Tetrahedron Lett. 1999, 40, 7695.
1751. Simpura, I.; Jevalainen, V. Tetrahedron 2001, 57, 9867.
1752. Simpura, I.; Nevalainen, V. Tetrahedron Lett. 2001, 42, 3905; Cavazzini, M.; Pozzi, G.; Quici, S.; Maillard, D.; Sinou, D. Chem. Commun. 2001, 1220.
1753. See Bur, S.K.; Padwa, A. Chem. Rev. 2004, 104, 2401.
1754. For a review, see Feldman, K.S. Tetrahedron 2006, 62, 5003. Also see Smith, L.H.S.; Coote, S.C.; Sneddon, H.F.; Procter, D.J. Angew. Chem. Int. Ed. 2010, 49, 5832.
1755. See De Lucchi, O.; Miotti, U.; Modena, G. Org. React. 1991, 40, 157; Warren, S. Chem. Ind. (London) 1980, 824; Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, pp. 154–162.
1756. See Sugihara, H.; Tanikaga, R.; Kaji, A. Synthesis 1978, 881.
1757. See Dilworth, B.M.; McKervey, M.A. Tetrahedron 1986, 42, 3731.
1758. Kita, Y.; Shibata, N.; Kawano, N.; Tohjo, T.; Fujimori, C.; Matsumoto, K. Tetrahedron Lett. 1995, 36, 115; Kita, Y.; Shibata, N.; Fukui, S.; Fujita, S. Tetrahedron Lett. 1994, 35, 9733.
1759. Wladislaw, B.; Marzorati, L.; Biaggio, F.C. J. Org. Chem. 1993, 58, 6132.
1760. See Kita, Y.; Shibata, N.; Yoshida, N.; Fukui, S.; Fujimori, C. Tetrahedron Lett. 1994, 35, 2569.
1761. Oae, S.; Kitao, T.; Kawamura, S.; Kitaoka, Y. Tetrahedron 1963, 19, 817.
1762. See Itoh, O.; Numata, T.; Yoshimura, T.; Oae, S. Bull. Chem. Soc. Jpn. 1983, 56, 266; Oae, S.; Itoh, O.; Numata, T.; Yoshimura, T. Bull. Chem. Soc. Jpn. 1983, 56, 270.
1763. See Marino, J.P. Top. Sulfur Chem. 1976, 1, 1.
1764. Sharma, A.K.; Swern, D. Tetrahedron Lett. 1974, 1503.
1765. See Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, pp. 154–156; Oae, S.; Numata, T. Isot. Org. Chem. 1980, 5, 45, p. 48; Wolfe, S.; Kazmaier, P.M. Can. J. Chem. 1979, 57, 2388, 2397; Russell, G.A.; Mikol, G.J. Mech. Mol. Migr. 1968, 1, 157.
1766. Kirpichenko, S.V.; Suslova, E.N.; Albanov, A.I.; Shainyan, B.A. Tetrahedron Lett. 1999, 40, 185.
1767. For a review, see Brown, E.V. Synthesis 1975, 358.
1768. See Mayer, R. in Oae, S. The Organic Chemistry of Sulfur, Plenum, NY, 1977, pp. 58–63; Lundstedt, T.; Carlson, R.; Shabana, R. Acta Chem. Scand. Ser. B 1987, 41, 157, and other papers in this series. See also, Kanyonyo, M.R.; Gozzo, A.; Lambert, D.M.; Lesieur, D.; Poupaert, J.H. Bull. Soc. Chim. Belg. 1997, 106, 39.
1769. See Asinger, F.; Offermanns, H. Angew. Chem. Int. Ed. 1967, 6, 907.
1770. Amupitan, J.O. Synthesis 1983, 730.
1771. Schroth, W.; Andersch, J. Synthesis 1989, 202.
1772. See Dutron-Woitrin, F.; Merényi, R.; Viehe, H.G. Synthesis 1985, 77.
1773. For a review, see Poupaert, J.H.; Bouinidane, K.; Renard, M.; Lambert, D.; Isa, M. Org. Prep. Proceed. Int. 2001, 33, 335.
1774. Higgins, S.D.; Thomas, C.B. J. Chem. Soc. Perkin Trans. 1 1982, 235. See also, Higgins, S.D.; Thomas, C.B. J. Chem. Soc. Perkin Trans. 1 1983, 1483.
1775. King, J.A.; McMillan, F.H. J. Am. Chem. Soc. 1946, 68, 632.
1776. See Asinger, F.; Saus, A.; Mayer, A. Monatsh. Chem. 1967, 98, 825.
1777. Asinger, F.; Halcour, K. Monatsh. Chem. 1964, 95, 24. See also, Nakova, E.P.; Tolkachev, O.N.; Evstigneeva, R.P. J. Org. Chem. USSR 1975, 11, 2660.
1778. Mayer, R. in Janssen, M.J. Organosulfur Chemistry, Wiley, NY, 1967, pp. 229–232.