March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 1. Localized Chemical Bonding
1.L. Bond Energies95
There are two kinds of bond energy. The energy necessary to cleave a bond to give the constituent radicals is called the dissociation energy (D). For example, D for H2O → HO + H is 118 kcal mol−1 (494 kJ mol−1). However, this is not taken as the energy of the O–H bond in water, since D for H–O → H + O is 100 kcal mol−1 (418 kJ mol−1). The average of these two values, 109 kcal mol−1 (456 kJ mol−1), is taken as the bond energy (E). In diatomic molecules, of course, D = E.
The D values may be easy or difficult to measure. They can be estimated by various techniques.96 When properly applied, “Pauling's original electronegativity equation accurately describes homolytic bond dissociation enthalpies of common covalent bonds, including highly polar ones, with an average deviation of (1.5 kcal mol−1 (~6.3 kJ mol−1) from literature values)”.97 Whether measured or calculated, there is no question as to what D values mean. With Evalues the matter is not so simple. For methane, the total energy of conversion from CH4 to C + 4H (at 0 K) is 393 kcal mol−1 (1644 kJ mol−1).98 Consequently, E for the C–H bond in methane is 98 kcal mol−1 (411 kJ mol−1) at 0 K. The more usual practice is not to measure the heat of atomization (i.e., the energy necessary to convert a compound to its atoms) directly, but to calculate it from the heat of combustion. Such a calculation is shown in Fig. 1.12.
Fig. 1.12 Calculation of the heat of atomization of ethane at 25 °C.

Heats of combustion are very accurately known for hydrocarbons.99 For methane, the value at 25 °C is 212.8 kcal mol−1 (890.4 kJ mol−1), which leads to a heat of atomization of 398.0 kcal mol−1 (1665 kJ mol−1) or a value of Efor the C–H bond at 25 °C of 99.5 kcal mol−1 (416 kJ mol−1). This method is fine for molecules like methane in which all the bonds are equivalent, but for more complicated molecules, assumptions must be made. Thus for ethane, the heat of atomization at 25 °C is 676.1 kcal mol−1 or 2829 kJ mol−1 (Fig. 1.12), and it must be decided how much of this energy is due to the C–C bond and how much to the six C–H bonds. Any assumption must be artificial, since there is no way of actually obtaining this information, and indeed the question has no real meaning. If the assumption is made that E for each of the C–H bonds is the same as E for the C–H bond in methane (99.5 kcal mol−1 or 416 kJ mol−1), then 6 × 99.5 (or 416) = 597.0 (or 2498), leaving 79.1 kcal mol−1 (331 kJ mol−1) for the C–C bond. However, a similar calculation for propane gives a value of 80.3 (or 336) for the C–C bond, and for isobutane, the value is 81.6 (or 341). A consideration of heats of atomization of isomers also illustrates the difficulty. The E values for the C–C bonds in pentane, isopentane, and neopentane, similarly calculated from heats of atomization, are (at 25 °C) 81.1, 81.8, and 82.4 kcal mol−1 (339, 342, 345 kJ mol−1), respectively, even though all of them have twelve C–H bonds and four C–C bonds.
These differences have been attributed to various factors caused by the introduction of new structural features. Thus isopentane has a tertiary carbon whose C–H bond does not have exactly the same amount of s character as the C–H bond in pentane, which for that matter contains secondary carbons not possessed by methane. It is known that D values, which can be measured, are not the same for primary, secondary, and tertiary C–H bonds (see Table 5.2). There is also the steric factor (see Sec. 4.Q). Hence it is certainly incorrect to use the value of 99.5 kcal mol−1 (416 kJ mol−1) from methane as the E value for all C–H bonds. Several empirical equations have been devised that account for these factors; the total energy can be computed100 if the proper set of parameters (one for each structural feature) is inserted. Of course, these parameters are originally calculated from the known total energies of some molecules that contain the structural feature.
Table 1.7 gives E values for various bonds.101–104 The values given are averaged over a large series of compounds. The literature contains charts that take hybridization into account (thus an sp3 C–H bond does not have the same energy as an sp2 C–H bond).105 Bond dissociation energies, both calculated and experientially determined, are constantly being refined. Improved values are available for the O–O bond of peroxides,106 the C–H bond in alkyl amines,107 the N–H bond in aniline derivatives,108 the N–H bond in protonated amines,109 the O–H bond in phenols,110 the C–H bond in alkenes,111 amides and ketones,112 and in CH2X2 and CH3X derivatives (X = COOR, C=O, SR, NO2, etc.),113 the O–H and S–H bonds of alcohols and thiols,114 and the C–Si bond of aromatic silanes.115 Solvent plays a role in the E values. When phenols bearing electron-releasing groups are in aqueous media, calculations show that the bond dissociation energies decrease due to hydrogen-bonding interactions with water molecules, while electron-withdrawing substituents on the phenol increase the bond dissociation energies.116
Table 1.7 Bond Energy (E) Values at 25 °C for Some Important Bond Typesa,b
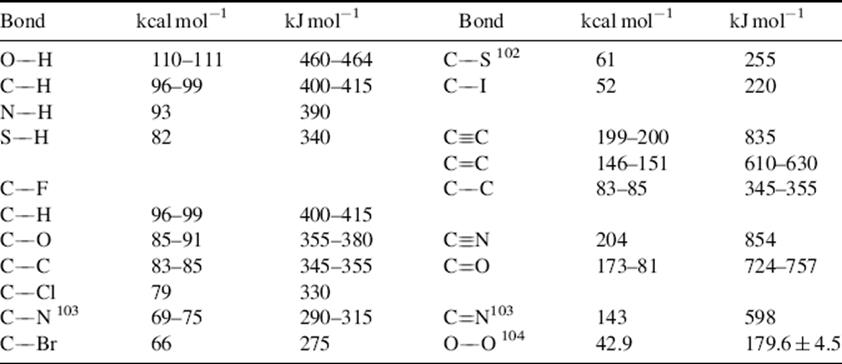
aThe E values are arranged within each group in order of decreasing strength. The values are averaged over a large series of compounds.
bSee Ref. 101.
[Reprinted with permission from Lovering E.G.; Laidler, K.J. Can. J. Chem. 1960, 38, 2259. Copyright © 1960 with permission from Canadian Science Publishing or its Licensors.]
Certain generalizations can be derived from the data in Table 1.7.
1. There is a correlation of bond strengths with bond distances. A comparison of Tables 1.5 and 1.7 shows that, in general, shorter bonds are stronger bonds. Since it is known that increasing s character shortens bonds (Sec. 1.J), it follows that bond strengths increase with increasing s character. Calculations show that ring strain has a significant effect on bond dissociation energy, particularly the C–H bond of hydrocarbons, because it forces the compound to adopt an undesirable hybridization.117
2. Bonds become weaker moving down the periodic table. Compare C–O and C–S, or the carbon–halogen bonds C–F, C–Cl, C–Br, C–I. This is a consequence of the first generalization, since bond distances must increase going down the periodic table because the number of inner electrons increases. However, note that “high-level ab initio molecular orbital calculations confirm that the effect of alkyl substituents on R–X bond dissociation energies varies according to the nature of X (the stabilizing influence of the ionic configurations to increase in the order Me < Et < i-Pr < t-Bu), accounting for the increase (rather than expected decrease) in the R–X bond dissociation energies with increasing alkylation in the R–OCH3, R–OH, and R–F molecules. This effect of X has been explained in terms of the increasing contribution of the ionic R+X− configuration for electronegative X substituents.”118
3. Double bonds are both shorter and stronger than the corresponding single bonds, but not twice as strong, because π overlap is < σ overlap. This means that a σ bond is stronger than a π bond. The difference in energy between a single bond, say C–C, and the corresponding double bond is the amount of energy necessary to cause rotation around the double bond.119
Calculations suggest that covalent bond strength and also equilibrium bond length are not determined by maximum overlap of the σ valence orbitals, as described in previous sections.120 Rather, orbital interactions, Pauli repulsion, and quasiclassical electrostatic attraction determine both.
Solvents are thought to play a role in bond dissociation energy of molecules, as noted for phenol above, and also for intermediates (see Chap 5). It has been assumed that the solvation enthalpies were small and they have been largely ignored in calculations involving various reactions. Solvent effects on the bond dissociation energy of a molecule may arise from the difference in solvation enthalpies between the molecule and the key intermediate. For radical reactions that involve polar molecules, the radical–solvent interaction may be larger.121
Notes
1. See Hoffmann, R.; Schleyer, P.v.R.; Schaefer, III, H.F. Angew. Chem. Int. Ed. (Engl.) 2008, 47, 7164.
2. This treatment of orbitals is simplified by necessity. For more detailed treatments of orbital theory, as applied to organic chemistry, see Matthews, P.S.C. Quantum Chemistry of Atoms and Molecules, Cambridge University Press, Cambridge, 1986; Clark, T. A Handbook of Computational Chemistry, Wiley, NY, 1985; Albright, T.A.; Burdett, J.K.; Whangbo, M. Orbital Interactions in Chemistry, Wiley, NY, 1985; MacWeeny, R.M. Coulson's Valence, Oxford University Press, Oxford, 1980; Murrell, J.N.; Kettle, S.F.A; Tedder, J.M. The Chemical Bond, Wiley, NY, 1978; Dewar, M.J.S.; Dougherty. R.C. The PMO Theory of Organic Chemistry, Plenum, NY, 1975; Zimmerman, H.E. Quantum Mechanics for Organic Chemists, Academic Press, NY, 1975; Borden, W.T. Modern Molecular Orbital Theory for Organic Chemists, Prentice-Hall, Englewood Cliffs, NJ, 1975.
3. When wave mechanical calculations are made according to the Schrödinger equation, the probability of finding the electron in a node is zero, but this treatment ignores relativistic considerations. When such considerations are applied, Dirac has shown that nodes do have a very small electron density: Powell, R.E. J. Chem. Educ. 1968, 45, 558. See also, Ellison, F.O.; Hollingsworth, C.A. J. Chem. Educ. 1976, 53, 767; McKelvey, D.R. J. Chem. Educ.1983, 60, 112; Nelson, P.G. J. Chem. Educ. 1990, 67, 643. For a general review of relativistic effects on chemical structures, see Pyykkö, P. Chem. Rev. 1988, 88, 563.
4. See Roothaan, C.C.J.; Weiss, A.W. Rev. Mod. Phys. 1960, 32, 194; Kolos, W.; Roothaan, C.C.J. Rev. Mod. Phys. 1960, 32, 219. For a review, see Clark, R.G.; Stewart, E.T. Q. Rev. Chem. Soc. 1970, 24, 95.
5. In this book, a pair of electrons in a bond is represented by a straight line.
6. Schwarz, W.H.E. Angew. Chem. Int. Ed. 2006, 45, 1508. For the ball-in-box model, see Pierrefixe, S.C.A.H.; Guerra, C.F.; Bickelhaupt, F.M. Chem. Eur. J. 2008, 14, 819; Pierrefixe, S.C.A.H.; Bickelhaupt, F.M. J. Phys. Chem. A. 2008, 112, 12816.
7. Bent, H.A. Chem. Rev. 1961, 61, 275, 277.
8. For an alternative representation, see Pauling, L. Theoretical Organic Chemistry, The Kekulé Symposium, Butterworth, London, 1959, pp. 2–5; Palke, W.E. J. Am. Chem. Soc. 1986, 108, 6543.
9. See Simonetta, M.; Gavezzotti, A., in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, Wiley, NY, 1978, pp. 1–56; Dale, J., in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 3–96.
10. For a review of metal–metal multiple bonds, see Cotton, F.A. J. Chem. Educ. 1983, 60, 713.
11. For discussions, see Schmidt, M.W.; Truong, P.N.; Gordon, M.S. J. Am. Chem. Soc. 1987, 109, 5217; Schleyer, P. von R.; Kost, D. J. Am. Chem. Soc. 1988, 110, 2105.
12. For double bonds between carbon and elements other than C, N, S, or O, see Jutzi, P. Angew. Chem. Int. Ed. 1975, 14, 232; Raabe, G.; Michl, J. Chem. Rev. 1985, 85, 419 (Si only); Wiberg, N. J. Organomet. Chem. 1984, 273, 141 (Si only); Gordon, M.S. Mol. Struct. Energ. 1986, 1, 101. For reviews of C=P and C![]() P bonds, see Regitz, M. Chem. Rev. 1990, 90, 191; Appel, R.; Knoll, F. Adv. Inorg. Chem. 1989, 33, 259; Markovski, L.N.; Romanenko, V.D. Tetrahedron 1989, 45, 6019.
P bonds, see Regitz, M. Chem. Rev. 1990, 90, 191; Appel, R.; Knoll, F. Adv. Inorg. Chem. 1989, 33, 259; Markovski, L.N.; Romanenko, V.D. Tetrahedron 1989, 45, 6019.
13. For Si=C bonds, see Fink, M.J.; DeYoung, D.J.; West, R.; Michl, J. J. Am. Chem. Soc. 1983, 105, 1070; Fink, M.J.; Michalczyk, M.J.; Haller, K.J.; West, R.; Michl, J. Organometallics 1984, 3, 793; West, R. Pure Appl. Chem.1984, 56, 163; Masamune, S.; Eriyama, Y.; Kawase, T. Angew. Chem. Int. Ed. 1987, 26, 584; Shepherd, B.D.; Campana, C.F.; West, R. Heteroat. Chem. 1990, 1, 1.
14. Michalczyk, M.J.; West, R.; Michl, J. J. Am. Chem. Soc. 1984, 106, 821, Organometallics 1985, 4, 826.
15. Miller, J.S.; Novoa, J.J. Acc. Chem. Res. 2007, 40, 189.
16. See Ballard, R.E. Photoelectron Spectroscopy and Molecular Orbital Theory, Wiley, NY, 1978; Rabalais, J.W. Principles of Ultraviolet Photoelectron Spectroscopy, Wiley, NY, 1977; Baker, A.D.; Betteridge, D. Photoelectron Spectroscopy, Pergamon, Elmsford, NY, 1972; Turner, D.W.; Baker, A.D.; Baker, C.; Brundle, C.R. High Resolution Molecular Photoelectron Spectroscopy, Wiley, NY, 1970. For reviews, see Westwood, N.P.C. Chem. Soc. Rev. 1989, 18, 317; Baker, C.; Brundle, C.R.; Thompson, M. Chem. Soc. Rev. 1972, 1, 355; Bock, H.; Ramsey, B.G. Angew. Chem. Int. Ed. 1973, 12, 734; Turner, D.W. Adv. Phys. Org. Chem. 1966, 4, 31. For the IUPAC descriptive classification of various electron spectroscopy techniques, see Porter, H.Q.; Turner, D.W. Pure Appl. Chem. 1987, 59, 1343.
17. The correlation is not perfect, but the limitations do not seriously detract from the usefulness of the method. The technique is not limited to vacuum UV radiation. Higher energy radiation can also be used.
18. From Brundle, C.R.; Robin, M.B., in Nachod, F.C.; Zuckerman, J.J. Determination of Organic Structures by Physical Methods, Vol. 3, Academic Press, NY, 1971, p. 18.
19. Brundle, C.R.; Robin, M.B.; Basch, H. J. Chem. Phys. 1970, 53, 2196; Baker, A.D.; Betteridge, D.; Kemp, N.R.; Kirby, R.E. J. Mol. Struct. 1971, 8, 75; Potts, A.W.; Price, W.C. Proc. R. Soc. London, Ser, A 1972, 326, 165.
20. A third band, at 290 eV, caused by the 1s electrons of carbon, can also be found if radiation of sufficiently high energy is used.
21. See Robinson, J.W., Practical Handbook of Spectroscopy, CRC Press, Boca Raton, FL, 1991, p. 178.
22. Novak, I.; Potts, A.W. Tetrahedron 1997, 53, 14713.
23. It has been argued that although the Lewis picture of two electrons making up a covalent bond may work well for organic compounds, it cannot be successfully applied to the majority of inorganic compounds: J![]() rgensen, C.K. Top. Curr. Chem. 1984, 124, 1.
rgensen, C.K. Top. Curr. Chem. 1984, 124, 1.
24. For a review concerning sulfur compounds with a valence shell larger than eight, see Salmond, W.G. Q. Rev. Chem. Soc. 1968, 22, 235.
25. For a collection of articles on this topic, see Sen, K.D.; J![]() rgensen, C.K. Electronegativity (Vol. 6 of Structure and Bonding), Springer, NY, 1987. For a review, see Batsanov, S.S. Russ. Chem. Rev. 1968, 37, 332.
rgensen, C.K. Electronegativity (Vol. 6 of Structure and Bonding), Springer, NY, 1987. For a review, see Batsanov, S.S. Russ. Chem. Rev. 1968, 37, 332.
26. Taken from Pauling, L. The Nature of the Chemical Bond, 3rd ed., Cornell University Press, Ithaca, NY, 1960, p. 93, except for the value for Na, which is from Sanderson, R.T. J. Am. Chem. Soc. 1983, 105, 2259; J. Chem. Educ. 1988, 65, 112, 223.
27. See Sanderson, R.T. J. Am. Chem. Soc. 1983, 105, 2259; J. Chem. Educ. 1988, 65, 112, 223.
28. See Huheey, J.E. Inorganic Chemistry, 3rd ed., Harper and Row, NY, 1983, pp. 146–148; Mullay, J., in Sen, K.D.; J![]() rgensen, C.K. Electronegativity (Vol. 6 of Structure and Bonding), Springer, NY, 1987, p. 9.
rgensen, C.K. Electronegativity (Vol. 6 of Structure and Bonding), Springer, NY, 1987, p. 9.
29. Hinze, J.; Jaffé, H.H. J. Am. Chem. Soc. 1962, 84, 540; Rienstra-Kiracofe, J.C.; Tschumper, G.S.; Schaefer, III, H.F.; Nandi, S.; Ellison, G.B. Chem. Rev. 2002, 102, 231.
30. Allen, L.C. J. Am. Chem. Soc. 1989, 111, 9003.
31. Walsh, A.D. Discuss. Faraday Soc. 1947, 2, 18; Bergmann, D.; Hinze, J., in Sen, K.D.; J![]() rgensen, C.K. Electronegativity (Vol. 6 of Structure and Bonding), Springer, NY, 1987, pp. 146–190.
rgensen, C.K. Electronegativity (Vol. 6 of Structure and Bonding), Springer, NY, 1987, pp. 146–190.
32. Inamoto, N.; Masuda, S. Chem. Lett. 1982, 1003. See also, Bratsch, S.G. J. Chem. Educ. 1988, 65, 223; Mullay, J. J. Am. Chem. Soc. 1985, 107, 7271; Zefirov, N.S.; Kirpichenok, M.A.; Izmailov, F.F.; Trofimov, M.I. Dokl. Chem. 1987, 296, 440; Boyd, R.J.; Edgecombe, K.E. J. Am. Chem. Soc. 1988, 110, 4182.
33. A magnetically anisotropic group is one that is not equally magnetized along all three axes. The most common such groups are benzene rings (see Sec. 2.I) and triple bonds.
34. This order is opposite to that expected from the field effect (Sec. 1.I). It is an example of the Baker–Nathan order (Sec. 2.M).
35. Moodie, R.B.; Connor, T.M.; Stewart, R. Can. J. Chem. 1960, 38, 626.
36. Williamson, K.L. J. Am. Chem. Soc. 1963, 85, 516; Laszlo, P.; Schleyer, P.v.R. J. Am. Chem. Soc. 1963, 85, 2709; Niwa, J. Bull. Chem. Soc. Jpn. 1967, 40, 2192.
37. See Exner, O. Dipole Moments in Organic Chemistry, Georg Thieme Publishers, Stuttgart, 1975; McClellan, A.L. Tables of Experimental Dipole Moments, Vol. 1, W.H. Freeman, San Francisco, 1963; Vol. 2, Rahara Enterprises, El Cerrito, CA, 1974.
38. For example, see Koudelka, J.; Exner, O. Collect. Czech. Chem. Commun. 1985, 50, 188, 200.
39. The values for toluene, nitrobenzene, and p-nitrotoluene are from MacClellan, A.L., Tables of Experimental Dipole Moments, Vol. 1, W.H. Freeman: San Francisco, 1963; Vol. 2, Rahara Enterprises, El Cerrito, CA, 1974. The values for phenol and p-cresol were determined by Goode, E.V.; Ibbitson, D.A. J. Chem. Soc. 1960, 4265.
40. Lide Jr., D.R.; Mann, D.E. J. Chem. Phys. 1958, 29, 914.
41. Muenter, J.S.; Laurie, V.W. J. Chem. Phys. 1966, 45, 855.
42. Actually, symmetrical tetrahedral molecules like methane do have extremely small dipole moments, caused by centrifugal distortion effects; these moments are so small that they can be ignored for all practical purposes. For CH4, μ is ~ 5.4 × 10−6D: Ozier, I. Phys. Rev. Lett. 1971, 27, 1329; Rosenberg, A.; Ozier, I.; Kudian, A.K. J. Chem. Phys. 1972, 57, 568.
43. Roberts, J.D.; Moreland, Jr., W.T. J. Am. Chem. Soc. 1953, 75, 2167.
44. This example is from Grubbs, E.J.; Fitzgerald, R.; Phillips, R.E.; Petty, R. Tetrahedron 1971, 27, 935.
45. See Schneider, H.; Becker, N. J. Phys. Org. Chem. 1989, 2, 214; Bowden, K.; Ghadir, K.D.F. J. Chem. Soc. Perkin Trans. 2 1990, 1333. Also see Exner, O.; Fiedler, P. Collect. Czech. Chem. Commun. 1980, 45, 1251; Li, Y.; Schuster, G.B. J. Org. Chem. 1987, 52, 3975.
46. There has been some question as to whether it is even meaningful to maintain the distinction between the two types of effect: see Grob, C.A. Helv. Chim. Acta 1985, 68, 882; Lenoir, D.; Frank, R.M. Chem. Ber. 1985, 118, 753; Sacher, E. Tetrahedron Lett. 1986, 27, 4683.
47. See also, Ceppi, E.; Eckhardt, W.; Grob, C.A. Tetrahedron Lett. 1973, 3627.
48. For a review of field and other effects of silicon-containing groups, see Bassindale, A.R.; Taylor. P.G., in Patai, S.; Rappoport, Z. The Chemistry of Organic Silicon Compounds, pt. 2, Wiley, NY, 1989, pp. 893–963.
49. See Levitt, L.S.; Widing, H.F. Prog. Phys. Org. Chem. 1976, 12, 119.
50. See Sebastian, J.F. J. Chem. Educ. 1971, 48, 97.
51. See Wahl, Jr., G.H.; Peterson, Jr., M.R. J. Am. Chem. Soc. 1970, 92, 7238; Minot, C.; Eisenstein, O.; Hiberty, P.C.; Anh, N.T. Bull. Soc. Chim. Fr. 1980, II-119.
52. Streitwieser, Jr., A.; Klein, H.S. J. Am. Chem. Soc. 1963, 85, 2759.
53. Bent, H.A. Chem. Rev. 1961, 61, 275, p. 281.
54. See Laurence, C.; Berthelot, M.; Lucon, M.; Helbert, M.; Morris, D.G.; Gal, J. J. Chem. Soc. Perkin Trans. 2 1984, 705.
55. For tables of bond distances and angles, see Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19 (follows p. 1914); Tables of Interatomic Distances and Configurations in Molecules and Ions Chem. Soc. Spec. Publ. No. 11, 1958; Interatomic Distances Supplement Chem. Soc. Spec. Publ. No. 18, 1965; Harmony, M.D.; Laurie, V.W.; Kuczkowski, R.L.; Schwendeman, R.H.; Ramsay, D.A.; Lovas, F.J.; Lafferty, W.J.; Maki, A.G. J. Phys. Chem. Ref. Data 1979, 8, 619–721. See Lathan, W.A.; Curtiss, L.A.; Hehre, W.J.; Lisle, J.B.; Pople, J.A. Prog. Phys. Org. Chem. 1974, 11, 175; Topsom, R.D. Prog. Phys. Org. Chem. 1987, 16, 85.
56. Burkert, U.; Allinger, N.L. Molecular Mechanics, ACS Monograph 177, American Chemical Society, Washington, 1982, pp. 6–9; Whiffen, D.H. Chem. Ber. 1971, 7, 57–61; Stals, J. Rev. Pure Appl. Chem. 1970, 20, 1, pp. 2–5.
57. Schleyer, P.v.R.; Bremer, M. Angew. Chem. Int. Ed. 1989, 28, 1226.
58. Lonsdale, K. Philos. Trans. R. Soc. London 1947, A240, 219.
59. Bartell, L.S.; Higginbotham, H.K. J. Chem. Phys. 1965, 42, 851.
60. Wagner, R.S.; Dailey, B.P. J. Chem. Phys. 1957, 26, 1588.
61. Iijima, T. Bull. Chem. Soc. Jpn. 1972, 45, 1291.
62. Tables of Interatomic Distances, Ref. 55.
63. Momany, F.A.; Bonham, R.A.; Druelinger, M.L. J. Am. Chem. Soc. 1963, 85, 3075. Also see, Lide, Jr., D.R.; Jen, M. J. Chem. Phys. 1963, 38, 1504.
64. Bonham, R.A.; Bartell, L.S.; Kohl, D.A. J. Am. Chem. Soc. 1959, 81, 4765.
65. Hilderbrandt, R.L.; Wieser, J.D. J. Mol. Struct. 1973, 15, 27.
66. Allen, F.H.; Kirby, A.J. J. Am. Chem. Soc. 1984, 106, 6197; Jones, P.G.; Kirby, A.J. J. Am. Chem. Soc. 1984, 106, 6207.
67. White, J.M.; Robertson, G.B. J. Org. Chem. 1992, 57, 4638.
68. Except where noted, values are from Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19 (follows p. 1914). In this source, values are given to three significant figures.
69. Kaupp, G.; Boy, J Angew. Chem. Int. Ed. 1997, 36, 48.
70. Ehrenberg, M. Acta Crystallogr. 1966, 20, 182.
71. Toda, F.; Tanaka, K.; Stein, Z.; Goldberg, I. Acta Crystallogr., Sect. C 1996, 52, 177.
72. Toda, F.; Tanaka, K.; Watanabe, M.; Taura, K.; Miyahara, I.; Nakai, T.; Hirotsu, K. J. Org. Chem. 1999, 64, 3102.
73. Tanaka, K.; Takamoto, N.; Tezuka, Y.; Kato, M.; Toda, F. Tetrahedron 2001, 57, 3761.
76. Costain, C.C.; Stoicheff, B.P. J. Chem. Phys. 1959, 30, 777.
77. For a full discussion of alkyne bond distances, see Simonetta, M.; Gavezzotti, A. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, Wiley, NY, 1978.
78. See Henry, B.R. Acc. Chem. Res. 1987, 20, 429.
79. Bartell, L.S.; Roth, E.A.; Hollowell, C.D.; Kuchitsu, K.; Young, Jr., J.E. J. Chem. Phys. 1965, 42, 2683.
80. For reviews of carbon–halogen bonds, see Trotter, J. in Patai, S. The Chemistry of the Carbon–Halogen Bond, pt. 1; Wiley, NY, 1973, pp. 49–62; Mikhailov, B.M. Russ. Chem. Rev. 1971, 40, 983.
81. Lide, Jr., D.R. Tetrahedron 1962, 17, 125.
82. Rajput, A.S.; Chandra, S. Bull. Chem. Soc. Jpn. 1966, 39, 1854.
74. Huntley, D.R.; Markopoulos, G.; Donovan, P.M.; Scott, L.T.; Hoffmann, R. Angew. Chem. Int. Ed. 2005, 44, 7549.
75. See Tanaka, M; Sekiguchi, A. Angew. Chem. Int. Ed. 2005, 44, 5821–5823.
83. Vannes, G.J.H.; Vos, A. Acta Crystallogr. Sect. B 1978, B34, 1947; Vannes, G.J.H.; Vos, A. Acta Crystallogr. Sect. B, 1979, B35, 2593; Mcmullan, R.K.; Kvick, A. Acta Crystallogr. Sect. B, 1992, B48, 726.
84. Jemmis, E.D.; Pathak, B.; King, R.B.; Schaefer, III, H.F. Chem. Commun. 2006, 2164.
85. Schwendeman, R.H.; Tobiason, F.L. J. Chem. Phys. 1965, 43, 201.
86. For a review of this concept, see Bingel, W.A.; Lüttke, W. Angew. Chem. Int. Ed. 1981, 20, 899.
87. This assumption has been challenged: see Pomerantz, M.; Liebman, J.F. Tetrahedron Lett. 1975, 2385.
88. Blukis, V.; Kasai, P.H.; Myers, R.J. J. Chem. Phys. 1963, 38, 2753.
89. Abrahams, S.C. Q. Rev. Chem. Soc. 1956, 10, 407.
90. Iijima, T.; Tsuchiya, S.; Kimura, M. Bull. Chem. Soc. Jpn. 1977, 50, 2564.
91. Lide, Jr., D.R. J. Chem. Phys. 1957, 27, 343.
92. Lide, Jr., D.R.; Mann, D.E. J. Chem. Phys. 1958, 28, 572.
93. An older theory holds that the bonding is indeed p2, and that the increased angles come from repulsion of the hydrogen or carbon atoms. See Laing, M., J. Chem. Educ. 1987, 64, 124.
94. See Blackburne, I.D.; Katritzky, A.R.; Takeuchi, Y. Acc. Chem. Res. 1975, 8, 300; Aaron, H.S.; Ferguson, C.P. J. Am. Chem. Soc. 1976, 98, 7013; Anet, F.A.L.; Yavari, I. J. Am. Chem. Soc. 1977, 99, 2794; Vierhapper, F.W.; Eliel, E.L. J. Org. Chem. 1979, 44, 1081; Gust, D.; Fagan, M.W. J. Org. Chem. 1980, 45, 2511. For other views, see Lambert, J.B.; Featherman, S.I. Chem. Rev. 1975, 75, 611; Breuker, K.; Kos, N.J.; van der Plas, H.C.; van Veldhuizen, B. J. Org. Chem. 1982, 47, 963.
95. Blanksby, S.J.; Ellison, G.B. Acc. Chem. Res. 2003, 36, 255. For reviews including methods of determination, see Wayner, D.D.M.; Griller, D. Adv. Free Radical Chem. (Greenwich, Conn.) 1990, 1, 159; Kerr, J.A. Chem. Rev.1966, 66, 465; Wiberg, K.B., in Nachod, F.C.; Zuckerman, J.J. Determination of Organic Structures by Physical Methods, Vol. 3, Academic Press, NY, 1971, pp. 207–245.
96. Cohen, N.; Benson, S.W. Chem. Rev. 1993, 93, 2419; Korth, H.-G.; Sicking, W. J. Chem. Soc. Perkin Trans. 2 1997, 715.
97. Matsunaga, N.; Rogers, D.W.; Zavitsas, A.A. J. Org. Chem, 2003, 68, 3158.
98. For the four steps, D values are 101–102, 88, 124, and 80 kcal mol−1 (423–427, 368, 519, and 335 kJ mol−1), respectively, though the middle values are much less reliable than the other two: Knox, B.E.; Palmer, H.B. Chem. Rev. 1961, 61, 247; Brewer, R.G.; Kester, F.L. J. Chem. Phys. 1964, 40, 812; Linevsky, M.J. J. Chem. Phys. 1967, 47, 3485.
99. See Cox, J.D.; Pilcher, G., Thermochemistry of Organic and Organometallic Compounds, Academic Press, NY, 1970; Domalski, E.S. J. Phys. Chem. Ref. Data 1972, 1, 221–277; Stull, D.R.; Westrum Jr., E.F.; Sinke, G.C. The Chemical Thermodynamics of Organic Compounds, Wiley, NY, 1969.
100. For a review, see Cox, J.D.; Pilcher, G. Thermochemistry of Organic and Organometallic Compounds, Academic Press, NY, 1970, pp. 531–597. See also, Gasteiger, J.; Jacob, P.; Strauss, U. Tetrahedron 1979, 35, 139.
101. These values, except where noted, are from Lovering, E.G.; Laidler, K.J. Can. J. Chem. 1960, 38, 2367; Levi, G.I.; Balandin, A.A. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1960, 149.
102. Grelbig, T.; Pötter, B.; Seppelt, K. Chem. Ber. 1987, 120, 815.
103. Bedford, A.F.; Edmondson, P.B.; Mortimer, C.T. J. Chem. Soc. 1962, 2927.
104. The average of the values obtained was DHo(O–O). dos Santos, R.M.B.; Muralha, V.S.F.; Correia, C.F.; Simões, J.A.M. J. Am. Chem. Soc. 2001, 123, 12670.
105. Cox, J.D.; Pilcher, G. Thermochemistry of Organic and Organometallic Compounds, Academic Press, NY, 1970, pp. 531–597; Cox, J.D. Tetrahedron 1962, 18, 1337.
106. Bach, R.D.; Ayala, P.Y.; Schlegel, H.B. J. Am. Chem. Soc. 1996, 118, 12758.
107. Wayner, D.D.M.; Clark, K.B.; Rauk, A.; Yu, D.; Armstrong, D.A. J. Am. Chem. Soc. 1997, 119, 8925. For the α C–H bond of tertiary amines, see Dombrowski, G.W.; Dinnocenzo, J.P.; Farid, S.; Goodman, J.L. Gould, I.R. J. Org. Chem. 1999, 64, 427.
108. Bordwell, F.G.; Zhang, X.-M.; Cheng, J.-P. J. Org. Chem. 1993, 58, 6410. See also, Li, Z.; Cheng, J.-P. J. Org. Chem. 2003, 68, 7350.
109. Liu, W.-Z.; Bordwell, F.G. J. Org. Chem. 1996, 61, 4778.
110. Lucarini, M.; Pedrielli, P.; Pedulli, G.F.; Cabiddu, S.; Fattuoni, C. J. Org. Chem. 1996, 61, 9259. For the O–H, E of polymethylphenols, see de Heer, M.I.; Korth, H.-G.; Mulder, P. J. Org. Chem. 1999, 64, 6969.
111. Zhang, X.-M. J. Org. Chem. 1998, 63, 1872.
112. Bordwell, F.G.; Zhang, X.-M.; Filler, R. J. Org. Chem. 1993, 58, 6067.
113. Brocks, J.J.; Beckhaus, H.-D.; Beckwith, A.L.J.; Rüchardt, C. J. Org. Chem. 1998, 63, 1935.
114. Hadad, C.M.; Rablen, P.R.; Wiberg, K.B. J. Org. Chem. 1998, 63, 8668.
115. Cheng, Y.-H.; Zhao, X.; Song, K.-S.; Liu, L.; Guo, Q.-X. J. Org. Chem. 2002, 67, 6638.
116. Guerra, M.; Amorati, R.; Pedulli, G.F. J. Org. Chem. 2004, 69, 5460.
117. Feng, Y.; Liu, L.; Wang, J.-T.; Zhao, S.-W.; Guo, Q.X. J. Org. Chem. 2004, 69, 3129; Song, K.-S.; Liu, L.; Guo, Q.X. Tetrahedron 2004, 60, 9909.
118. Coote, M.L.; Pross, A.; Radom, L. Org. Lett. 2003, 5, 4689.
119. See Miller, S.I. J. Chem. Educ. 1978, 55, 778.
120. Krapp, A.; Bickelhaupt, F.M.; Frenking, G. Chem.: Eur. J. 2006, 12, 9196.
121. Borges dos Santos, R.M.; Costa Cabral, B.J.; Martinho Simões, J.A. Pure Appl. Chem. 2007, 79, 1369.