March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 2. Delocalized Chemical Bonding
2.I. Aromaticity98
In the 19th century, it was recognized that aromatic compounds99 differ greatly from unsaturated aliphatic compounds,100 but for many years chemists were hard pressed to arrive at a mutually satisfactory definition of aromatic character.101 Qualitatively, there has never been real disagreement. Definitions include statements that aromatic compounds are characterized by a special stability and that they undergo substitution reactions more easily than addition reactions. These definitions are vague, however, and not easily applied to borderline cases. Definitions of aromaticity102 must encompass molecules ranging from polycyclic conjugated hydrocarbons,103 to heterocyclic compounds104 of various ring sizes, to reactive intermediates. In 1925, Armit and Robinson105 recognized that the aromatic properties of the benzene ring are related to the presence of a closed loop of electrons, the aromatic sextet (aromatic compounds are thus the archetypal examples of delocalized bonding), but determining whether rings other than the benzene ring possessed such a loop remained difficult. With the advent of magnetic techniques, most notably NMR, it is possible to determine experimentally whether or not a compound has a closed ring of electrons. Aromaticity can now be defined as the ability to sustain an induced ring current. A compound with this ability is called diatropic. Although this definition also has its flaws,106 it is the one most commonly accepted today. There are several methods of determining whether a compound can sustain a ring current, but the most important one is based on NMR chemical shifts.107 Water-soluble calix[4]resorcarenes (see Sec. 3.C.ii) have been developed as an enantioselective NMR shift reagents for aromatic compounds.108 In order to understand this, it is necessary to remember that, as a general rule, the value of the chemical shift of a proton in an NMR spectrum depends on the electron density of its bond; the greater the density of the electron cloud surrounding or partially surrounding a proton, the more upfield is its chemical shift (a lower value of δ). However, this rule has several exceptions; one is for protons in the vicinity of an aromatic ring. When an external magnetic field is imposed upon an aromatic ring (as in an NMR instrument), the closed loop of aromatic electrons circulates in a diamagnetic ring current, which generates a field of its own (ring current; known as magnetic anisotropy). As seen in Fig. 2.7, this induced field curves around and in the area of the proton is parallel to the external field, so the field “seen” by the aromatic protons is greater than it would have been in the absence of the diamagnetic ring current. The protons are moved downfield (to higher δ) compared to where they would be if electron density were the only factor. Thus ordinary alkene hydrogen atoms are found at ~ 5–6 δ, while the hydrogen atoms of benzene rings are located at 7–8 δ. However, if there were protons located above or within the ring, they would be subjected to a decreased field and should appear at lower δ values than normal CH2 groups (normal δ for CH2 is ~ 1–2). The NMR spectrum of [10]paracyclophane (48A) showed that this was indeed the case,109 and the CH2 peaks were shifted to lower δ the closer they were to the middle of the chain. This fact that a portion of the methylene chain is positioned directly over the benzene ring is easier to see in the molecular model 48B, and those protons are subject to the anisotropy shift.
Fig. 2.7 Ring current in benzene.
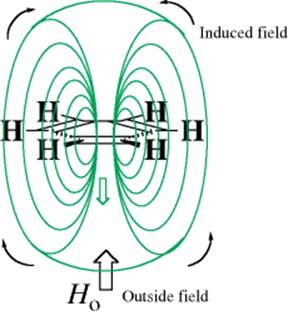
It follows from this analysis that aromaticity can be determined from an NMR spectrum. If the protons attached to the ring are shifted downfield from the normal alkene region, it can be concluded that the molecule is diatropic and hence aromatic. In addition, if the compound has protons above or within the ring (see an example of the latter in Sec. 2.K.vi), then these will be shifted upfield if the compound is diatropic. However, local framework effects are important to aromaticity, and it has been argued that downfield chemical shifts of arene hydrogen atoms are not reliable indicators of aromaticity.110 One drawback to this method is that it cannot be applied to compounds that have no protons in either category (e.g, the dianion of squaric acid see Sec. 2.L). Unfortunately, 13C NMR is of no help here, since these spectra do not show ring currents.111
Bickelhaupt has argued that “double-bond delocalization” actually refers to bond-length equalization. The major effect is argued to be optimal sigma overlap, where the π electrons force the bonds to a somewhat shorter distance. For antiaromatic systems, the π electrons are said to have a stronger localizing drive.111
It has been shown that when the nucleus independent chemical shifts for a set of aromatic and antiaromatic hydrocarbons are summed, there is a linear relationship with the magnetic susceptibility exaltation (the difference between the measured magnetic susceptibility of a compound and a calculated value based on group additivity tables) for neutral, cationic, and monoanionic species.112 Aromatic and antiaromatic dianions show a similar relationship, but with a different slope.112
It appears that there are both energetic and magnetic criteria for aromaticity. The so-called circuit resonance energy113 is an important quantity that connects energetic and magnetic criteria of aromaticity. It is defined as a contribution of each cyclic path in a polycyclic π-system to the aromatic stabilization energy. The individual circuit contributions to aromaticity from the magnetic response of a polycyclic system have been determined, and named circuit resonance energies, with the same sign and essentially the same magnitude as the corresponding cyclic conjugation energy defined by Bosanac and by Gutman.114 Ring-current diamagnetism was taken as the tendency of a cyclic π system to retain aromatic stabilization energy of the individual circuits.
Antiaromatic systems exhibit a paramagnetic ring current,115 which causes protons on the outside of the ring to be shifted upfield while any inner protons are shifted downfield, in sharp contrast to a diamagnetic ring current, which causes shifts in the opposite directions (see above). Compounds that sustain a paramagnetic ring current are called paratropic and are prevalent in four and eight-electron systems. As with aromaticity, antiaromaticity should be at a maximum when the molecule is planar and when bond distances are equal. The diamagnetic and paramagnetic effects of the ring currents associated with aromatic and antiaromatic compounds (i.e., shielding and deshielding of nuclei) can be measured by a simple and efficient criterion known as nucleus independent chemical shift (NICS).116 The aromatic–antiaromatic ring currents reflect the extra π-effects that the molecules experience. The unique near zero value of NICS at the cyclobutadiene ring center is due to cancellation by large and opposite anisotropic components.117
Apart from the experimental NMR techniques, there are at least four theoretical models for aromaticity that have been compared and evaluated for predictive ability.118 The Hess–Schaad model119 is good for predicting aromatic stability of benzenoid hydrocarbons, but does not predict reactivity. The Herndon model120 is also good for predicting aromatic stability, but is unreliable for benzenoidicity and does not predict reactivity. The conjugated-circuit model121 is very good for predicting aromatic stability but not reactivity, and the hardness model122 is best for predicting kinetic stability. Delocalization energy of π electrons has been used as an index for aromaticity in polycyclic aromatic hydrocarbons.123 The claims for linear relationships between aromaticity and energetics, geometries, and magnetic criteria were said to be invalid for any representative set of heteroaromatics in which the number of heteroatoms varies.124
It should be emphasized that the old and new definitions of aromaticity are not necessarily parallel. If a compound is diatropic, and therefore aromatic under the new definition, it is more stable than the canonical form of lowest energy, but this does not mean that it will be stable to air, light, or common reagents, since this stability is determined not by the resonance energy, but by the difference in free energy between the molecule and the transition states for the reactions involved. These differences may be quite small, even if the resonance energy is large. A unified theory has been developed that relates ring currents, resonance energies, and aromatic character.125 Note that aromaticity varies in magnitude relatively and sometimes absolutely with the molecular environment, which includes the polarity of the medium.126
The vast majority of aromatic compounds have a closed loop of six electrons in a ring (the aromatic sextet), and those compounds will be considered first.127 Note that a “formula periodic table” for the benzenoid polyaromatic hydrocarbons has been developed.128
2.I.i. Six-Membered Rings
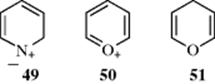
Not only is the benzene ring aromatic, but so are many heterocyclic analogues in which one or more heteroatoms replace carbon in the ring.129 When nitrogen is the heteroatom, there is a sextet and there is an unshared pair on the nitrogen that does not participate in the aromaticity. Therefore, derivatives (e.g., N-oxides or pyridinium ions) are still aromatic. There are more significant canonical forms (e.g., 49) for nitrogen heterocycles than for benzene. Where oxygen or sulfur is the heteroatom, it must be present in its ionic form (50) in order to possess the valence of 3 demanded for participation in such a system. Thus, pyran (51) is not aromatic, but the pyrylium ion (50) is.130
In systems of fused six-membered aromatic rings,131 the principal canonical forms are usually not all equivalent. Naphthalene, (52) has a central double bond and is thus different from the other two canonical forms, which are equivalent to each other.132 For naphthalene, these are the only forms that can be drawn
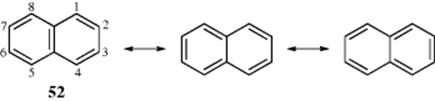
without consideration of Dewar forms or those with charge separation.133 If it is assumed that the three forms contribute equally, the 1,2-bond has more double-bond character than the 2,3-bond. Molecular orbital calculations show bond orders of 1.724 and 1.603, respectively (cf. benzene, 1.667). In agreement with these predictions, the 1,2 and 2,3 bond distances are 1.36 and 1.415 Å, respectively,134 and ozone (19–09 and 15–58) preferentially attacks the 1,2-bond.135 This nonequivalency of bonds, called partial bond fixation,136 is found in nearly all fused aromatic systems. Note that a strained naphthalene derivative has been prepared in which one benzene ring is fused to two bicyclic systems. In this new naphthalene derivative, one six-membered ring has equalized bond lengths, but the other ring has alternating bond lengths.137 The aromaticity of cation, anion, and ion–radical derivatives of naphthalene and other arenes has also been calculated.138 A six-membered ring with a circle is often used to indicate an aromatic system, but Kekulé structures having the C=C units rather than a circle are used most often in this book. This statement is made here because one circle can be used for benzene, but it would be misleading to use two circles for naphthalene, for example, because that would imply 12 aromatic electrons, when naphthalene has only 10.139
In phenanthrene, where the 9,10-bond is a single bond in only one of five canonical forms (53), bond fixation is significant and this bond is readily attacked by many reagents:140 It has been observed that increased steric crowding leads to an increase in Dewar-benzene-type structures.141 In general, there is a good correlation between bond distances in fused aromatic compounds and bond orders. Another experimental quantity that correlates well with the bond order of a given bond in an aromatic system is the NMR coupling constant for coupling between the hydrogen atoms on the two carbons of the bond.142
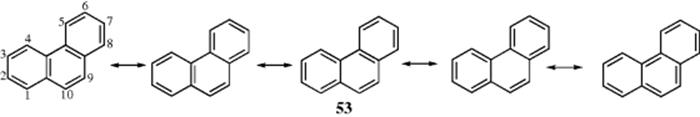
The resonance energies of fused systems increase as the number of principal canonical forms increases, as predicted by rule 6 (Sec. 2.E).143 Thus, for benzene, naphthalene, anthracene, and phenanthrene, for which can be drawn, respectively, two, three, four, and five principal canonical forms, the resonance energies are, respectively, 36, 61, 84, and 92 kcal mol−1 (152, 255, 351, and 385 kJ mol−1), calculated from heat of combustion data.144 Note that when phenanthrene, which has a total resonance energy of 92 kcal mol−1 (385 kJ mol−1), loses the 9,10-bond by attack of a reagent (e.g., ozone or bromine), two complete benzene rings remain, each with 36 kcal mol−1 (152 kJ mol−1) that would be lost if those rings were similarly attacked. The fact that anthracene undergoes many reactions across the 9,10-positions can be explained in a similar manner. Resonance energies for fused systems can be estimated by counting canonical forms.145 Calculations offer complementary evidence for the repulsive character of the H-H interactions in phenanthrene's bay region.146
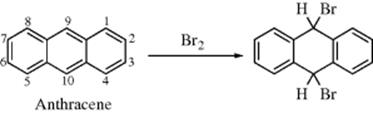
Not all fused systems can be fully aromatic. Thus for phenalene (54) there is no way to distribute double bonds so that each carbon has one single and one double bond.147 However, phenalene is acidic and reacts with potassium methoxide to give the corresponding anion (55), which is completely aromatic. So are the corresponding radical and cation, in which the resonance energies are the same (see Sec. 2.I.iii).148
Molecules that contain fused rings (e.g., phenanthrene or anthracene) are generally referred to as linear or angular polyacenes. Acenes are a class of organic compounds and polycyclic aromatic hydrocarbons made up of linearly fused benzene rings. In a fused system, with a focus on each individual ring, there are not six electrons for each ring.149 In naphthalene, for example, if one ring is to have six, the other must have only four. The greater reactivity of the ring system of naphthalene compared with benzene has been explained by regarding one of the naphthalene rings as aromatic and the other as a butadiene system.150 This effect can become extreme, as in the case of triphenylene.151 For this compound, there are eight canonical forms like 56, in which none of the three bonds marked a is a double bond and only one form (57) in which at least one of them is double. Thus the molecule behaves as if the 18 electrons were distributed so as to give each of the outer rings a sextet, while the middle ring is “empty”. Since none of the outer rings need share any electrons with an adjacent ring, they are as stable as benzene. Triphenylene, unlike most fused aromatic hydrocarbons, does not dissolve in concentrated sulfuric acid and has a low reactivity.152 This phenomenon, whereby some rings in fused systems give up part of their aromaticity to adjacent rings, is called annellation and can be demonstrated by UV spectra,131 as well as chemical reactivity. In general, an increase of size of both linear and angular polyacenes is associated with a substantial decrease in their aromaticity, with a greater decrease for the linear polyacenes.153 Note that molecular loops and belts can be made that involve acenes.154
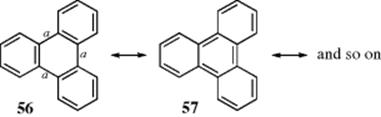
2.I.ii. Five, Seven, and Eight-Membered Rings
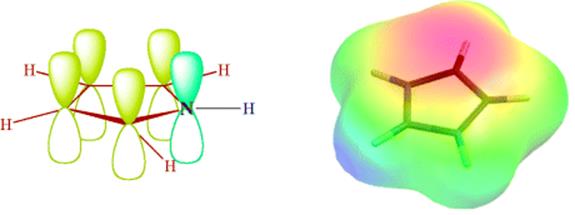
Aromatic sextets can also be present in five- and seven-membered rings. If a five-membered ring has two double bonds, and the fifth atom possesses an unshared pair of electrons, as in pyrrole, the ring has five p orbitals that can overlap to create five new orbitals: three bonding and two antibonding. There are six electrons for these orbitals: the four p orbitals of the double bonds each contribute one and the filled orbital contributes
the other two. The six electrons occupy the bonding orbitals and constitute an aromatic sextet, illustrated in Fig. 2.8. The electron potential map of pyrrole in Fig. 2.8 shows the aromatic cloud (dark area near the top of the model), indicative of significant electron density. The heterocyclic compounds pyrrole, thiophene, and furan are the most important examples of this kind of aromaticity, although furan has a lower degree of aromaticity when compared to the other two.155 Resonance energies for these three compounds are, respectively, 21, 29, and 16 kcal mol−1 (88, 121, and 67 kJ mol−1).156 The aromaticity can also be shown by canonical forms, (e.g., for pyrrole):
Fig. 2.8 Overlap of five p orbitals in pyrrole and the electron potential map of pyrrole.
In contrast to pyridine, the unshared pair in canonical structure A in pyrrole is needed for the aromatic sextet. Since the electron pair is not available for donation, pyrrole is a much weaker base than pyridine.
The fifth atom may be carbon rather than a heteroatom, if carbon has an unshared pair (as in an anion). Cyclopentadiene is known to react with a suitable base, and loss of a proton to give a carbanion that is aromatic and therefore quite stable, although it is reactive to alkylating agents and electrophilic reagents. Due to formation of the stable cyclopentadienyl anion, cyclopentadiene (pKa ~ 16) is approximately as strong an acid as water. The cyclopentadienide ion is sometimes represented as in 58, although more commonly one of the canonical forms is used. Resonance in this ion is greater than in pyrrole, thiophene, and furan, since all five
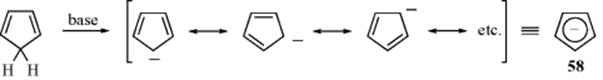
forms are equivalent, and the resonance energy for 58 has been estimated to be 24-27 kcal mol−1 (100–113 kJ mol−1).157 All five carbons are equivalent, as demonstrated by labeling the starting compound with 14C and finding all positions equally labeled when cyclopentadiene was regenerated.158 As expected for an aromatic system, 58 is diatropic159 and aromatic substitutions (see Chapters 11 and 13) on it have been successfully carried out.160 Average bond order has been proposed as a parameter to evaluate the aromaticity of such rings, but there is poor correlation with nonaromatic and antiaromatic systems.161 A model that relies on calculating relative aromaticity from appropriate molecular fragments has also been developed.162 Bird163 devised the aromatic index (IA, or aromaticity index), which is a statistical evaluation of the extent of ring bond order, and has been used as a criterion of aromaticity. Another bond-order index was proposed by Pozharskii,164 which builds on the work of Fringuelli et al.165 Absolute hardness (see Sec. 8.E), calculated from molecular refractions for a range of aromatic and heteroaromatic compounds, shows good linear correlation with aromaticity.166 Indene and fluorene are also acidic (pKa ~ 20 and 23, respectively), but less so than cyclopentadiene, since annellation causes the electrons to be less available to the five-membered ring. On the other hand, the acidity of 1,2,3,4,5-pentakis(trifluoromethyl)cyclopentadiene (59) is greater than that of nitric acid,167 because of the electron-withdrawing effects of the trifluoromethyl groups (see Sec. 8.F). Modifications of the Bird163 and Pozharskii164 systems have been introduced that are particularly useful for five-membered ring heterocycles.168 Recent work introduced a new local aromaticity measure, defined as the mean of Bader's electron delocalization index (DI)169 of para-related carbon atoms in six-membered rings.170 Bond resonance energy has been used as an indicator of local aromaticity.171 The relative merits of several aromaticity indices has been discussed.172
As seen above, relative acidity can be used to study the aromatic character of the resulting conjugate base of a given compound. In sharp contrast to cyclopentadiene (see Sec. 2.I.ii) is cycloheptatriene (60), which has no unusual acidity. This would be hard to explain without the aromatic sextet theory, since, on the basis of resonance forms or a simple consideration of orbital overlaps, 61 should be as stable as the cyclopentadienyl anion (58). This eight electron system is antiaromatic, however. While 61 has been prepared in solution,173 it is less stable than 58 and far less stable than 62, in which 60 has lost not a proton but the equivalent of a hydride ion. The six double-bond electrons of 62 overlap with the empty orbital on the seventh carbon and there is a sextet of electrons covering seven carbon atoms. The cycloheptatrienyl cation (known as the tropylium ion, 62) is quite stable,174 but are generally formed from the corresponding halide rather than by loss of a hydride. Tropylium bromide (63), which could be completely covalent if the electrons of the bromine were sufficiently attracted to the ring, is actually better viewed as an ionic compound.175 Many substituted tropylium ions have been prepared to probe the aromaticity, structure, and reactivity of such systems.176 As with 58, the equivalence of the carbon atoms in 62 has been demonstrated by isotopic labeling.177 The aromatic cycloheptatrienyl cations C7Me7+ and C7Ph7+ are known,178 although their coordination complexes with transition metals have been problematic, possibly because they assume a boat-like rather than a planar conformation179
Tropone (64) is another seven-membered ring that shows some aromatic character. This molecule would have an aromatic sextet if the two C=O electrons stayed away from the ring and resided near the electronegative oxygen atom. In fact, tropones are stable compounds, and tropolones (65) are found in nature.180 However, analyses of dipole moments, NMR spectra, and X-ray diffraction measurements show that tropones and tropolones display appreciable bond alternations.181 These molecules must be regarded as essentially non-aromatic, although some have aromatic character. Tropolones readily undergo aromatic substitution, emphasizing that the old and the new definitions of aromaticity are not always parallel. It is known that 65 is acidic (pKa ~ 6.7),182 in large part because the resulting anion has aromatic character. Indeed, 65 is considered to be a vinylogous carboxylic acid. In sharp contrast to 64, cyclopentadienone (66) has been isolated only in an Ar matrix < 38 K.183 Above this temperature it dimerizes. Many earlier attempts to prepare it were unsuccessful.184 As in 64, the electronegative oxygen atom draws electron density to itself, but in this case it leaves only four electrons and the molecule is unstable. Some derivatives of 66 have been prepared.145
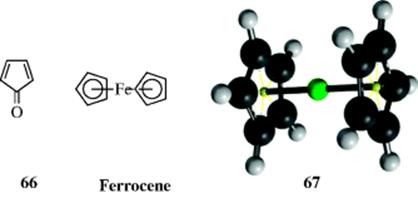
The metallocenes (also called sandwich compounds) constitute another type of five-membered aromatic compound in which two cyclopentadienide rings form a sandwich around a metal. The best known of these is ferrocene, where the η5-coordination of the two cyclopentadienyl rings to iron is apparent in the 3D model 67. Other metallocenes have been prepared with Co, Ni, Cr, Ti, V, and many other metals.185 As a reminder (see Sec. 2.C), the η terminology refers to π-donation of electrons to the metal (η3 for π-allyl systems, η6 for coordination to a benzene ring, etc.), and η5 refers to donation of five π-electrons to the iron. Ferrocene is quite stable, subliming >100°C and unchanged at 400°C. The two rings rotate freely.186 Many aromatic substitutions (Chapter 11) have been carried out on metallocenes.187 Metallocenes containing two metal atoms and three cyclopentadienyl rings have also been prepared and are known as triple-decker sandwiches.188 Even tetradecker, pentadecker, and hexadecker sandwiches have been reported.189
The bonding in ferrocene may be looked upon in simplified MO terms as follows.190 Each of the cyclopentadienide rings has five molecular orbitals:three filled bonding and two empty antibonding orbitals (Sec. 2.I.ii). The outer shell of the Fe atom possesses nine atomic orbitals, that is, one 4s, three 4p, and five 3d orbitals. The six filled orbitals of the two cyclopentadienide rings overlap with the s, three p, and two of the d orbitals of the Fe to form 12 new orbitals, six of which are bonding. These six orbitals make up two ring–metal triple bonds. In addition, further bonding results from the overlap of the empty antibonding orbitals of the rings with additional filled d orbitals of the iron. All told, there are 18 electrons (10 of which may be considered to come from the rings and 8 from iron in the zero oxidation state) in nine orbitals; six of these are strongly bonding and three weakly bonding or nonbonding.
The tropylium ion has an aromatic sextet spread over seven carbon atoms. An analogous ion, with the sextet spread over eight carbon atoms, is the 1,3,5,7-tetramethylcyclooctatetraene dictation (68). This ion, which is stable in solution at −50°C, is diatropic and approximately planar. The dication 68 is not stable above about −30°C.191
2.I.iii. Other Systems Containing Aromatic Sextets
Simple resonance theory predicts that pentalene (69), azulene (70), and heptalene (71) should be aromatic, although no nonionic canonical form can have a double bond at the ring junction. Molecular orbital calculations show that azulene should be stable but not the other two. This finding is borne out by experiment. Heptalene has been prepared,192 but reacts readily with oxygen, acids, and bromine, is easily hydrogenated, and polymerizes on standing. Analysis of its NMR spectrum shows that it is not planar.193 The 3,8-dibromo and 3,8-dicarbomethoxy derivatives of 71 are stable in air at room temperature, but are not diatropic.194 A number of methylated heptalenes and dimethyl 1,2-heptalenedicarboxylates have also been prepared and are stable non-aromatic compounds.195 Pentalene has not been prepared,196 but the hexaphenyl197 and 1,3,5-tri-tert-butyl derivatives198 are known. The former is air sensitive in solution. The latter is stable, but X-ray diffraction and photoelectron spectral data show bond alternation.199 Pentalene and its methyl and dimethyl derivatives have been formed in solution, but they dimerize before they can be isolated.200 Many other attempts to prepare these two systems have failed.
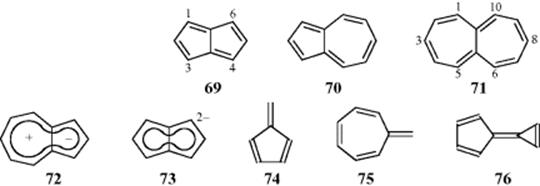
In sharp contrast to 69 and 71, azulene (70) is a blue solid, is quite stable, and many of its derivatives are known.201 Azulene readily undergoes aromatic substitution. Azulene may be regarded as a combination of 58 and 62 and, indeed, possesses a dipole moment of 0.8 D (see 72).202 Interestingly, if two electrons are added to pentalene, a stable dianion (73) results.203 It can be concluded that an aromatic system of electrons will be spread over two rings only if 10 electrons (not 8 or 12) are available for aromaticity. [n,m]-Fulvalenes (n ≠ m, where fulvalene is 74), as well as azulene are known to shift their π-electrons due to the influence of dipolar aromatic resonance structures.204However, calculations showed that dipolar resonance structures contribute only 5% to the electronic structure of heptafulvalene (75), although 22–31% to calicene (76).205 Based on Baird's theory,206 these molecules are influenced by aromaticity in both the ground and excited states, therefore acting as aromatic “chameleons.” This premise was confirmed in work by Ottosson and co-workers.204 Aromaticity indexes for various substituted fulvalene compounds has been reported.207